
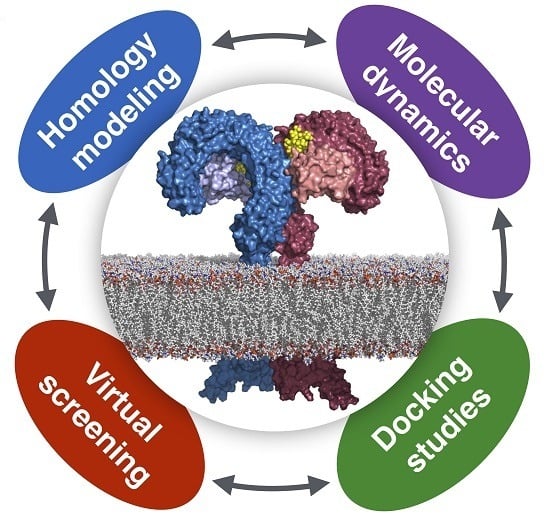
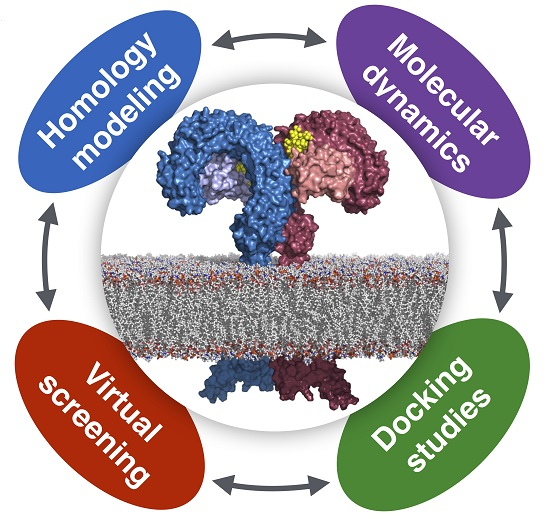
Computational Approaches to Toll-Like Receptor 4 Modulation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
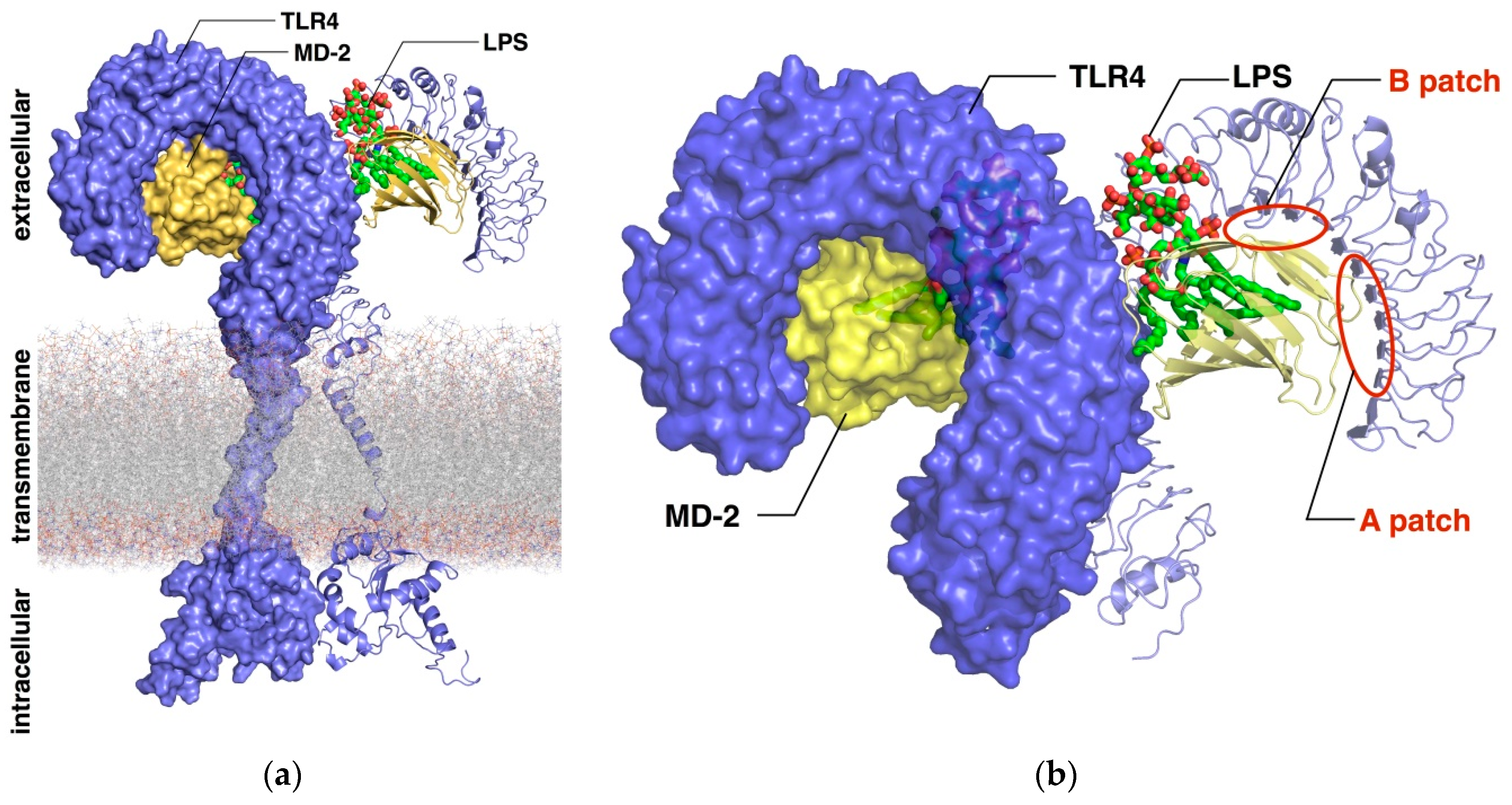
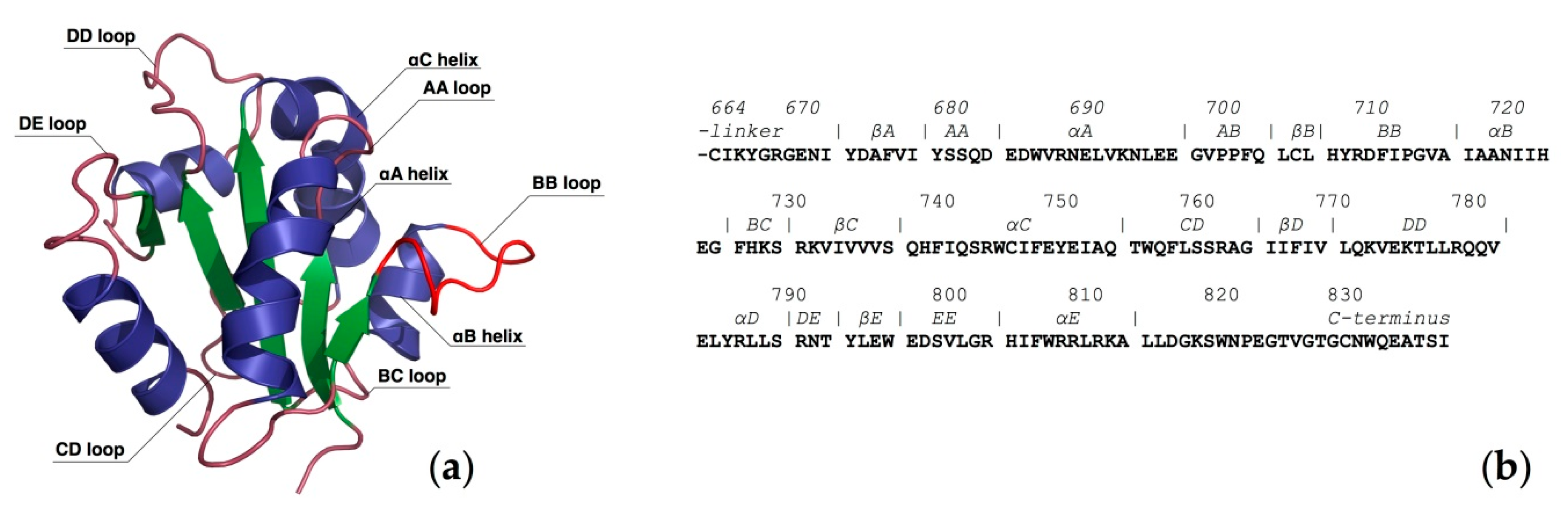
2. Computational Studies of the TLR4/MD-2 Ectodomain
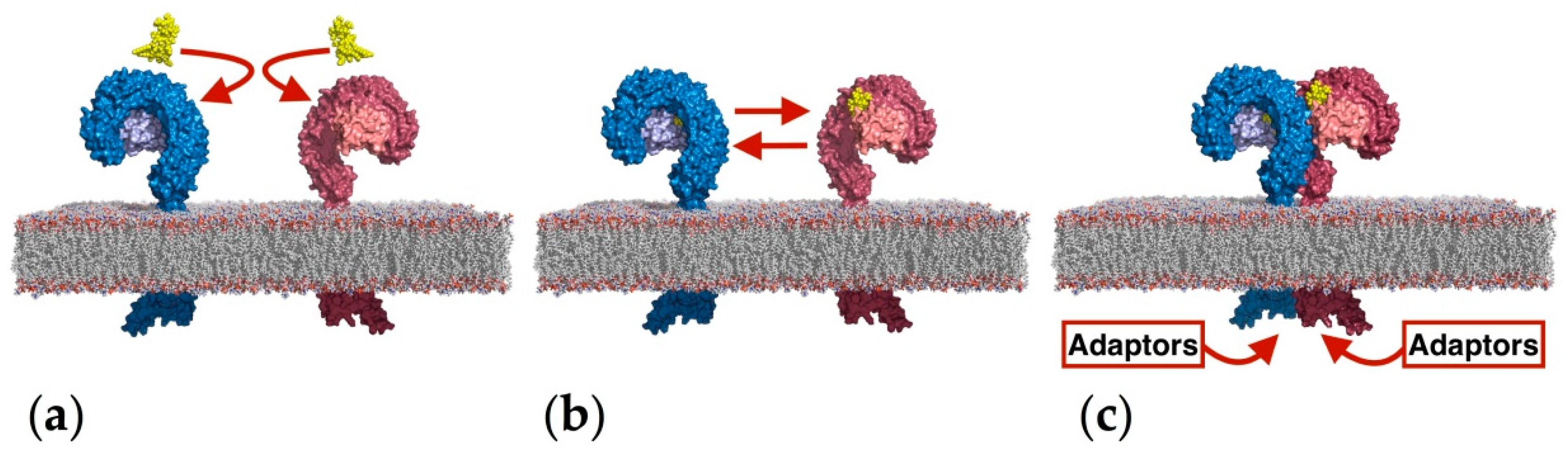
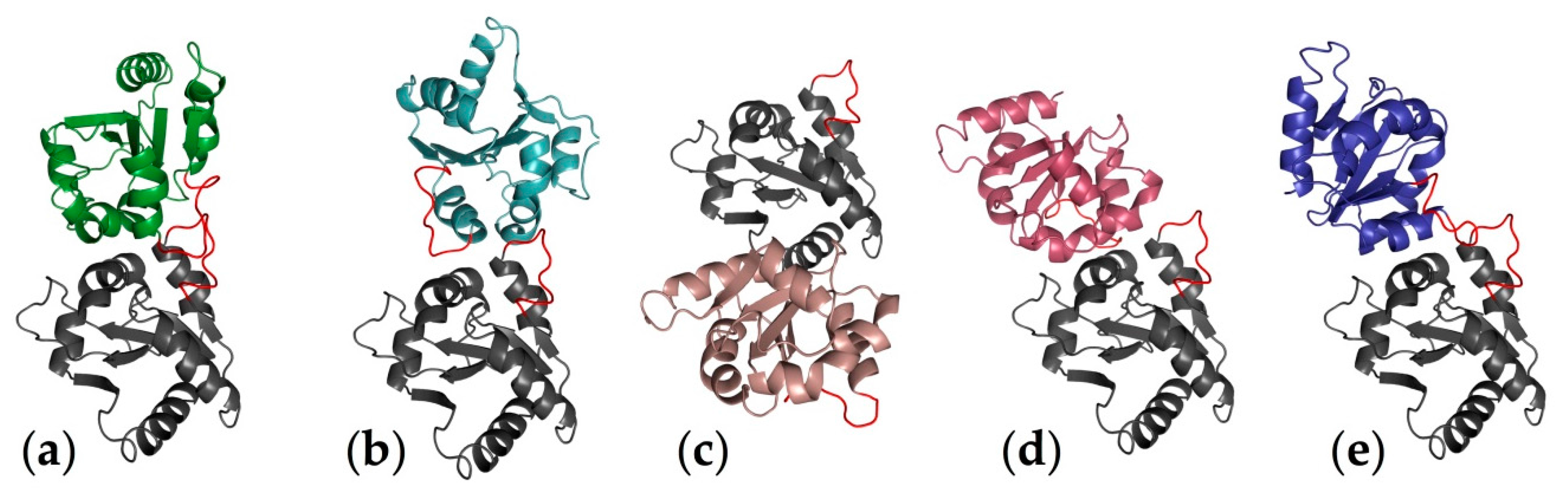
2.1. Computational Perspectives of the TLR4/MD-2/Ligand Recognition Process and Dimerization Event
2.2. Computational Strategies to Study Mutant TLR4 and MD-2 Proteins
2.3. The Binding Mode of TLR4 Modulators: Computational Approaches
2.3.1. Synthetic LPS Mimetics
2.3.2. Computational Studies of Natural LPSs
2.3.3. Computational Studies of Non-LPS-Like TLR4 Modulators
2.3.4. Computational Studies of Proteins as TLR4 Modulators
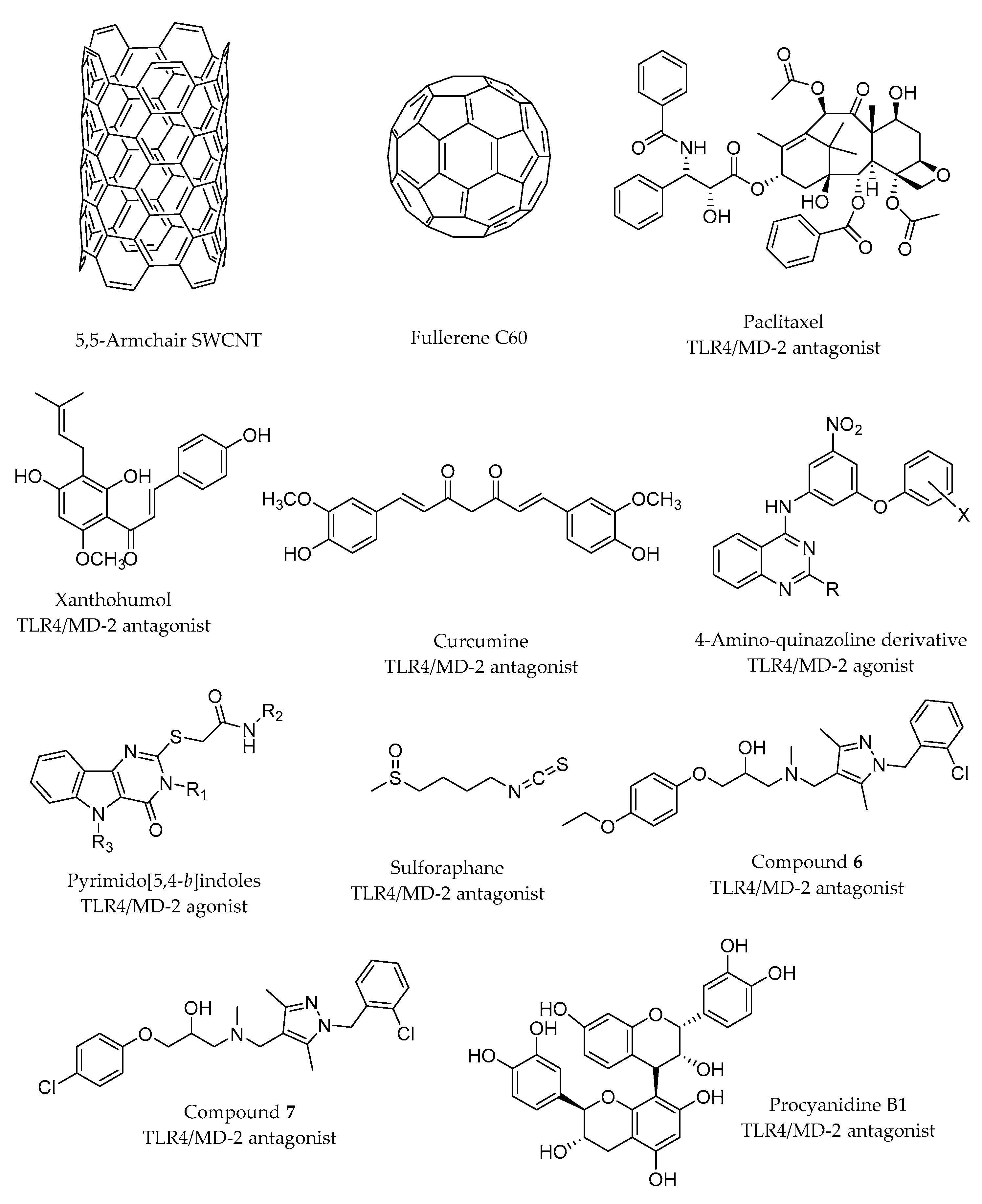
2.3.5. Computational Studies of Fullerenes, Nanotubes and Dendrimers as TLR4 Ligands
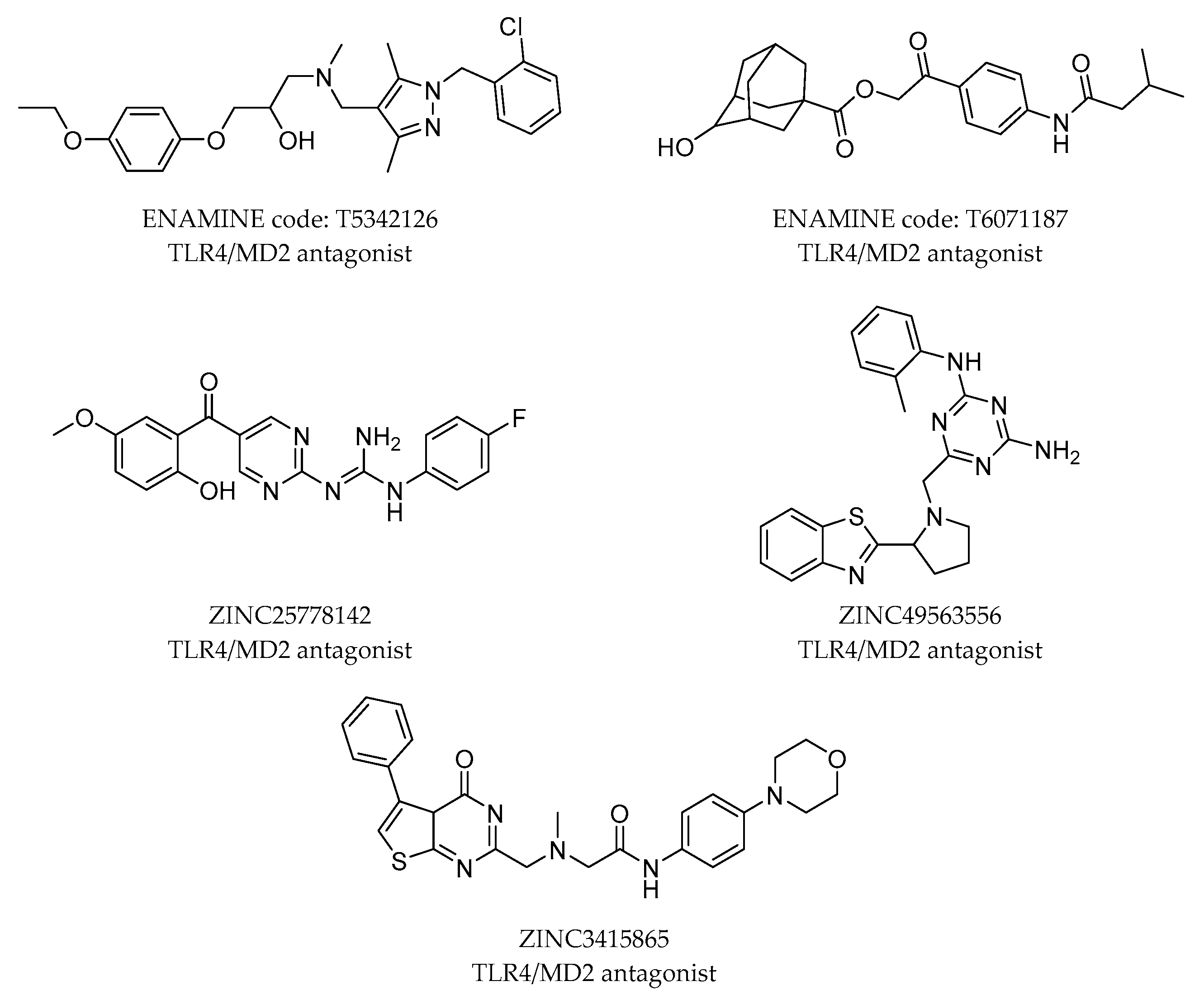
2.4. Virtual Screening Strategies in the Search of Novel TLR4 Modulators
3. Computational Studies on the Intracellular Domain of TLR4
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
Abbreviations
| Ara4N | 4-amino-arabinose |
| 5,5-armchair SWCNT | 5,5-single-walled carbon nanotube |
| AnxA2 | Annexin A2 |
| APBS | Adaptive Poisson–Boltzmann solver |
| CD14 | Cluster of differentiation 14 |
| CNT | Carbon nanotubes |
| CPK | Corey-Pauling-Koltun |
| FA | Fatty acid |
| H-bond | Hydrogen bond |
| HEK293 | Human embryonic kidney |
| h/mMD-2 | Human/mouse MD-2 |
| h/mTLR4 | Human/mouse TLR4 |
| HTS | High throughput screening |
| IRF-3 | Interferon regulatory factor 3 |
| LPS | Lipopolysaccharide |
| Mal | MyD88 adapter-like (also termed TIRAP) |
| MD Simulations | Molecular dynamics simulations |
| MD-2 | Myeloid differentiation factor 2 |
| MyD88 | Myeloid differentiation primary response gene 88 |
| MM-GBSA | Molecular mechanics-generalized Born surface area |
| MM-ISMSA | Molecular mechanics-implicit solvent model surface area |
| NMR | Nuclear magnetic resonance |
| PAMP | Pathogen- associated molecular pattern |
| PO4 | Phosphate group |
| PTX | Paclitaxel |
| RMSD | Root mean square deviation |
| RsLA | Rhodobacter sphaeroides lipid A |
| SAR | Structure-activity relationship |
| SASA | Solvent accessible surface area |
| SEAP | Secreted embryonic alkaline phosphatase |
| SFN | Sulforaphane |
| TIR | Toll/Interleukininterleukin-1 receptor |
| TLR | Toll-like receptor |
| TRAM | TRIF-related adaptor molecule |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| VS | Virtual screening |
| WT | Wild-type |
References
- Beutler, B. TLR4 as the mammalian endotoxin sensor. Curr. Top. Microbiol. Immunol. 2002, 270, 109–120. [Google Scholar] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Klett, J.; Reeves, J.; Oberhauser, N.; Perez-Regidor, L.; Martin-Santamaria, S. Modulation of Toll-like receptor 4. Insights from X-ray crystallography and molecular modeling. Curr. Top. Med. Chem. 2014, 14, 2672–2683. [Google Scholar] [CrossRef] [PubMed]
- Schletter, J.; Heine, H.; Ulmer, A.J.; Rietschel, E.T. Molecular mechanisms of endotoxin activity. Arch. Microbiol. 1995, 164, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Lien, E.; Golenbock, D.T. MD-2-mediated ionic interactions between lipid A and TLR4 are essential for receptor activation. J. Biol. Chem. 2010, 285, 8695–8702. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.; Holst, O.; di Lorenzo, F.; Callaghan, M.; Nurisso, A.; D’Errico, G.; Zamyatina, A.; Peri, F.; Berisio, R.; Jerala, R.; et al. Chemistry of lipid A: At the heart of innate immunity. Chem. Eur. J. 2015, 21, 500–519. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, F.; Kubik, Ł.; Oblak, A.; Lorè, N.I.; Cigana, C.; Lanzetta, R.; Parrilli, M.; Hamad, M.A.; de Soyza, A.; Silipo, A. Activation of human Toll-like receptor 4 (TLR4)· myeloid differentiation factor 2 (MD-2) by hypoacylated lipopolysaccharide from a clinical isolate of burkholderia cenocepacia. J. Biol. Chem. 2015, 290, 21305–21319. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, N.; Evans-Atkinson, T.; Hajjar, A.M.; Fernandez, R.C. Bordetella pertussis lipid A recognition by Toll-like receptor 4 and MD-2 is dependent on distinct charged and uncharged interfaces. J. Biol. Chem. 2015, 290, 13440–13453. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U.; Fukase, K.; Miyake, K.; Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. USA 2012, 109, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Resman, N.; Vašl, J.; Oblak, A.; Pristovšek, P.; Gioannini, T.L.; Weiss, J.P.; Jerala, R. Essential roles of hydrophobic residues in both MD-2 and Toll-like receptor 4 in activation by endotoxin. J. Biol. Chem. 2009, 284, 15052–15060. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U.; Fukase, K.; Miyake, K.; Satow, Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 2007, 316, 1632–1634. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Alexander, C.; Zaehringer, U. Reviewing and identifying amino acids of human, murine, canine and equine TLR4/MD-2 receptor complexes conferring endotoxic innate immunity activation by LPS/lipid A, or antagonistic effects by eritoran, in contrast to species-dependent modulation by lipid IVa. Comput. Struct. Biotechnol. J. 2013, 5, 1–13. [Google Scholar]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Lozano-Aponte, J.; Figueroa-Vazquez, V.; Yunes-Rojas, J.A.; Zähringer, U.; Alexander, C. Three-dimensional mapping of differential amino acids of human, murine, canine and equine TLR4/MD-2 receptor complexes conferring endotoxic activation by lipid A, antagonism by eritoran and species-dependent activities of lipid IVa in the mammalian lps sensor system. Comput. Struct. Biotechnol. J. 2013, 7, 1–11. [Google Scholar]
- Peri, F.; Calabrese, V. Toll-like receptor 4 (TLR4) modulation by synthetic and natural compounds: An update. J. Med. Chem. 2014, 57, 3612–3622. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A. Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Garate, J.A.; Oostenbrink, C. Lipid A from lipopolysaccharide recognition: Structure, dynamics and cooperativity by molecular dynamics simulations. Proteins Struct. Funct. Bioinf. 2013, 81, 658–674. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, M.L.; Woods, R.J. From agonist to antagonist: Structure and dynamics of innate immune glycoprotein MD-2 upon recognition of variably acylated bacterial endotoxins. Mol. Immunol. 2011, 49, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Paramo, T.; Piggot, T.J.; Bryant, C.E.; Bond, P.J. The structural basis for endotoxin-induced allosteric regulation of the Toll-like receptor 4 (TLR4) innate immune receptor. J. Biol. Chem. 2013, 288, 36215–36225. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Phillips, R.L.; Zhang, D.; Teghanemt, A.; Weiss, J.P.; Gioannini, T.L. NMR studies of hexaacylated endotoxin bound to wild-type and F126A mutant MD-2 and MD-2 TLR4 ectodomain complexes. J. Biol. Chem. 2012, 287, 16346–16355. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar, C.; Costa, M.G.; Verli, H. Dynamics on human Toll-like receptor 4 complexation to MD-2: The coreceptor stabilizing function. Proteins Struct. Funct. Bioinf. 2015, 83, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Panneerselvam, S.; Shah, M.; Choi, S. Insights into the species-specific TLR4 signaling mechanism in response to Rhodobacter sphaeroides lipid A detection. Sci. Rep. 2015, 5, 7657. [Google Scholar] [CrossRef] [PubMed]
- Slivka, P.F.; Shridhar, M.; Lee, G.i.; Sammond, D.W.; Hutchinson, M.R.; Martinko, A.J.; Buchanan, M.M.; Sholar, P.W.; Kearney, J.J.; Harrison, J.A. A peptide antagonist of the TLR4-MD2 interaction. ChemBioChem 2009, 10, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.; Misura, K.M.; Baker, D. Protein structure prediction using rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock 4 and AuutoDockTools 4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Klett, J.; Nunez-Salgado, A.; Dos Santos, H.G.; Cortes-Cabrera, A.; Perona, A.; Gil-Redondo, R.; Abia, D.; Gago, F.; Morreale, A. Mm-ismsa: An ultrafast and accurate scoring function for protein-protein docking. J. Chem. Theory Comput. 2012, 8, 3395–3408. [Google Scholar] [CrossRef] [PubMed]
- Vašl, J.; Oblak, A.; Peternelj, T.T.; Klett, J.; Martín-Santamaría, S.; Gioannini, T.L.; Weiss, J.P.; Jerala, R. Molecular basis of the functional differences between soluble human versus murine MD-2: Role of Val135 in transfer of lipopolysaccharide from CD14 to MD-2. J. Immunol. 2016, 196, 2309–2318. [Google Scholar] [PubMed]
- Barochia, A.; Solomon, S.; Cui, X.; Natanson, C.; Eichacker, P.Q. Eritoran tetrasodium (e5564) treatment for sepsis: Review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 2011, 7, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Cighetti, R.; Ciaramelli, C.; Sestito, S.E.; Zanoni, I.; Kubik, Ł.; Ardá-Freire, A.; Calabrese, V.; Granucci, F.; Jerala, R.; Martín-Santamaría, S. Modulation of CD14 and TLR4⋅MD-2 activities by a synthetic lipid A mimetic. ChemBioChem 2014, 15, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Glide & Impact software. Available online: http://www.schrodinger.com (accessed on 26 July 2016).
- Garate, J.A.; Stöckl, J.; Fernández-Alonso, M.D.C.; Artner, D.; Haegman, M.; Oostenbrink, C.; Jiménez-Barbero, J.; Beyaert, R.; Heine, H.; Kosma, P. Anti-endotoxic activity and structural basis for human MD-2·TLR4 antagonism of tetraacylated lipid A mimetics based on βglcn (1↔1) αglcn scaffold. Innate Immun. 2015, 21, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.; Calabrese, V.; Baruffa, C.; Gioannini, T.; Weiss, J.; Peri, F. The cationic amphiphile 3,4-bis(tetradecyloxy)benzylamine inhibits LPS signaling by competing with endotoxin for CD14 binding. Biochem. Pharmacol. 2010, 80, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Ciaramelli, C.; Calabrese, V.; Sestito, S.E.; Pérez-Regidor, L.; Klett, J.; Oblak, A.; Jerala, R.; Piazza, M.; Martín-Santamaría, S.; Peri, F. Glycolipid-based TLR4 modulators and fluorescent probes: Rational design, synthesis, and biological properties. Chem. Biol. Drug Des. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. Castp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinform. 2014, 47. [Google Scholar] [CrossRef]
- Walsh, C.; Gangloff, M.; Monie, T.; Smyth, T.; Wei, B.; McKinley, T.J.; Maskell, D.; Gay, N.; Bryant, C. Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J. Immunol. 2008, 181, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.L.; Gangloff, M.; Walsh, C.M.; Spring, D.R.; Gay, N.J.; Bryant, C.E. Identification of key residues that confer rhodobacter sphaeroides LPS activity at horse TLR4/MD-2. PLoS ONE 2014, 9, e98776. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Akashi, S.; Shimazu, R.; Yoshida, T.; Miyake, K.; Nishijima, M. Mouse Toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by taxol. J. Biol. Chem. 2000, 275, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Manthey, C.L.; Qureshi, N.; Stutz, P.L.; Vogel, S.N. Lipopolysaccharide antagonists block taxol-induced signaling in murine macrophages. J. Exp. Med. 1993, 178, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Resman, N.; Gradišar, H.; Vašl, J.; Keber, M.M.; Pristovšek, P.; Jerala, R. Taxanes inhibit human TLR4 signaling by binding to MD-2. FEBS Lett. 2008, 582, 3929–3934. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, S.M.; Liu, J.; Clayton, J.L.; Stephens, D.S.; Snyder, J.P. Paclitaxel binding to human and murine MD-2. J. Biol. Chem. 2008, 283, 27916–27926. [Google Scholar] [CrossRef] [PubMed]
- Sybyl-x Suite. Available online: https://www.certara.com/software/molecular-modeling-and-simulation/sybyl-x-suite/ (accessed on 26 July 2016).
- Fu, W.; Chen, L.; Wang, Z.; Zhao, C.; Chen, G.; Liu, X.; Dai, Y.; Cai, Y.; Li, C.; Zhou, J. Determination of the binding mode for anti-inflammatory natural product xanthohumol with myeloid differentiation protein 2. Drug Des. Dev. Ther. 2016, 10, 455–463. [Google Scholar]
- Wang, Z.; Chen, G.; Chen, L.; Liu, X.; Fu, W.; Zhang, Y.; Li, C.; Liang, G.; Cai, Y. Insights into the binding mode of curcumin to MD-2: Studies from molecular docking, molecular dynamics simulations and experimental assessments. Mol. Biosyst. 2015, 11, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.; Hayashi, T.; Mathewson, R.D.; Nour, A.; Hayashi, Y.; Yao, S.; Tawatao, R.I.; Crain, B.; Tsigelny, I.F.; Kouznetsova, V.L.; et al. Identification of substituted pyrimido[5,4-b]indoles as selective Toll-like receptor 4 ligands. J. Med. Chem. 2013, 56, 4206–4223. [Google Scholar] [CrossRef] [PubMed]
- Nour, A.; Hayashi, T.; Chan, M.; Yao, S.; Tawatao, R.I.; Crain, B.; Tsigelny, I.F.; Kouznetsova, V.L.; Ahmadiiveli, A.; Messer, K. Discovery of substituted 4-aminoquinazolines as selective Toll-like receptor 4 ligands. Bioorg. Med. Chem. Lett. 2014, 24, 4931–4938. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.E.; Park, Z.-Y.; Kim, N.D.; Lee, J.Y. Sulforaphane inhibits the engagement of LPS with TLR4/MD2 complex by preferential binding to Cys133 in MD2. Biochem. Biophys. Res. Commun. 2013, 434, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Koo, J.E.; Seo, Y.J.; Tyagi, N.; Jeong, E.; Choi, J.; Lim, K.M.; Park, Z.Y.; Lee, J.Y. Suppression of Toll-like receptor 4 activation by caffeic acid phenethyl ester is mediated by interference of LPS binding to MD2. Br. J. Pharmacol. 2013, 168, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Bevan, D.E.; Martinko, A.J.; Loram, L.C.; Stahl, J.A.; Taylor, F.R.; Joshee, S.; Watkins, L.R.; Yin, H. Selection, preparation, and evaluation of small-molecule inhibitors of Toll-like receptor 4. ACS Med. Chem. Lett. 2010, 1, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Byun, E.B.; Sung, N.Y.; Byun, E.H.; Song, D.S.; Kim, J.K.; Park, J.H.; Song, B.S.; Park, S.H.; Lee, J.W.; Byun, M.W.; et al. The procyanidin trimer C1 inhibits LPS-induced MAPK and NF-κB signaling through TLR4 in macrophages. Int. Immunopharmacol. 2013, 15, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Terra, X.; Palozza, P.; Fernandez-Larrea, J.; Ardevol, A.; Blade, C.; Pujadas, G.; Salvado, J.; Arola, L.; Blay, M.T. Procyanidin dimer B1 and trimer C1 impair inflammatory response signalling in human monocytes. Free Radic. Res. 2011, 45, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Triebel, S.; Anke, T.; Richling, E.; Erkel, G. Influence of apple polyphenols on inflammatory gene expression. Mol. Nutr. Food Res. 2009, 53, 1263–1280. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Li, R.; Li, N.; Zhang, J.; Li, Y.; Gong, P.; Gao, D.; Liu, H.; Zhang, Y. Anti-inflammatory effect of procyanidin B1 on LPS-treated THP1 cells via interaction with the TLR4–MD-2 heterodimer and p38 MAPK and NF-κB signaling. Mol. Cell. Biochem. 2015, 407, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, A.; Tomita, T.; Ohto, U.; Takemura, K.; Kitao, A.; Akashi-Takamura, S.; Miyake, K.; Maru, Y. Eritoran inhibits S100A8-mediated TLR4/MD-2 activation and tumor growth by changing the immune microenvironment. Oncogene 2016, 35, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, L.; Weng, Z. Zdock: An initial-stage protein-docking algorithm. Proteins 2003, 52, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, M.; Guo, Q.; Li, R.; Li, G.; Tan, S.; Li, X.; Wei, Y.; Wu, M. Annexin A2 binds to endosomes and negatively regulates TLR4-triggered inflammatory responses via the TRAM-TRIF pathway. Sci. Rep. 2015, 5, 15859. [Google Scholar] [CrossRef] [PubMed]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using swiss-model workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Anbanandam, A.; Rodgers, K.K. Structure of a TLR4-interacting SPA4 peptide. RSC Adv. 2015, 5, 27431–27438. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, H. When carbon nanotubes encounter the immune system: Desirable and undesirable effects. Adv. Drug Deliv. Rev. 2013, 65, 2120–2126. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Ma, Q. Advances in mechanisms and signaling pathways of carbon nanotube toxicity. Nanotoxicology 2015, 9, 658–676. [Google Scholar] [CrossRef] [PubMed]
- Turabekova, M.; Rasulev, B.; Theodore, M.; Jackman, J.; Leszczynska, D.; Leszczynski, J. Immunotoxicity of nanoparticles: A computational study suggests that CNTs and C60 fullerenes might be recognized as pathogens by Toll-like receptors. Nanoscale 2014, 6, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Barata, T.; Teo, I.; Lalwani, S.; Simanek, E.; Zloh, M.; Shaunak, S. Computational design principles for bioactive dendrimer based constructs as antagonists of the TLR4-MD-2-LPS complex. Biomaterials 2011, 32, 8702–8711. [Google Scholar] [CrossRef] [PubMed]
- Joce, C.; Stahl, J.A.; Shridhar, M.; Hutchinson, M.R.; Watkins, L.R.; Fedichev, P.O.; Yin, H. Application of a novel in silico high-throughput screen to identify selective inhibitors for protein-protein interactions. Bioorg. Med. Chem. Lett. 2010, 20, 5411–5413. [Google Scholar] [CrossRef] [PubMed]
- Enamine database. Available online: http://www.enamine.net (accessed on 26 July 2016).
- Svajger, U.; Brus, B.; Turk, S.; Sova, M.; Hodnik, V.; Anderluh, G.; Gobec, S. Novel Toll-like receptor 4 (TLR4) antagonists identified by structure- and ligand-based virtual screening. Eur. J. Med. Chem. 2013, 70, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. Zinc: A free tool to discover chemistry for biology. J. Chem. Inform. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.B.; Park, H.H. Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis Int. J. Program. Cell Death 2015, 20, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tao, X.; Shen, B.; Horng, T.; Medzhitov, R.; Manley, J.L.; Tong, L. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 2000, 408, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.; Chaffois, C.; Gaillard, C.; Saulnier, V.; Merck, E.; Tancredi, S.; Guiet, C.; Brière, F.; Vlach, J.; Lebecque, S. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J. Immunol. 2005, 174, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Tochio, H.; Kato, Z.; Orii, K.E.; Li, A.; Kimura, T.; Hiroaki, H.; Kondo, N.; Shirakawa, M. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 10260–10265. [Google Scholar] [CrossRef] [PubMed]
- Loiarro, M.; Sette, C.; Gallo, G.; Ciacci, A.; Fantò, N.; Mastroianni, D.; Carminati, P.; Ruggiero, V. Peptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of nf-κb. J. Biol. Chem. 2005, 280, 15809–15814. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Georgel, P.; Li, C.; Choe, J.; Crozat, K.; Rutschmann, S.; Du, X.; Bigby, T.; Mudd, S.; Sovath, S. Details of Toll-like receptor: Adapter interaction revealed by germ-line mutagenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 10961–10966. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Lu, J.; Zhou, W.; Shen, Y. Structural insights into TIR domain specificity of the bridging adaptor mal in TLR4 signaling. PLoS ONE 2012, 7, e34202. [Google Scholar] [CrossRef] [PubMed]
- Dunne, A.; Ejdebäck, M.; Ludidi, P.L.; O’Neill, L.A.; Gay, N.J. Structural complementarity of Toll/interleukin-1 receptor domains in toll-like receptors and the adaptors Mal and MyD88. J. Biol. Chem. 2003, 278, 41443–41451. [Google Scholar] [CrossRef] [PubMed]
- Kubarenko, A.; Frank, M.; Weber, A. Structure–function relationships of Toll-like receptor domains through homology modelling and molecular dynamics. Biochem. Soc. Trans. 2007, 35, 1515–1518. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wei, T.; Stark, R.W.; Jamitzky, F.; Heckl, W.M.; Anders, H.J.; Lech, M.; Rössle, S.C. Inhibition of toll-like receptors TLR4 and 7 signaling pathways by SIGIRR: A computational approach. J. Struct. Biol. 2010, 169, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Miguel, R.N.; Wong, J.; Westoll, J.F.; Brooks, H.J.; O′Neill, L.A.; Gay, N.J.; Bryant, C.E.; Monie, T.P. A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS ONE 2007, 2, e788. [Google Scholar]
- Basith, S.; Manavalan, B.; Govindaraj, R.G.; Choi, S. In silico approach to inhibition of signaling pathways of Toll-like receptors 2 and 4 by ST2L. PLoS ONE 2011, 6, e23989. [Google Scholar] [CrossRef] [PubMed]
- Guven-Maiorov, E.; Keskin, O.; Gursoy, A.; VanWaes, C.; Chen, Z.; Tsai, C.-J.; Nussinov, R. The architecture of the TIR domain signalosome in the Toll-like receptor-4 signaling pathway. Sci. Rep. 2015, 5, 13128. [Google Scholar] [CrossRef] [PubMed]
- Bovijn, C.; Ulrichts, P.; de Smet, A.-S.; Catteeuw, D.; Beyaert, R.; Tavernier, J.; Peelman, F. Identification of interaction sites for dimerization and adapter recruitment in Toll/interleukin-1 receptor (TIR) domain of Toll-like receptor 4. J. Biol. Chem. 2012, 287, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pandey, K.; Rathore, Y.S.; Sagar, A.; Pattnaik, U.B.K.; Ashish. A communication network within the cytoplasmic domain of Toll-like receptors has remained conserved during evolution. J. Biomol. Struct. Dyn. 2014, 32, 694–700. [Google Scholar] [CrossRef] [PubMed]


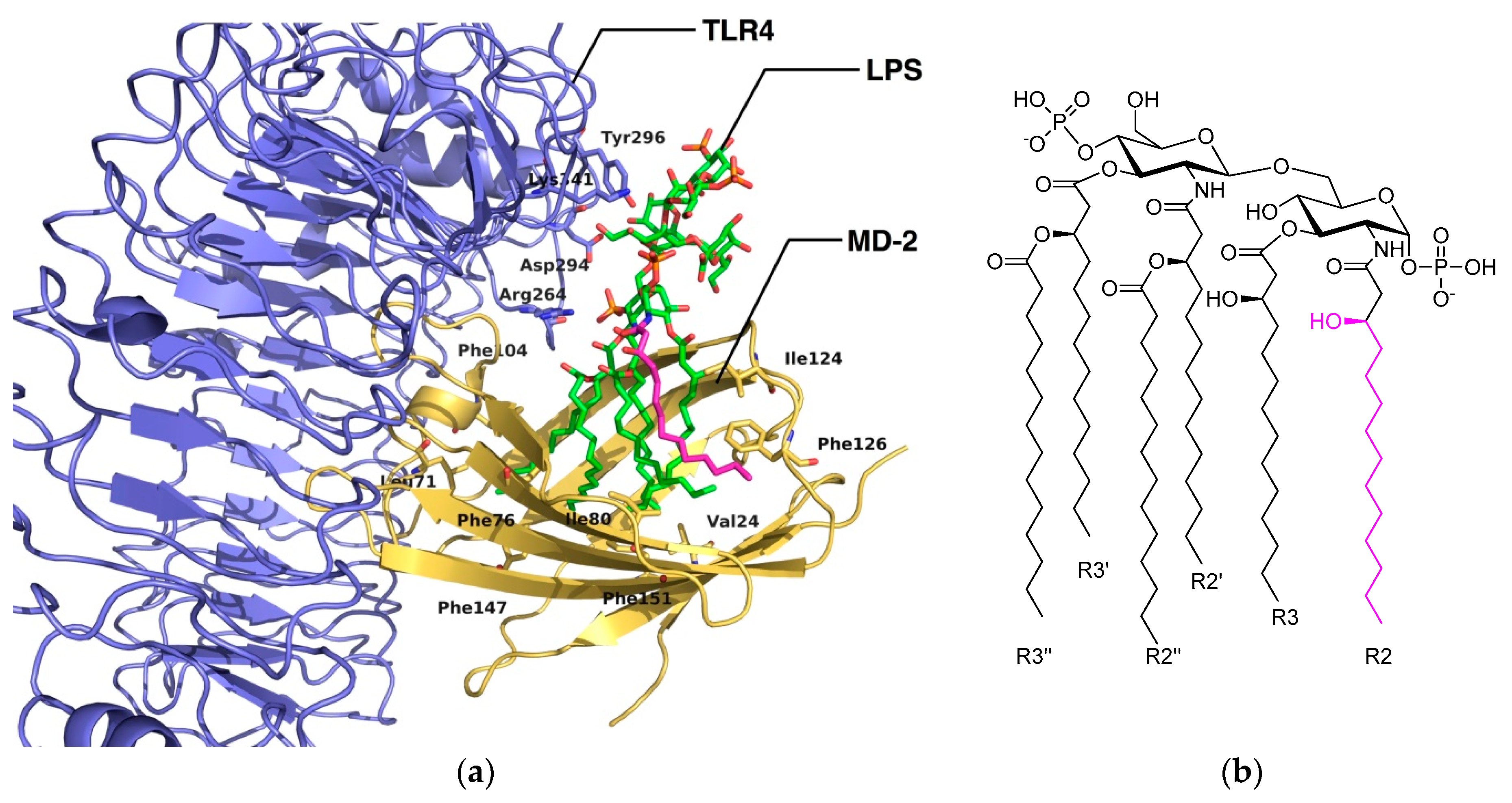
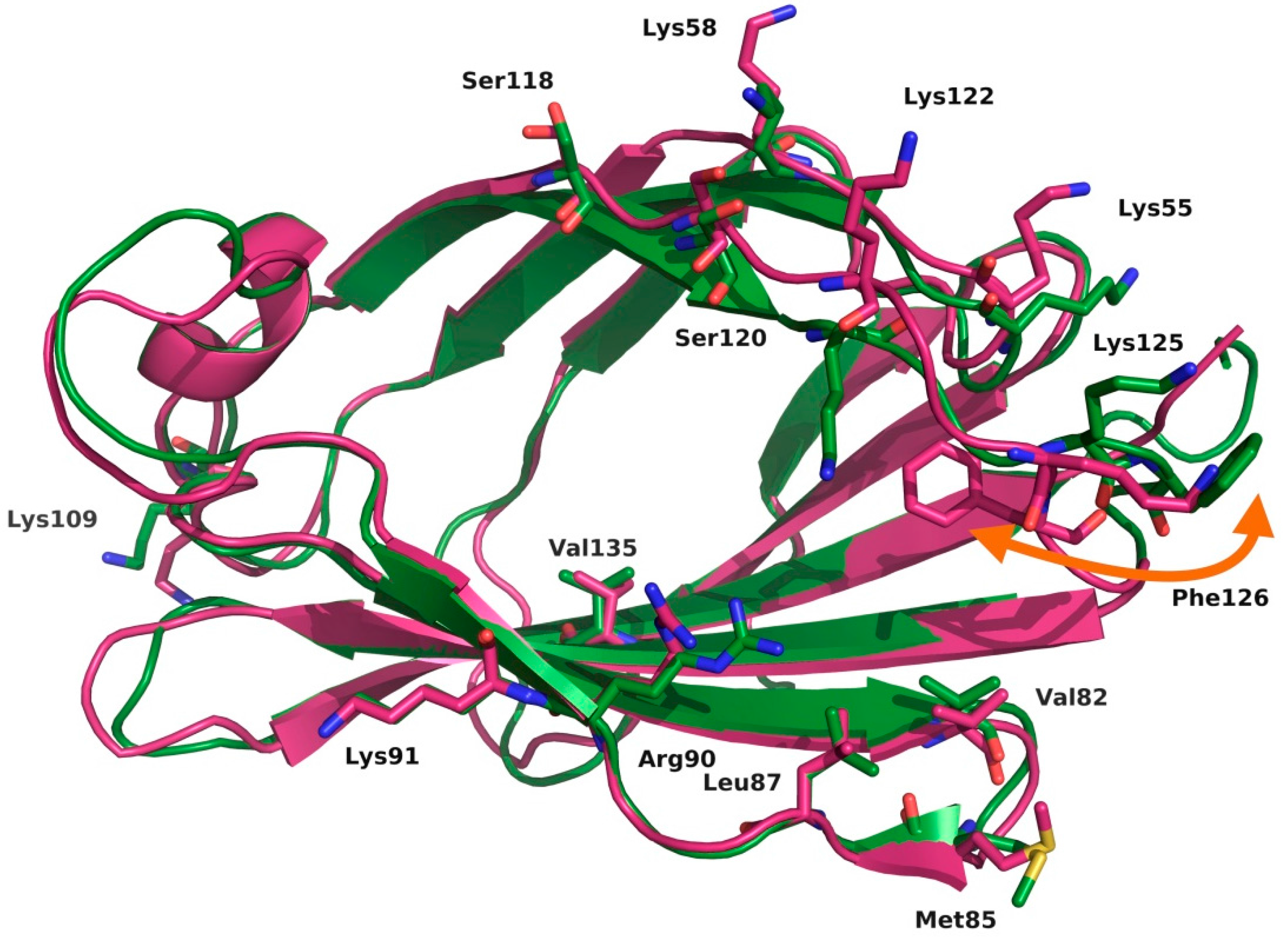
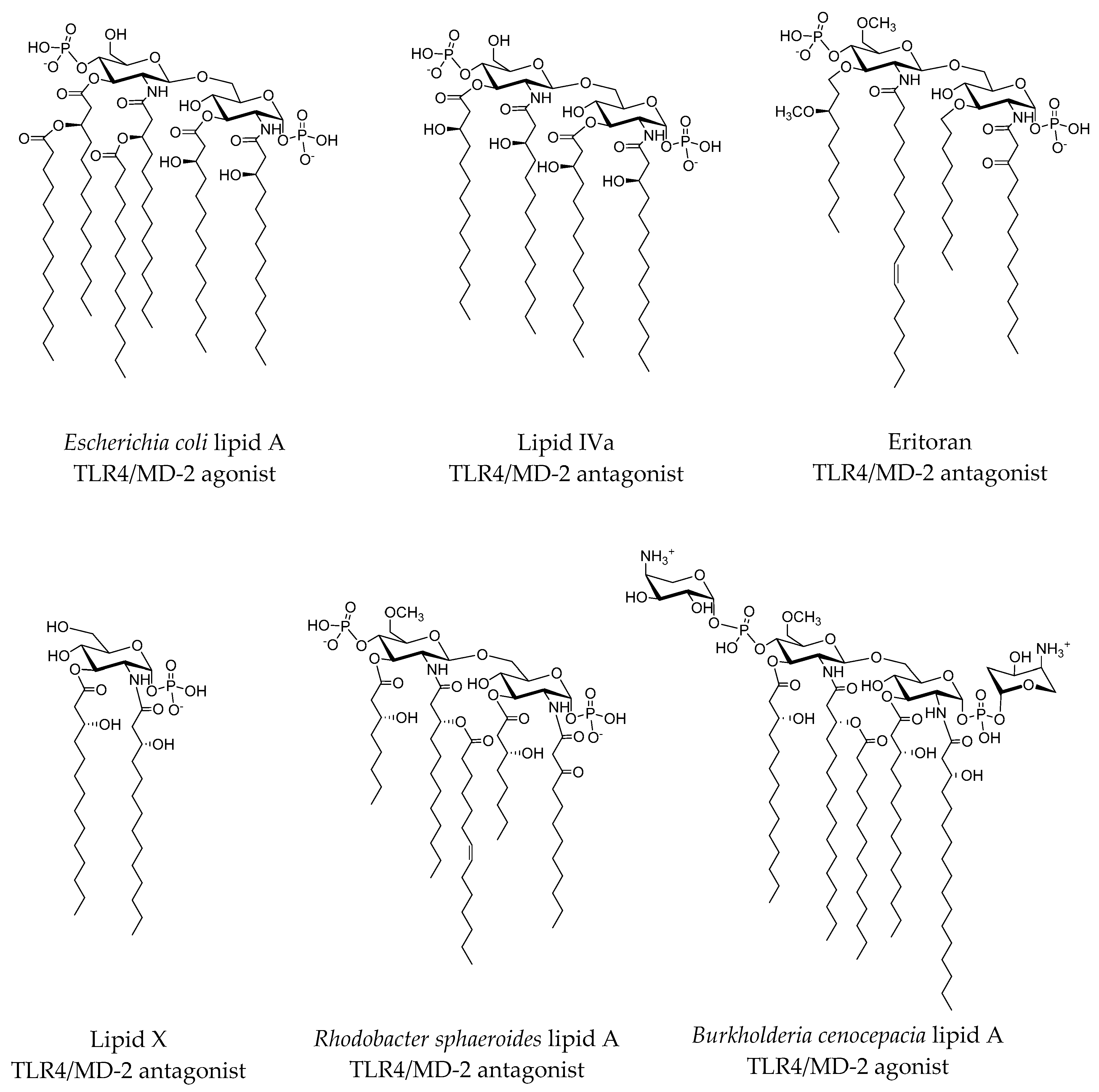
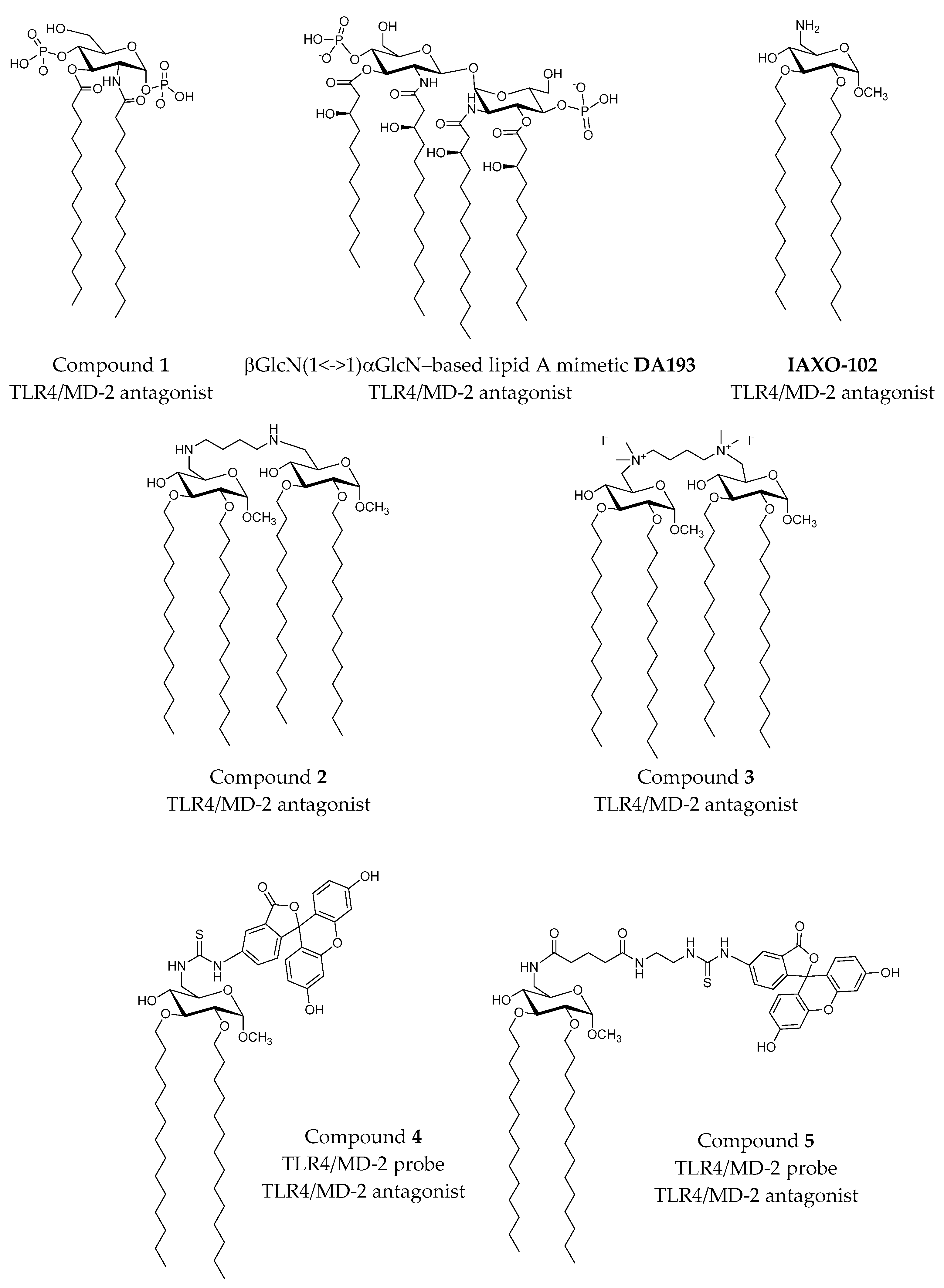

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billod, J.-M.; Lacetera, A.; Guzmán-Caldentey, J.; Martín-Santamaría, S. Computational Approaches to Toll-Like Receptor 4 Modulation. Molecules 2016, 21, 994. https://doi.org/10.3390/molecules21080994
Billod J-M, Lacetera A, Guzmán-Caldentey J, Martín-Santamaría S. Computational Approaches to Toll-Like Receptor 4 Modulation. Molecules. 2016; 21(8):994. https://doi.org/10.3390/molecules21080994
Chicago/Turabian StyleBillod, Jean-Marc, Alessandra Lacetera, Joan Guzmán-Caldentey, and Sonsoles Martín-Santamaría. 2016. "Computational Approaches to Toll-Like Receptor 4 Modulation" Molecules 21, no. 8: 994. https://doi.org/10.3390/molecules21080994