
Immobilization of Glycoside Hydrolase Families GH1, GH13, and GH70: State of the Art and Perspectives

 ,
,
Abstract
:1. Introduction
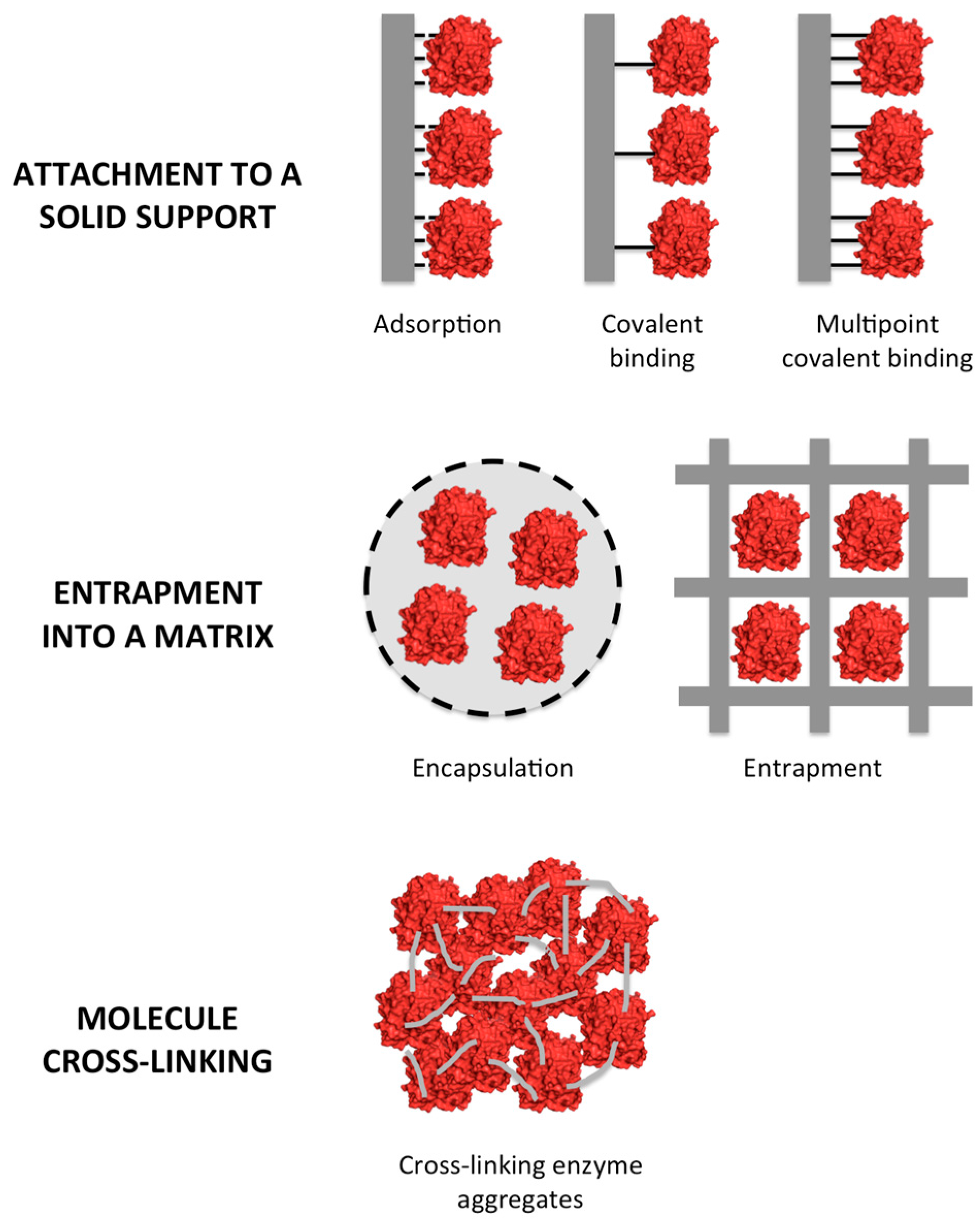
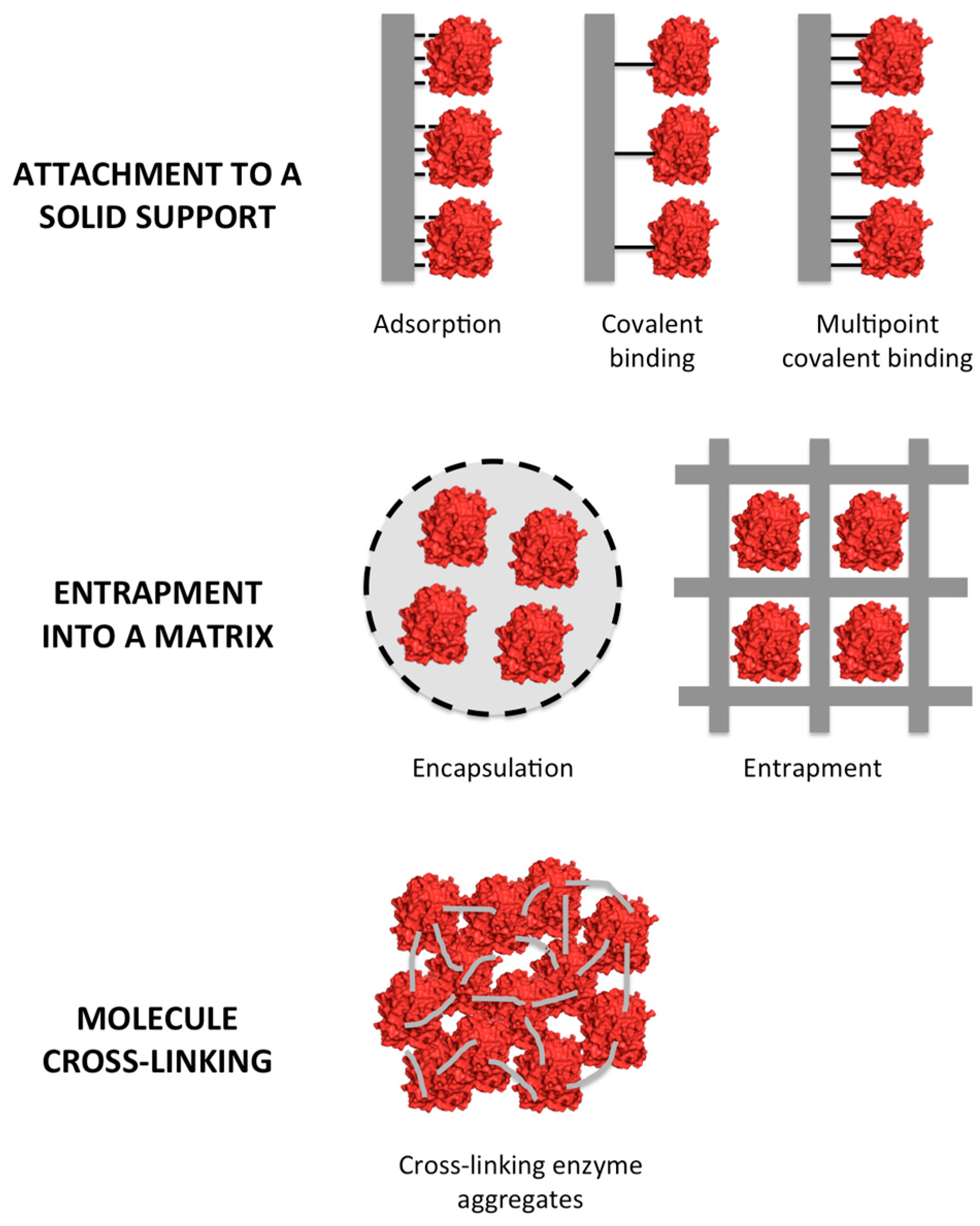
2. Types of Enzyme Immobilization
2.1. Enzyme Attachment to a Solid Support
2.2. Entrapment
2.3. Cross-Linking of Enzymes
3. The GH1 Family of Enzymes
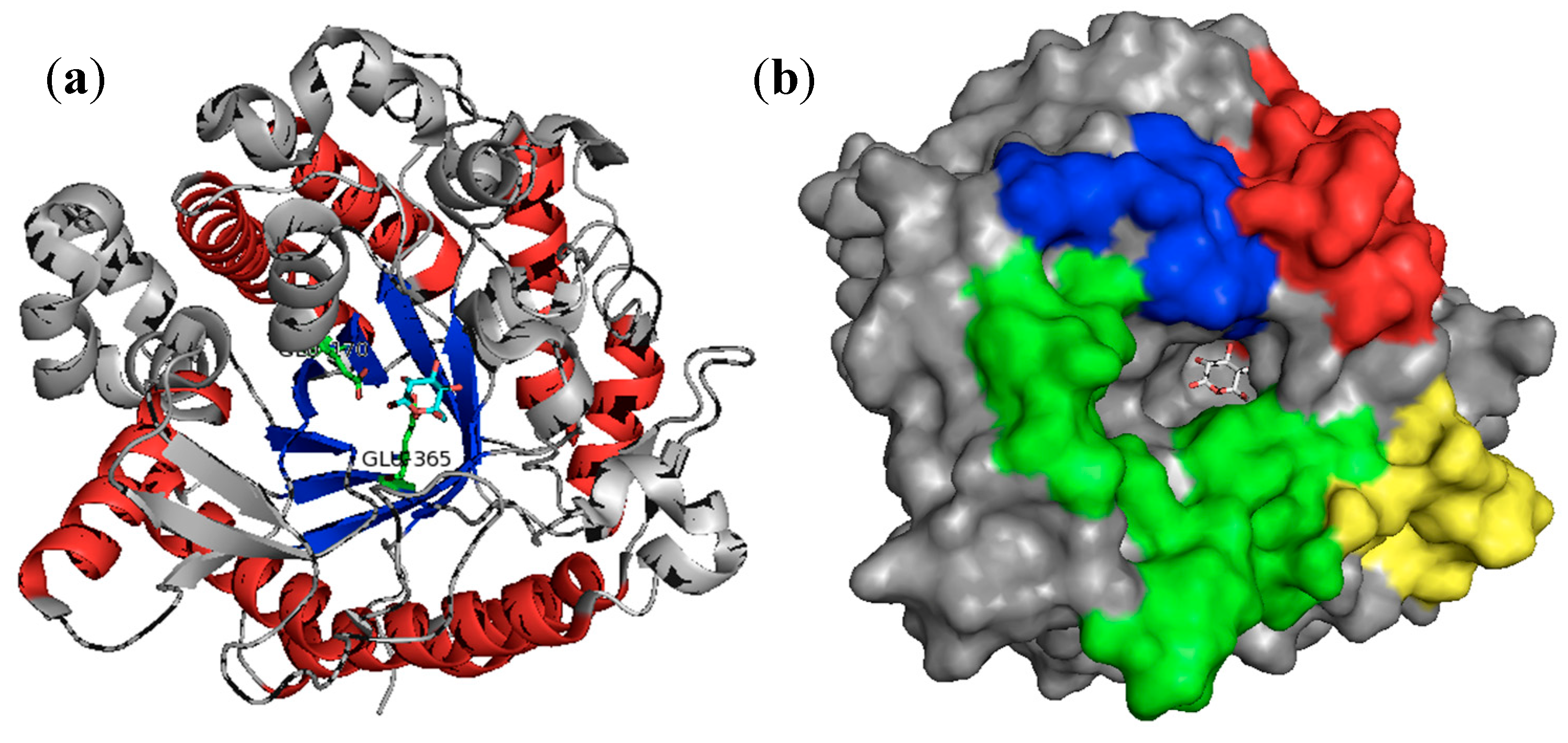
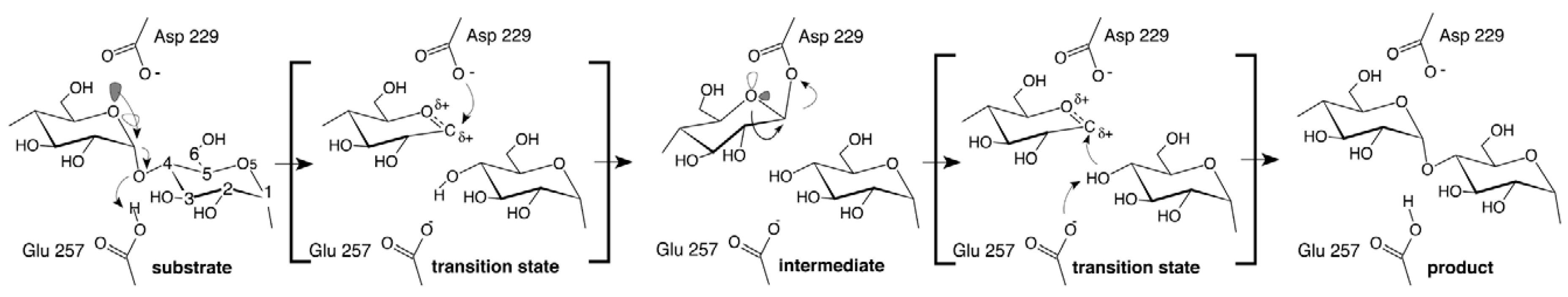
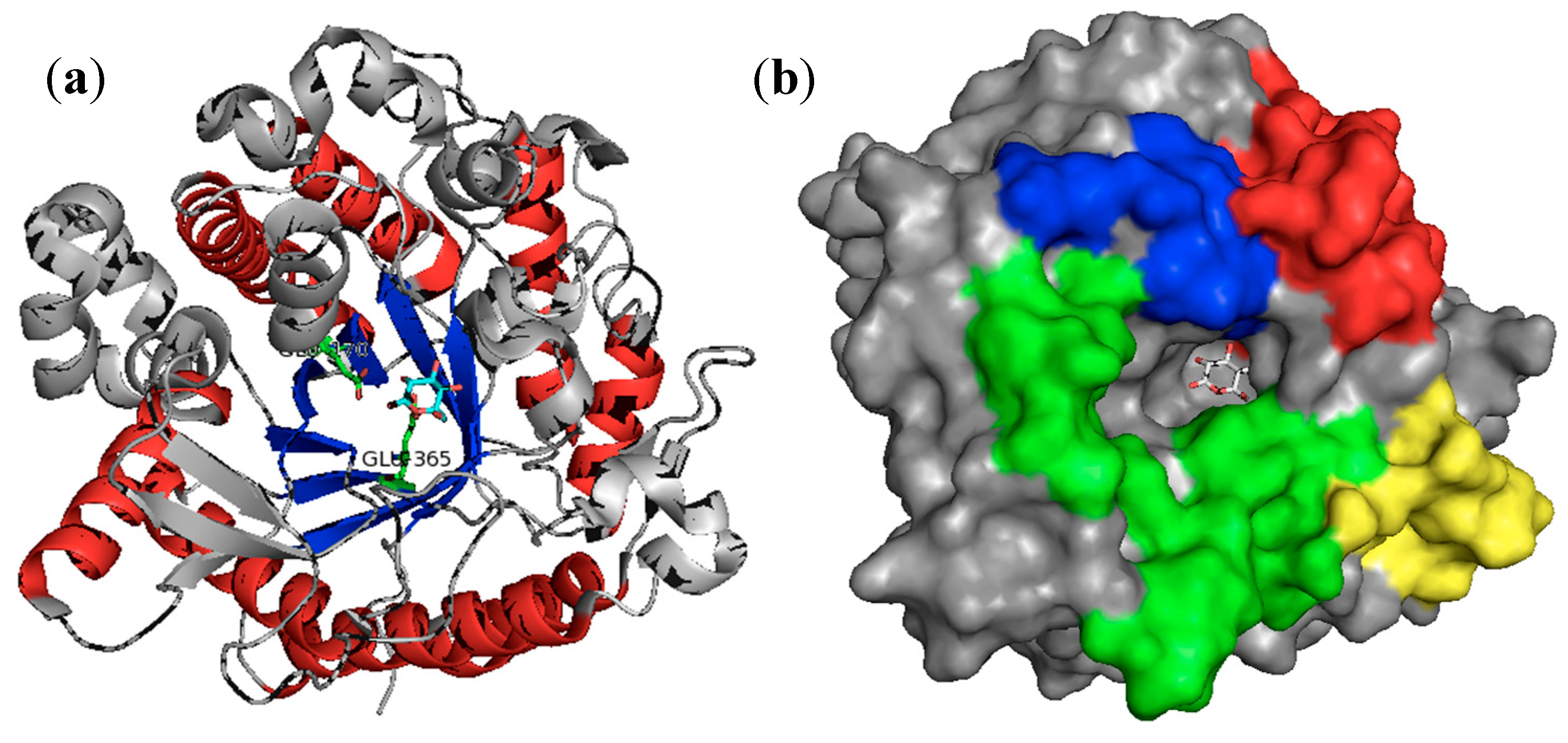
3.1. β-Glucosidases Features
3.2. β-Glucosidases Immobilization
3.2.1. Immobilization on Chitosan Particles
3.2.2. Covalent Immobilization of β-Glucosidases on other Supports
3.2.3. Immobilization of β-Glucosidases by Adsorption
3.2.4. Immobilization of β-Glucosidases by Entrapment
4. The GH13 Family of Enzymes
4.1. α-Amylases
4.1.1. Immobilization of α-Amylases
- Immobilization of α-amylases on insoluble supports:
- CLEAs of α-amylases:
- Other approaches for α-amylase immobilization:
4.2. Cyclodextrin Glycosyltransferase
4.2.1. Immobilization of CGTases
5. The GH 70 Family of Enzymes
5.1. GH 70 Enzymes and Their Characteristics
5.2. Immobilization of Dextransucrase
5.2.1. Immobilization of Dextransucrase by Entrapment
5.2.2. Immobilization of Dextransucrase by Covalent Immobilization
5.2.3. Immobilization of Dextransucrase by Adsorption
5.2.4. New Approaches in Dextransucrase Immobilization
6. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davies, G.J.; Gloster, T.M.; Henrissat, B. Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol. 2005, 15, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Enzyme nomenclature. Available online: http://www.chem.qmul.ac.uk/iubmb/enzyme/ (accessed on 13 August 2016).
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Henrissat, B. Glycoside hydrolases and glycosyltransferases: Families and functional modules. Curr. Opin. Struct. Biol. 2001, 11, 593–600. [Google Scholar] [CrossRef]
- Thuan, N.H.; Sohng, J.K. Recent biotechnological progress in enzymatic synthesis of glycosides. J. Ind. Microbiol. Biotechnol. 2013, 40, 1329–1356. [Google Scholar] [CrossRef] [PubMed]
- Vuong, T.V.; Wilson, D.B. Glycoside hydrolases: Catalytic base/nucleophile diversity. Biotechnol. Bioeng. 2010, 107, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Sulzenbacher, G.; Bourne, Y. Glycosyltransferases, glycoside hydrolases: Surprise, surprise! Curr. Opin. Struct. Biol. 2008, 18, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Bojarová, P.; Křen, V. Glycosidases: A key to tailored carbohydrates. Trends Biotechnol. 2009, 27, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, B.; Monsan, P.; Fauré, R.; O’Donohue, M.J. Glycosynthesis in a waterworld: New insight into the molecular basis of transglycosylation in retaining glycoside hydrolases. Biochem. J. 2015, 467, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Galan, C.; Berenguer-Murcia, A.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of different enzyme immobilization strategies to improve enzyme performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef]
- Min, K.; Yoo, Y.J. Recent progress in nanobiocatalysis for enzyme immobilization and its application. Biotechnol. Bioprocess Eng. 2014, 19, 553–567. [Google Scholar] [CrossRef]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.; Jordaan, J. Advances in enzyme immobilisation. Biotechnol. Lett. 2009, 31, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Cao, L. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 2005, 9, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; van Langen, L.; Sheldon, R.A. Immobilised enzymes: Carrier-bound or carrier-free? Curr. Opin. Biotechnol. 2003, 14, 387–394. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Polizzi, K.M.; Bommarius, A.S.; Broering, J.M.; Chaparro-Riggers, J.F. Stability of biocatalysts. Curr. Opin. Chem. Biol. 2007, 11, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.A.; Husain, Q. Potential applications of enzymes immobilized on/in nano materials: A review. Biotechnol. Adv. 2012, 30, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R. Coupling chemical modification and immobilization to improve the catalytic performance of enzymes. Adv. Synth. Catal. 2011, 353, 2216–2238. [Google Scholar] [CrossRef]
- Rueda, N.; dos Santos, J.C.S.; Ortiz, C.; Torres, R.; Barbosa, O.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R. Chemical modification in the design of immobilized enzyme biocatalysts: Drawbacks and opportunities. Chem. Rec. 2016, 1436–1455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, J.; Liu, Z. Enhanced activity of immobilized or chemically modified enzymes. ACS Catal. 2015, 5, 4503–4513. [Google Scholar] [CrossRef]
- Hernandez, K.; Fernandez-Lafuente, R. Control of protein immobilization: Coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance. Enzyme Microb. Technol. 2011, 48, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernandez-Lafuente, R. Amination of enzymes to improve biocatalyst performance: Coupling genetic modification and physicochemical tools. RSC Adv. 2014, 4, 38350–38374. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Strategies for the one-step immobilization-purification of enzymes as industrial biocatalysts. Biotechnol. Adv. 2015, 33, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lafuente, R. Stabilization of multimeric enzymes: Strategies to prevent subunit dissociation. Enzyme Microb. Technol. 2009, 45, 405–418. [Google Scholar] [CrossRef]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the support properties for immobilization or purification of enzymes. ChemCatChem 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Barbosa, O.; Torres, R.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Heterofunctional supports in enzyme immobilization: From traditional immobilization protocols to opportunities in tuning enzyme properties. Biomacromolecules 2013, 14, 2433–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Galan, C.; Barbosa, O.; Hernandez, K.; Dos Santos, J.C.S.; Rodrigues, R.C.; Fernandez-Lafuente, R. Evaluation of styrene-divinylbenzene beads as a support to immobilize lipases. Molecules 2014, 19, 7629–7645. [Google Scholar] [CrossRef] [PubMed]
- An, N.; Zhou, C.H.; Zhuang, X.Y.; Tong, D.S.; Yu, W.H. Immobilization of enzymes on clay minerals for biocatalysts and biosensors. Appl. Clay Sci. 2015, 114, 283–296. [Google Scholar] [CrossRef]
- Carlsson, N.; Gustafsson, H.; Thörn, C.; Olsson, L.; Holmberg, K.; Åkerman, B. Enzymes immobilized in mesoporous silica: A physical-chemical perspective. Adv. Colloid Interface Sci. 2014, 205, 339–360. [Google Scholar] [CrossRef] [PubMed]
- Asanomi, Y.; Yamaguchi, H.; Miyazaki, M.; Maeda, H. Enzyme-immobilized microfluidic process reactors. Molecules 2011, 16, 6041–6059. [Google Scholar] [CrossRef] [PubMed]
- Poppe, J.K.; Fernandez-Lafuente, R.; Rodrigues, R.C.; Ayub, M.A.Z. Enzymatic reactors for biodiesel synthesis: Present status and future prospects. Biotechnol. Adv. 2015, 33, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fuentes, M.; Betancor, L.; Grazu, V.; López-Gallego, F.; Pessela, B.C.C.; Hidalgo, A.; Fernández-Lorente, G.; Fernández-Lafuente, R.; et al. Glyoxyl agarose: A fully inert and hydrophilic support for immobilization and high stabilization of proteins. Enzyme Microb. Technol. 2006, 39, 274–280. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Fernández-Lorente, G.; Pedroche, J.; Fernández-Lafuente, R.; Guisan, J.M.; Tam, A.; Daminati, M. Epoxy sepabeads: A novel epoxy support for stabilization of industrial enzymes via very intense multipoint covalent attachment. Biotechnol. Prog. 2002, 18, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Jaeger, K.E. Overexpression, immobilization and biotechnological application of pseudomonas lipases. Chem. Phys. Lipids 1998, 93, 3–14. [Google Scholar] [CrossRef]
- Krajewska, B. Application of chitin- and chitosan-based materials for enzyme immobilizations: A review. Enzyme Microb. Technol. 2004, 35, 126–139. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Schoevaart, R.; Van Langen, L.M. Cross-linked enzyme aggregates (cleas): A novel and versatile method for enzyme immobilization (a review). Biocatal. Biotransform. 2005, 23, 141–147. [Google Scholar] [CrossRef]
- Cui, J.D.; Jia, S.R. Optimization protocols and improved strategies of cross-linked enzyme aggregates technology: Current development and future challenges. Crit. Rev. Biotechnol. 2015, 35, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.; Barbosa, O.; Rodrigues, R.C.; Fernandez-Lafuente, R.; Torres, R.; Ortiz, C. Optimized preparation of calb-cleas by response surface methodology: The necessity to employ a feeder to have an effective crosslinking. J. Mol. Catal. B Enzym. 2012, 80, 7–14. [Google Scholar] [CrossRef]
- Dal Magro, L.; Hertz, P.F.; Fernandez-Lafuente, R.; Klein, M.P.; Rodrigues, R.C. Preparation and characterization of a combi-cleas from pectinases and cellulases: A potential biocatalyst for grape juice clarification. RSC Adv. 2016, 6, 27242–27251. [Google Scholar] [CrossRef]
- Shah, S.; Sharma, A.; Gupta, M.N. Preparation of cross-linked enzyme aggregates by using bovine serum albumin as a proteic feeder. Anal. Biochem. 2006, 351, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Characteristic features and biotechnological applications of cross-linked enzyme aggregates (cleas). Appl. Microbiol. Biotechnol. 2011, 92, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Pletschke, B.I. Magnetic cross-linked enzyme aggregates (cleas): A novel concept towards carrier free immobilization of lignocellulolytic enzymes. Enzyme Microb. Technol. 2014, 61–62, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Nadar, S.S.; Rathod, V.K. Magnetic macromolecular cross linked enzyme aggregates (cleas) of glucoamylase. Enzyme. Microb. Technol. 2016, 83, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Engineering a more sustainable world through catalysis and green chemistry. J. R. Soc. Interface 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.I.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The carbohydrate-active enzymes database (cazy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef]
- Jeng, W.Y.; Wang, N.C.; Lin, M.H.; Lin, C.T.; Liaw, Y.C.; Chang, W.J.; Liu, C.I.; Liang, P.H.; Wang, A.H.J. Structural and functional analysis of three β-glucosidases from bacterium Clostridium cellulovorans, fungus Trichoderma reesei and termite Neotermes koshunensis. J. Struct. Biol. 2011, 173, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Trimbur, D.; Graham, R.; Warren, R.A.J.; Withers, S.G. Identification of the acid/base catalyst in agrobacterium faecalis β- glucosidase by kinetic analysis of mutants. Biochemistry 1995, 34, 14554–14562. [Google Scholar] [CrossRef] [PubMed]
- Thongpoo, P.; McKee, L.S.; Araújo, A.C.; Kongsaeree, P.T.; Brumer, H. Identification of the acid/base catalyst of a glycoside hydrolase family 3 (gh3) β-glucosidase from Aspergillus niger asku28. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Divakar, S. Glycosidases. In Enzymatic Transformation; Divakar, S., Ed.; Springer: New Delhi, India, 2013; p. 284. [Google Scholar]
- Jenkins, J.; Lo Leggio, L.; Harris, G.; Pickersgill, R. B-glucosidase, β-galactosidase, family a cellulases, family f xylanases and two barley glycanases form a superfamily of enzymes wit 8-fold β/α architecture and with two conserved glutamates near the carboxy-terminal ends of β-strands four and seven. FEBS Lett. 1995, 362, 281–285. [Google Scholar] [CrossRef]
- Henrissat, B.; Callebaut, I.; Fabrega, S.; Lehn, P.; Mornon, J.P.; Davies, G. Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc. Natl. Acad. Sci. USA 1995, 92, 7090–7094. [Google Scholar] [CrossRef] [PubMed]
- Nijikken, Y.; Tsukada, T.; Igarashi, K.; Samejima, M.; Wakagi, T.; Shoun, H.; Fushinobu, S. Crystal structure of intracellular family 1 β-glucosidase bgl1a from the basidiomycete Phanerochaete chrysosporium. FEBS Lett. 2007, 581, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, Y.; Mishra, S.; Bisaria, V.S. Microbial β-glucosidases: Cloning, properties, and applications. Crit. Rev. Biotechnol. 2002, 22, 375–407. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J.R.K.; Esen, A. B-glucosidases. Cell. Mol. Life Sci. 2010, 67, 3389–3405. [Google Scholar] [CrossRef] [PubMed]
- Marana, S.R. Molecular basis of substrate specificity in family 1 glycoside hydrolases. IUBMB Life 2006, 58, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kempton, J.B.; Withers, S.G. Mechanism of Agrobacterium β-glucosidase: Kinetic studies. Biochemistry 1992, 31, 9961–9969. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.J.; Wilson, K.S.; Henrissat, B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997, 321, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, L.M.F.; Marana, S.R. The role in the substrate specificity and catalysis of residues forming the substrate aglycone-binding site of a β-glycosidase. FEBS J. 2008, 275, 2536–2547. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, W.; Miki, H.; Nankai, H.; Sato, N.; Kawai, S.; Murata, K. Molecular cloning of two genes for β-d-glucosidase in Bacillus sp. Gl1 and identification of one as a gellan-degrading enzyme. Arch. Biochem. Biophys. 1998, 360, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, R.K.; Singhania, R.R.; Mathew, G.M.; Pandey, A. Cellulase production using biomass feed stock and its application in lignocellulose saccharification for bio-ethanol production. Renew. Energ. 2009, 34, 421–424. [Google Scholar] [CrossRef]
- Su, E.; Xia, T.; Gao, L.; Dai, Q.; Zhang, Z. Immobilization of β-glucosidase and its aroma-increasing effect on tea beverage. Food Bioprod. Process. 2010, 88, 83–89. [Google Scholar] [CrossRef]
- Kuhad, R.C.; Gupta, R.; Singh, A. Microbial cellulases and their industrial applications. Enzyme Res. 2011, 2011, 280696. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.A.; El-Shayeb, N.M.A.; Hashem, A.M.; Saleh, S.A.; Abdel-Fattah, A.F. Biochemical studies on immobilized fungal β-glucosidase. Braz. J. Chem. Eng. 2013, 30, 747–758. [Google Scholar] [CrossRef]
- Srinivasan, V.R.; Bumm, M.W. Isolation and immobilization of β d glucosidase from Alcaligenes faecalis. Biotechnol. Bioeng. 1974, 16, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Gallifuoco, A.; D’Ercole, L.; Alfani, F.; Cantarella, M.; Spagna, G.; Pifferi, P.G. On the use of chitosan-immobilized β-glucosidase in wine-making: Kinetics and enzyme inhibition. Process Biochem. 1998, 33, 163–168. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Wu, T.; Tang, X.; Pan, S. Optimal immobilization of β-glucosidase into chitosan beads using response surface methodology. Electron. J. Biotechnol. 2013, 16. [Google Scholar] [CrossRef]
- Desai, J.D.; Ray, R.; Desai, A. Immobilization of β-glucosidase from Scytalidium lignicola on chitosan. J. Ferment. Technol. 1986, 64, 255–258. [Google Scholar] [CrossRef]
- Bissett, F.; Sternberg, D. Immobilization of Aspergillus beta-glucosidase on chitosan. Appl. Environ. Microbiol. 1978, 35, 750–755. [Google Scholar] [PubMed]
- Chang, M.Y.; Juang, R.S. Use of chitosan-clay composite as immobilization support for improved activity and stability of β-glucosidase. Biochem. Eng. J. 2007, 35, 93–98. [Google Scholar] [CrossRef]
- Chang, J.; Lee, Y.S.; Fang, S.J.; Park, D.J.; Choi, Y.L. Hydrolysis of isoflavone glycoside by immobilization of β-glucosidase on a chitosan-carbon in two-phase system. Int. J. Biol. Macromol. 2013, 61, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Wang, J.; Lu, C.; Xu, Y.; Sun, Z. Immobilized β-glucosidase on magnetic chitosan microspheres for hydrolysis of straw cellulose. Process Biochem. 2013, 48, 683–687. [Google Scholar] [CrossRef]
- Ghose, T.K.; Sachdev, R.K. Kinetics of immobilized β-glucosidase for hydrolysis of cellobiose to glucose. J. Mol. Catal. 1979, 6, 99–109. [Google Scholar] [CrossRef]
- Caldini, C.; Bonomi, F.; Pifferi, P.G.; Lanzarini, G.; Galante, Y.M. Kinetic and immobilization studies on fungal glycosidases for aroma enhancement in wine. Enzyme. Microb. Technol. 1994, 16, 286–291. [Google Scholar] [CrossRef]
- Lei, S.; Xu, Y.; Fan, G.; Xiao, M.; Pan, S. Immobilization of naringinase on mesoporous molecular sieve mcm-41 and its application to debittering of white grapefruit. Appl. Surf. Sci. 2011, 257, 4096–4099. [Google Scholar] [CrossRef]
- Agrawal, R.; Verma, A.K.; Satlewal, A. Application of nanoparticle-immobilized thermostable β-glucosidase for improving the sugarcane juice properties. Innov. Food Sci. Emerg. 2016, 33, 471–482. [Google Scholar] [CrossRef]
- Song, J.; Imanaka, H.; Imamura, K.; Kajitani, K.; Nakanishi, K. Development of a highly efficient indigo dyeing method using indican with an immobilized β-glucosidase from Aspergillus niger. J. Biosci. Bioeng. 2010, 110, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Spagna, G.; Barbagallo, R.N.; Pifferi, P.G.; Blanco, R.M.; Guisan, J.M. Stabilization of a β-glucosidase from Aspergillus niger by binding to an amine agarose gel. J. Mol. Catal. B Enzym. 2000, 11, 63–69. [Google Scholar] [CrossRef]
- Jung, Y.R.; Shin, H.Y.; Song, Y.S.; Kim, S.B.; Kim, S.W. Enhancement of immobilized enzyme activity by pretreatment of β-glucosidase with cellobiose and glucose. J. Ind. Eng. Chem. 2012, 18, 702–706. [Google Scholar] [CrossRef]
- Aguado, J.; Dolores Romero, M.; Rodríguez, L.; Calles, J.A. Thermal deactivation of free and immobilized β-glucosidase from penicillium funiculosum. Biotechnol. Prog. 1995, 11, 104–106. [Google Scholar] [CrossRef]
- Vieira, M.F.; Vieira, A.M.S.; Zanin, G.M.; Tardioli, P.W.; Mateo, C.; Guisán, J.M. B-glucosidase immobilized and stabilized on agarose matrix functionalized with distinct reactive groups. J. Mol. Catal. B Enzym. 2011, 69, 47–53. [Google Scholar] [CrossRef]
- Chen, K.I.; Lo, Y.C.; Liu, C.W.; Yu, R.C.; Chou, C.C.; Cheng, K.C. Enrichment of two isoflavone aglycones in black soymilk by using spent coffee grounds as an immobiliser for β-glucosidase. Food Chem. 2013, 139, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.L.; Chaudhary, R.; Tsuzuki, T.; Barrow, C.J.; Puri, M. Immobilization of β-glucosidase on a magnetic nanoparticle improves thermostability: Application in cellobiose hydrolysis. Bioresour. Technol. 2013, 135, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Saquing, C.D.; Sarin, P.K.; Kelly, R.M.; Khan, S.A. Nanofibrous membranes for single-step immobilization of hyperthermophilic enzymes. J. Membr. Sci. 2015, 472, 251–260. [Google Scholar] [CrossRef]
- Reshmi, R.; Sugunan, S. Improved biochemical characteristics of crosslinked β-glucosidase on nanoporous silica foams. J. Mol. Catal. B: Enzym. 2013, 85–86, 111–118. [Google Scholar]
- Zhang, J.; Wang, D.; Pan, J.; Wang, J.; Zhao, H.; Li, Q.; Zhou, X. Efficient resveratrol production by immobilized β-glucosidase on cross-linked chitosan microsphere modified by l-lysine. J. Mol. Catal. B Enzym. 2014, 104, 29–34. [Google Scholar] [CrossRef]
- Bon, E.; Freire, D.; Fonseca Mendez, M.; Ferreira Soares, V. Immobilization of aspergillus niger β-d-glucosidase on aminated chitin and alumina/alginate. Biomass 1986, 11, 291–299. [Google Scholar] [CrossRef]
- Tan, I.S.; Lee, K.T. Immobilization of β-glucosidase from Aspergillus niger on κ-carrageenan hybrid matrix and its application on the production of reducing sugar from macroalgae cellulosic residue. Bioresour. Technol. 2014, 184, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.; Durante, M.; Pifferi, P.G.; Spagna, G.; Bianchi, G. Immobilization of β-glucosidase from a commercial preparation. Part 1. A comparative study of natural supports. Process Biochem. 1996, 31, 281–285. [Google Scholar] [CrossRef]
- González-Pombo, P.; Fariña, L.; Carrau, F.; Batista-Viera, F.; Brena, B.M. A novel extracellular β-glucosidase from issatchenkia terricola: Isolation, immobilization and application for aroma enhancement of white muscat wine. Process Biochem. 2011, 46, 385–389. [Google Scholar] [CrossRef]
- Khan, S.; Lindahl, S.; Turner, C.; Karlsson, E.N. Immobilization of thermostable β-glucosidase variants on acrylic supports for biocatalytic processes in hot water. J. Mol. Catal. B Enzym. 2012, 80, 28–38. [Google Scholar] [CrossRef]
- Tu, M.; Zhang, X.; Kurabi, A.; Gilkes, N.; Mabee, W.; Saddler, J. Immobilization of β-glucosidase on eupergit c for lignocellulose hydrolysis. Biotechnol. Lett. 2006, 28, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Iborra, J.L.; Castellar, M.R.; Cánovas, M.; Manjón, A. Analysis of a packed-bed reactor for hydrolysis of picrocrocin by immobilized β-glucosidase. Enzyme. Microb. Technol. 1993, 15, 780–784. [Google Scholar] [CrossRef]
- Roy, S.K.; Raha, S.K.; Dey, S.K.; Chakrabarty, S.L. Immobilization of β-glucosidase from myceliophthora thermophila d-14. Enzyme. Microb. Technol. 1989, 11, 431–435. [Google Scholar] [CrossRef]
- Ferner, M.J.; Müller, G.; Schumann, C.; Kampeis, P.; Ulber, R.; Raddatz, H. Immobilisation of glycosidases from commercial preparation on magnetic beads. Part 1. Characterisation of immobilised glycosidases with a particular emphasis on β-glucosidase. J. Mol. Catal. B Enzym. 2016, 123, 23–28. [Google Scholar] [CrossRef]
- Wei, C.; Lu, Q.; Ouyang, J.; Yong, Q.; Yu, S. Immobilization of β-glucosidase on mercaptopropyl-functionalized mesoporous titanium dioxide. J. Mol. Catal. B Enzym. 2013, 97, 303–310. [Google Scholar] [CrossRef]
- Borges, D.G.; Baraldo Junior, A.; Farinas, C.S.; de Lima Camargo Giordano, R.; Tardioli, P.W. Enhanced saccharification of sugarcane bagasse using soluble cellulase supplemented with immobilized β-glucosidase. Bioresour. Technol. 2014, 167, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Dekker, R.F.H. Application of a magnetic immobilized β-glucosidase in the enzymatic saccharification of steam-exploded lignocellulosic residues. Appl. Biochem. Biotechnol. 1990, 23, 25–39. [Google Scholar] [CrossRef]
- Matthijs, G.; Schacht, E. Comparative study of methodologies for obtaining β-glucosidase immobilized on dextran-modified silica. Enzyme Microb. Technol. 1996, 19, 601–605. [Google Scholar] [CrossRef]
- Hirsh, S.L.; Nosworthy, N.J.; Kondyurin, A.; Dos Remedios, C.G.; McKenzie, D.R.; Bilek, M.M.M. Linker-free covalent thermophilic β-glucosidase functionalized polymeric surfaces. J. Mater. Chem. 2011, 21, 17832–17841. [Google Scholar] [CrossRef]
- Nosworthy, N.J.; Kondyurin, A.; Bilek, M.M.M.; McKenzie, D.R. Ion implantation treatment of beads for covalent binding of molecules: Application to bioethanol production using thermophilic beta-glucosidase. Enzyme Microb. Technol. 2014, 54, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhou, X. Synthesis and properties of a novel crosslinked chitosan resin modified by l-lysine. React. Funct. Polym. 2008, 68, 1281–1289. [Google Scholar] [CrossRef]
- Singh, R.K.; Zhang, Y.W.; Nguyen, N.P.T.; Jeya, M.; Lee, J.K. Covalent immobilization of β-1,4-glucosidase from Agaricus arvensis onto functionalized silicon oxide nanoparticles. Appl. Microbiol. Biotechnol. 2011, 89, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Petro, M.; Svec, F.; Frechet, J.M.J. Immobilization of trypsin onto ‘molded’ macroporous poly(glycidyl methacrylate-co-ethylene dimethacrylate) rods and use of the conjugates as bioreactors and for affinity chromatography. Biotechnol. Bioeng. 1996, 49, 355–363. [Google Scholar] [CrossRef]
- Varavinit, S.; Chaokasem, N.; Shobsngob, S. Covalent immobilization of a glucoamylase to bagasse dialdehyde cellulose. World J. Microbiol. Biotechnol. 2001, 17, 721–725. [Google Scholar] [CrossRef]
- Karagulyan, H.K.; Gasparyan, V.K.; Decker, S.R. Immobilization of fungal β-glucosidase on silica gel and kaolin carriers. Appl. Biochem. Biotechnol. 2008, 146, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Pan, G.; Li, L.; Quan, G.; Ding, C.; Luo, A. Adsorption, immobilization, and activity of β-glucosidase on different soil colloids. J. Colloid Interface Sci. 2010, 348, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.S.; Wang, J.B.; Yao, S.J. High production of β-glucosidase from a marine aspergillus niger immobilized on towel gourd vegetable sponges. Chin. Chem. Lett. 2015, 26, 1011–1015. [Google Scholar] [CrossRef]
- Gueguen, Y.; Chemardin, P.; Pien, S.; Arnaud, A.; Galzy, P. Enhancement of aromatic quality of muscat wine by the use of immobilized β-glucosidase. J. Biotechnol. 1997, 55, 151–156. [Google Scholar] [CrossRef]
- Riccio, P.; Rossano, R.; Vinella, M.; Domizio, P.; Zito, F.; Sansevrino, F.; D’Elia, A.; Rosi, I. Extraction and immobilization in one step of two β-glucosidases released from a yeast strain of Debaryomyces hansenii. Enzyme Microb. Technol. 1999, 24, 123–129. [Google Scholar] [CrossRef]
- Venardos, D.; Klei, H.E.; Sundstrom, D.W. Conversion of cellobiose to glucose using immobilized β-glucosidase reactors. Enzyme Microb. Technol. 1980, 2, 112–116. [Google Scholar] [CrossRef]
- Sardar, M.; Agarwal, R.; Kumar, A.; Gupta, M.N. Noncovalent immobilization of enzymes on an enteric polymer eudragit s-100. Enzyme Microb. Technol. 1997, 20, 361–367. [Google Scholar] [CrossRef]
- Gargouri, M.; Smaali, I.; Maugard, T.; Legoy, M.D.; Marzouki, N. Fungus β-glycosidases: Immobilization and use in alkyl-β- glycoside synthesis. J. Mol. Catal. B Enzym. 2004, 29, 89–94. [Google Scholar] [CrossRef]
- Tyagi, R.; Gupta, M.N. Noncovalent and reversible immobilization of chemically modified amyloglucosidase and beta-glucosidase on deae-cellulose. Process Biochem. 1994, 29, 443–448. [Google Scholar] [CrossRef]
- Da Silva, T.M.; Pessela, B.C.; Da Silva, J.C.R.; Lima, M.S.; Jorge, J.A.; Guisan, J.M.; Polizeli, M.D.L.T.M. Immobilization and high stability of an extracellular β-glucosidase from Aspergillus japonicus by ionic interactions. J. Mol. Catal. B Enzym. 2014, 104, 95–100. [Google Scholar] [CrossRef]
- Lasanta, C.; Caro, I.; Pérez, L. B-glucosidase immobilization on ion exchange resins for using as aromatic enhancement of wines. In Proceeding of the 9th International Conference on Chemical and Process Engineering, Rome, Italy, 10–13 May 2009; ICheaP-9, Italian Association of Chemical Engineering. Volume 17, pp. 897–902.
- Chen, T.; Yang, W.; Guo, Y.; Yuan, R.; Xu, L.; Yan, Y. Enhancing catalytic performance of β-glucosidase via immobilization on metal ions chelated magnetic nanoparticles. Enzyme. Microb. Technol. 2014, 63, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ducret, A.; Carrière, J.F.; Trani, M.; Lortie, R. Enzymatic synthesis of octyl glucoside catalyzed by almond β-glucosidase in organic media. Can. J. Chem. 2002, 80, 653–656. [Google Scholar] [CrossRef]
- Basso, A.; Ducret, A.; Gardossi, L.; Lortie, R. Synthesis of octyl glucopyranoside by almond β-glucosidase adsorbed onto celite r-640®. Tetrahedron Lett. 2002, 43, 2005–2008. [Google Scholar] [CrossRef]
- Mazzuca, S.; Giorno, L.; Spadafora, A.; Mazzei, R.; Drioli, E. Immunolocalization of β-glucosidase immobilized within polysulphone capillary membrane and evaluation of its activity in situ. J. Membr. Sci. 2006, 285, 152–158. [Google Scholar] [CrossRef]
- Mazzei, R.; Giorno, L.; Piacentini, E.; Mazzuca, S.; Drioli, E. Kinetic study of a biocatalytic membrane reactor containing immobilized β-glucosidase for the hydrolysis of oleuropein. J. Membr. Sci. 2009, 339, 215–223. [Google Scholar] [CrossRef]
- Ong, E.; Gilkes, N.R.; Miller, R.C., Jr.; Antony, R.; Warren, J.; Kilburn, D.G. Enzyme immobilization using a cellulose-binding domain: Properties of a β-glucosidase fusion protein. Enzyme Microb. Technol. 1991, 13, 59–65. [Google Scholar] [CrossRef]
- Kierstan, M.; McHale, A.; Coughlan, M.P. The production of ethanol from cellobiose using immobilized β-glucosidase coentrapped with yeast in alginate gels. Biotechnol. Bioeng. 1982, 24, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Busto, M.D.; Ortega, N.; Perez-Mateos, M. Studies of microbial β-d-glucosidase immobilized in alginate gel beads. Process Biochem. 1995, 30, 421–426. [Google Scholar]
- Maitan-Alfenas, G.P.; de A. Lage, L.G.; de Almeida, M.N.; Visser, E.M.; de Rezende, S.T.; Guimarães, V.M. Hydrolysis of soybean isoflavones by Debaryomyces hansenii ufv-1 immobilised cells and free β-glucosidase. Food Chem. 2014, 146, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Keerti; Gupta, A.; Kumar, V.; Dubey, A.; Verma, A.K. Kinetic characterization and effect of immobilized thermostable β-glucosidase in alginate gel beads on sugarcane juice. ISRN Biochem. 2014, 2014, 178498. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; Meyer, A.S. Enzymatic cellulose hydrolysis: Enzyme reusability and visualization of β-glucosidase immobilized in calcium alginate. Molecules 2014, 19, 19390–19406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, N.; Busto, M.D.; Perez-Mateos, M. Optimisation of β-glucosidase entrapment in alginate and polyacrylamide gels. Bioresour. Technol. 1998, 64, 105–111. [Google Scholar] [CrossRef]
- El-Ghonemy, D.H. Immobilization and characterization of a thermostable β-glucosidase from Aspergillus terreus nrrl 265. J. Microb. Biotechnol. Food Sci. 2015, 4, 287–291. [Google Scholar] [CrossRef]
- Magalhães, D.B.; Miguez da Rocha-Leão, M.H. Immobilization of β glucosidase aggregates in calcium alginate. Biomass Bioenergy 1991, 1, 213–216. [Google Scholar] [CrossRef]
- Ang, S.S.; Salleh, A.B.; Bakar, F.A.; Yusof, N.A.; Heng, L.Y. Characteristics of β-glucosidase production by Paecilomyces variotii and its potencial application in bioassay system for boric acid determination. Afr. J. Biotechnol. 2012, 11, 3394–3401. [Google Scholar]
- Heichal-Segal, O.; Rappoport, S.; Braun, S. Immobilization in alginate-silicate sol-gel matrix protects β-glucosidase against thermal and chemical denaturation. Nat. Biotechnol. 1995, 13, 798–800. [Google Scholar] [CrossRef]
- Nagatomo, H.; Matsushita, Y.I.; Sugamoto, K.; Matsui, T. Preparation and properties of gelatin-immobilized β-glucosidase from pyrococcus furiosus. Biosci. Biotechnol. Biochem. 2005, 69, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Figueira, J.A.; Dias, F.F.G.; Sato, H.H.; Fernandes, P. Screening of supports for the immobilization of β-glucosidase. Enzyme Res. 2011, 2011, 642460. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, L.; Alfani, F.; Cantarella, M. Stability and activity of immobilized hydrolytic enzymes in two-liquid-phase systems: Acid phosphatase, β-glucosidase, and β-fructofuranosidase entrapped in poly(2-hydroxyethyl methacrylate) matrices. Enzyme Microb. Technol. 1993, 15, 861–867. [Google Scholar] [CrossRef]
- Javed, M.R.; Buthe, A.; Rashid, M.H.; Wang, P. Cost-efficient entrapment of β-glucosidase in nanoscale latex and silicone polymeric thin films for use as stable biocatalysts. Food Chem. 2016, 190, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Vila-Real, H.; Alfaia, A.J.; Rosa, J.N.; Gois, P.M.P.; Rosa, M.E.; Calado, A.R.T.; Ribeiro, M.H. A-rhamnosidase and β-glucosidase expressed by naringinase immobilized on new ionic liquid sol-gel matrices: Activity and stability studies. J. Biotechnol. 2011, 152, 147–158. [Google Scholar] [CrossRef] [PubMed]
- van der Maarel, M.J.E.C.; van der Veen, B.; Uitdehaag, J.C.M.; Leemhuis, H.; Dijkhuizen, L. Properties and applications of starch-converting enzymes of the α-amylase family. J. Biotechnol. 2002, 94, 137–155. [Google Scholar] [CrossRef]
- Kötzler, M.P.; Hancock, S.M.; Withers, S.G. Glycosidases: Functions, families and folds. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2001. [Google Scholar]
- MacGregor, E.A.; Janeček, Š.; Svensson, B. Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim. Biophys. Acta 2001, 1546, 1–20. [Google Scholar] [CrossRef]
- Uitdehaag, J.C.M.; van der Veen, B.A.; Dijkhuizen, L.; Dijkstra, B.W. Catalytic mechanism and product specificity of cyclodextrin glycosyltransferase, a prototypical transglycosylase from the α-amylase family. Enzyme Microb. Technol. 2002, 30, 295–304. [Google Scholar] [CrossRef]
- Lairson, L.L.; Withers, S.G. Mechanistic analogies amongst carbohydrate modifying enzymes. Chem. Commun. 2004, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Francis, F.; Sabu, A.; Nampoothiri, K.M.; Ramachandran, S.; Ghosh, S.; Szakacs, G.; Pandey, A. Use of response surface methodology for optimizing process parameters for the production of α-amylase by Aspergillus oryzae. Biochem. Eng. J. 2003, 15, 107–115. [Google Scholar] [CrossRef]
- Gangadharan, D.; Sivaramakrishnan, S.; Nampoothiri, K.M.; Sukumaran, R.K.; Pandey, A. Response surface methodology for the optimization of alpha amylase production by Bacillus amyloliquefaciens. Bioresour. Technol. 2008, 99, 4597–4602. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.S.; Rodríguez, M.R.; Guerra, N.P.; Rosés, R.P. Amylase production by aspergillus niger in submerged cultivation on two wastes from food industries. J. Food Eng. 2006, 73, 93–100. [Google Scholar] [CrossRef]
- Crabb, W.D.; Shetty, J.K. Commodity scale production of sugars from starches. Curr. Opin. Microbiol. 1999, 2, 252–256. [Google Scholar] [CrossRef]
- Gupta, R.; Gigras, P.; Mohapatra, H.; Goswami, V.K.; Chauhan, B. Microbial α-amylases: A biotechnological perspective. Process Biochem. 2003, 38, 1599–1616. [Google Scholar] [CrossRef]
- Kirk, O.; Borchert, T.V.; Fuglsang, C.C. Industrial enzyme applications. Curr. Opin. Biotechnol. 2002, 13, 345–351. [Google Scholar] [CrossRef]
- Jaiswal, N.; Prakash, O.; Talat, M.; Hasan, S.H.; Pandey, R.K. A-amylase immobilization on gelatin: Optimization of process variables. J. Genet. Eng. Biotechnol. 2012, 10, 161–167. [Google Scholar] [CrossRef]
- Chakraborty, S.; Jana, S.; Gandhi, A.; Sen, K.K.; Zhiang, W.; Kokare, C. Gellan gum microspheres containing a novel α-amylase from marine nocardiopsis sp. Strain b2 for immobilization. Int. J. Biol. Macromol. 2014, 70, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Swarnalatha, V.; Aluri Esther, R.; Dhamodharan, R. Immobilization of α-amylase on gum acacia stabilized magnetite nanoparticles, an easily recoverable and reusable support. J. Mol. Catal. B Enzym. 2013, 96, 6–13. [Google Scholar] [CrossRef]
- Guo, H.; Tang, Y.; Yu, Y.; Xue, L.; Qian, J.-Q. Covalent immobilization of α-amylase on magnetic particles as catalyst for hydrolysis of high-amylose starch. Int. J. Biol. Macromol. 2016, 87, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Baskar, G.; Afrin Banu, N.; Helan Leuca, G.; Gayathri, V.; Jeyashree, N. Magnetic immobilization and characterization of α-amylase as nanobiocatalyst for hydrolysis of sweet potato starch. Biochem. Eng. J. 2015, 102, 18–23. [Google Scholar] [CrossRef]
- Soleimani, M.; Khani, A.; Najafzadeh, K. A-amylase immobilization on the silica nanoparticles for cleaning performance towards starch soils in laundry detergents. J. Mol. Catal. B Enzym. 2012, 74, 1–5. [Google Scholar] [CrossRef]
- Talekar, S.; Ghodake, V.; Ghotage, T.; Rathod, P.; Deshmukh, P.; Nadar, S.; Mulla, M.; Ladole, M. Novel magnetic cross-linked enzyme aggregates (magnetic cleas) of alpha amylase. Bioresour. Technol. 2012, 123, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; Kumari, A.; Chauhan, G.S.; Chauhan, K. Modified chitosan microspheres in non-aggregated amylase immobilization. Int. J. Biol. Macromol. 2014, 66, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Kayastha, A.M. Immobilization of soybean (glycine max) α-amylase onto chitosan and amberlite mb-150 beads: Optimization and characterization. J. Mol. Catal. B Enzym. 2011, 69, 8–14. [Google Scholar] [CrossRef]
- Singh, K.; Kayastha, A.M. Optimal immobilization of α-amylase from wheat (Triticum aestivum) onto deae-cellulose using response surface methodology and its characterization. J. Mol. Catal. B Enzym. 2014, 104, 75–81. [Google Scholar] [CrossRef]
- Shukla, R.J.; Singh, S.P. Structural and catalytic properties of immobilized α-amylase from laceyella sacchari tsi-2. Int. J. Biol. Macromol. 2016, 85, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Veesar, I.A.; Solangi, I.B.; Memon, S. Immobilization of α-amylase onto a calix[4]arene derivative: Evaluation of its enzymatic activity. Bioorg. Chem. 2015, 60, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, A.M.; Mitidieri, S.; Fernandes, K.F. Immobilisation of α-amylase from aspergillus niger onto polyaniline. Food Bioprod. Process. 2011, 89, 300–306. [Google Scholar] [CrossRef]
- Häring, D.; Schreier, P. Cross-linked enzyme crystals. Curr. Opin. Chem. Biol. 1999, 3, 35–38. [Google Scholar] [CrossRef]
- Gupta, M.N.; Raghava, S. Enzyme stabilization via cross-linked enzyme aggregates. In Enzyme Stabilization and Immobilization: Methods and Protocols; Minteer, D.S., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 133–145. [Google Scholar]
- Nadar, S.S.; Muley, A.B.; Ladole, M.R.; Joshi, P.U. Macromolecular cross-linked enzyme aggregates (m-cleas) of α-amylase. Int. J. Biol. Macromol. 2016, 84, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Talekar, S.; Joshi, G.; Chougle, R.; Nainegali, B.; Desai, S.; Joshi, A.; Kambale, S.; Kamat, P.; Haripurkar, R.; Jadhav, S.; et al. Preparation of stable cross-linked enzyme aggregates (cleas) of nadh-dependent nitrate reductase and its use for silver nanoparticle synthesis from silver nitrate. Catal. Commun. 2014, 53, 62–66. [Google Scholar] [CrossRef]
- Torabizadeh, H.; Tavakoli, M.; Safari, M. Immobilization of thermostable α-amylase from Bacillus licheniformis by cross-linked enzyme aggregates method using calcium and sodium ions as additives. J. Mol. Catal. B Enzym. 2014, 108, 13–20. [Google Scholar] [CrossRef]
- Prajapati, V.D.; Jani, G.K.; Khanda, S.M. Pullulan: An exopolysaccharide and its various applications. Carbohydr. Polym. 2013, 95, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Bender, H.; Lehmann, J.; Wallenfels, K. Pullulan, ein extracelluläres glucan von Pullularia pullulans. Biochim. Biophys. Acta 1959, 36, 309–316. [Google Scholar] [CrossRef]
- Talekar, S.; Pandharbale, A.; Ladole, M.; Nadar, S.; Mulla, M.; Japhalekar, K.; Pattankude, K.; Arage, D. Carrier free co-immobilization of alpha amylase, glucoamylase and pullulanase as combined cross-linked enzyme aggregates (combi-cleas): A tri-enzyme biocatalyst with one pot starch hydrolytic activity. Bioresour. Technol. 2013, 147, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Tüzmen, N.; Kalburcu, T.; Denizli, A. A-amylase immobilization onto dye attached magnetic beads: Optimization and characterization. J. Mol. Catal. B Enzym. 2012, 78, 16–23. [Google Scholar] [CrossRef]
- Gashtasbi, F.; Ahmadian, G.; Noghabi, K.A. New insights into the effectiveness of alpha-amylase enzyme presentation on the bacillus subtilis spore surface by adsorption and covalent immobilization. Enzyme Microb. Technol. 2014, 64–65, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Homaei, A.; Saberi, D. Immobilization of α-amylase on gold nanorods: An ideal system for starch processing. Process Biochem. 2015, 50, 1394–1399. [Google Scholar] [CrossRef]
- Stam, M.R.; Danchin, E.G.J.; Rancurel, C.; Coutinho, P.M.; Henrissat, B. Dividing the large glycoside hydrolase family 13 into subfamilies: Towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 2006, 19, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Svensson, B. Protein engineering in the α-amylase family: Catalytic mechanism, substrate specificity, and stability. Plant Mol. Biol. 1994, 25, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.M.; Kalk, K.H.; van der Veen, B.A.; Dijkhuizen, L.; Dijkstra, B.W. The cyclization mechanism of cyclodextrin glycosyltransferase (cgtase) as revealed by a gamma-cyclodextrin-cgtase complex at 1.8-angstrom resolution. J. Biol. Chem. 1999, 274, 34868–34876. [Google Scholar] [CrossRef] [PubMed]
- Tonkova, A. Bacterial cyclodextrin glucanotransferase. Enzyme. Microb. Technol. 1998, 22, 678–686. [Google Scholar] [CrossRef]
- Alves-Prado, H.F.; Hilário, E. Seleção de microrganismos produtores de ciclodextrina glicosiltransferase (cgtase), produção e caracterização da enzima. Braz. J. Food Technol. 2002, 5, 189–196. [Google Scholar]
- Biwer, A.; Antranikian, G.; Heinzle, E. Enzymatic production of cyclodextrins. Appl. Microbiol. Biotechnol. 2002, 59, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Schulz, G.E. Structure of cyclodextrin glycosyltransferase refined at 2.0-a resolution. J. Mol. Biol. 1991, 217, 737–750. [Google Scholar] [CrossRef]
- Terada, Y.; Yanase, M.; Takata, H.; Takaha, T.; Okada, S. Cyclodextrins are not the major cyclic alpha-1,4-glucans produced by the initial action of cyclodextrin glucanotransferase on amylose. J. Biol. Chem. 1997, 272, 15729–15733. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Kato, T.; Nakamura, N.; Horikoshi, K. Spectrophotometric determination of cyclization activity of β-cyclodextrin-forming cyclomaltodextrin glucanotransferase. J. Jpn. Soc. Starch Sci. 1987, 34, 45–48. [Google Scholar] [CrossRef]
- Makela, M.J.; Korpela, T.K. Determination of the catalytic activity of cyclomaltodextrin glucanotransferase by maltotriose-methylorange assay. J. Biochem. Biophys. Methods 1988, 15, 307–318. [Google Scholar] [CrossRef]
- Van der Veen, B.A.; van Alebeek, G.; Uitdehaag, J.C.M.; Dijkstra, B.W.; Dijkhuizen, L. The three transglycosylation reactions catalyzed by cyclodextrin glycosyltransferase from Bacillus circulans (strain 251) proceed via different kinetic mechanisms. Eur. J. Biochem. 2000, 267, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Toshima, N.; Uenoyama, S. Inclusion complex-formation of cyclodextrin ith large dye molecule. Polym. J. 1981, 13, 607–610. [Google Scholar] [CrossRef]
- Szejtli, J. Utilization of cyclodextrins in industrial products and processes. J. Mater. Chem. 1997, 7, 575–587. [Google Scholar] [CrossRef]
- Szente, L.; Szejtli, J. Cyclodextrins as food ingredients. Trends Food Sci. Technol. 2004, 15, 137–142. [Google Scholar] [CrossRef]
- Qi, Q.S.; Zimmermann, W. Cyclodextrin glucanotransferase: From gene to applications. Appl. Microbiol. Biotechnol. 2005, 66, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Astray, G.; Gonzalez-Barreiro, C.; Mejuto, J.C.; Rial-Otero, R.; Simal-Gandara, J. A review on the use of cyclodextrins in foods. Food Hydrocoll. 2009, 23, 1631–1640. [Google Scholar] [CrossRef]
- Hedges, A.R. Industrial applications of cyclodextrins. Chem. Rev. 1998, 98, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, M.; Wang, F.; Gu, Z.; Du, G.; Wu, J.; Chen, J. Gamma-cyclodextrin: A review on enzymatic production and applications. Appl. Microbiol. Biotechnol. 2007, 77, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Marques, H.M.C. A review on cyclodextrin encapsulation of essential oils and volatiles. Flavour Fragr. J. 2010, 25, 313–326. [Google Scholar] [CrossRef]
- Van der Veen, B.A.; Uitdehaag, J.C.M.; Dijkstra, B.W.; Dijkhuizen, L. Engineering of cyclodextrin glycosyltransferase reaction and product specificity. Biochim. Biophys. Acta 2000, 1543, 336–360. [Google Scholar] [CrossRef]
- Kelly, R.M.; Dijkhuizen, L.; Leemhuis, H. The evolution of cyclodextrin glucanotransferase product specificity. Appl. Microbiol. Biotechnol. 2009, 84, 119–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Song, B.; Yue, Y.; Chao, Y.; Qian, S. Site-saturation mutagenesis of central tyrosine 195 leading to diverse product specificities of an α-cyclodextrin glycosyltransferase from Paenibacillus sp. 602–1. J. Biotechnol. 2014, 170, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Sobral, K.A.; Rodrigues, R.O.; de Oliveira, R.; de Moraes, F.; Zanin, G. Immobilization of cyclodextringlycosyltransferase (cgtase) from Bacillus firmus in commercial chitosan. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 383–386. [Google Scholar] [CrossRef]
- Mazzer, C.; Ferreira, L.R.; Rodella, J.R.T.; Moriwaki, C.; Matioli, G. Cyclodextrin production by Bacillus firmus strain 37 immobilized on inorganic matrices and alginate gel. Biochem. Eng. J. 2008, 41, 79–86. [Google Scholar] [CrossRef]
- Martín, M.T.; Plou, F.J.; Alcalde, M.; Ballesteros, A. Immobilization on eupergit c of cyclodextrin glucosyltransferase (cgtase) and properties of the immobilized biocatalyst. J. Mol. Catal. B Enzym. 2003, 21, 299–308. [Google Scholar] [CrossRef]
- Sobral, K.A.; Rodrigues, R.O.; Oliveira, R.D.; Olivo, J.E.; de Moraes, F.F.; Zanin, G.M. Evaluation of supports and methods for immobilization of enzyme cyclodextringlycosyltransferase. Appl. Biochem. Biotechnol. 2003, 105, 809–819. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, M.S.; Ryoo, J.J.; Lee, K.P.; Shin, H.D.; Kim, S.H.; Lee, Y.H. Immobilization of a cyclodextrin glucanotransferase (cgtase) onto polyethylene film with a carboxylic acid group and production of cyclodextrins from corn starch using cgtase-immobilized pe film. J. Appl. Polym. Sci. 2002, 85, 2451–2457. [Google Scholar] [CrossRef]
- Abdel-Naby, M.A. Immobilization of Paenibacillus macerans nrrl b-3186 cyclodextrin glucosyltransferase and properties of the immobilized enzyme. Process Biochem. 1999, 34, 399–405. [Google Scholar] [CrossRef]
- Norman, B.E.; Jorgensen, S.T. Thermoanaerobacter sp. Cgtase: Its properties and application. J. Jpn. Soc. Starch Sci. 1992, 39, 101–108. [Google Scholar] [CrossRef]
- Tardioli, P.W.; Zanin, G.M.; de Moraes, F.F. Characterization of thermoanaerobacter cyclomaltodextrin glucanotransferase immobilized on glyoxyl-agarose. Enzyme Microb. Technol. 2006, 39, 1270–1278. [Google Scholar] [CrossRef]
- Schöffer, J.d.N.; Klein, M.P.; Rodrigues, R.C.; Hertz, P.F. Continuous production of β-cyclodextrin from starch by highly stable cyclodextrin glycosyltransferase immobilized on chitosan. Carbohydr. Polym. 2013, 98, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Matte, C.R.; Nunes, M.R.; Benvenutti, E.V.; Schöffer, J.d.N.; Ayub, M.A.Z.; Hertz, P.F. Characterization of cyclodextrin glycosyltransferase immobilized on silica microspheres via aminopropyltrimethoxysilane as a “spacer arm”. J. Mol. Catal. B Enzym. 2012, 78, 51–56. [Google Scholar] [CrossRef]
- Nakamura, N.; Horikoshi, K. Production of schardinger β-dextrin by soluble and immobilized cyclodextrin glycosyltransferase of an alkalophilic Bacillus sp. Biotechnol. Bioeng. 1977, 19, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Lee, S.-H.; Shin, H.-D. Performance of column type bioreactor packed with immobilized cyclodextrin glucanotransferase for cyclodextrin production. J. Microbiol. Biotechnol. 1991, 1, 63–69. [Google Scholar]
- Tardioli, P.W.; Zanin, G.M.; de Moraes, F.F. Production of cyclodextrins in a fluidized-bed reactor using cyclodextrin-glycosyl-transferase. Appl. Biochem. Biotechnol. 2000, 84–86, 1003–1019. [Google Scholar] [CrossRef]
- Rha, C.S.; Lee, D.H.; Kim, S.G.; Min, W.K.; Byun, S.G.; Kweon, D.H.; Han, N.S.; Seo, J.H. Production of cyclodextrin by poly-lysine fused bacillus macerans cyclodextrin glycosyltransferase immobilized on cation exchanger. J. Mol. Catal. B Enzym. 2005, 34, 39–43. [Google Scholar] [CrossRef]
- Kweon, D.H.; Kim, S.G.; Han, N.S.; Lee, J.H.; Chung, K.M.; Seo, J.H. Immobilization of bacillus macerans cyclodextrin glycosyltransferase fused with poly-lysine using cation exchanger. Enzyme Microb. Technol. 2005, 36, 571–578. [Google Scholar] [CrossRef]
- Robyt, J.F.; Walseth, T.F. The mechanism of acceptor reactions of Leuconostoc mesenteroides b-512f dextransucrase. Carbohydr. Res. 1978, 60, 433–445. [Google Scholar] [CrossRef]
- Henrissat, B.; Davies, G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. [Google Scholar] [CrossRef]
- Monsan, P.; Potocki de Montalk, G.; Sarçabal, P.; Remaud-Siméon, M.; Willemot, R.M. Glucansucrases: Efficient tools for the synthesis of oligosaccharides of nutritional interest. Prog. Biotechnol. 2000, 17, 115–122. [Google Scholar]
- MacGregor, E.A.; Jespersen, H.M.; Svensson, B. A circularly permuted α-amylase-type α/β-barrel structure in glucan-synthesizing glucosyltransferases. FEBS Lett. 1996, 378, 263–266. [Google Scholar] [CrossRef]
- Kato, C.; Nakano, Y.; Lis, M.; Kuramitsu, H.K. Molecular genetic analysis of the catalytic site of Streptococcus mutans glucosyltransferases. Biochem. Biophys. Res. Commun. 1992, 189, 1184–1188. [Google Scholar] [CrossRef]
- Monchois, V.; Remaud-Simeon, M.; Russell, R.R.B.; Monsan, P.; Willemot, R.M. Characterization of Leuconostoc mesenteroides nrrl b-512f dextransucrase (dsrs) and identification of amino-acid residues playing a key role in enzyme activity. Appl. Microbiol. Biotechnol. 1997, 48, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Funane, K.; Ishii, T.; Ono, H.; Kobayashi, M. Changes in linkage pattern of glucan products induced by substitution of lys residues in the dextransucrase. FEBS Lett. 2005, 579, 4739–4745. [Google Scholar] [CrossRef] [PubMed]
- Funane, K.; Mizuno, K.; Takahara, H.; Kobayashi, M. Gene encoding a dextransucrase-like protein in leuconostoc mesenteroides nrrl b-512f. Biosci. Biotechnol. Biochem. 2000, 64, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Konishi, N.; Torii, Y.; Yamamoto, T.; Miyagi, A.; Ohta, H.; Fukui, K.; Hanamoto, S.; Matsuno, H.; Komatsu, H.; Kodama, T.; et al. Structure and enzymatic properties of genetically truncated forms of the water-insoluble glucan-synthesizing glucosyltransferase from Streptococcus sobrinus. J. Biochem. 1999, 126, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Monchois, V.; Willemot, R.M.; Monsan, P. Glucansucrases: Mechanism of action and structure-function relationships. FEMS Microbiol. Rev. 1999, 23, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Moulis, C.; Joucla, G.; Harrison, D.; Fabre, E.; Potocki-Veronese, G.; Monsan, P.; Remaud-Simeon, M. Understanding the polymerization mechanism of glycoside-hydrolase family 70 glucansucrases. J. Biol. Chem. 2006, 281, 31254–31267. [Google Scholar] [CrossRef] [PubMed]
- Ebert, K.H.; Schenk, G.; Scholz, R. Untersuchungen über das schicksal von dextran im organismus von meerschweinchen. Clin. Chem. Lab. Med. 1968, 6, 435–441. [Google Scholar] [CrossRef]
- Buchholz, K.; Monsan, P. Dextransucrase. In Handbook of Food Enzymology; Dekker: New York, NY, USA, 2003; pp. 589–603. [Google Scholar]
- Binder, T.P.; Côté, G.L.; Robyt, J.F. Disproportionation reactions catalyzed by leuconostoc and streptococcus glucansucrases. Carbohydr. Res. 1983, 124, 275–286. [Google Scholar] [CrossRef]
- Robyt, J.F. Mechanism and action of glucansucrases. Prog. Biotechnol. 1996, 12, 1–22. [Google Scholar]
- Lee, M.S.; Cho, S.K.; Eom, H.J.; Kim, S.Y.; Kim, T.J.; Han, N.S. Optimized substrate concentrations for production of long-chain isomaltooligosaccharides using dextransucrase of Leuconostoc mesenteroides b-512f. J. Microbiol. Biotechnol. 2008, 18, 1141–1145. [Google Scholar] [PubMed]
- Rabelo, M.C.; Honorato, T.L.; Gonçalves, L.R.B.; Pinto, G.A.S.; Rodrigues, S. Enzymatic synthesis of prebiotic oligosaccharides. Appl. Biochem. Biotechnol. 2006, 133, 31–40. [Google Scholar] [CrossRef]
- Côté, G.L.; Leathers, T.D. A method for surveying and classifying Leuconostoc spp. Glucansucrases according to strain-dependent acceptor product patterns. J. Ind. Microbiol. Biotechnol. 2005, 32, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Day, D.F. Optimization of oligosaccharide synthesis from cellobiose by dextransucrase. Appl. Biochem. Biotechnol. 2008, 148, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, I.M.; Rabelo, M.C.; Rodrigues, S. Cashew juice containing prebiotic oligosaccharides. J. Food Sci. Technol. 2012, 51, 2078–2084. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, F.A.; Kügler, J.; Chakravarthula, R.R.; Jördening, H.J. Co-immobilization of dextransucrase and dextranase for the facilitated synthesis of isomalto-oligosaccharides: Preparation, characterization and modeling. Biotechnol. Bioeng. 2008, 100, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Neely, W.B.; Nott, J. Dextransucrase, an induced enzyme from leuconostoc mesenteroides. Biochemistry 1962, 1, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Matsuda, K. Structural characteristics of dextrans synthesized by dextransucrases from Leuconostoc mesenteroides nrrl b-1299. Agric. Biol. Chem. 1977, 41, 1931–1937. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, D.; Kim, D.W.; Moon, Y.Y.; Robyt, J.F. Cloning of a dextransucrase gene (fmcmds) from a constitutive dextransucrase hyper-producing Leuconostoc mesenteroides b-512fmcm developed using vuv. Biotechnol. Lett. 2000, 22, 421–425. [Google Scholar] [CrossRef]
- Otgonbayar, G.E.; Eom, H.J.; Kim, B.S.; Ko, J.H.; Han, N.S. Mannitol production by leuconostoc citreum kacc 91348p isolated from kimchi. J. Microbiol. Biotechnol. 2011, 21, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.W.; Robyt, J.F. Stabilization of dextransucrase from leuconostoc mesenteroides nrrl b-512f by nonionic detergents, poly(ethylene glycol) and high-molecular-weight dextran. Biochim. Biophys. Acta 1984, 785, 89–96. [Google Scholar] [CrossRef]
- Berensmeier, S.; Ergezinger, M.; Bohnet, M.; Buchholz, K. Design of immobilised dextransucrase for fluidised bed application. J. Biotechnol. 2004, 114, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Eggleston, G.; Côté, G.L. Oligosaccharides in Food and Agriculture; American Chemical Society: Washington, DC, USA, 2003; Volume 849, p. 269. [Google Scholar]
- Koepsell, H.J.; Tsuchiya, H.M.; Hellman, N.N.; Kazenko, A.; Hoffman, C.A.; Sharpe, E.S.; Jackson, R.W. Enzymatic synthesis of dextran: Acceptor specificity and chain initiation. J. Biol. Chem. 1953, 200, 793–801. [Google Scholar] [PubMed]
- Ruas-Madiedo, P.; Hugenholtz, J.; Zoon, P. An overview of the functionality of exopolysaccharides produced by lactic acid bacteria. Int. Dairy J. 2002, 12, 163–171. [Google Scholar] [CrossRef]
- De Vuyst, L.; Degeest, B. Heteropolysaccharides from lactic acid bacteria. FEMS Microbiol. Rev. 1999, 23, 153–177. [Google Scholar] [CrossRef] [PubMed]
- Madhukumar, M.S.; Muralikrishna, G. Fermentation of xylo-oligosaccharides obtained from wheat bran and bengal gram husk by lactic acid bacteria and bifidobacteria. J. Food Sci. Technol. 2012, 49, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Roberfroid, M.B. Dietary modulation of the human colonic microbiota: Introducing the concept of prebiotics. J. Nutr. 1995, 125, 1401–1412. [Google Scholar] [PubMed]
- Valette, P.; Pelenc, V.; Djouzi, Z.; Andrieux, C.; Paul, F.; Monsan, P.; Szyli, O. Bioavailability of new synthesised glucooligosaccharides in the intestinal tract of gnotobiotic rats. J. Sci. Food Agric. 1993, 62, 121–127. [Google Scholar] [CrossRef]
- Fernando, W.M.A.D.B.; Flint, S.; Zou, M.; Brennan, C.S.; Ranaweera, K.K.D.S.; Bamunuarachchi, A. The effect of rice fibre fractions on the growth of co-cultures of probiotics. J. Food Sci. Technol. 2011, 48, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.H.; Cho, J.Y.; Seo, Y.S.; Woo, H.J.; Kim, H.K.; Kim, G.J.; Jhon, D.Y.; Kim, D. Production of a low calorie mandarin juice by enzymatic conversion of constituent sugars to oligosaccharides and prevention of insoluble glucan formation. Biotechnol. Lett. 2015, 37, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.H.; Seo, Y.S.; Cho, J.Y.; Lee, S.; Kim, G.J.; Yoon, J.W.; Ahn, S.H.; Hwang, K.H.; Park, J.S.; Jang, T.S.; et al. Synthesis of oligosaccharide-containing orange juice using glucansucrase. Biotechnol. Bioprocess Eng. 2015, 20, 447–452. [Google Scholar] [CrossRef]
- Reischwitz, A.; Reh, K.D.; Buchholz, K. Unconventional immobilization of dextransucrase with alginate. Enzyme Microb. Technol. 1995, 17, 457–461. [Google Scholar] [CrossRef]
- Berensmeier, S.; Jördening, H.J.; Buchholz, K. Isomaltose formation by free and immobilized dextransucrase. Biocatal. Biotransform. 2006, 24, 280–290. [Google Scholar] [CrossRef]
- Gómez de Segura, A.; Alcalde, M.; Bernabé, M.; Ballesteros, A.; Plou, F.J. Synthesis of methyl α-d-glucooligosaccharides by entrapped dextransucrase from Leuconostoc mesenteroides b-1299. J. Biotechnol. 2006, 124, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Alcalde, M.; Plou, F.J.; de Gómez Segura, A.; Remaud-Simeon, M.; Willemot, R.M.; Monsan, P.; Ballesteros, A. Immobilization of native and dextran-free dextransucrases from Leuconostoc mesenteroides nrrl b-512f for the synthesis of glucooligosaccharides. Biotechnol. Technol. 1999, 13, 749–755. [Google Scholar] [CrossRef]
- Kothari, D.; Baruah, R.; Goyal, A. Immobilization of glucansucrase for the production of gluco-oligosaccharides from Leuconostoc mesenteroides. Biotechnol. Lett. 2012, 34, 2101–2106. [Google Scholar] [CrossRef] [PubMed]
- Tanriseven, A.; Doǧan, Ş. Production of isomalto-oligosaccharides using dextransucrase immobilized in alginate fibres. Process Biochem. 2002, 37, 1111–1115. [Google Scholar] [CrossRef]
- Parlak, M.; Ustek, D.; Tanriseven, A. A novel method for covalent immobilization of dextransucrase. J. Mol. Catal. B Enzym. 2013, 89, 52–60. [Google Scholar] [CrossRef]
- Dols-Lafargue, M.; Willemot, R.M.; Monsan, P.F.; Remaud-Simeon, M. Reactor optimization for α-1,2 glucooligosaccharide synthesis by immobilized dextransucrase. Biotechnol. Bioeng. 2001, 75, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Gómez de Segura, A.; Alcalde, M.; Yates, M.; Rojas-Cervantes, M.L.; López-Cortés, N.; Ballesteros, A.; Plou, F.J. Immobilization of dextransucrase from Leuconostoc mesenteroides nrrl b-512f on eupergit c supports. Biotechnol. Prog. 2004, 20, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Dols, M.; Simeon, M.R.; Willemot, R.M.; Vignon, M.R.; Monsan, P.F. Structural characterization of the maltose acceptor-products synthesized by Leuconostoc mesenteroides nrrl b-1299 dextransucrase. Carbohydr. Res. 1997, 305, 549–559. [Google Scholar] [CrossRef]
- Monsan, P.; Lopez, A. On the production of dextran by free and immobilized dextransucrase. Biotechnol. Bioeng. 1981, 23, 2027–2037. [Google Scholar] [CrossRef]
- Ölçer, Z.; Tanriseven, A. Co-immobilization of dextransucrase and dextranase in alginate. Process Biochem. 2010, 45, 1645–1651. [Google Scholar] [CrossRef]
- Dols, M.; Remaud-Simeon, M.; Willemot, R.M.; Vignon, M.; Monsan, P. Characterization of the different dextransucrase activities excreted in glucose, fructose, or sucrose medium by Leuconostoc mesenteroides nrrl b-1299. Appl. Environ. Microbiol. 1998, 64, 1298–1302. [Google Scholar] [PubMed]
- Gómez de Segura, A.; Alcalde, M.; Plou, F.J.; Remaud-Simeon, M.; Monsan, P.; Ballesteros, A. Encapsulation in lentikats of dextransucrase from leuconostoc mesenteroides nrrl b-1299, and its effect on product selectivity. Biocatal. Biotransform. 2003, 21, 325–331. [Google Scholar] [CrossRef]
- Kubik, C.; Sikora, B.; Bielecki, S. Immobilization of dextransucrase and its use with soluble dextranase for glucooligosaccharides synthesis. Enzyme Microb. Technol. 2004, 34, 555–560. [Google Scholar] [CrossRef]
- Huguet, M.L.; Neufeld, R.J.; Dellacherie, E. Calcium-alginate beads coated with polycationic polymers: Comparison of chitosan and deae-dextran. Process Biochem. 1996, 31, 347–353. [Google Scholar] [CrossRef]
- Quirasco, M.; Remaud-Simeon, M.; Monsan, P.; López-Munguía, A. Experimental behavior of a whole cell immobilized dextransucrase biocatalyst in batch and packed bed reactors. Bioprocess Eng. 1999, 20, 289–295. [Google Scholar] [CrossRef]
- Tanriseven, A.; Robyt, J.F. Interpretation of dextransucrase inhibition at high sucrose concentrations. Carbohydr. Res. 1993, 245, 97–104. [Google Scholar] [CrossRef]
- Goulas, A.K.; Cooper, J.M.; Grandison, A.S.; Rastall, R.A. Synthesis of isomaltooligosaccharides and oligodextrans in a recycle membrane bioreactor by the combined use of dextransucrase and dextranase. Biotechnol. Bioeng. 2004, 88, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Auh, J.H.; Yoon, H.G.; Kim, M.J.; Park, J.H.; Hong, S.S.; Kang, M.H.; Kim, T.J.; Moon, T.W.; Kim, J.W.; et al. Cooperative action of α-glucanotransferase and maltogenic amylase for an improved process of isomaltooligosaccharide (imo) production. J. Agric. Food Chem. 2002, 50, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Fanchini Terrasan, C.R.; Trobo-Maseda, L.; Moreno-Pérez, S.; Carmona, E.C.; Pessela, B.C.; Guisan, J.M. Co-immobilization and stabilization of xylanase, β-xylosidase and α-l-arabinofuranosidase from Penicillium janczewskii for arabinoxylan hydrolysis. Process Biochem. 2016, 51, 614–623. [Google Scholar] [CrossRef]
- Goulas, A.K.; Fisher, D.A.; Grimble, G.K.; Grandison, A.S.; Rastall, R.A. Synthesis of isomaltooligosaccharides and oligodextrans by the combined use of dextransucrase and dextranase. Enzyme. Microb. Technol. 2004, 35, 327–338. [Google Scholar] [CrossRef]
- Robyt, J.F.; Kimble, B.K.; Walseth, T.F. The mechanism of dextransucrase action: Direction of dextran biosynthesis. Arch. Biochem. Biophys. 1974, 165, 634–640. [Google Scholar] [CrossRef]
- Robyt, J.F.; Taniguchi, H. The mechanism of dextransucrase action: Biosynthesis of branch linkages by acceptor reactions with dextran. Arch. Biochem. Biophys. 1976, 174, 129–135. [Google Scholar] [CrossRef]
- Robyt, J.F.; Corrigan, A.J. The mechanism of dextransucrase action: Activation of dextransucrase from Streptococcus mutans omz 176 by dextran and modified dextran and the nonexistence of the primer requirement for the synthesis of dextran. Arch. Biochem. Biophys. 1977, 183, 726–731. [Google Scholar] [CrossRef]
- Kaboli, H.; Reilly, P. Immobilization and properties of Leuconostoc mesenteroides dextransucrase. Biotechnol. Bioeng. 1980, 22, 1055–1069. [Google Scholar] [CrossRef]
- Monsan, P.; Paul, F.; Auriol, D.; Lopez, A. Dextran synthesis using immobilized leuconostoc mesenteroides dextransucrase. Methods Enzymol. 1987, 136, 239–254. [Google Scholar]
- Robyt, J.F.; Yoon, S.H.; Mukerjea, R. Dextransucrase and the mechanism for dextran biosynthesis. Carbohydr. Res. 2008, 343, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Kraemer, D.M. Eupergit® c, a carrier for immobilization of enzymes of industrial potential. J. Mol. Catal. B Enzym. 2000, 10, 157–176. [Google Scholar] [CrossRef]
- Mateo, C.; Fernández-Lorente, G.; Abian, O.; Fernández-Lafuente, R.; Guisán, J.M. Multifunctional epoxy supports: A new tool to improve the covalent immobilization of proteins. The promotion of physical adsorptions of proteins on the supports before their covalent linkage. Biomacromolecules 2000, 1, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Grazú, V.; Pessela, B.C.C.; Montes, T.; Palomo, J.M.; Torres, R.; López-Gallego, F.; Fernández-Lafuente, R.; Guisán, J.M. Advances in the design of new epoxy supports for enzyme immobilization-stabilization. Biochem. Soc. Trans. 2007, 35, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Willemot, R.M. Étude de la Dextrane-Saccharase de Leuconostoc mesenteroides nrrl b-512f. Ph.D. Thesis, Institut National des Sciences Appliquées, Toulouse, France, 1993. [Google Scholar]
- Hashem, A.M.; Gamal, A.A.; Hassan, M.E.; Hassanein, N.M.; Esawy, M.A. Covalent immobilization of Enterococcus faecalis esawy dextransucrase and dextran synthesis. Int. J. Biol. Macromol. 2016, 82, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Katiyar, S.S. Chemical modification of dextransucrase from leuconostoc mesenteroides nrrl b-512f by pyridoxal 5′-phosphate: Evidence of the presence of an essential lysine residue at the active site. Biochem. Mol. Biol. Int. 1998, 44, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Katiyar, S.S. Studies on the inactivation of leuconostoc mesenteroides nrrl b-512f dextransucrase by o-phthalaldehyde: Evidence for the presence of an essential lysine residue at the active site. J. Enzym. Inhib. 1998, 13, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Prabhu, K.A. Immobilization and properties of dextransucrase from Leuconostoc mesenteroides culture, lm1. J. Gen. Appl. Microbiol. 1995, 41, 399–407. [Google Scholar] [CrossRef]
- Kobs, S.F. Acceptor activity of affinity-immobilized dextransucrase from Streptococcus sanguis atcc 10558. Carbohydr. Res. 1991, 211, 337–342. [Google Scholar] [CrossRef]
- Chang, H.N.; Ghim, Y.S.; Cho, Y.R.; Landis, D.A.; Reilly, P.J. Immobilization of Leuconostoc mesenteroides dextransucrase to porous phenoxyacetyl cellulose beads. Biotechnol. Bioeng. 1982, 24, 1928. [Google Scholar] [CrossRef] [PubMed]
- Fabre, E.; Bozonnet, S.; Arcache, A.; Willemot, R.M.; Vignon, M.; Monsan, P.; Remaud-Simeon, M. Role of the two catalytic domains of dsr-e dextransucrase and their involvement in the formation of highly α-1,2 branched dextran. J. Bacteriol. 2005, 187, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Vujičić-Žagar, A.; Pijning, T.; Kralj, S.; López, C.A.; Eeuwema, W.; Dijkhuizen, L.; Dijkstra, B.W. Crystal structure of a 117 kda glucansucrase fragment provides insight into evolution and product specificity of gh70 enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21406–21411. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, S.; Shimamura, T.; Weyand, S.; Kawarasaki, Y.; Misaka, T.; Abe, K.; Kobayashi, T.; Cameron, A.D.; Iwata, S. Crystal structure of glucansucrase from the dental caries pathogen streptococcus mutans. J. Mol. Biol. 2011, 408, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Brison, Y.; Pijning, T.; Malbert, Y.; Fabre, É.; Mourey, L.; Morel, S.; Potocki-Véronèse, G.; Monsan, P.; Tranier, S.; Remaud-Siméon, M.; et al. Functional and structural characterization of α-(1→2) branching sucrase derived from dsr-e glucansucrase. J. Biol. Chem. 2012, 287, 7915–7924. [Google Scholar] [CrossRef] [PubMed]
- Pijning, T.; Vujičić-Žagar, A.; Kralj, S.; Dijkhuizen, L.; Dijkstra, B.W. Structure of the α-1,6/α-1,4-specific glucansucrase gtfa from Lactobacillus reuteri 121. Acta Crystallogr. 2012, 68, 1448–1454. [Google Scholar]
- Brison, Y.; Malbert, Y.; Czaplicki, G.; Mourey, L.; Remaud-Simeon, M.; Tranier, S. Structural insights into the carbohydrate-binding ability of an α-(1→2) branching sucrase from glycoside-hydrolase family 70. J. Biol. Chem. 2016, 291, 7527–7540. [Google Scholar] [CrossRef] [PubMed]
- Funane, K.; Ookura, T.; Kobayashi, M. Glucan binding regions of dextransucrase from Leuconostoc mesenteroides nrrl b-512f. Biosci. Biotechnol. Biochem. 1998, 62, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.

{kind=link}
{kind=link}
{kind=link}
| Reactive Group | Immobilization Support | Reference |
|---|---|---|
| Glutaraldehyde | pore glass particles | [81] |
| chitosan and alginate beads | [70,76] | |
| Bentonite, celite, silica gel, and Nylon | [82] | |
| mesoporous silica MCM-41 | [83] | |
| SiO2 nanoparticles | [84] | |
| Chitin, Loofa, Sawdust coarse, Sawdust fine, Sponge, Stainless steel, Pumice, Wool and agar, agarose and sodium alginate | [72] | |
| Chitopearl beads BCW-3001 | [85] | |
| amine agarose gel | [86] | |
| silica gel | [87] | |
| nylon powder | [88] | |
| chitosan–clay composite | [78] | |
| agarose matrix | [89] | |
| chitosan | [74,75,77] | |
| magnetic chitosan microspheres | [80] | |
| chitosan-carbon beads | [79] | |
| Spent coffee grounds | [90] | |
| iron oxide magnetic nanoparticle | [91] | |
| polyvinylalcohol (PVA) nanofibrous membranes | [92] | |
| mesocellular silica foams | [93] | |
| l-lysine and glutaraldehyde | chitosan microspheres | [94] |
| Hexamethylenediamine and glutaraldehyde | chitin (IME-C) and calcium alginate (IME-A) | [95] |
| Polyelectrolytes (PEI) and glutaraldehyde | Kappa-carrageenan beads | [96] |
| APTMS and glutaraldehyde | cellulose PEI, alpha-alumina, gamma-alumina and chitosan | [97] |
| Epoxy | Eupergit C 250L | [98] |
| polyacrylic matrices supports (Eupergit® C, Eupergit® C250L, and cryogel) | [99] | |
| Eupergit C | [100] | |
| Nylon-hydrazide | nylon pellets | [101] |
| CNBr | sepharose gel beads 4B | [102] |
| Carbodiimide | magnetic beads | [103] |
| Mercaptopropyl-functionalized | Mesoporous titanium dioxide | [104] |
| Aldehyde groups | glyoxyl–agarose | [105] |
| Polyethyleneimine and glutaraldehyde | Magnetite (PAM) and (TiO2)-coated magnetite (TAM) | [106] |
| Dextran dialdehyde and β-glucosidase-dextran conjugates | silica and aminopropylsilica | [107] |
| Plasma immersion ion implantation (PIII) | polystyrene films | [108] |
| polyethylene granules | [109] |
| Reactive Group | Immobilization Support | Reference |
|---|---|---|
| Physical adsorption | kaolin | [114] |
| soil colloidal particles | [115] | |
| towel gourd vegetable sponges | [116] | |
| Cation Exchanger | Duolite A-568 resin | [117] |
| hydroxyapatite (HTP) | [118] | |
| resin Amberlite DP-1 | [119] | |
| Eudragit S-100 | [120] | |
| polyacrylic resin | [105] | |
| Anion Exchanger | DEAE-sepharose | [121] |
| DEAE-cellulose | [122,123] | |
| Anion Exchanger and Macroporous | different ion exchange resins | [124] |
| Metal Ionic Binding | Magnetic Fe3O4 nanoparticles coupled with agarose | [125] |
| Magnetic Fe3O4 nanoparticles | [75] | |
| Hydrophobic polyaromatic | Amberlite XAD-4 resin | [126] |
| Celite R-640 | [127] | |
| Physically immobilized by crossflow ultrafiltration | 30 kDa cut-off capillary polysulphone membranes | [128] |
| capillary membranes of polysulphone | [129] | |
| Not declared | cellulosic adsorbents: dewaxed, absorbent cotton, CF1 cellulose, Avicel™ PH-101, and Cellufine | [130] |
| Immobilization Support | Reference |
|---|---|
| Calcium alginate beads | [70,131,132,133,134,137,139] |
| Calcium alginate beads and alumina | [95] |
| Calcium alginate beads and glutaraldehyde | [135,138] |
| Calcium alginate beads in tetramethoxy-ortho-silicate (TMOS) and hexane | [140] |
| Calcium alginate and polyacrylamide gel | [136] |
| Polyacrylamide gel | [102] |
| Gelatin gel | [141] |
| Calcium alginate beads, gelatin, polyvinyl alcohol- (PVA-) based matrices (Lentikats), and sol-gel | [142] |
| Hydrogels of poly(2-hydroxyethyl methacrylate) | [143] |
| Nanoscale polymeric materials (polyurethane, latex and silicone) | [144] |
| Ionic liquid sol–gel matrices | [145] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graebin, N.G.; Schöffer, J.D.N.; Andrades, D.D.; Hertz, P.F.; Ayub, M.A.Z.; Rodrigues, R.C. Immobilization of Glycoside Hydrolase Families GH1, GH13, and GH70: State of the Art and Perspectives. Molecules 2016, 21, 1074. https://doi.org/10.3390/molecules21081074
Graebin NG, Schöffer JDN, Andrades DD, Hertz PF, Ayub MAZ, Rodrigues RC. Immobilization of Glycoside Hydrolase Families GH1, GH13, and GH70: State of the Art and Perspectives. Molecules. 2016; 21(8):1074. https://doi.org/10.3390/molecules21081074
Chicago/Turabian StyleGraebin, Natália G., Jéssie Da N. Schöffer, Diandra De Andrades, Plinho F. Hertz, Marco A. Z. Ayub, and Rafael C. Rodrigues. 2016. "Immobilization of Glycoside Hydrolase Families GH1, GH13, and GH70: State of the Art and Perspectives" Molecules 21, no. 8: 1074. https://doi.org/10.3390/molecules21081074