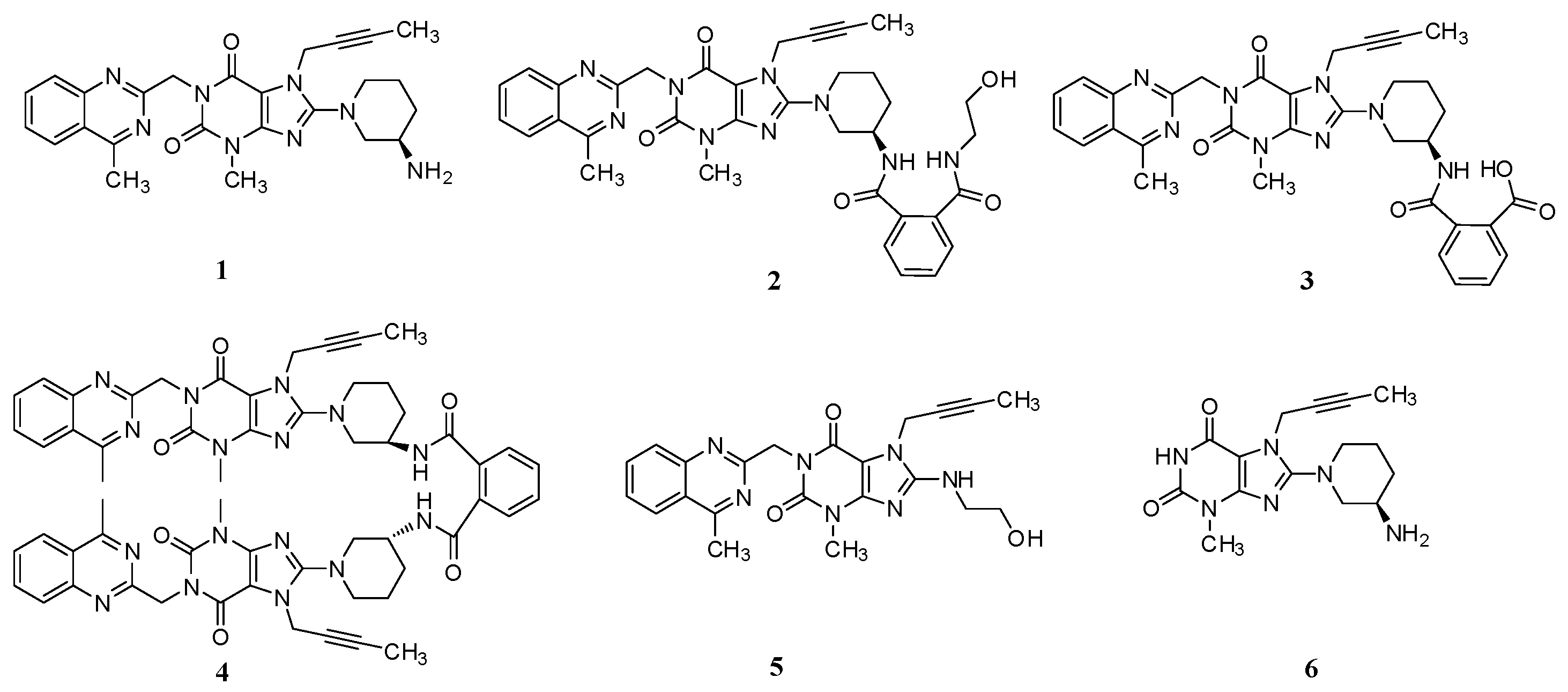
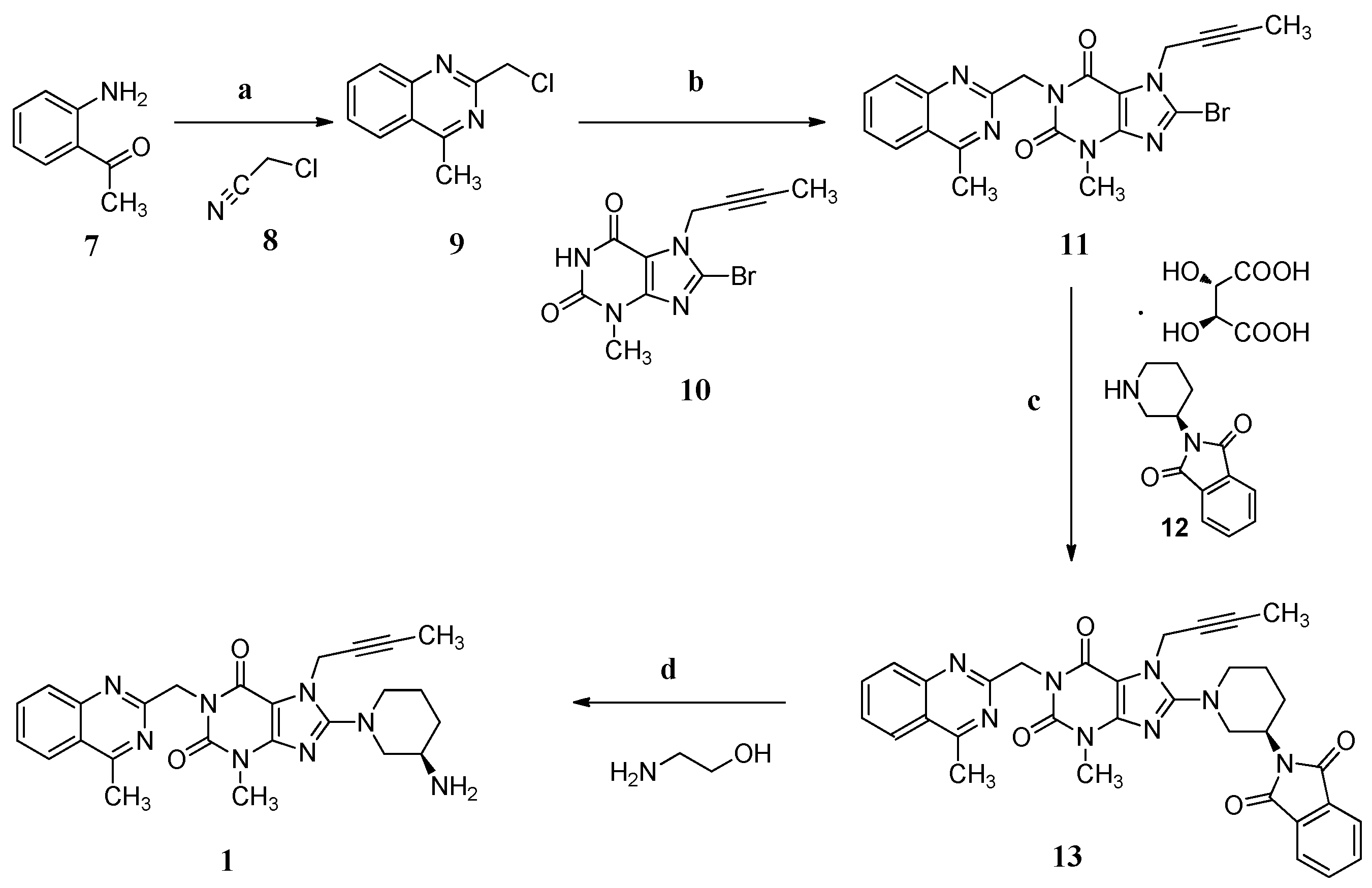
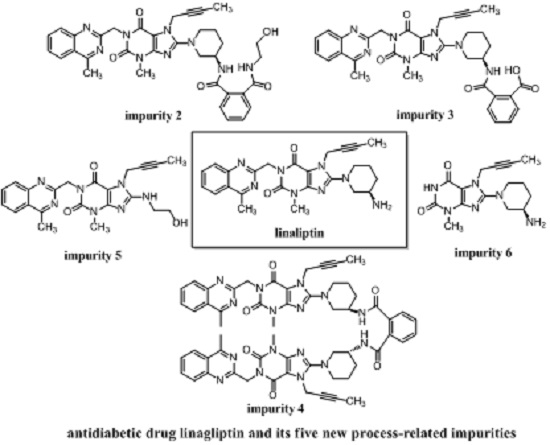
General Information
The compound
11 (chemical purity 98.36%),
13 (chemical purity 97.92%; chiral purity 99.97%) and
1 (chemical purity 98.35%; chiral purity 99.97%) were prepared according to the literature procedure [
4]. Other materials, solvents and reagents were of commercial origin and used without additional operations.
The 1H- and 13C-NMR spectra were recorded on a Bruker Arance III 400 MHz spectrometer. The solvents used were DMSO-d6 or CDCl3. The 1H-NMR chemical shift values were reported as δ ppm relative to tetramethylsilane (TMS) and the 13C-NMR chemical shift values were reported on δ ppm relative to DMSO-d6 or CDCl3. DEPT spectra revealed the presence of methyl and methine groups as positive peaks and methylene as negative peaks. The IR spectra were recorded in the solid state as KBr dispersion using a NICOLET 670 FT-IR spectrophotometer. Mass spectra and high resolution mass spectrum were recorded on Agilent 6120B series single quadrupole LC-MS and Q-Tof micro YA019 instrument. Melting points were measured on a WRS-1B apparatus. The specific rotation was calculated from an optical rotation measurement performed on the Autopol IV, serial number 80799 (Rudolph, Hackettstown, NJ, USA) at the wavelength of 589 nm (D line of a sodium lamp), at 20 °C.
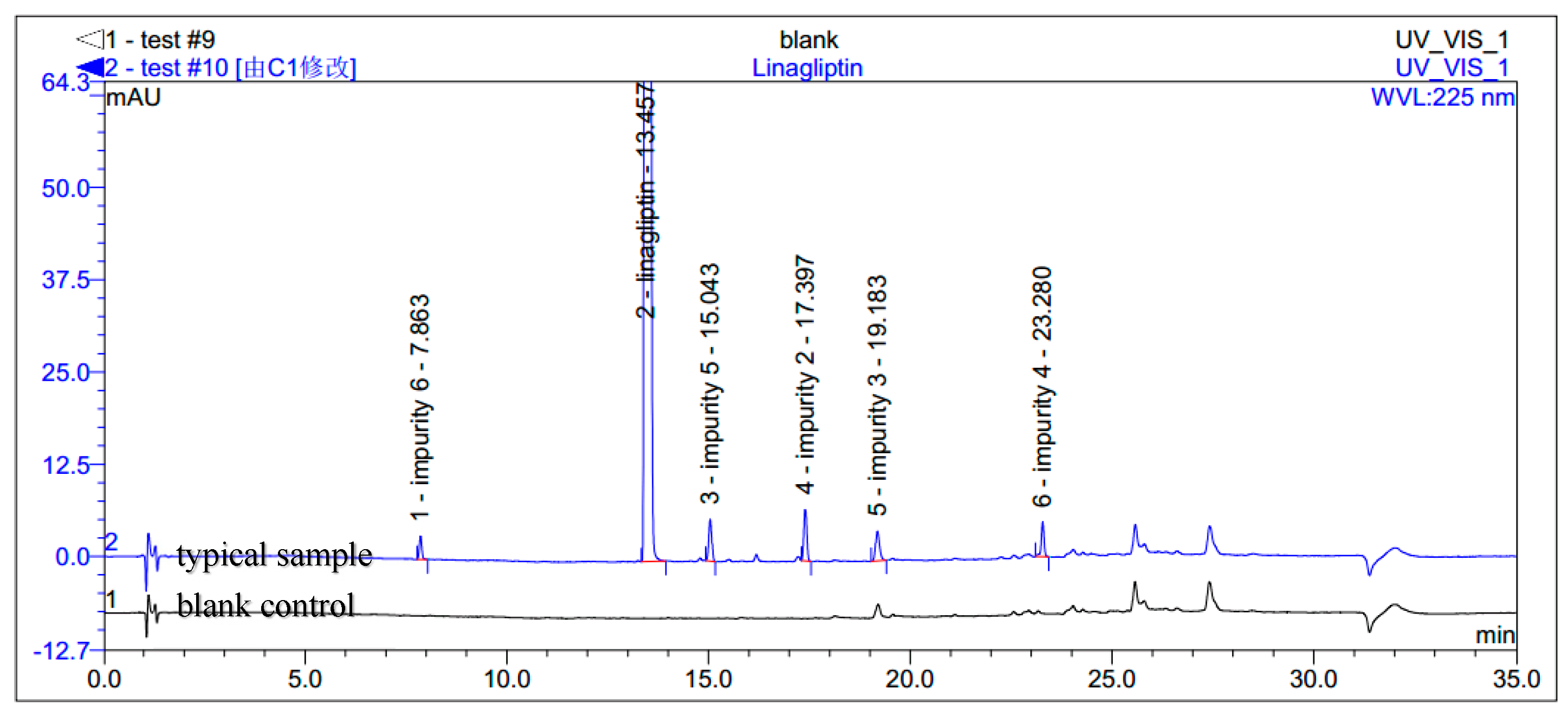
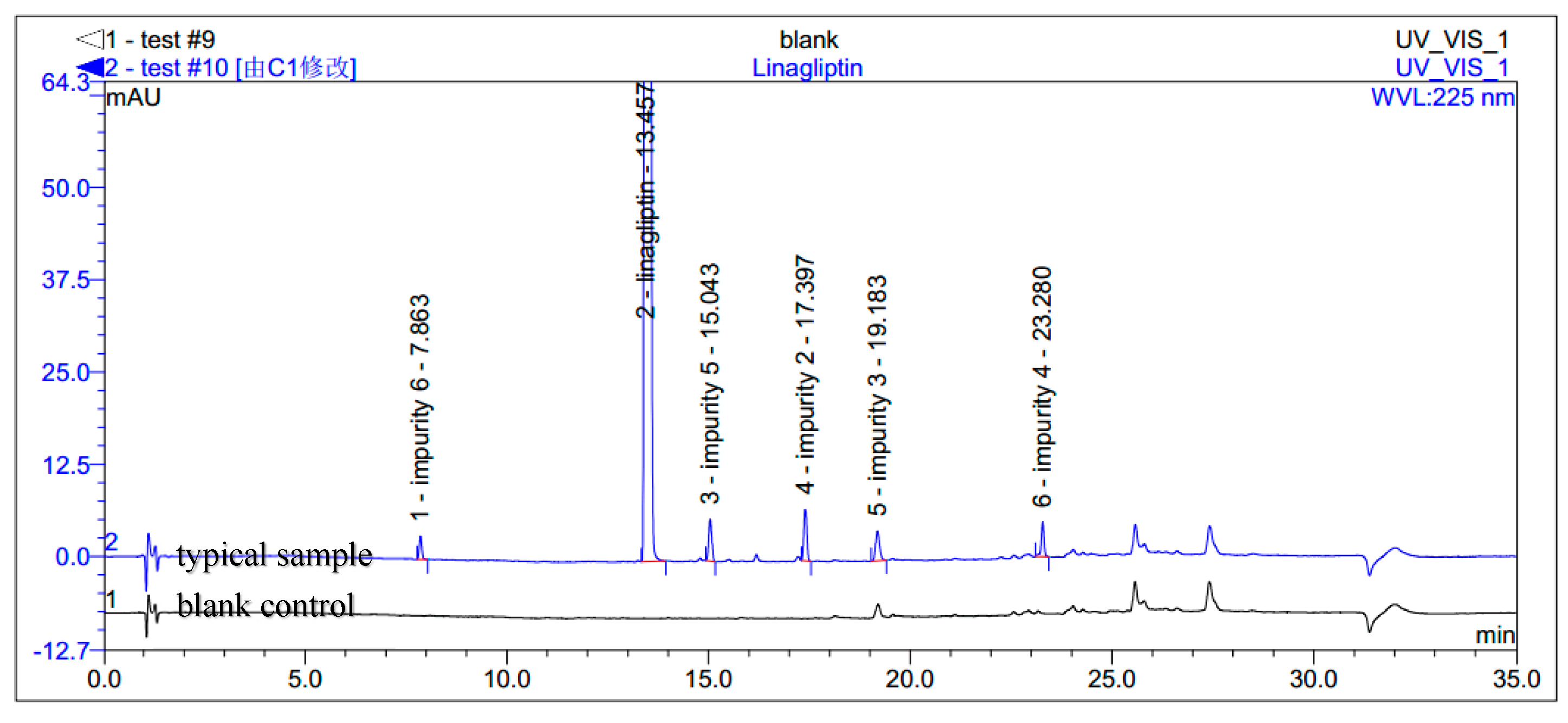
The HPLC analyses were recorded on a Dionex UItiMate 3000 HPLC instrument using Agilent Eclipse XDB C18 column (150 mm × 4.6 mm, 5 µm) in a thermostated column heater at 55 °C. The mobile phases consisting of A (0.1% methanoic acid, pH 2.5) and B (acetonitrile) were used with the gradient mode at the flow rate of 1.5 mL/min. The UV detection at 225 nm was used. Initial gradient starts with 5% of B and at 18 min it was set to 40%. The ratio had been set to 70% at 30 min and at 30.1 min it was 5%, which continued up to 35 min. The samples were diluted in acetonitrile with a concentration of 0.5 mg/mL. The injection volume was 5 µL. Limit of Detection (LOD) was 0.10 μg/mL or 0.02% for impurities 2, 3, 4, 5 and 0.15 μg/mL or 0.03% for impurity 6. Limit of Quantity (LOQ) was 0.25 μg/mL or 0.05% for impurities 2, 3, 4, 5 and 0.33 μg/mL or 0.07% for impurity 6.
This HPLC method further subjected for LC-MS. Samples were run in Electro-Spray Ionization positive mode (ESI+) and 12 L/min nebulizer gas flow rate. The fragmentor voltage was 70 V and capillary voltage was maintained at 3.0 kV. The drying gas temperature was at 350 °C.
The detailed spectral data (IR,
1H-NMR,
13C-NMR, MS, HRMS) of the compounds
2–
6 as well as the HPLC chromatogram of the compounds
1–
6 are provided in the
Supplementary Materials.
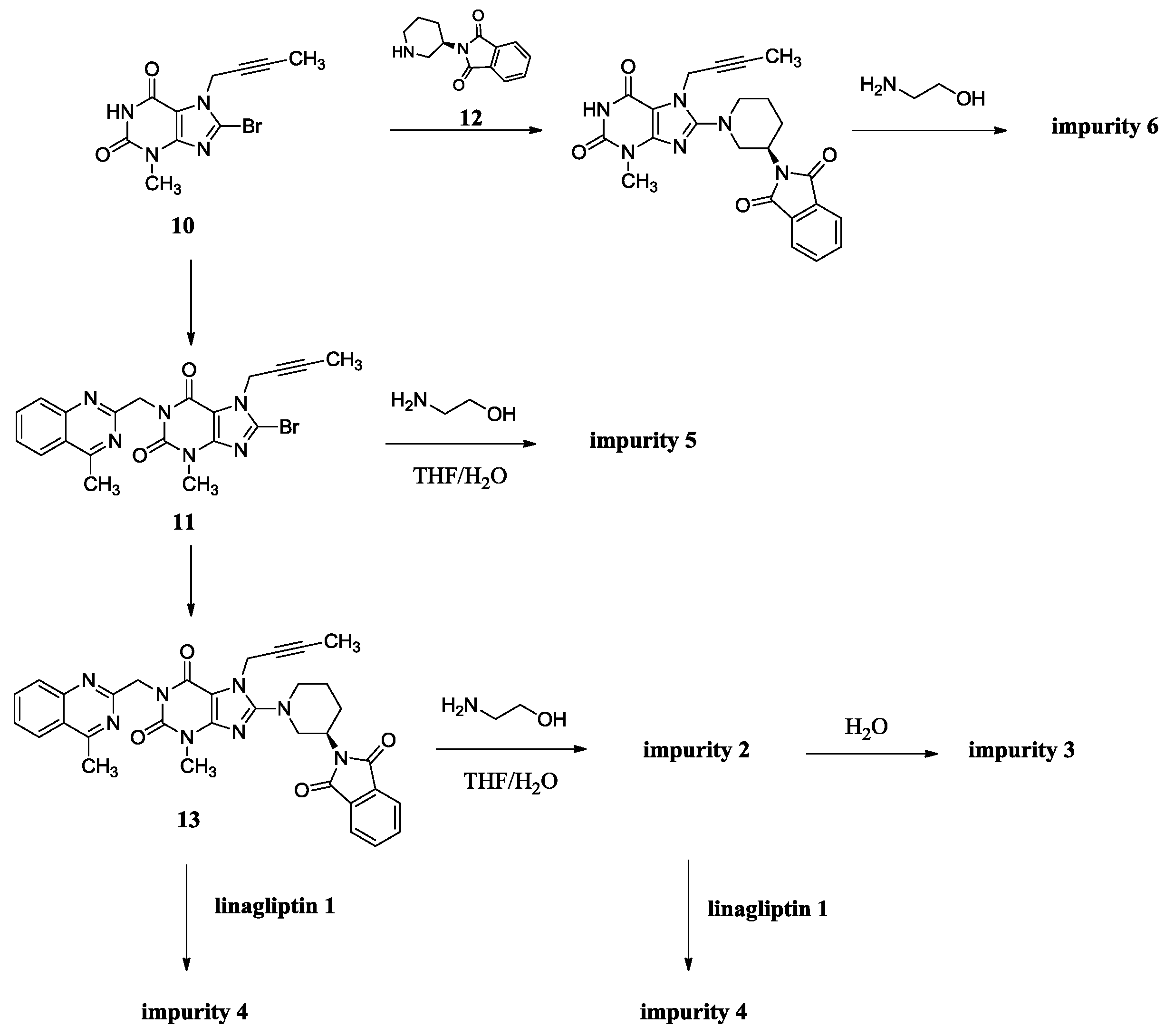
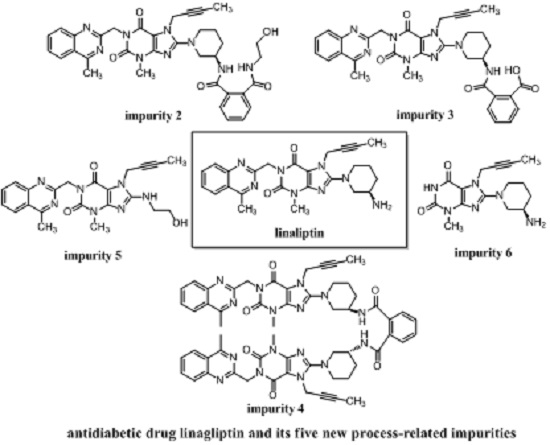
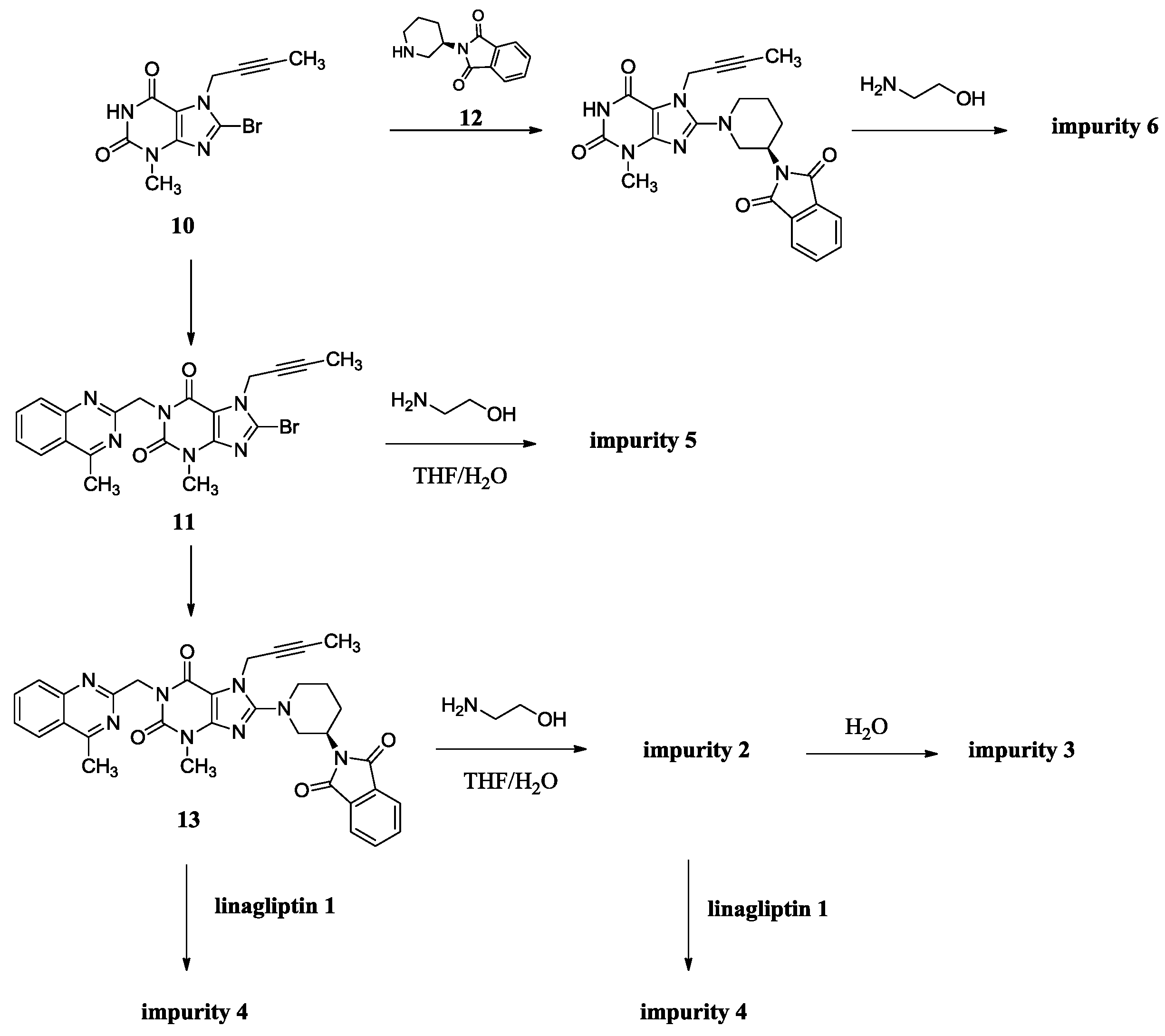
(R)-N1-(1-(7-(But-2-yn-1-yl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)piperidin-3-yl)-N2-(2-hydroxyethyl)phthalamide (2). To a stirred solution of compound 13 (5.0 g, 0.008 mol) in CH2Cl2 (50.0 mL) was added ethanolamine (1.5 g, 0.024 mol) and maintained the reaction mass at room temperature for 24 h. Then added 15 mL H2O stirring for 30 min to dilute ethanolamine. The isolated solid was collected by filtration and washed with 10 mL H2O and 20 mL CH2Cl2. Then dried to yield 2 as a light yellow solid (1.5 g, 27.3%), HPLC purity 97.52%; m.p. 189–192 °C; −32.567 (c = 1 g/100 mL, DMSO); IR (KBr) νmax 3473, 3263 (amide N-Hν, O-Hν); 3062 (aromatic C-Hν); 2937, 2850 (C-Hν); 2230 (C≡Cν); 1698, 1662 (C=Oν); 1522 (aromatic C=Cν, aromatic C=Nν); 1440, 1400 (C-Hδ); 1228 (C-Oν); 763 (aromatic C-Hγ) cm−1; 1H-NMR (400 MHz, CDCl3) δ 8.35 (d, J = 6.6 Hz, 1H), 8.04 (d, J = 8.2 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.82–7.76 (m, 1H), 7.64–7.52 (m, 3H), 7.47 (m, 2H), 6.95 (t, J = 5.6 Hz, 1H), 5.56 (s, 2H), 4.89 (q, J = 2.0 Hz, 2H), 4.31 (s, 1H), 3.78 (m, 2H), 3.70 (m, 2H), 3.60 (m, 2H), 3.52 (m, 2H), 3.40 (m, 1H), 3.23 (s, 3H), 2.91 (s, 3H), 2.17–2.10 (m, 1H), 1.92 (m, 2H), 1.75 (not resolved, 4H); 13C-NMR (100 MHz, DMSO-d6) δ 169.29, 168.75, 168.46, 161.44, 156.37, 153.80, 151.41, 149.51, 148.03, 136.58, 136.51, 134.51, 129.80, 129.67, 128.33, 128.26, 128.02, 127.57, 126.13, 122.94, 103.90, 81.74, 74.21, 60.22, 53.84, 50.47, 46.13, 46.08, 42.55, 35.90, 29.90, 29.59, 23.53, 22.01, 3.55; HRMS (ESI) m/z 664.3019 (calcd for C35H38N9O5, 664.2996 [M + H]+).
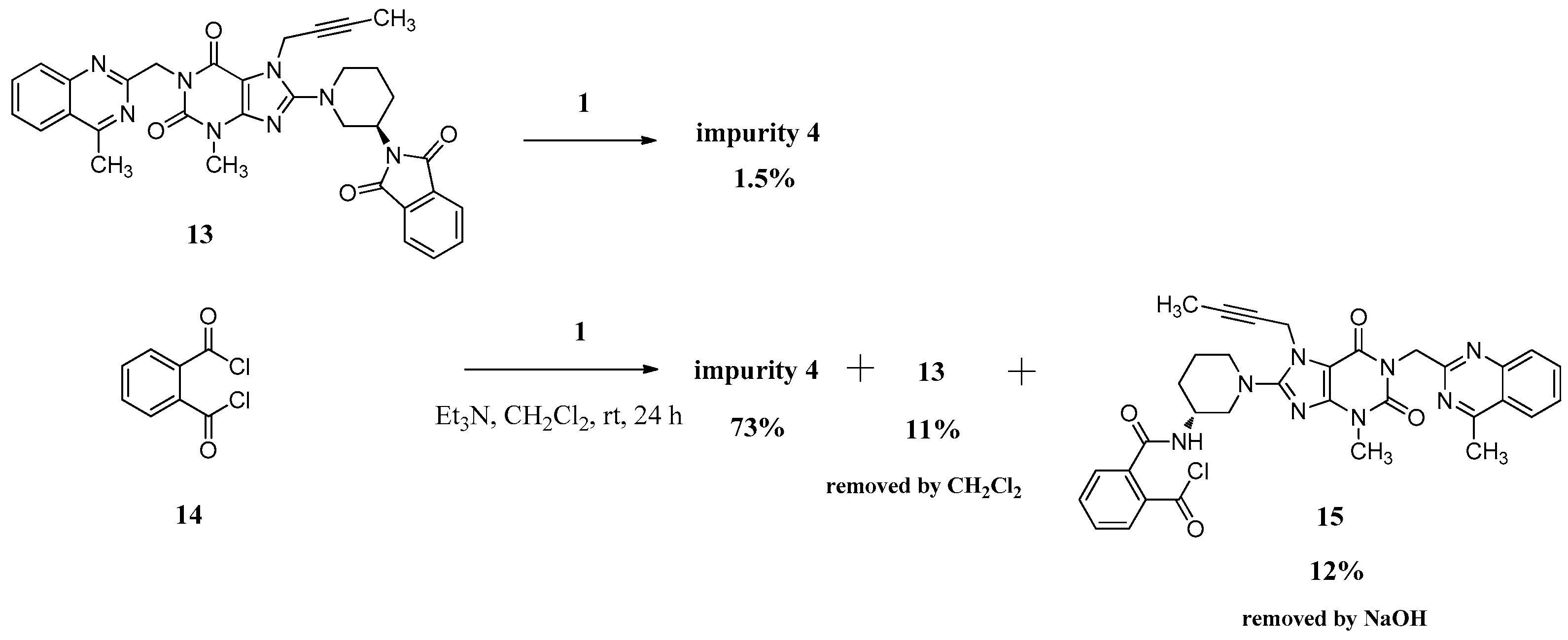
(R)-2-((1-(7-(But-2-yn-1-yl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)piperidin-3-yl)carbamoyl)benzoic acid (3). To a stirred solution of compound 13 (5.0 g, 0.008 mol) in CH2Cl2 (50.0 mL) was added aqueous sodium hydroxide solution (1.0 g of NaOH in 50.0 mL of H2O) and tetra-n-butylammonium bromide (0.27 g, 0.0008 mol). Then maintained the reaction mass at rt for 72 h. Then water phase was collected and washed with CH2Cl2 (25.0 mL) for three times. To the stirred water phase was added 1 mol/L aq. HCl (25.0 mL) and maintained at rt for 30 min. The isolated solid was collected by filtration and washed with water (20.0 mL) and CH2Cl2 (20.0 mL). Then dried to yield 3 as a light yellow solid (3.2 g, 62.2%), HPLC purity 97.34%; m.p. 144–148 °C;
−49.700 (c = 1 g/100 mL, DMSO); IR (KBr) νmax: 3527 (amide N-Hν); 3255 (acid O-Hν); 3068 (aromatic C-Hν); 2944, 2856 (C-Hν); 2224 (C≡Cν); 1700, 1654 (C=Oν); 1573, 1519 (aromatic C=Cν, aromatic C=Nν); 1440, 1400 (C-Hδ); 763 (aromatic C-Hγ) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 12.86(s, 1H), 8.35 (d, J = 7.8 Hz, 1H), 8.25 (d, J = 8.2 Hz, 1H), 7.96–7.89 (m, 1H), 7.85–7.78 (m, 2H), 7.68 (t, J = 7.6 Hz, 1H), 7.59 (td, J = 7.5, 1.1 Hz, 1H), 7.51 (td, J = 7.6, 1.1 Hz, 1H), 7.41 (d, J = 7.4 Hz, 1H), 5.34 (s, 2H), 4.92 (q, J = 2.1 Hz, 2H), 4.10–4.01 (m, 1H), 3.82 (m, 1H), 3.63 (m, 1H), 3.40 (s, not resolved, CH3 and water), 3.14–2.98 (m, 2H), 2.89 (s, 3H), 2.02–1.86 (m, 2H), 1.76 (not resolved, 4H), 1.59 (m, 1H); 13C-NMR (100 MHz, DMSO-d6) δ 169.44, 169.28, 168.79, 161.43, 156.46, 153.79, 151.40, 149.50, 148.06, 137.82, 134.51, 133.49, 130.68, 129.64, 129.51, 128.35, 128.32, 127.57, 126.12, 122.93, 103.88, 81.70, 74.26, 53.98, 50.43, 46.06, 46.01, 35.92, 29.91, 29.56, 23.56, 22.01, 3.56; HRMS (ESI) m/z 621.2599 (calcd for C33H33N8O5, 621.2574 [M + H]+).
N,N-bis((R)-1-(7-(But-2-yn-1-yl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)piperidin-3-yl)phthalamide (4). Phthaloyl dichloride (1.0 g, 0.005 mol) was added slowly to a stirred solution of 1 (4.8 g, 0.010 mol) and triethylamine (5.0 g, 0.05 mol) in CH2Cl2 (50.0 mL) at rt and refluxed for 24 h. Then the mixture was cooled to rt and 1 mol/L aq. NaOH (30 mL) was added, and maintained for 2 h. The isolated solid was collected by filtration and washed with water (20.0 mL), CH2Cl2 (20.0 mL) and methanol (20.0 mL). Then dried to yield 4 as a light yellow solid (3.5 g, 66.1%), HPLC purity 99.15%; = −18.133 (c = 1 g/100 mL, CHCl3); IR (KBr) νmax 3243 (amide N-Hν); 3068 (aromatic C-Hν); 2945, 2858 (C-Hν); 2224 (C≡Cν); 1700, 1662 (C=Oν); 1634, 1563, 1519 (aromatic C=Cν, aromatic C=Nν); 1440, 1400 (C-Hδ); 763 (aromatic C-Hγ) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 7.6 Hz, 1H), 8.22 (d, J = 8.1 Hz, 1H), 7.92–7.85 (m, 1H), 7.79 (d, J = 8.3 Hz, 1H), 7.67–7.62 (m, 1H), 7.50 (m, 2H), 5.30 (s, 2H), 4.90 (s, 2H), 4.10–4.00 (m, 1H), 3.81 (d, J = 9.0 Hz, 1H), 3.65 (m, 1H), 3.36 (s, 3H), 3.15–2.99 (m, 2H), 2.88 (s, 3H), 1.98–1.84 (m, 2H), 1.74 (not resolved, 4H), 1.65–1.55 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ 168.68, 168.52, 161.01, 155.73, 154.36, 151.71, 149.89, 147.29, 135.04, 133.23, 130.18, 128.80, 128.38, 126.71, 124.83, 123.10, 104.60, 81.53, 73.01, 53.57, 51.11, 46.32, 45.83, 35.63, 29.54, 28.76, 21.74, 21.67, 3.60; HRMS (ESI) m/z 1075.4800 (calcd for C58H59N16O6, 1075.4803 [M + H]+).
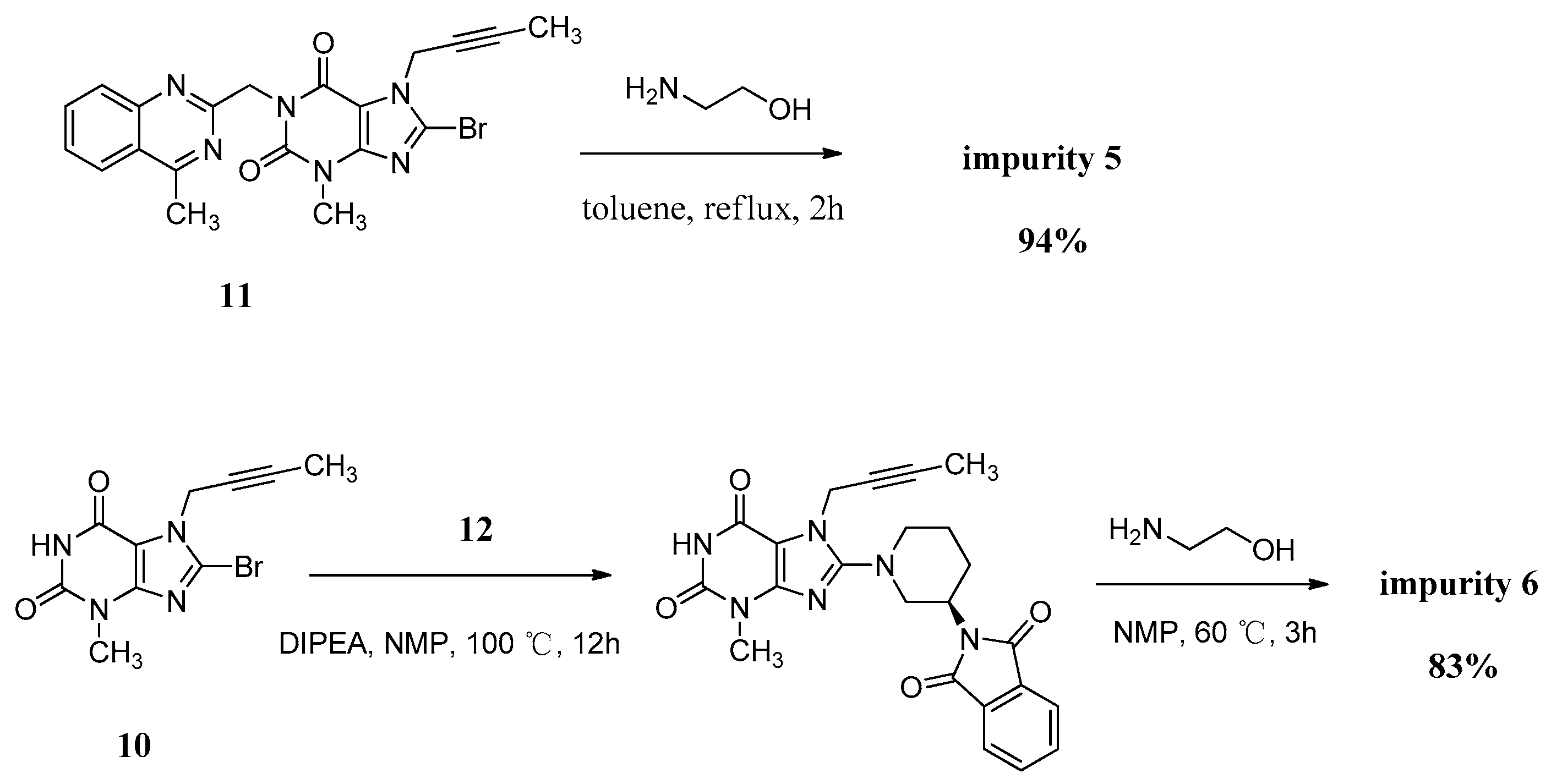
7-(But-2-yn-1-yl)-8-((2-hydroxyethyl)amino)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-1H-purine-2,6(3H,7H)-dione (5). Ethanolamine (8.0 g, 0.13 mol) was added to a stirred solution of 11 (2.0 g, 0.0044 mol) in toluene (32.0 mL) at reflux temperature and maintained for 2 h. Then the mixture was cooled to rt and 20 mL H2O was added, and stirred for 30 min. The isolated solid was collected by filtration and washed with toluene (10.0 mL). Then dried to yield 5 as a white solid (1.8 g, 94.2%), HPLC purity 99.61%; m.p. 238–239 °C; IR (KBr) νmax 3446, 3364 (N-Hν, O-Hν); 2936, 2867 (C-Hν); 1702, 1652 (C=Oν); 1619, 1581, 1540 (aromatic C=Cν, aromatic C=Nν); 1448, 1397 (C-Hδ); 1225 (C-Oν); 764 (aromatic C-Hγ) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 8.25 (d, J = 8.0 Hz, 1H), 7.95–7.88 (m, 1H), 7.82 (d, J = 8.2 Hz, 1H), 7.72–7.62 (m, 1H), 7.23 (t, J = 5.6 Hz, 1H), 5.31 (s, 2H), 4.89 (q, J = 2.2 Hz, 2H), 4.78 (t, J = 5.5 Hz, 1H), 3.61 (t, J = 6.0 Hz, 2H), 3.46 (t, J = 6.0 Hz, 2H), 3.39 (s, 3H), 2.89 (s, 3H), 1.77 (t, J = 2.1 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 169.19, 161.68, 154.38, 152.99, 151.50, 149.54, 149.52, 134.45, 128.31, 127.51, 126.11, 122.92, 101.31, 81.06, 74.27, 60.20, 45.89, 45.56, 33.13, 29.80, 21.99, 3.57; HRMS (ESI) m/z 434.1937 (calcd for C22H24N7O3, 434.1941 [M+H]+).
(R)-8-(3-Aminopiperidin-1-yl)-7-(but-2-yn-1-yl)-3-methyl-1H-purine-2,6(3H,7H)-dione (6). Diisopropylethylamine (9.8 g, 0.076 mol) was added to a stirred solution of 10 (5.0 g, 0.017 mol) and 12 (9.6 g, 0.025 mol) in NMP (50 mL) and maintained the reaction mass at 100 °C for 13 h. Then ethanolamine (10.2 g, 0.17 mol) was added at 65 °C and stirred for 4 h at 65 °C. The isolated solid was collected by filtration and washed with NMP (30 mL). Then dried to yield 6 as a off-white solid (4.4g, 82.7%), HPLC purity 99.36%; m.p. 299–302 °C; −3.467 (c = 1 g/100 mL, DMSO); IR (KBr) νmax 3115, 3074 (primary amine N-Hν); 3020 (imide N-Hν); 2947, 2793 (C-Hν); 2242 (C≡Cν); 1706 (C=Oν); 1660 (C=Oν, primary amine N-Hδ); 1610, 1519 (C=Cν, C=Nν); 1445, 1384 (C-Hδ) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1H), 8.27 (s, 2H), 4.92 (q, J = 2.4 Hz, 2H), 3.65 (m, 1H), 3.48–3.40 (m, 1H), 3.33 (incompletely resolved, 4H), 3.15 (m, 2H), 2.00 (m, 1H), 1.96–1.85 (m, 1H), 1.81 (t, J = 2.2 Hz, 3H), 1.67 (m, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 154.97, 153.99, 150.76, 148.37, 103.93, 81.20, 73.75, 51.43, 50.33, 46.09, 35.16, 28.49, 27.27, 21.85, 3.14; HRMS (ESI) m/z 317.1714 (calcd for C15H20N6O2, 317.1721 [M + H]+).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}