
X-ray Single Crystal Structure, DFT Calculations and Biological Activity of 2-(3-Methyl-5-(pyridin-2’-yl)-1H-pyrazol-1-yl) Ethanol

 ,
,  and
and
Abstract
:1. Introduction
2. Results
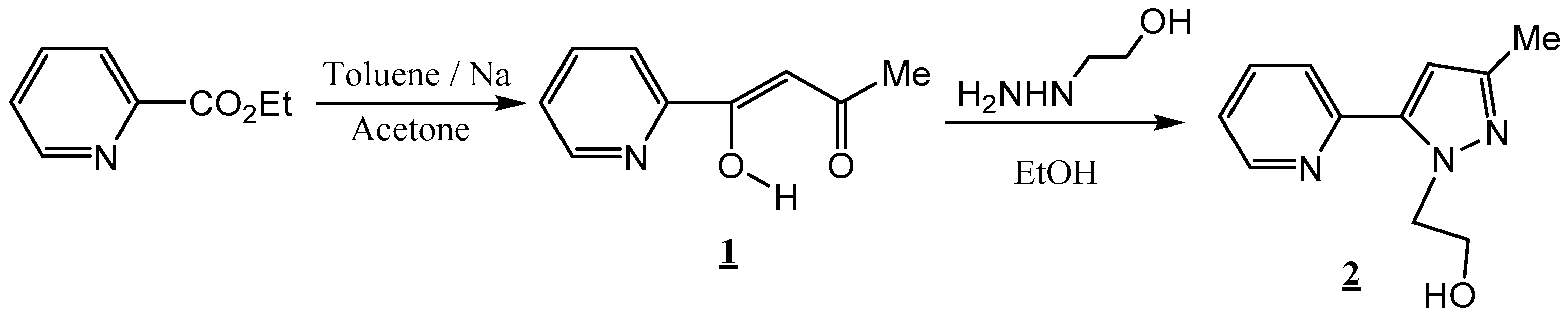
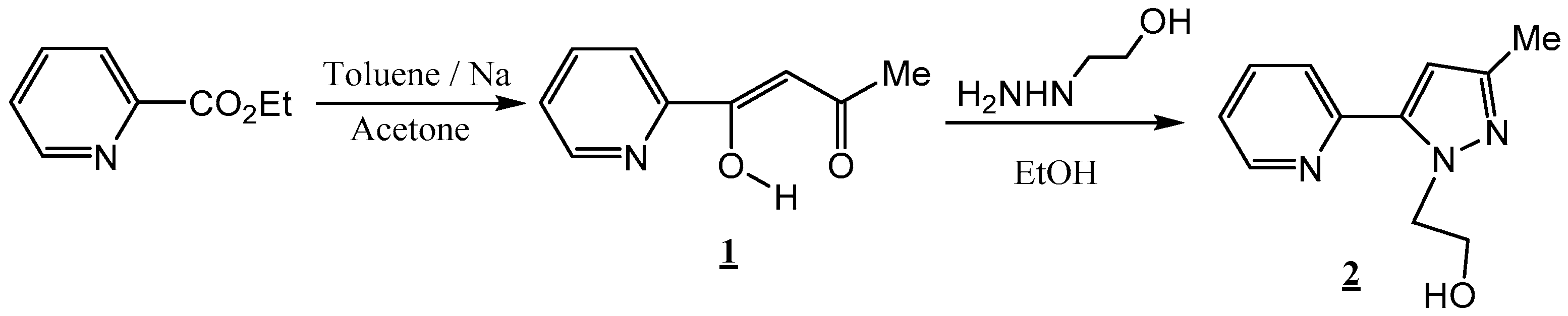
2.1. Chemistry
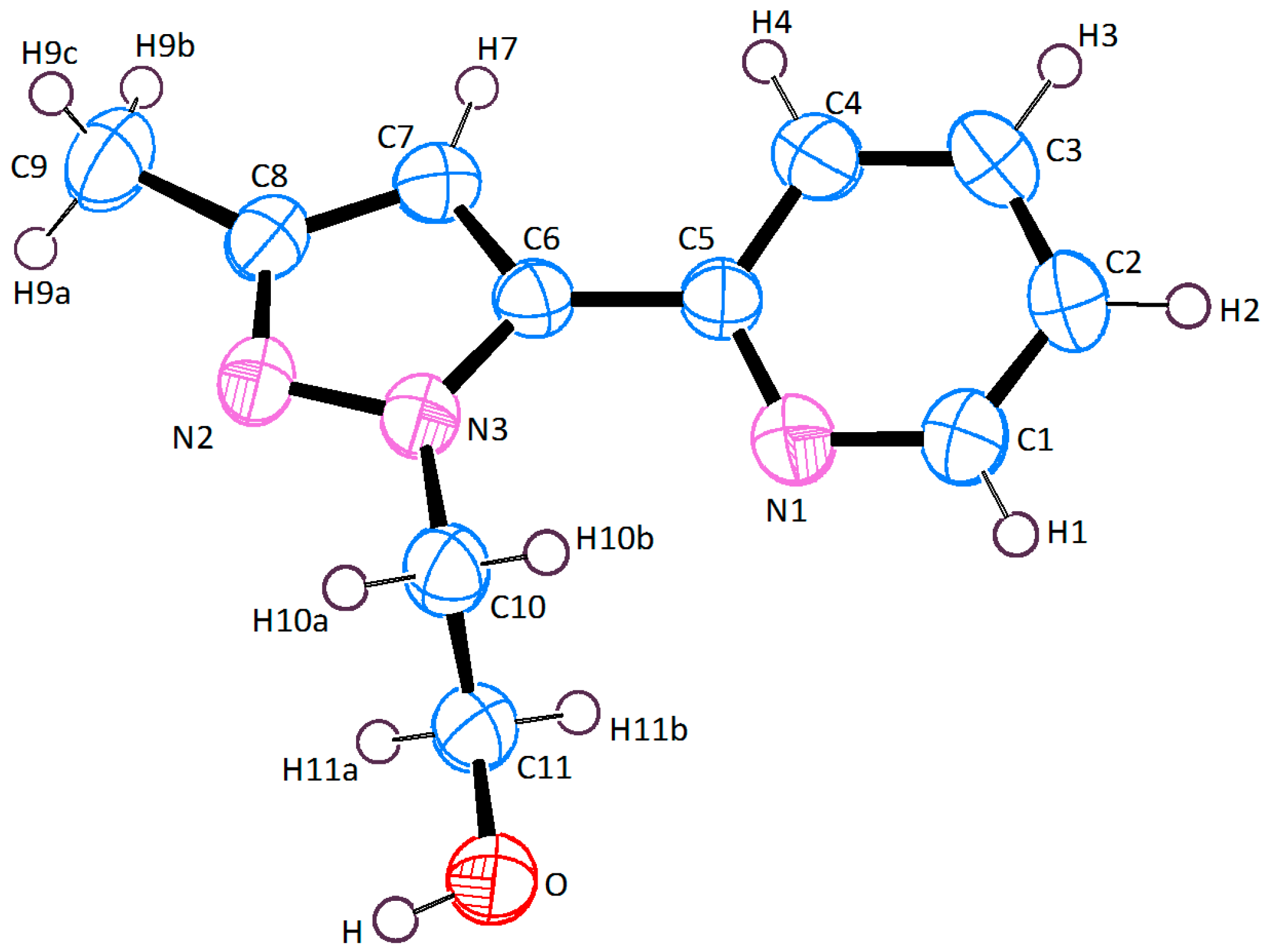
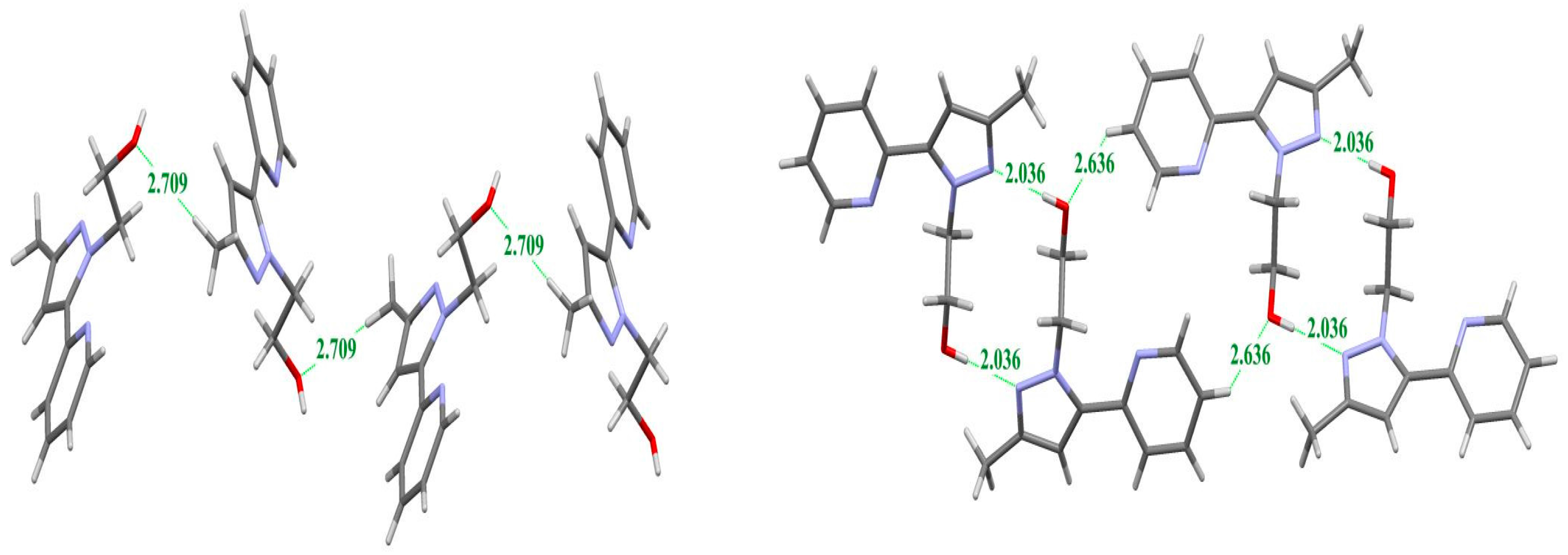
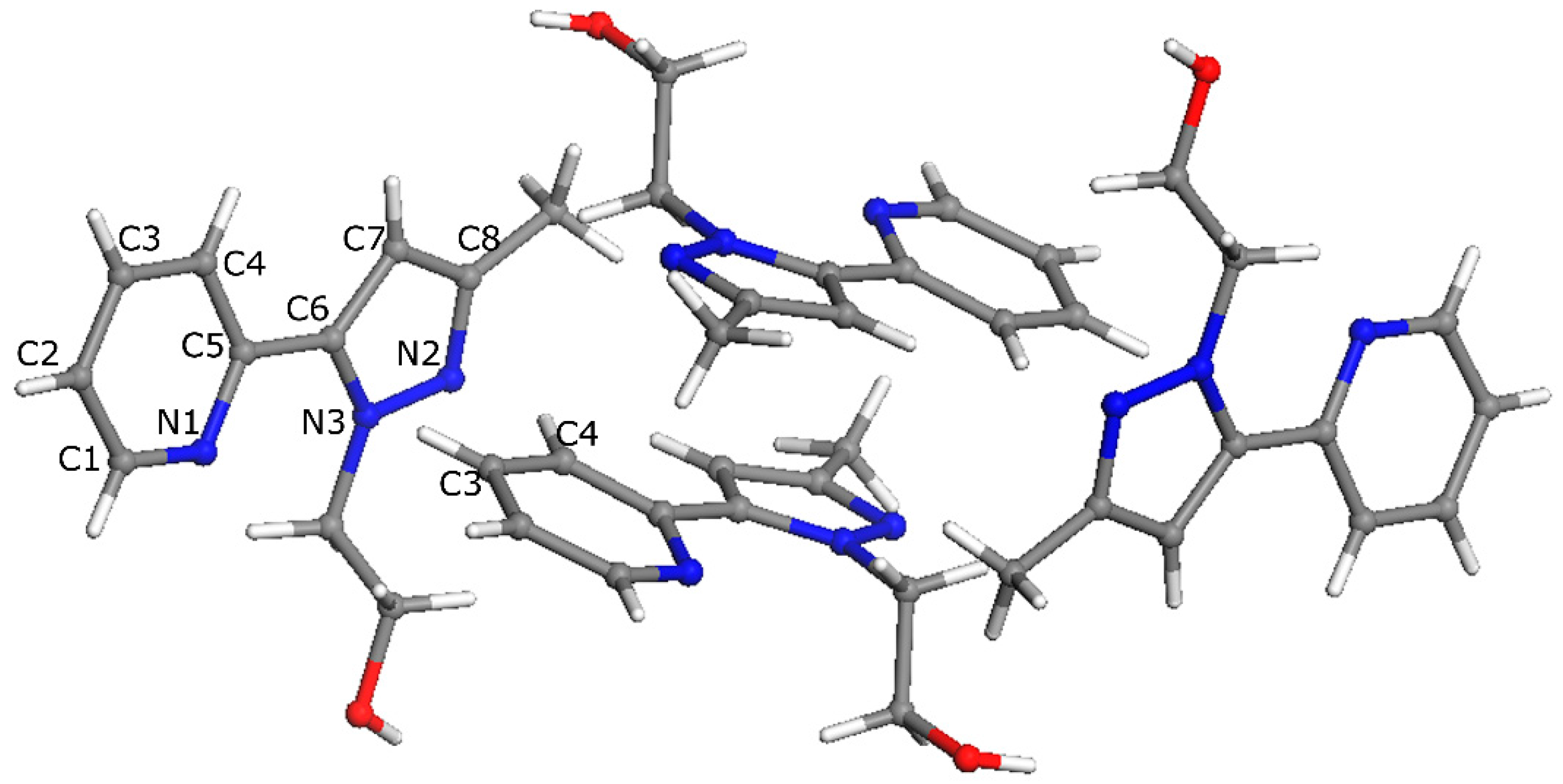
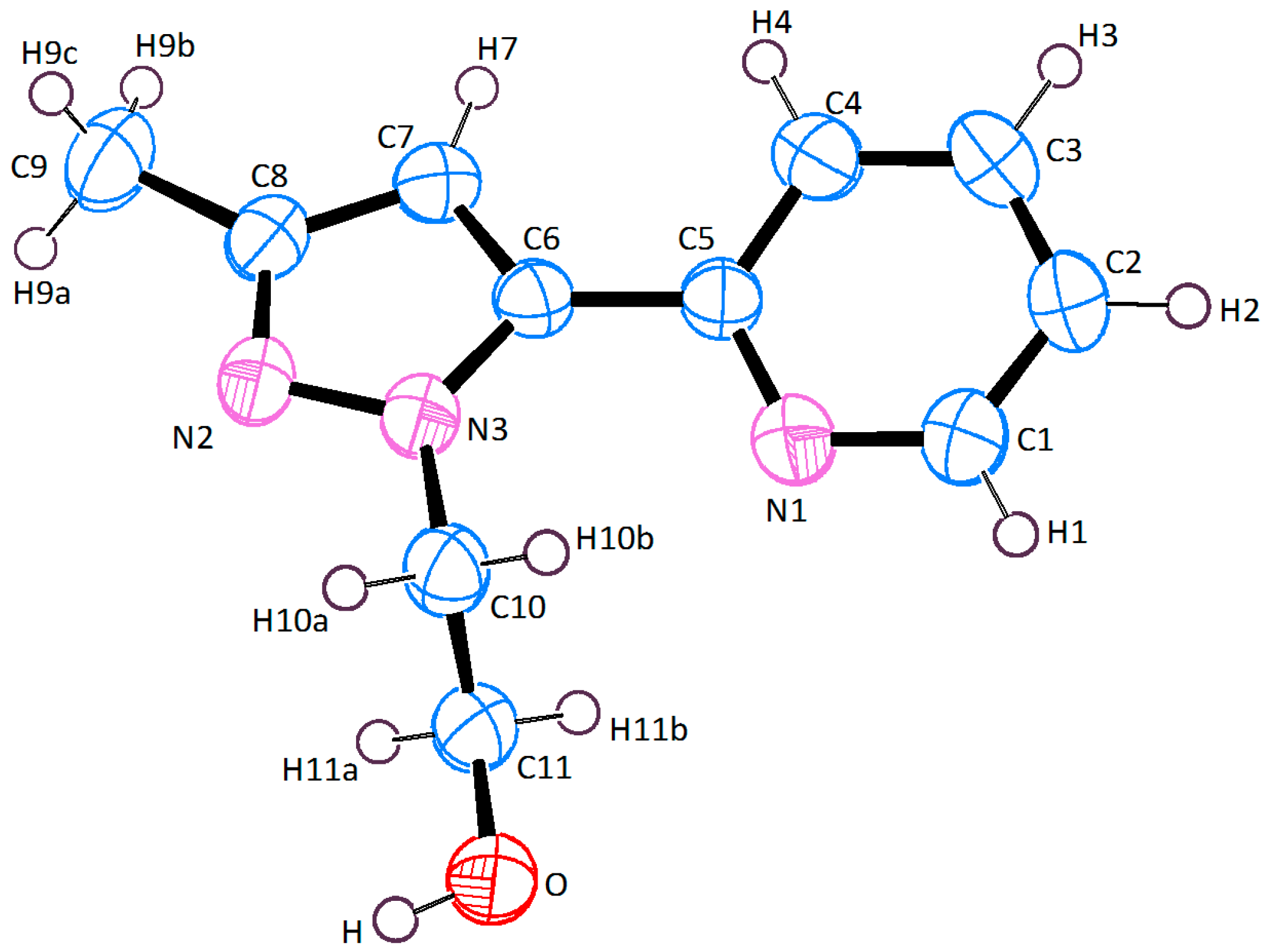
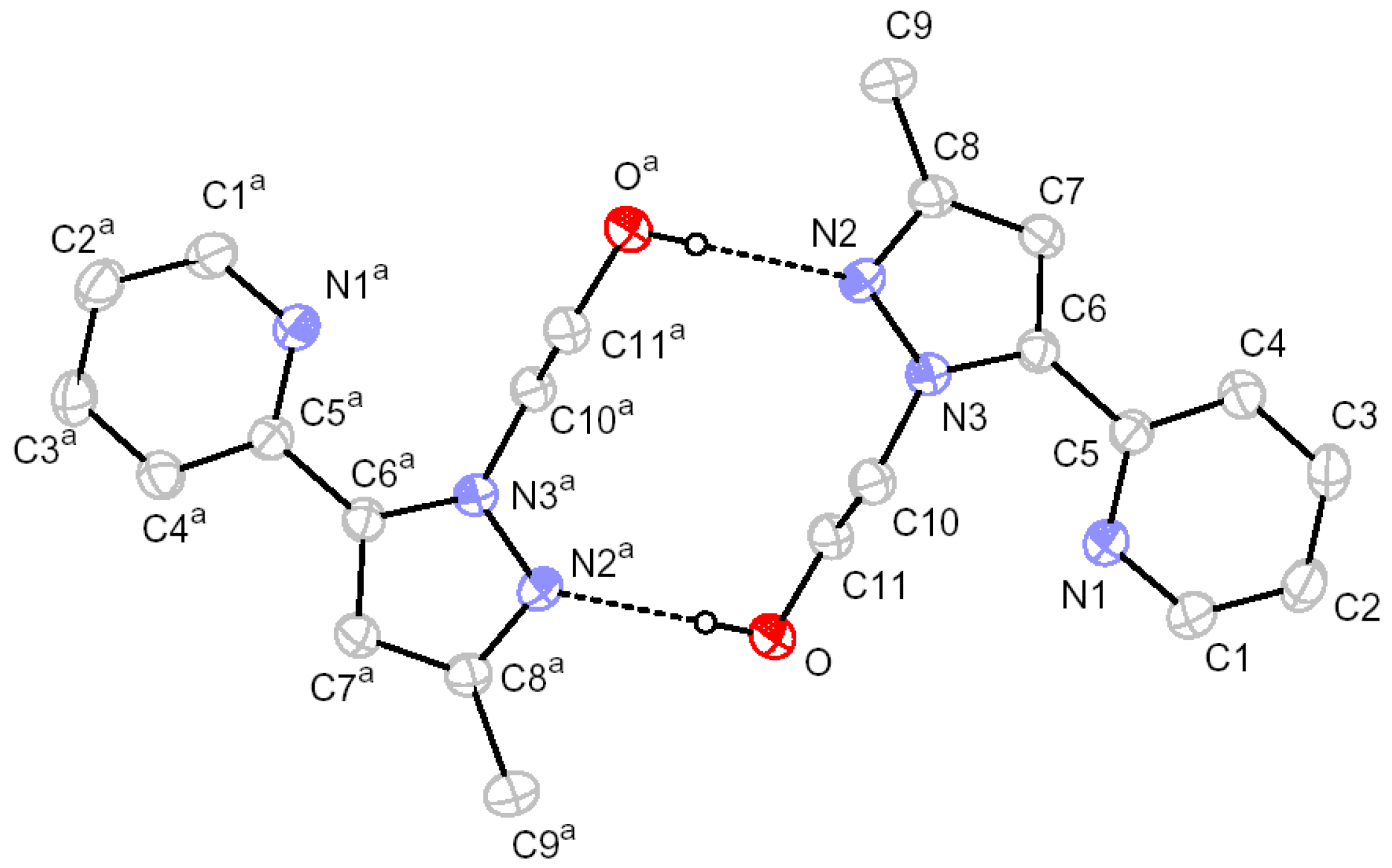
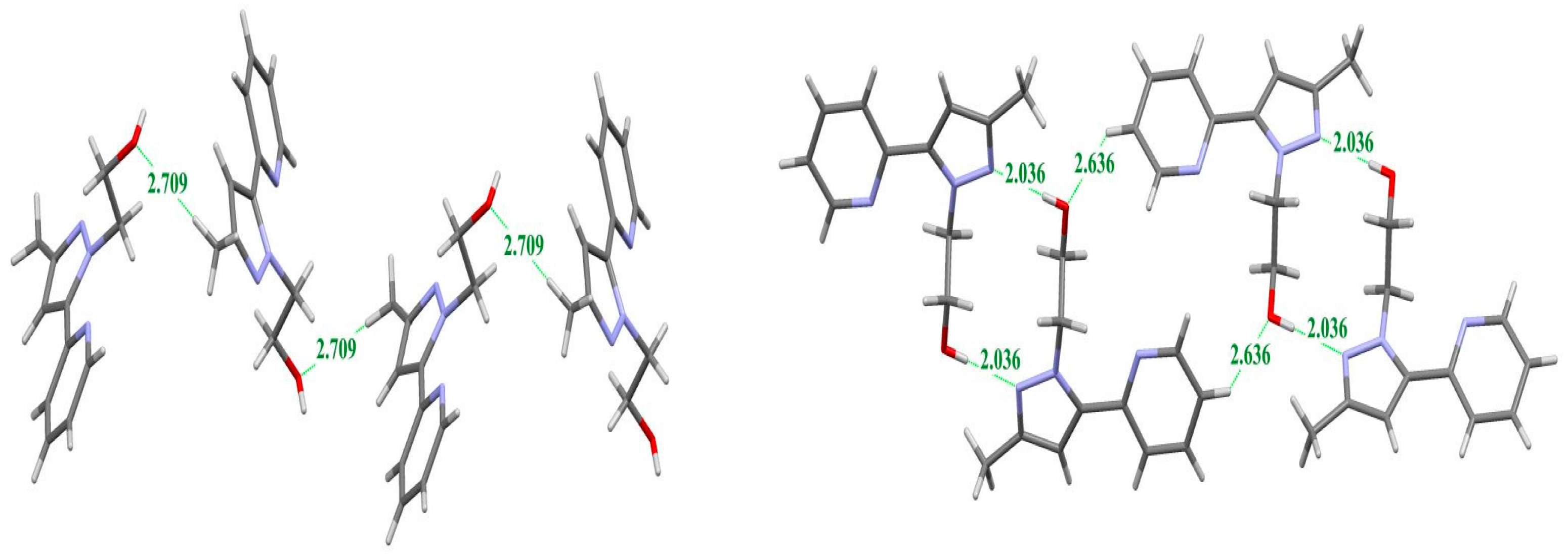
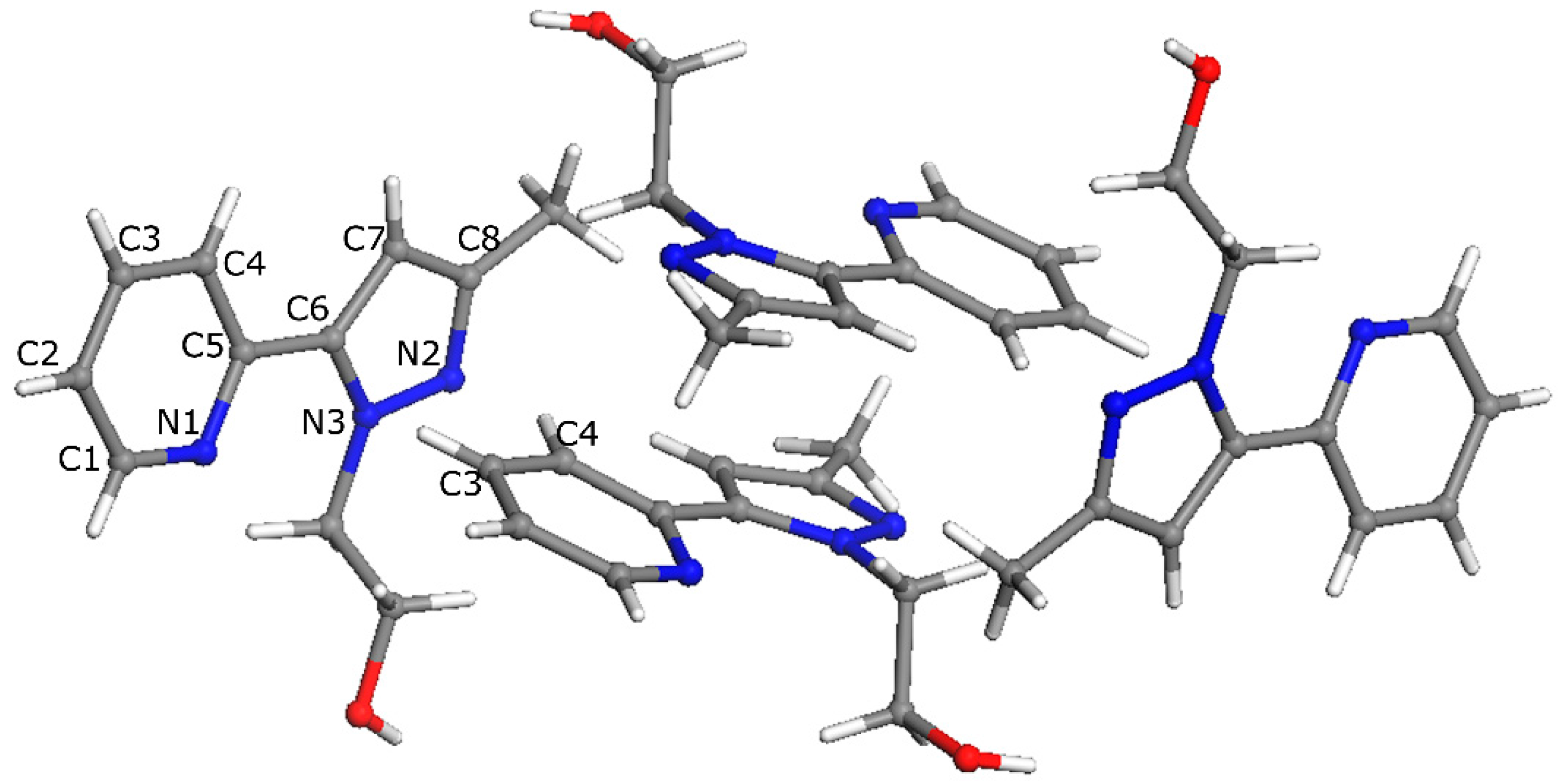
2.2. X-ray Crystal Structure Description
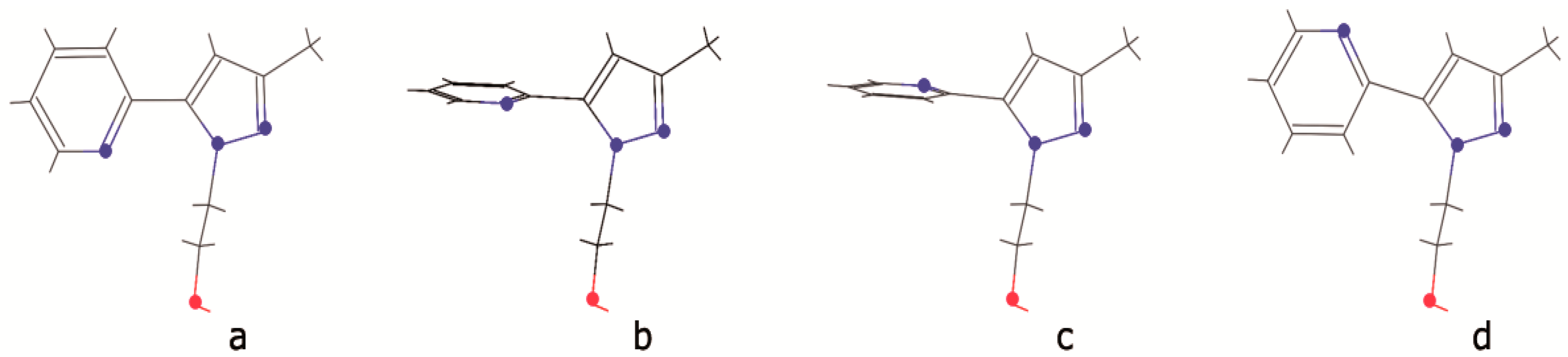
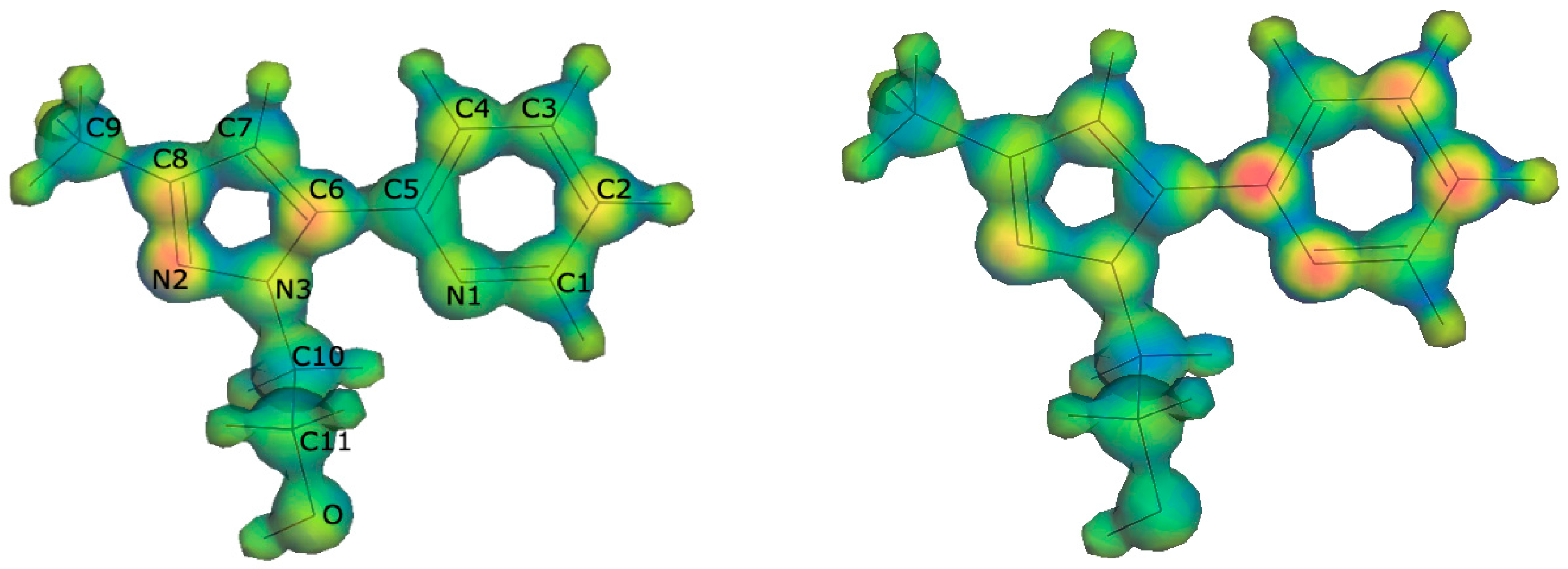
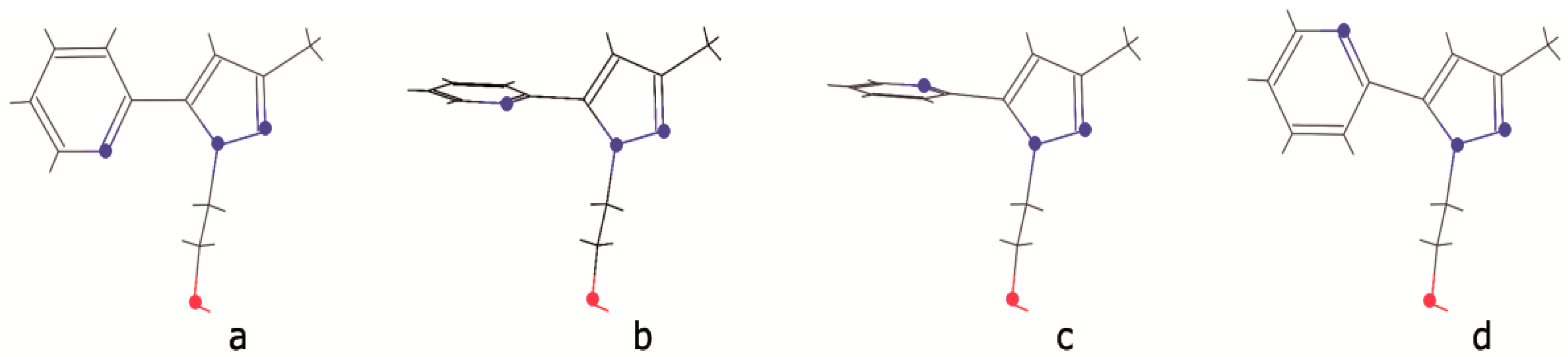
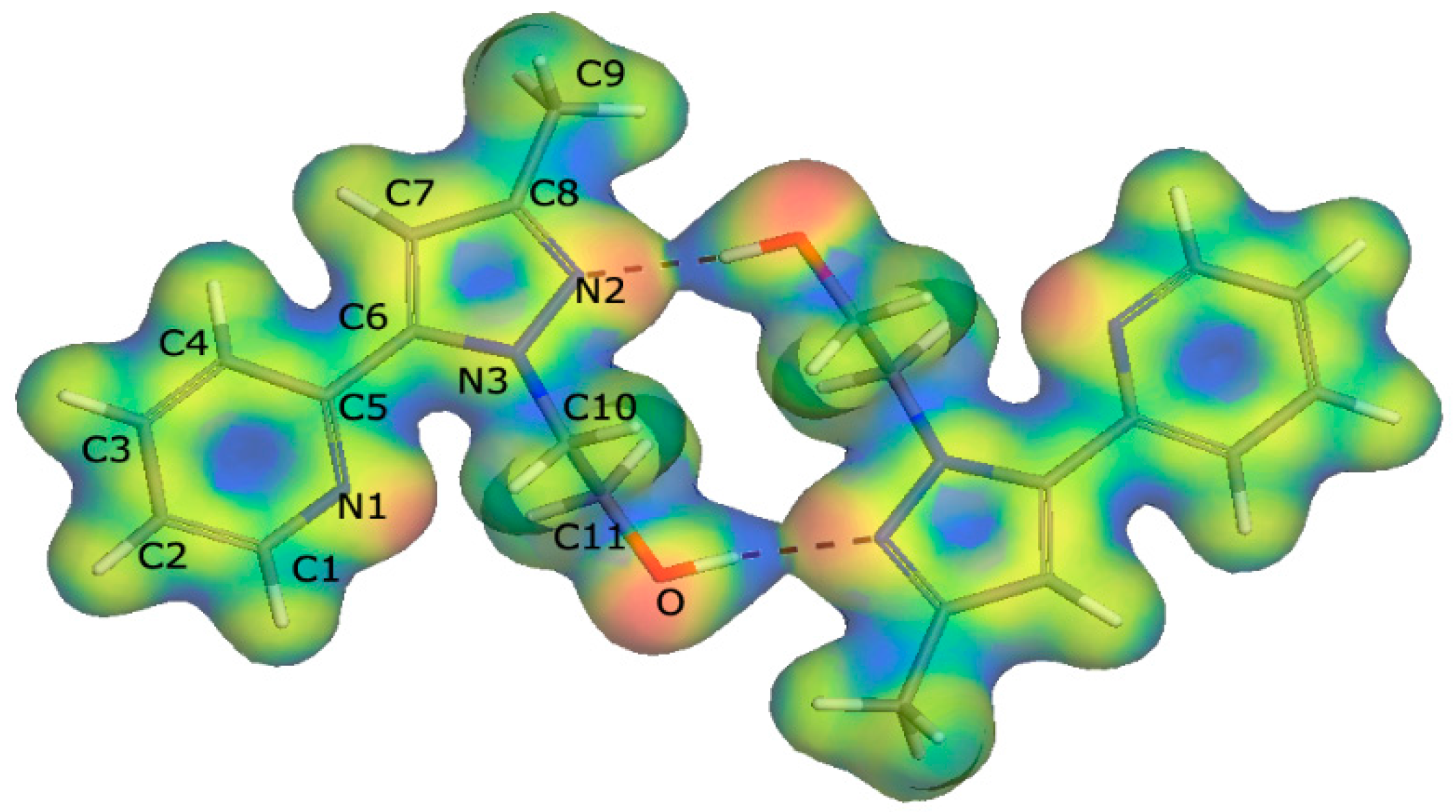
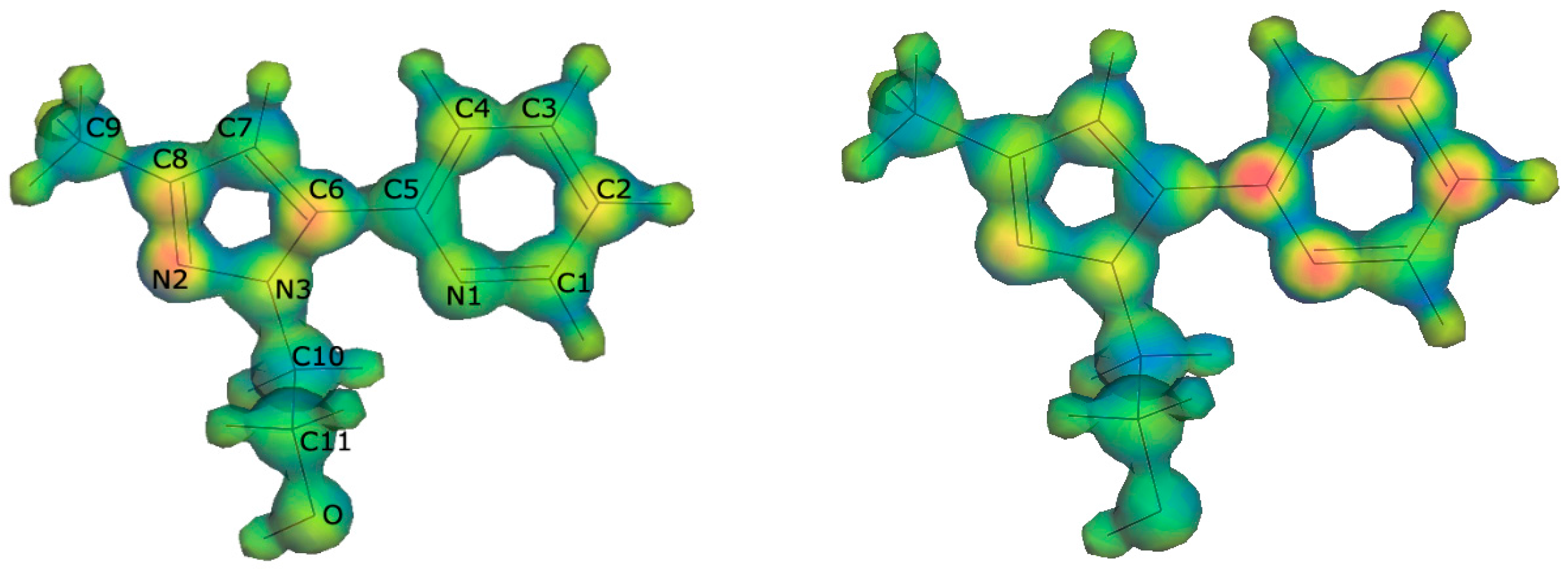
2.3. DFT Calculations
2.4. Biological Activity
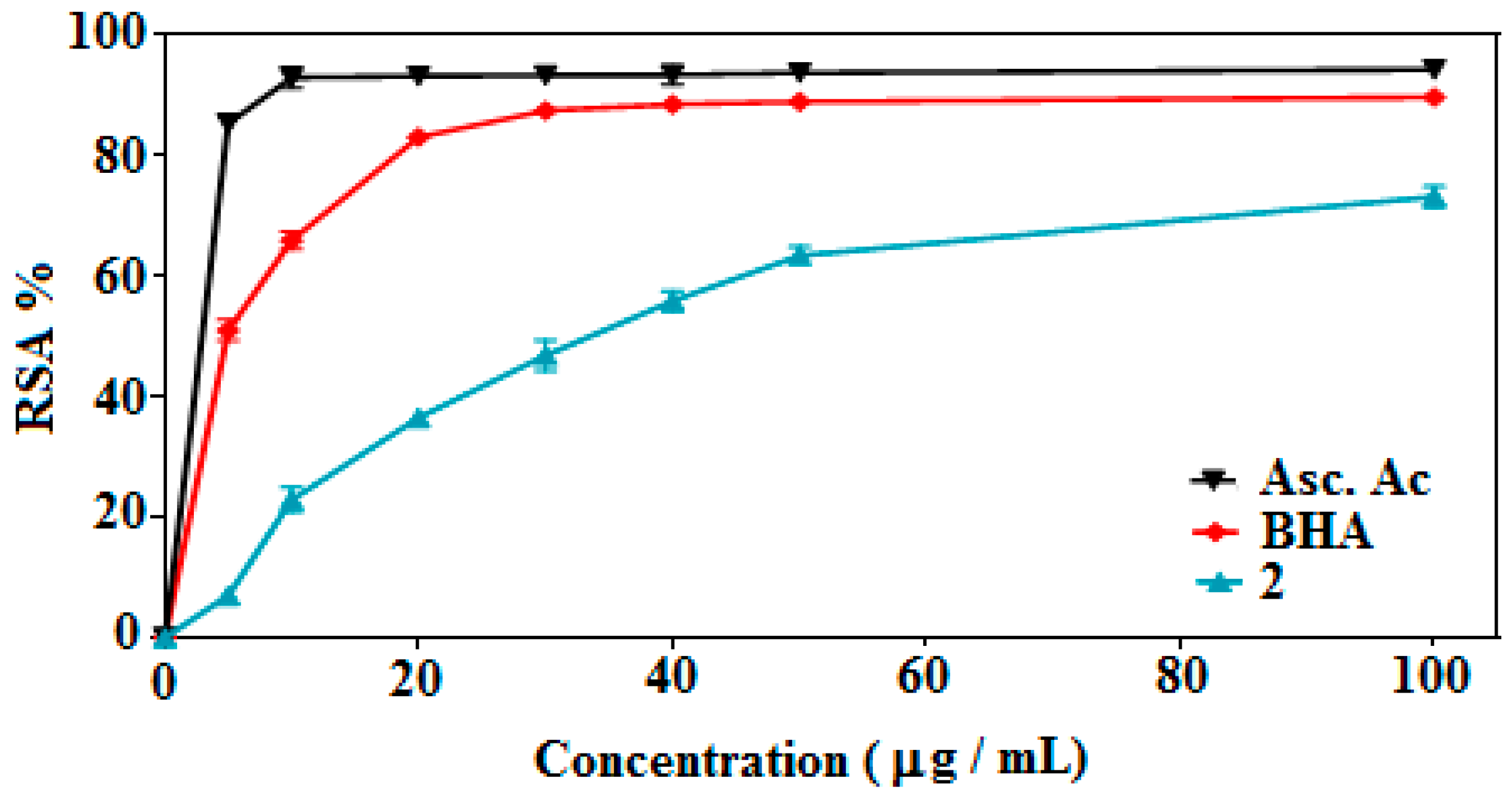
2.4.1. DPPH Radical Scavenging Activity
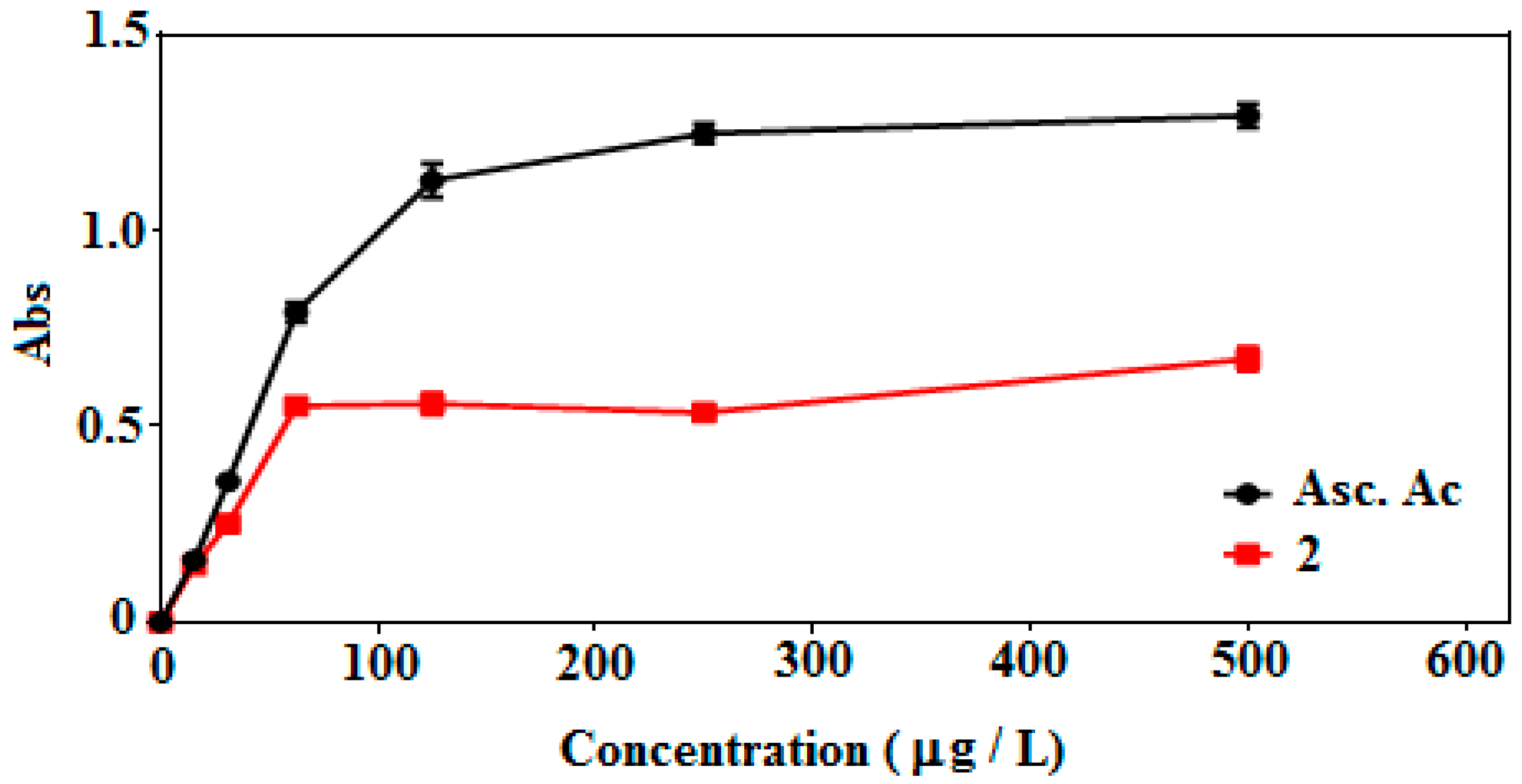
2.4.2. Ferric Reducing Antioxidant Power (FRAP) Assay
3. Experimental Section
3.1. General Information
3.2. Synthesis of 2-(3-Methyl-5-(pyridin-2-yl)-1H-pyrazol-1-yl)ethan-1-ol (2)
3.3. X-ray Crystallographic Analysis
3.4. DPPH Radical Scavenging Activity
3.5. Ferric Reducing Antioxidant Power (FRAP) Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dowling, C.; Murphy, V.J.; Parkin, G. Bis(pyrazolylethyl) Ether Ligation to Zinc and Cobalt: Meridional vs. Facial Coordination and the Suitability of Such Ligands in Providing a NNO Donor Set for Modeling Bioinorganic Aspects of Zinc Chemistry. Inorg. Chem. 1996, 35, 2415–2420. [Google Scholar] [PubMed]
- Haanstra, W.J.; Driessen, W.L.; Van Roon, M.; Stoffels, A.L.E.; Reedijk, J. Co-ordination compounds with the N2S-donor ligand 1,5-bis(3,5-dimethylpyrazol-1-yl)-3-thiapentane. J. Chem. Soc. Dalton Trans. 1992, 3, 481–486. [Google Scholar] [CrossRef]
- Driessen, W.L.; Wiesmeijer, W.G.R.; Schipper-Zablotskaja, M.; de Graaff, R.A.G.; Reedijk, J.; Reedijk, J. Transition metal coordination compounds of two pyrazole-substituted ammonia ligands. X-ray structure of [Biss(1-pyrazolylmethyl)aminecobalt(II)] bis(nitrate). Inorg. Chim. Acta 1989, 162, 233–238. [Google Scholar] [CrossRef]
- Pennings, Y.C.M.; Driessen, W.L.; Reedijk, J. Copper(I) coordination compounds with two bidentate chelating pyrazole ligands. X-ray crystal structures of DI-μ-chloro-bis[[N,N-bis(1-pyrazolylmethyl)aminoethane]copper(I)] and [bis(N,N-bis(1-pyrazolylmethyl)aminoethane]copper(I) triflate. Polyhedron 1988, 7, 2583–2589. [Google Scholar] [CrossRef]
- Veldhuis, J.B.; Driessen, W.L.; Reedijk, J. A pyrazole derivative of aminoethane as a tridentate chelating ligand towards transition metals. The X-ray structure of [N,N-bis(pyrazol-1-ylmethyl)aminoethane]dibromocopper(II). Chem. Soc. Dalton Trans. 1986, 3, 537–541. [Google Scholar] [CrossRef]
- Blonk, H.L.; Driessen, W.L.; Reedijk, J. Transition-metal co-ordination compounds of a novel aniline-based pyrazole derivative. X-Ray crystal structures of [N,N-bis(3,5-dimethylpyrazol-1-ylmethyl) aminobenzene]-dichlorocobalt(II) and -dibromocopper(II). Chem. Soc. Dalton Trans. 1985, 8, 1699–1705. [Google Scholar] [CrossRef]
- Mukherjee, R. Coordination chemistry with pyrazole-based chelating ligands: Molecular structural aspects. Coord. Chem. Rev. 2000, 203, 151–218. [Google Scholar] [CrossRef]
- Trofimenko, S. Recent advances in poly(pyrazolyl)borate (scorpionate) chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Trofimenko, S. The Coordination Chemistry of Pyrazole-Derived Ligands. Prog. Inorg. Chem. 1986, 34, 115–210. [Google Scholar]
- Bouabdallah, I.; M’barek, L.A.; Zyad, A.; Ramadan, A.; Zidane, I.; Melhaoui, A. Anticancer effect of three pyrazole derivatives. Nat. Prod. Res. 2006, 20, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Janus, S.L.; Magdif, A.Z.; Erik, B.P.; Claus, N.N. Synthesis of triazenopyrazole derivatives as potential inhibitors of HIV-1. Monatsh. Chem. 1999, 130, 1167–1174. [Google Scholar]
- Tewari, A.K.; Mishra, A. Synthesis and anti-inflammatory activities of N4,N5-disubstituted-3-methyl-1H-pyrazolo[3,4-c]pyridazines. Bioorg. Med. Chem. 2001, 9, 715–718. [Google Scholar] [CrossRef]
- Wustrow, D.J.; Capiris, T.; Rubin, R.; Knobelsdorf, J.A.; Akunne, H.; Davis, M.D.; MacKenzie, R.; Pugsley, T.A.; Zoski, K.T.; Heffner, T.G.; et al. Pyrazolo[1,5-a]pyrimidine CRF-1 receptor antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 2067–2070. [Google Scholar] [CrossRef]
- Pimerova, E.V.; Voronina, E.V. Antimicrobial Activity of Pyrazoles and Pyridazines Obtained by Intreraction of 4-Aryl-3-arylhydrazono-2,4-dioxobutanoic Acids and Their Esters with Hydrazines. Pharm. Chem. J. 2001, 35, 602–604. [Google Scholar] [CrossRef]
- Campo, J.A.; Cano, M.; Heras, J.V.; Lagunas, M.C.; Perles, J.; Pinilla, E.; Torres, M.R. Copper Complexes with New Pyridylpyrazole Based Ligands. Helv. Chim. Acta 2002, 85, 1079–1095. [Google Scholar] [CrossRef]
- Halcrow, M.A.; Mc Innes, E.J.L.; Mabbs, F.E.; Scowen, I.J.; Mc Partlin, M.; Powell, H.R.; Davies, J.E. Syntheses, structures and electrochemistry of [CuL1(LR)]BF4 [L1 = 3-{2,5-dimethoxy phenyl)-1-(2-pyridyl)pyrazole; LR = tris(3-arylpyrazolyl)hydroborate] and [CuL12][BF4]2. Effects of graphitic interactions on the stability of an aryl radical cation. J. Chem. Soc. Dalton Trans. 1997, 21, 4025–4035. [Google Scholar] [CrossRef]
- Mayoral, M.J.; Torralba, M.C.; Cano, M.; Campo, J.A.; Heras, J.V. Pyridylpyrazole derivatives. A new type of mesogenic bidentate ligands inducing mesomorphism on their related PdX2 complexes. Inorg. Chem. Commun. 2003, 6, 626–629. [Google Scholar] [CrossRef]
- Steel, P.J.; Lahousse, F.; Lerner, D.; Marzin, C. New ruthenium(II) complexes with pyridylpyrazole ligands. Photosubstitution and proton, carbon-13, and ruthenium-99 NMR structural studies. Inorg. Chem. 1983, 22, 1488–1493. [Google Scholar] [CrossRef]
- Mao, M.; Li, Y.; Liu, Q.; Xiong, L.; Zhang, X.; Li, Z. Synthesis and biological evaluation of novel N-pyridylpyrazole derivatives containing 1,2,3-triazole moieties. J. Pesticide Sci. 2015, 40, 138–142. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, G.; Dong, W.; Shi, Y.; Li, B.; Wang, S.; Li, Z. Design, synthesis, and biological activities of novel heterocyclic derivatives containing N-pyridylpyrazole. Der Pharm. Chem. 2014, 240–249. [Google Scholar]
- Solomons, K.R.H.; Lieberman, H.E.; Groundwater, P.W.; Hibbs, D.E.; Hursthouse, M.B. Molecular modelling and biological evaluation of a series of hydroxylated benzylideneanilines and benzylamines designed as tyrosine kinase inhibitors. Anti-Cancer Drug Des. 1997, 12, 635–647. [Google Scholar]
- Luo, X.Y.; Zhao, J.Z.; Lin, Y.J.; Liu, Z.Q. Antioxidative Effect of Schiff Bases with o-Hydroxybenzylidene Groupon Free Radical Induced Hemolysis of Human Red Blood Cell. Chem. Res. Chin. Univ. 2002, 18, 287–289. [Google Scholar]
- Patel, K.M.; Patel, K.N.; Patel, N.H.; Patel, M.N. Synthesis, characterization, and antimicrobial activities of some transition metal complexes with a tridentate dibasic Schiff base and bidentate 2,2′-bipyridylamine. Inorg. Met-Org. Chem. 2001, 31, 239–246. [Google Scholar] [CrossRef]
- Radi, S.; Tighadouini, S.; Feron, O.; Riant, O.; Bouakka, M.; Benabbes, R.; Mabkhot, Y.N. Synthesis of Novel β-Keto-enol Derivatives Tethered Pyrazole, Pyridine and Furan as New Potential Antifungal and Anti-Breast Cancer Agents. Molecules 2015, 20, 20186–20194. [Google Scholar] [CrossRef] [PubMed]
- Radi, S.; Attayibat, A.; Ramdani, A.; Bacquet, M. New functionalised C,C-pyridylpyrazoles. Synthesis and cation binding properties. J. Chem. Res. 2009, 2, 72–74. [Google Scholar] [CrossRef]
- Strotmeyer, K.P.; Fritsky, I.O.; Ott, R.; Pritzkow, H.; Krämer, R. Evaluating the Conformational Role of an Allosteric Cu II Ion in Anion Recognition and Catalysis by a Tricopper Complex. Supramol. Chem. 2003, 15, 529–547. [Google Scholar] [CrossRef]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef]
- Weber, J.; Roch, M.; Williams, A.F. Molecular electrostatic potentials and electron deformation densities of chromium(V), molybdenum(VI) and niobium(V) tetraperoxo complexes. Chem. Phys. Let. 1986, 123, 246–253. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Berger, B. Using conceptual density functional theory to rationalize regioselectivity: A case study on the nucleophilic ring-opening of activated aziridines. Comput. Theor. Chem. 2013, 1010, 11–18. [Google Scholar] [CrossRef]
- Khaled, K.F. Experimental, density function theory calculations and molecular dynamics simulations to investigate the adsorption of some thiourea derivatives on iron surface in nitric acid solutions. Appl. Surf. Sci. 2010, 256, 6753–6763. [Google Scholar] [CrossRef]
- Koubský, T.; Kalvoda, L. Application of ab-initio molecular electronic structure calculations of radiolytic and hydrolytic stabilities of prospective extractants. J. Radioanal. Nucl. Chem. 2015, 304, 227–235. [Google Scholar] [CrossRef]
- Lakbaibi, Z.; Abou El Makarim, H.; Tabyaoui, M.; El Hajbi, A. Theoretical study of the formation of α-chloroglycidic esters in aliphatic series using the quantum DFT method with B3LYP/6-311G (d,p). Intern. J. Innov. Appl. Stud. 2014, 7, 602–616. [Google Scholar]
- Cedillo, A.; Contreras, R. A Local Extension of the Electrophilicity Index Concept. J. Mex. Chem. Soc. 2012, 56, 257–260. [Google Scholar]
- Domingo, L.R.; Pérez, P.; Saez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748–769. [Google Scholar] [CrossRef] [PubMed]
- Sudha, G.; Priya, M.S.; Shree, R.I.; Vadivukkarasi, S. In vitro free radical scavenging activity of raw pepino fruit (Solanum muricatum Aiton). Int. J. Curr. Pharm. Res. 2011, 3, 137–140. [Google Scholar]
- Oxford Diffraction. CrysAlis CCD and CrysAlis RED. Version 171; Oxford Diffraction Ltd.: Abingdon, Oxfordshire, UK, 2004. [Google Scholar]
- The SHELX Homepage. Available online: http://shelx.uni-ac.gwdg.de/SHELX/ (accessed on 5 August 2016).
- Farrugia, L.J. WinGX, ORTEP for Windows, an update. J. Appl, Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Moure, A.; Franco, D.; Sineiro, J.; Domínguez, H.; Núnez, M.J.; Lema, J.M. Evaluation of Extracts from Gevuina avellana Hulls as Antioxidants. J. Agric. Food Chem. 2000, 48, 3890–3897. [Google Scholar] [CrossRef] [PubMed]
- Oyaizu, M. Studies on products of browning reaction: Antioxidative activities of products of browning reaction prepared from glucosamine. Jpn. J. Nutr. 1986, 44, 307–315. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.





| CCDC Deposition Number | 1487543 |
|---|---|
| Molecular Formula | C11H13N3O |
| Molecular Weight | 203.11 |
| Crystal System | Monoclinic |
| Space Group | C2/c |
| a (Å) | 18.837(2) |
| b (Å) | 12.344(2) |
| c (Å) | 9.3011(10) |
| α (°) | 90 |
| β (°) | 101.939(10) |
| γ (°) | 90 |
| V (Å3) | 2116.0(5) |
| Z | 8 |
| Dcalc (g·cm−3) | 1.276 |
| Crystal Dimension (mm) | 0.17 × 0.28 × 0.34 |
| μ (mm−1) | 0.085 |
| Tmin/Tmax | 0.972/0.986 |
| Measured Reflections | 15317 |
| −24 ,24 | |
| Indices Range (h, k, l) | −16, 16 |
| 12, 12 | |
| θ Limit (°) | 3.30–26.14 |
| Unique Reflections | 2114 |
| Observed Reflections (I > 2σ(I)) | 1616 |
| Parameters | 136 |
| Goodness of Fit on F2 | 1.033 |
| R1,wR2 [I > 2σ(I)] | 0.0498, 0.1363 |
| RX | Molecule | Dimer | Solid | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| exp | BLYP | PW91 | PWC | BLYP | PW91 | PWC | BLYP | PW91 | PWC | |
| C8-N2 | 1.330 | 1.347 | 1.343 | 1.334 | 1.349 | 1.344 | 1.334 | 1.349 | 1.346 | 1.338 |
| C1-N1 | 1.331 | 1.345 | 1.339 | 1.328 | 1.344 | 1.338 | 1.327 | 1.344 | 1.339 | 1.331 |
| N1-C5 | 1.338 | 1.357 | 1.351 | 1.338 | 1.356 | 1.349 | 1.337 | 1.356 | 1.349 | 1.456 |
| C5-C6 | 1.472 | 1.470 | 1.462 | 1.447 | 1.472 | 1.463 | 1.449 | 1.471 | 1.466 | 1.340 |
| N3-C6 | 1.359 | 1.383 | 1.375 | 1.362 | 1.381 | 1.372 | 1.359 | 1.379 | 1.371 | 1.360 |
| N3-N2 | 1.358 | 1.367 | 1.352 | 1.333 | 1.369 | 1.354 | 1.334 | 1.370 | 1.357 | 1.341 |
| N1-N3 | 2.910 | 2.965 | 2.948 | 2.893 | 2.964 | 2.941 | 2.883 | 2.935 | 2.931 | 2.919 |
| C10-C11 | 1.506 | 1.540 | 1.530 | 1.510 | 1.545 | 1.536 | 1.516 | 1.537 | 1.530 | 1.518 |
| C11-O | 1.411 | 1.444 | 1.429 | 1.405 | 1.430 | 1.415 | 1.390 | 1.433 | 1.419 | 1.399 |
| N3-C10 | 1.459 | 1.472 | 1.459 | 1.438 | 1.476 | 1.462 | 1.441 | 1.471 | 1.460 | 1.441 |
| C6-C5-N1 | 118.38 | 118.630 | 118.660 | 118.790 | 118.730 | 118.760 | 118.670 | 118.710 | 118.840 | 118.990 |
| C5-C6-N3 | 125.66 | 126.13 | 126.04 | 125.48 | 126.19 | 126.06 | 125.24 | 125.67 | 125.84 | 125.88 |
| C4-C5-C6 | 119.80 | 119.98 | 119.75 | 119.66 | 119.81 | 119.64 | 119.66 | 119.62 | 119.34 | 119.28 |
| C4-C5-N1 | 121.80 | 121.41 | 121.57 | 121.54 | 121.41 | 121.59 | 121.67 | 121.66 | 121.81 | 121.73 |
| C7-C6-N3 | 105.93 | 105.54 | 105.52 | 105.63 | 105.90 | 105.91 | 106.17 | 105.90 | 105.87 | 106.03 |
| C6-N3-C10 | 131.12 | 131.09 | 130.57 | 129.98 | 131.30 | 130.88 | 130.56 | 130.90 | 130.52 | 130.08 |
| C11-C10-N3 | 111.50 | 112.00 | 111.82 | 111.33 | 112.59 | 112.43 | 111.91 | 111.91 | 111.72 | 111.70 |
| O-C11-C10 | 111.03 | 111.08 | 111.23 | 111.56 | 110.74 | 110.90 | 110.96 | 111.04 | 111.41 | 111.69 |
| C4-C5-C6-N3 | 169.13 | 160.52 | 160.42 | 168.07 | 162.30 | 163.80 | 170.12 | 169.73 | 169.00 | 165.83 |
| N3-C10-C11-O | 177.15 | 176.40 | 176.30 | 174.34 | 174.52 | 174.60 | 174.08 | 178.46 | 177.17 | 176.39 |
| N1-C5-C6-N3 | 11.80 | 20.44 | 20.47 | 12.27 | 18.39 | 16.86 | 10.29 | 10.92 | 11.8 | 14.92 |
| C10-N1 (intra) | 2.919 | 3.006 | 2.974 | 2.869 | 3.002 | 2.960 | 2.866 | 2.939 | 2.927 | 2.909 |
| intramolecular | 2.382 | 2.455 | 2.420 | 2.334 | 2.452 | 2.409 | 2.328 | 2.360 | 2.330 | 2.270 |
| C10-H10-N1 | 114.35 | 109.84 | 109.83 | 107.84 | 109.74 | 109.64 | 108.04 | 111.42 | 112.47 | 114.81 |
| O-N2 (inter) | 2.855 | 2.927 | 2.862 | 2.703 | 2.840 | 2.817 | 2.760 | |||
| intermolecular | 2.036 | 1.941 | 1.875 | 1.700 | 1.846 | 1.823 | 1.751 | |||
| O-H-N2 | 178.99 | 173.02 | 173.74 | 174.67 | 177.11 | 175.76 | 175.65 | |||
| C2-O (inter) | 3.327 | 3.242 | 3.278 | 3.314 | ||||||
| intermolecular | 2.636 | 2.405 | 2.429 | 2.410 | ||||||
| C2-H2-O | 131.63 | 132.93 | 133.87 | 137.64 | ||||||
| Etot/molecule (Ha) | 666.46 | 666.44 | 660.95 | 666.44 | 666.48 | 660.98 | 666.47 | 666.49 | 661.02 | |
| Binding E (kcal/mol) | 3061.86 | 3187.32 | 3557.69 | 3068.01 | 3195.53 | 3571.68 | 3063.73 | 3202.79 | 3596.28 |
| Atom | Mulliken | Hirshfeld | Mulliken | Hirshfeld |
|---|---|---|---|---|
| C8 | 0.108 | 0.092 | ||
| C6 | 0.166 | 0.126 | ||
| C5 | 0.235 | 0.174 | ||
| C4 | 0.108 | 0.078 | ||
| C3 | 0.122 | 0.102 | ||
| C2 | 0.136 | 0.097 | 0.238 | 0.160 |
| N1 | 0.163 | 0.149 | ||
| N3 | 0.084 | 0.094 | 0.094 | 0.088 |
| N2 | 0.197 | 0.177 | 0.113 | 0.096 |
| O | 0.129 | 0.114 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Scavenging Ability (%, Mean ± SD), Concentration (µg/mL) | ||||||
|---|---|---|---|---|---|---|---|
| 5.0 | 10.0 | 20.0 | 30.0 | 40.0 | 50.0 | 100.0 | |
| 2 | 6.9 ± 1.4 | 22.8 ± 1.9 | 36.5 ± 0.8 | 46.8 ± 2.5 | 55.9 ± 1.6 | 63.4 ± 1.4 | 73.2 ± 1.7 |
| BHA | 51.0 ± 1.6 | 65.9 ± 1.5 | 83.1 ± 0.9 | 87.5 ± 0.1 | 88.5 ± 1.0 | 89.0 ± 1.2 | 89.7 ± 0.7 |
| Asc. acid | 85.3 ± 1.3 | 93.0 ± 1.7 | 93.2 ± 1.2 | 93.4 ± 1.1 | 93.4 ± 1.5 | 93.8 ± 1.0 | 94.3 ± 1.0 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radi, S.; Attayibat, A.; El-Massaoudi, M.; Salhi, A.; Eddike, D.; Tillard, M.; Mabkhot, Y.N. X-ray Single Crystal Structure, DFT Calculations and Biological Activity of 2-(3-Methyl-5-(pyridin-2’-yl)-1H-pyrazol-1-yl) Ethanol. Molecules 2016, 21, 1020. https://doi.org/10.3390/molecules21081020
Radi S, Attayibat A, El-Massaoudi M, Salhi A, Eddike D, Tillard M, Mabkhot YN. X-ray Single Crystal Structure, DFT Calculations and Biological Activity of 2-(3-Methyl-5-(pyridin-2’-yl)-1H-pyrazol-1-yl) Ethanol. Molecules. 2016; 21(8):1020. https://doi.org/10.3390/molecules21081020
Chicago/Turabian StyleRadi, Smaail, Ahmed Attayibat, Mohamed El-Massaoudi, Amin Salhi, Driss Eddike, Monique Tillard, and Yahia N. Mabkhot. 2016. "X-ray Single Crystal Structure, DFT Calculations and Biological Activity of 2-(3-Methyl-5-(pyridin-2’-yl)-1H-pyrazol-1-yl) Ethanol" Molecules 21, no. 8: 1020. https://doi.org/10.3390/molecules21081020