
Identification of the Structural Features of Guanine Derivatives as MGMT Inhibitors Using 3D-QSAR Modeling Combined with Molecular Docking



Abstract
:
1. Introduction
2. Results and Discussion
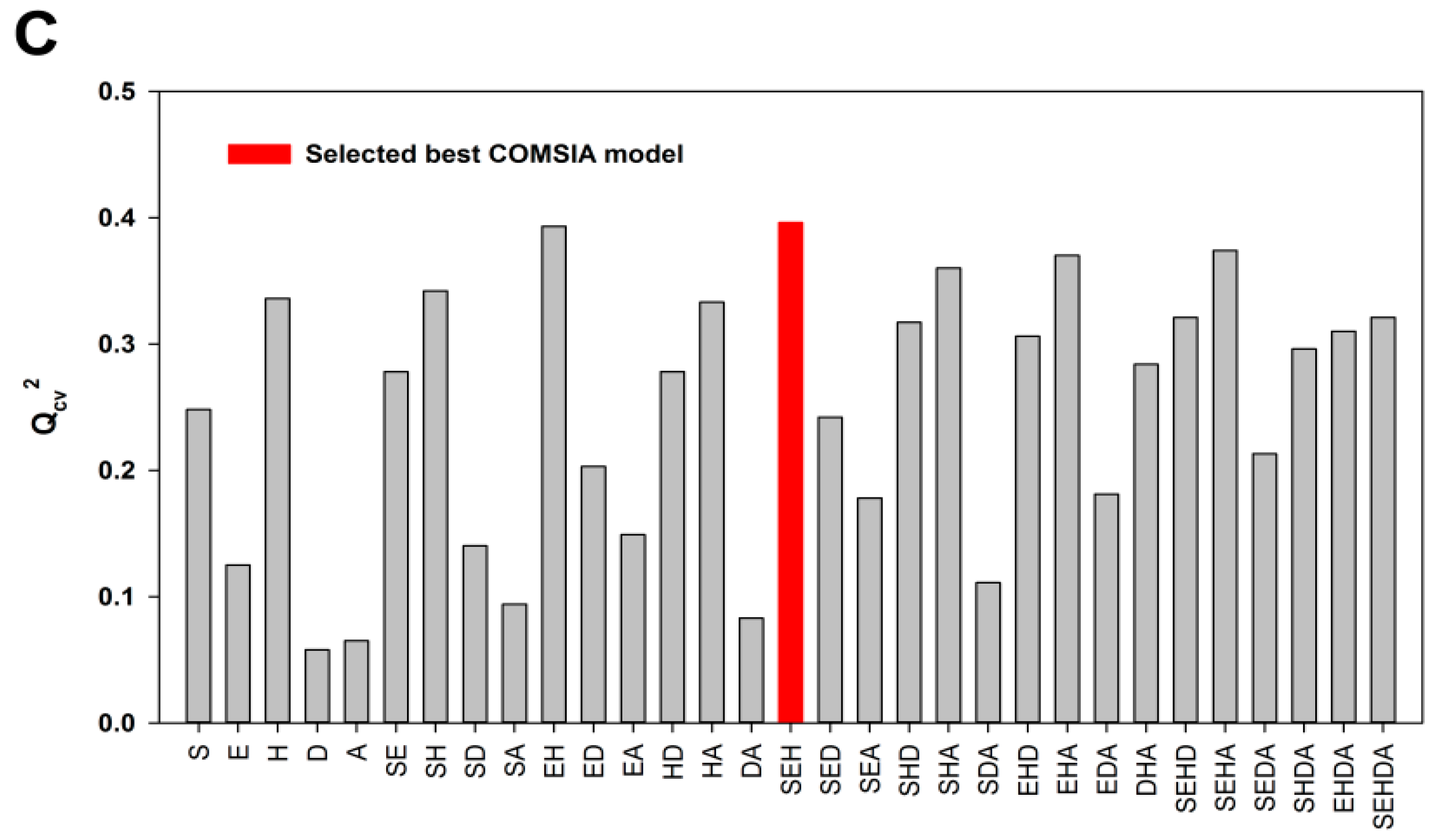
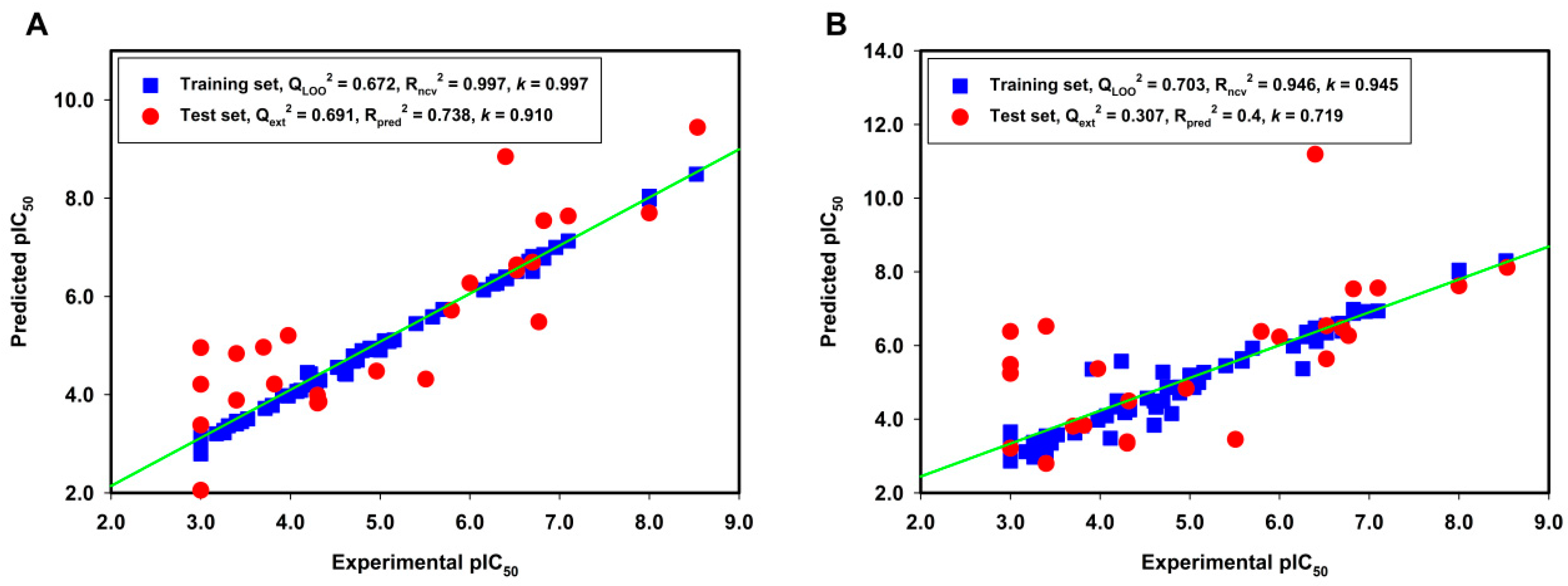
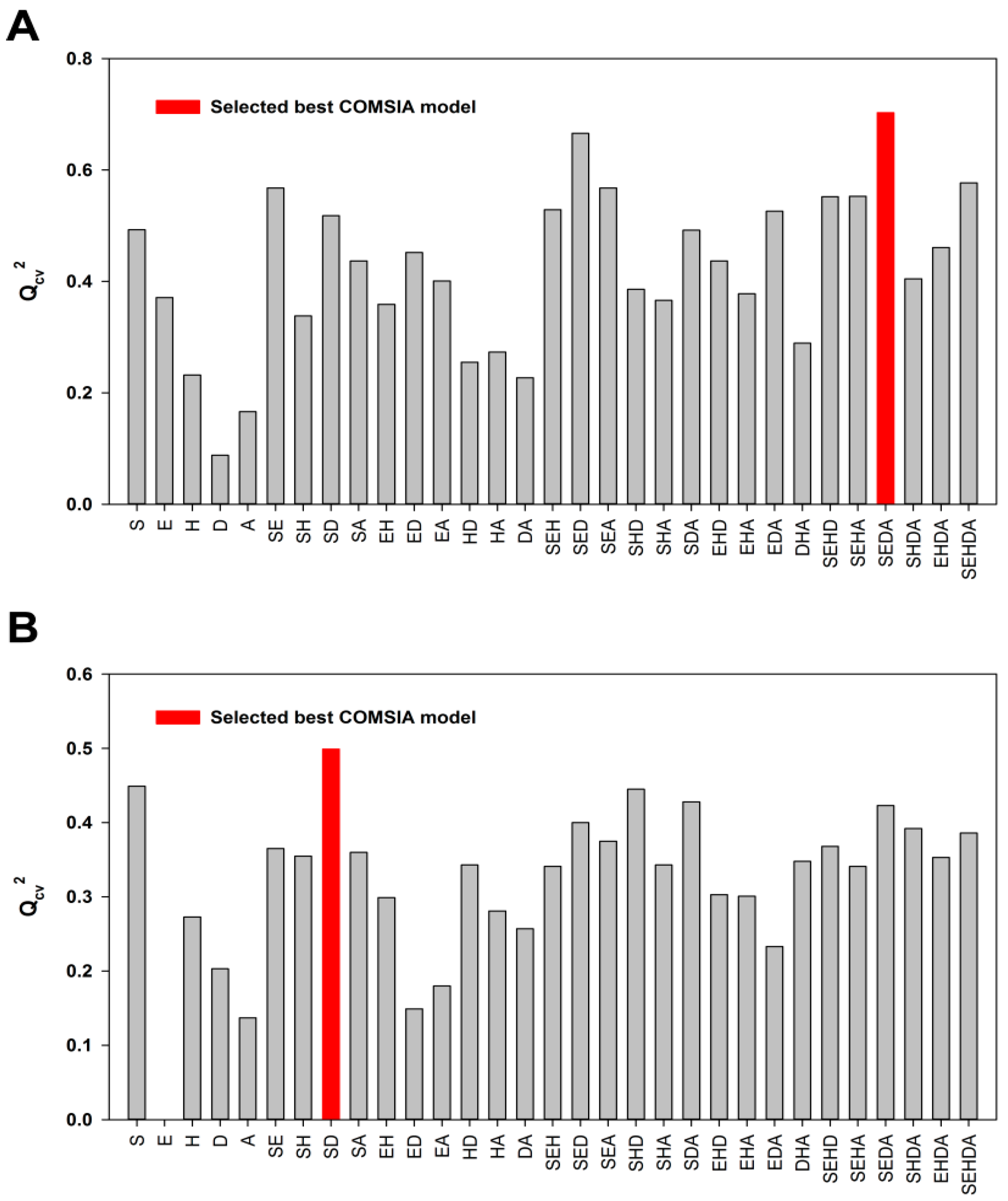
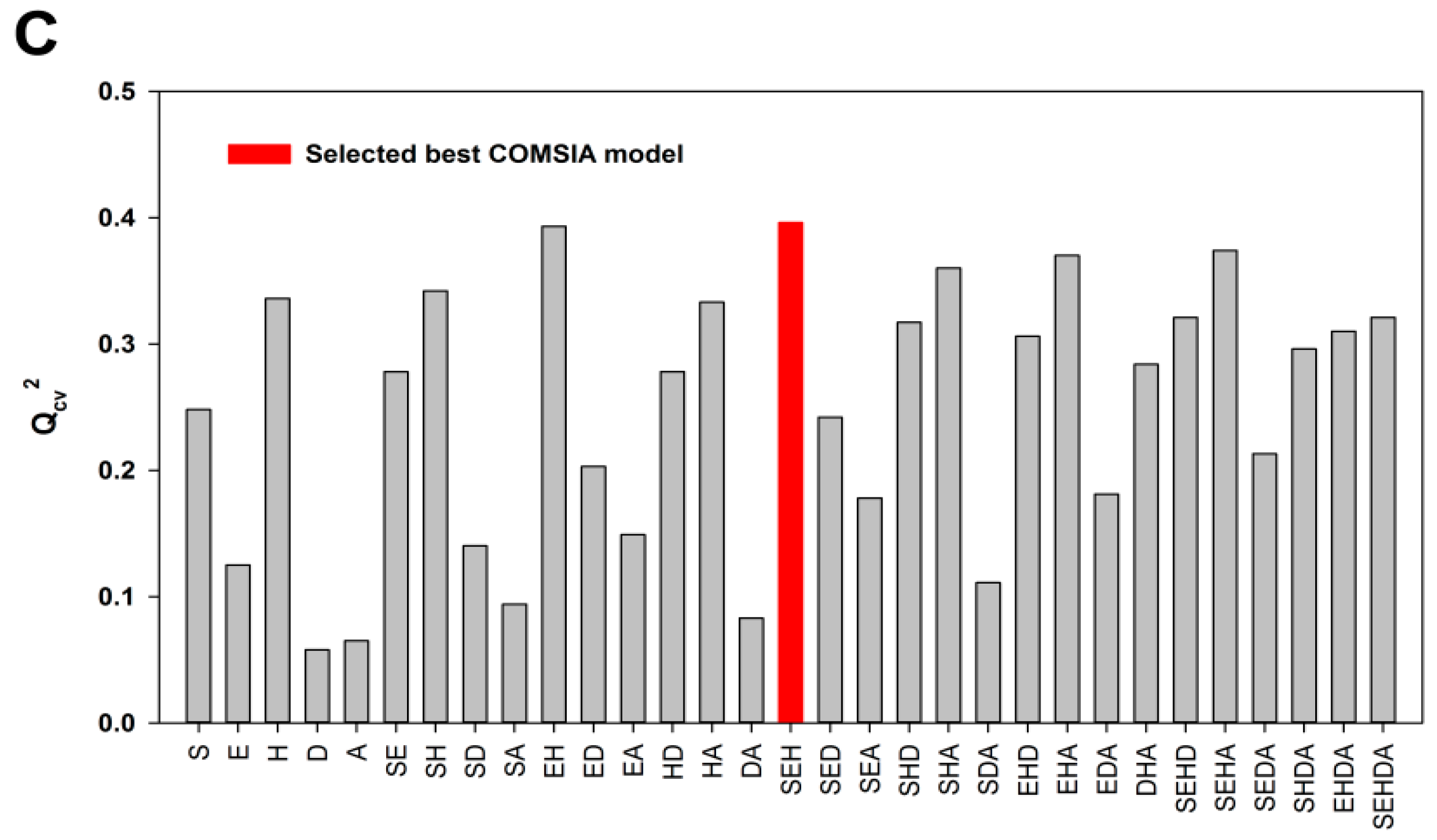
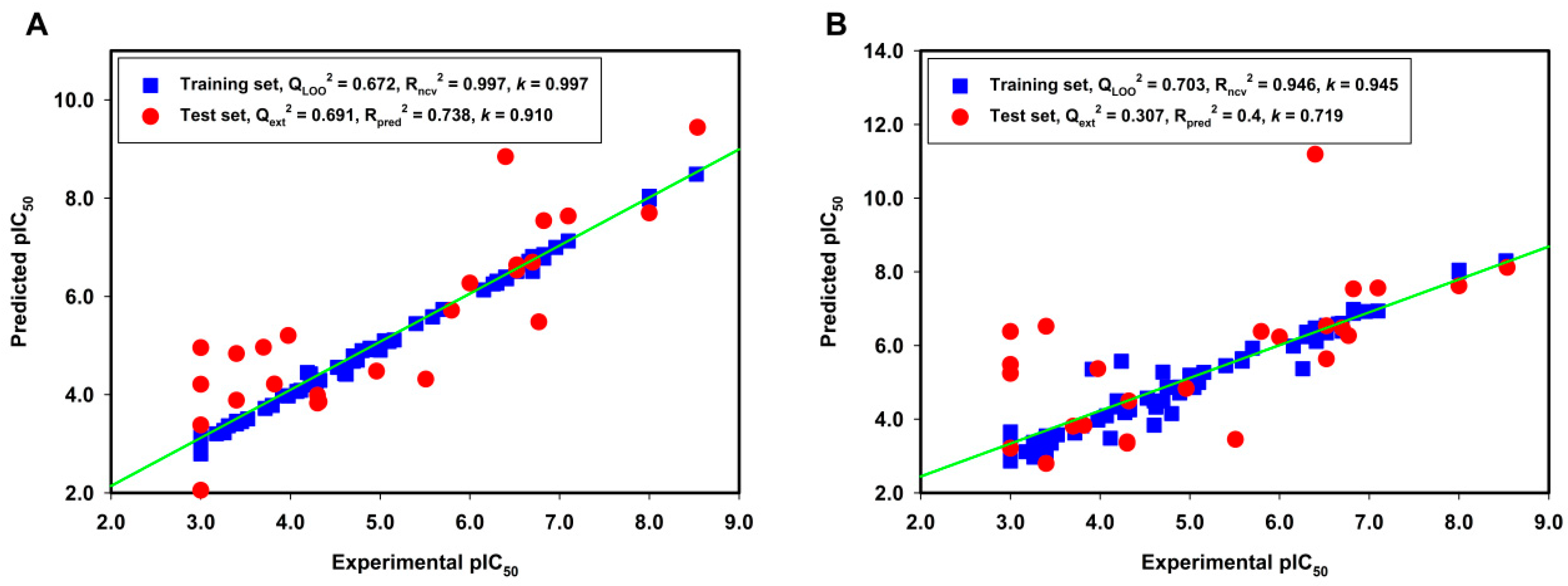
2.1. Model Validation
2.2. External Test Set Validation and Y-Randomization Test
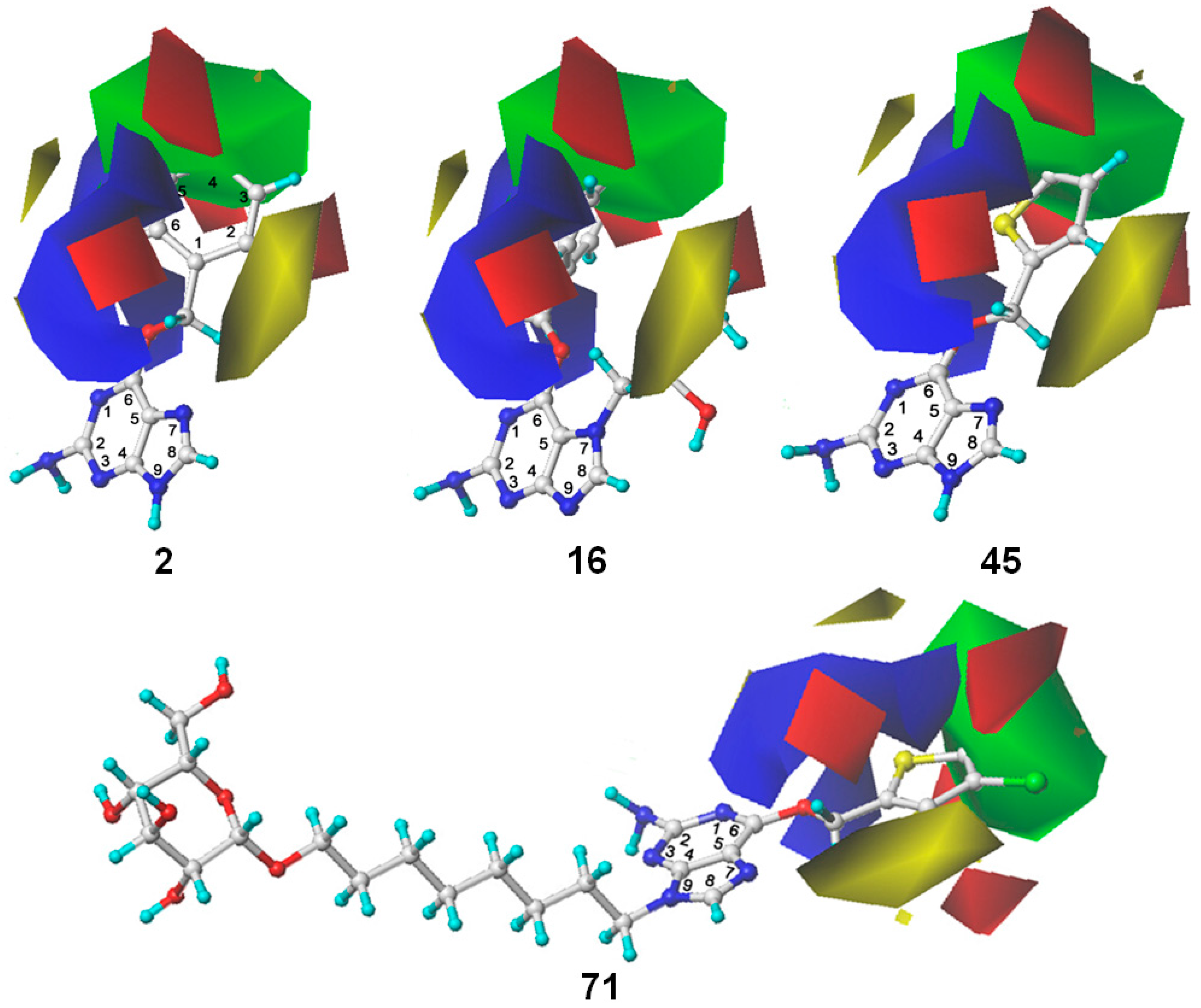
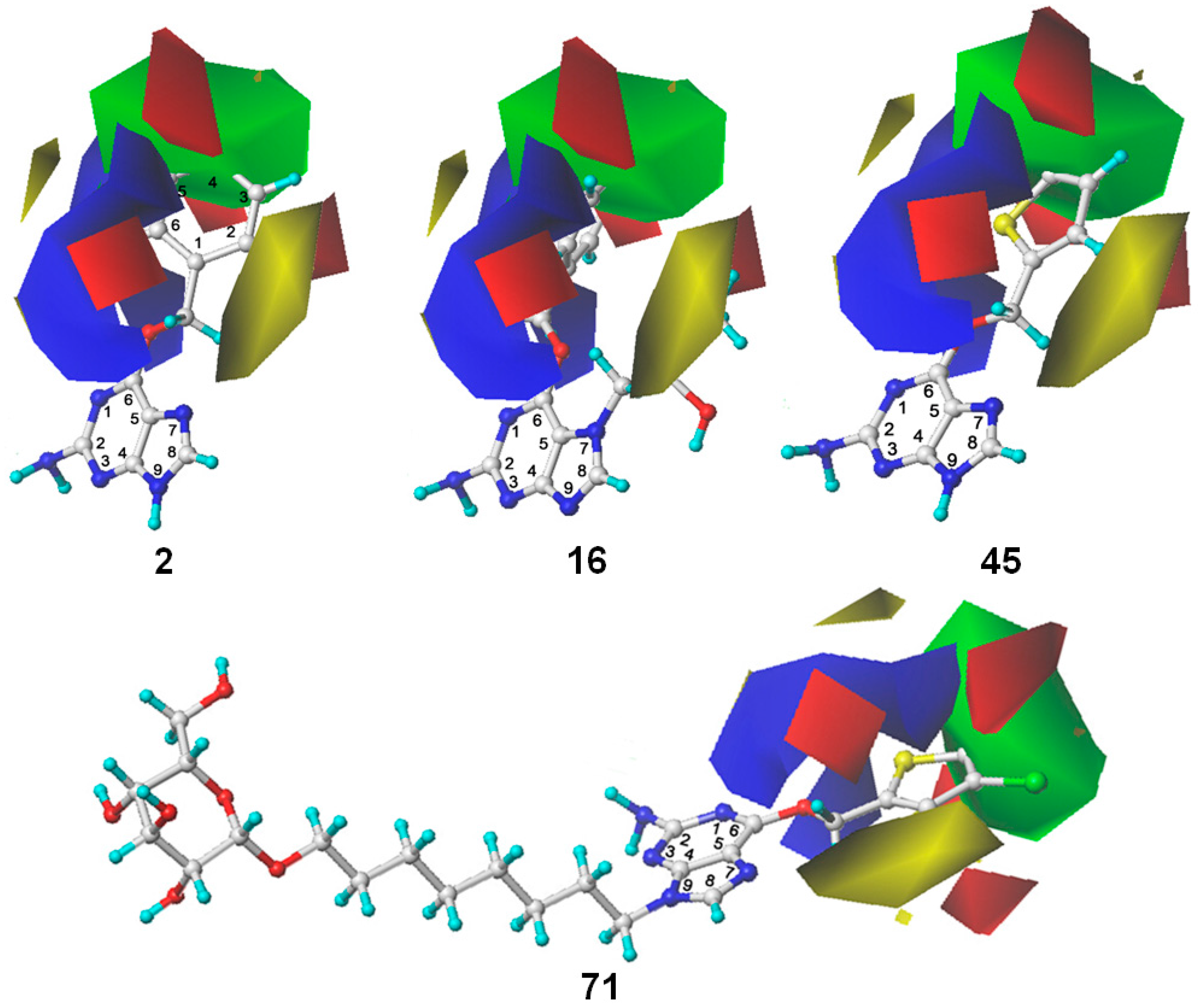
2.3. 3D Contour Map Analysis
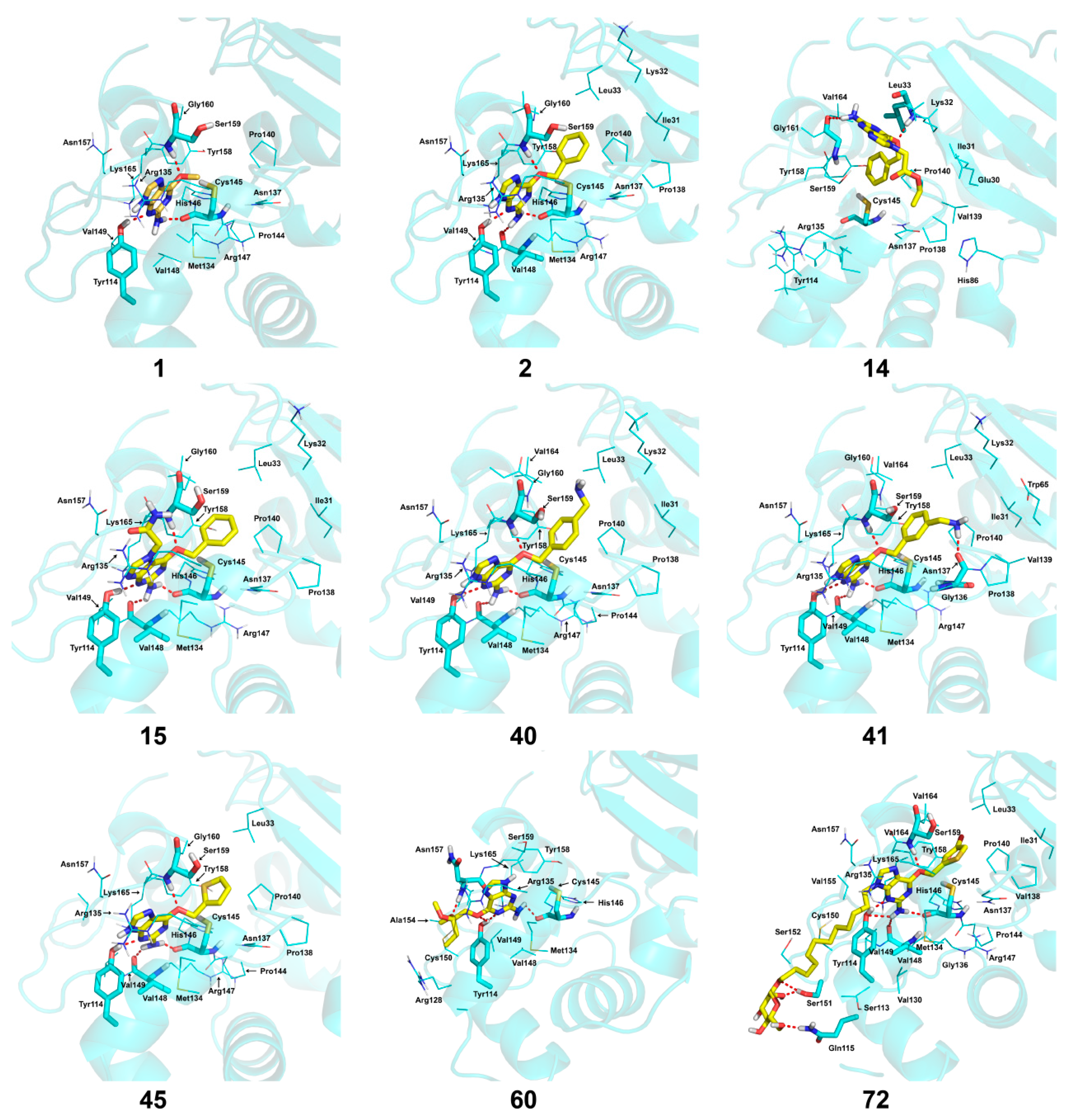
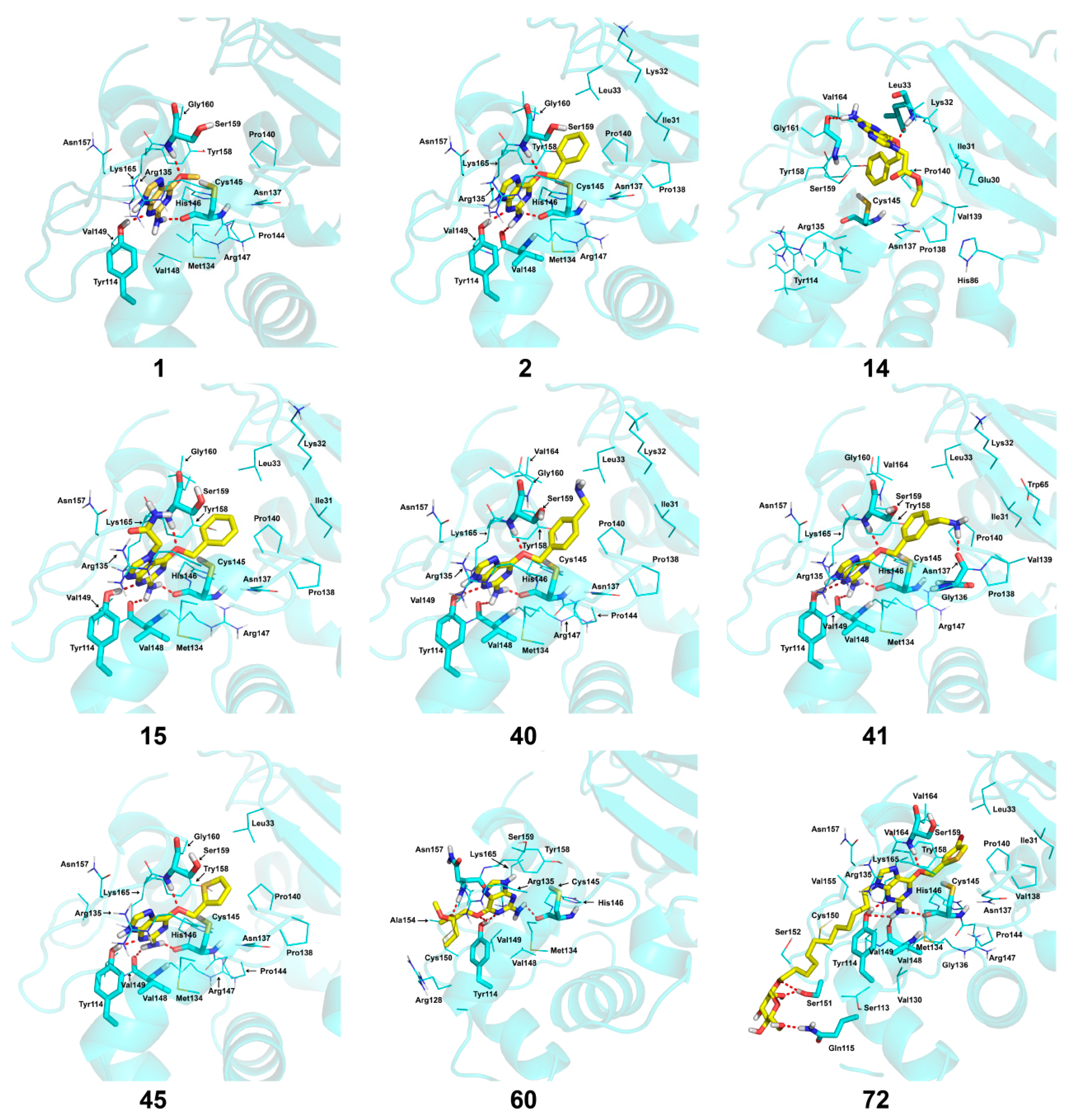
2.4. Docking Analysis
3. Experimental Section
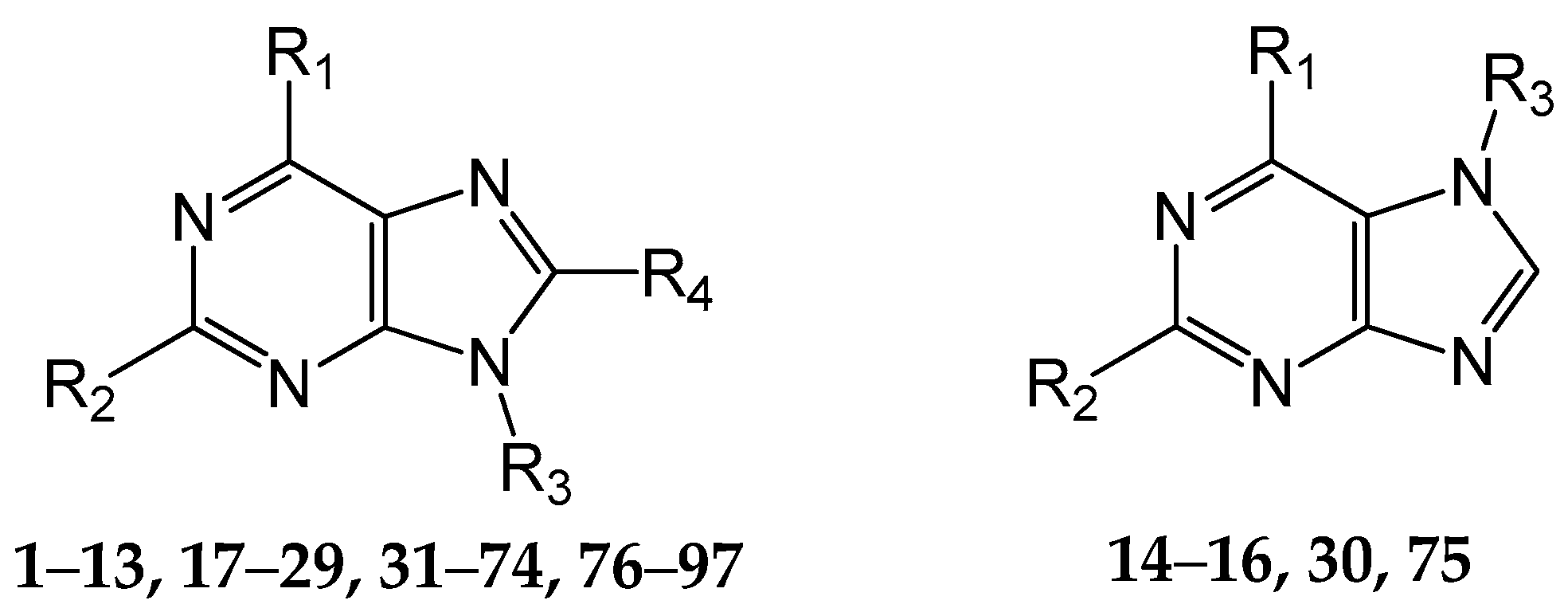
3.1. Data Set
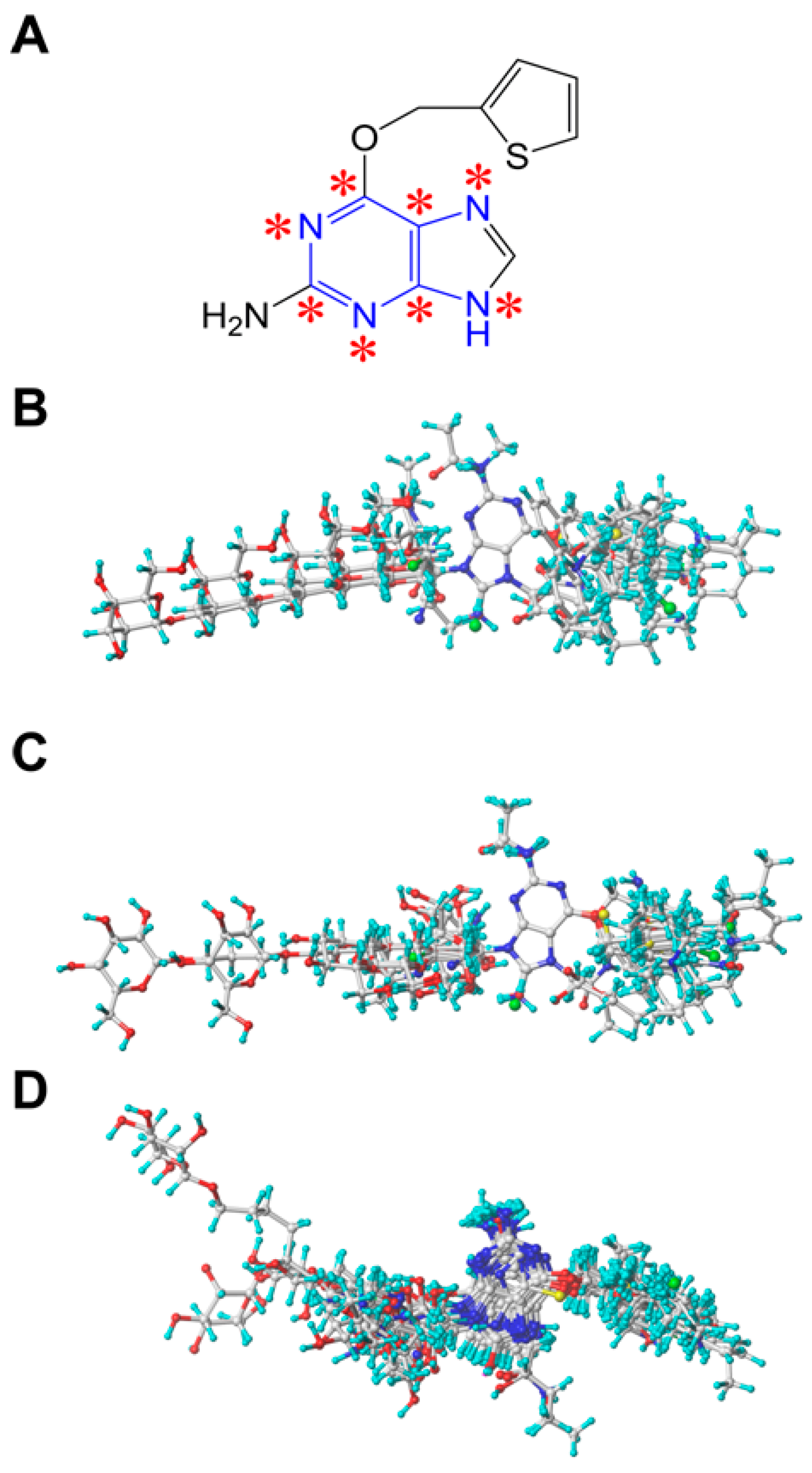
3.2. Molecular Modeling and Alignment
3.3. 3D-QSAR Studies
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hegi, M.E.; Liu, L.L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-Methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O6-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Zhao, L.J.; Zhong, R.G. The induction and repair of DNA interstrand crosslinks and implications in cancer chemotherapy. Anti Cancer Agents Med. Chem. 2016, 16, 221–246. [Google Scholar]
- Pegg, A.E. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 2011, 24, 618–639. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Naumann, S.C.; Roos, W.P.; Jost, E.; Belohlavek, C.; Lennerz, V.; Schmidt, C.W.; Christmann, M.; Kaina, B. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: Response related to MGMT, MMR, DSBs, and p53. Br. J. Cancer 2009, 100, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Srivenugopal, K.S.; Yuan, X.H.; Friedman, H.S.; AliOsman, F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry 1996, 35, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Xu-Welliver, M.; Pegg, A.E. Degradation of the alkylated form of the DNA repair protein, O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002, 23, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Philip, S.; Swaminathan, S.; Kuznetsov, S.G.; Kanugula, S.; Biswas, K.; Chang, S.; Loktionova, N.A.; Haines, D.C.; Kaldis, P.; Pegg, A.E. Degradation of BRCA2 in alkyltransferase-mediated DNA repair and its clinical implications. Cancer Res. 2008, 68, 9973–9981. [Google Scholar] [CrossRef] [PubMed]
- Belanich, M.; Pastor, M.; Randall, T.; Guerra, D.; Kibitel, J.; Alas, L.; Li, B.; Citron, M.; Wasserman, P.; White, A.; et al. Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res. 1996, 56, 783–788. [Google Scholar] [PubMed]
- Gerson, S.L. Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. 2002, 20, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Zhu, Y.L.; Shyam, K.; Penketh, P.G.; Baumann, R.P.; Sartorelli, A.C. Quantitative relationship between guanine O6-alkyl lesions produced by Onrigin™ and tumor resistance by O6-alkylguanine-DNA alkyltransferase. Biochem. Pharmacol. 2010, 80, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Zhao, L.J.; Fan, T.J.; Li, S.S.; Zhong, R.G. Investigations on the effect of O6-benzylguanine on the formation of dG-dC interstrand cross-links induced by chloroethylnitrosoureas in human glioma cells using stable isotope dilution high-performance liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2014, 27, 1253–1262. [Google Scholar] [PubMed]
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.E.; Mitchell, R.B.; Mummert, C.; Moschel, R.C.; Pegg, A.E. Effect of O6-benzylguanine analogs on sensitivity of human tumor cells to the cytotoxic effects of alkylating agents. Cancer Res. 1991, 51, 3367–3372. [Google Scholar] [PubMed]
- Moschel, R.C.; McDougall, M.G.; Dolan, M.E.; Stine, L.; Pegg, A.E. Structural features of substituted purine derivatives compatible with depletion of human O6-alkylguanine-DNA alkyltransferase. J. Med. Chem. 1992, 35, 4486–4491. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.Y.; McDougall, M.G.; Dolan, M.E.; Swenn, K.; Pegg, A.E.; Moschel, R.C. Substituted O6-benzylguanine derivatives and their inactivation of human O6-alkylguanine-DNA alkyltransferase. J. Med. Chem. 1994, 37, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.Y.; Swenn, K.; Kanugula, S.; Dolan, M.E.; Pegg, A.E.; Moschel, R.C. 8-Substituted O6-benzylguanine, substituted 6(4)-(benzyloxy)pyrimidine, and related derivatives as inactivators of human O6-alkylguanine-DNA alkyltransferase. J. Med. Chem. 1995, 38, 359–365. [Google Scholar] [CrossRef] [PubMed]
- McElhinney, R.S.; Donnelly, D.J.; McCormick, J.E.; Kelly, J.; Watson, A.J.; Rafferty, J.A.; Elder, R.H.; Middleton, M.R.; Willington, M.A.; McMurry, T.B.H.; et al. Inactivation of O6-alkylguanine-DNA alkyltransferase. 1. Novel O6-(hetarylmethyl)guanines having basic rings in the side chain. J. Med. Chem. 1998, 41, 5265–5271. [Google Scholar] [CrossRef] [PubMed]
- Terashima, I.; Kohda, K. Inhibition of human O6-alkylguanine-DNA alkyltransferase and potentiation of the cytotoxicity of chloroethylnitrosourea by 4(6)-(benzyloxy)-2,6(4)-diamino-5-(nitro or nitroso)pyrimidine derivatives and analogues. J. Med. Chem. 1998, 41, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.J.; Arris, C.E.; Bleasdale, C.; Boyle, F.T.; Calvert, A.H.; Curtin, N.J.; Dalby, C.; Kanugula, S.; Lembicz, N.K.; Newell, D.R.; et al. Resistance-modifying agents. 8. Inhibition of O6-alkylguanine-DNA alkyltransferase by O6-alkenyl-, O6-cycloalkenyl-, and O6-(2-oxoalkyl)guanines and potentiation of temozolomide cytotoxicity in vitro by O6-(1-cyclopentenylmethyl)guanine. J. Med. Chem. 2000, 43, 4071–4083. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, J.; Hull, W.E.; von der Lieth, C.W.; Eichhorn, U.; Kliem, H.C.; Kaina, B.; Wiessler, M. Monosaccharide-linked inhibitors of O6-methylguanine-DNA methyltransferase (MGMT): Synthesis, molecular modeling, and structure-activity relationships. J. Med. Chem. 2001, 44, 4050–4061. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.E.; Loktionova, N.A.; Pegg, A.E.; Moschel, R.C. 2-amino-O4-benzylpteridine derivatives: Potent inactivators of O6-alkylguanine-DNA alkyltransferase. J. Med. Chem. 2004, 47, 3887–3891. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.P.; Loktionova, N.A.; Pegg, A.E.; Moschel, R.C. β-Glucuronidase-cleavable prodrugs of O6-benzylguanine and O6-benzyl-2′-deoxyguanosine. J. Med. Chem. 2005, 48, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Pauly, G.T.; Loktionova, N.A.; Fang, Q.M.; Vankayala, S.L.; Guida, W.C.; Pegg, A.E. Substitution of aminomethyl at the meta-position enhances the inactivation of O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. J. Med. Chem. 2008, 51, 7144–7153. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Sosman, J.; Gerson, S.L.; Liu, L.L.; Dolan, E.; Lin, S.; Vokes, E.E. Phase II trial of the O6-alkylguanine DNA alkyltransferase inhibitor O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea in advanced melanoma. Clin. Cancer Res. 2005, 11, 7861–7865. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E.; Gururangan, S.; Geyer, J.R.; McLendon, R.E.; Poussaint, T.Y.; Wallace, D.; Balis, F.M.; Berg, S.L.; Packer, R.J.; Goldman, S.; et al. A phase II study of O6-benzylguanine and temozolomide in pediatric patients with recurrent or progressive high-grade gliomas and brainstem gliomas: A Pediatric Brain Tumor Consortium study. J. Neuro Oncol. 2012, 106, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Ranson, M.; Middleton, M.R.; Bridgewater, J.; Lee, S.M.; Dawson, M.; Jowle, D.; Halbert, G.; Waller, S.; McGrath, H.; Gumbrell, L.; et al. Lomeguatrib, a potent inhibitor of O6-alkylguanine-DNA-alkyltransferase: Phase I safety, pharmacodynamic, and pharmacokinetic trial and evaluation in combination with temozolomide in patients with advanced solid tumors. Clin. Cancer Res. 2006, 12, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, R.L.; Dolan, M.E.; Bertucci, D.; Ewesuedo, R.B.; Vogelzang, N.J.; Mani, S.; Wilson, L.R.; Ratain, M.J. Phase I clinical and pharmacological study of O6-benzylguanine followed by carmustine in patients with advanced cancer. Clin. Cancer Res. 2000, 6, 3025–3031. [Google Scholar] [PubMed]
- Thareja, S. Steroidal 5α-reductase inhibitors: A comparative 3D-QSAR study review. Chem. Rev. 2015, 115, 2883–2894. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Wei, X.; Zhang, R.S. CoMFA and CoMSIA 3D-QSAR studies on quionolone caroxylic acid derivatives inhibitors of HIV-1 integrase. Eur. J. Med. Chem. 2010, 45, 3413–3419. [Google Scholar] [CrossRef] [PubMed]
- Moonsamy, S.; Dash, R.C.; Soliman, M.E.S. Integrated computational tools for identification of CCR5 antagonists as potential HIV-1 entry inhibitors: Homology modeling, virtual screening, molecular dynamics simulations and 3D QSAR analysis. Molecules 2014, 19, 5243–5265. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Wang, J.; Wang, R.; Lin, Y.; Hu, Y.; Wang, Y.Q.; Shu, M.; Lin, Z.H. Combined pharmacophore modeling, 3D-QSAR, homology modeling and docking studies on CYP11B1 inhibitors. Molecules 2015, 20, 1014–1030. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular-field analysis (CoMFA). 1. effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indexes in a comparative-analysis (CoMSIA) of drug molecules to correlate and predict their biological-activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U. Comparative Molecular Similarity Index Analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J. Comput. Aid. Mol. Des. 1999, 13, 1–10. [Google Scholar] [CrossRef]
- Bohm, M.; Sturzebecher, J.; Klebe, G. Three-dimensional quantitative structure-activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J. Med. Chem. 1999, 42, 458–477. [Google Scholar] [PubMed]
- Doytchinova, I.A.; Flower, D. A comparative molecular similarity index index analysis (CoMSIA) study identifies an HLA-A2 binding supermotif. J. Comput. Aid. Mol. Des. 2002, 16, 535–544. [Google Scholar] [CrossRef]
- Balasubramanian, P.K.; Balupuri, A.; Gadhe, C.G.; Cho, S.J. 3D QSAR modeling study on 7-aminofuro [2,3-c] pyridine derivatives as TAK1 inhibitors using CoMFA and CoMSIA. Med. Chem. Res. 2015, 24, 2347–2365. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.W.; Zhang, L.Z.; Zhan, P.; Liu, X.Y. 3D-QSAR and docking studies on piperidine-substituted diarylpyrimidine analogues as HIV-1 reverse transcriptase inhibitors. Med. Chem. Res. 2015, 24, 3314–3326. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Wibley, J.E.A.; Pegg, A.E.; Moody, P.C.E. Crystal structure of the human O6-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000, 28, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Woo, T.T.; Luu, K.X.; Noll, D.M.; Clarke, N.D.; Pegg, A.E.; Tainer, J.A. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 2004, 11, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Cramer, R.D.; Vanopdenbosch, N. Validation of the general-purpose tripos 5.2 force-field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Powell, M.J.D. Restart procedures for conjugate gradient method. Math. Progr. 1977, 12, 241–254. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Wang, F.F.; Yang, W.; Shi, Y.H.; Le, G.W. Structural analysis of selective agonists of thyroid hormone receptor beta using 3D-QSAR and molecular docking. J. Taiwan Inst. Chem. Eng. 2015, 49. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wold, S.; Ruhe, A.; Wold, H.; Dunn, W.J. The collinearity problem in linear-regression. The partial least-squares (PLS) approach to generalized inverses. SIAM J. Sci. Stat. Comput. 1984, 5, 735–743. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.Y.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aid. Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters 1 | Ligand-Based Alignment | DFT Optimization-Based Alignment | Docking-Based Alignment | |||
|---|---|---|---|---|---|---|
| CoMFA | CoMSIA 2 | CoMFA | CoMSIA 2 | CoMFA | CoMSIA 2 | |
| Qcv2 | 0.672 | 0.703 | 0.498 | 0.499 | 0.164 | 0.396 |
| ONC | 8 | 13 | 4 | 4 | 4 | 5 |
| Rncv2 | 0.997 | 0.946 | 0.717 | 0.695 | 0.696 | 0.763 |
| SEE | 0.089 | 0.384 | 0.814 | 0.846 | 0.845 | 0.751 |
| F value | 1096.142 | 77.545 | 42.483 | 38.108 | 38.273 | 42.425 |
| Field Distribution (%) | ||||||
| Steric | 55.3 | 25.8 | 66.5 | 54.7 | 48.0 | 14.9 |
| Electrostatic | 44.7 | 45.0 | 33.5 | – | 52.0 | 46.0 |
| Hydrophobic | – | – | – | – | – | 39.1 |
| HBD | – | 13.4 | – | 45.3 | – | – |
| HBA | – | 15.8 | – | – | – | – |
| Compounds | Experimental pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Predicted pIC50 | Residues | Predicted pIC50 | Residues | ||
| Training Set | |||||
| 1 | 3.46 | 3.45 | 0.01 | 3.35 | 0.11 |
| 2 | 6.70 | 6.51 | 0.19 | 6.39 | 0.31 |
| 3 | 6.70 | 6.61 | 0.09 | 6.43 | 0.27 |
| 4 | 6.70 | 6.81 | −0.12 | 6.60 | 0.10 |
| 5 | 5.70 | 5.73 | −0.03 | 5.93 | −0.23 |
| 6 | 5.00 | 4.91 | 0.09 | 5.19 | −0.19 |
| 7 | 5.05 | 5.09 | −0.05 | 4.86 | 0.19 |
| 8 | 4.70 | 4.68 | 0.02 | 4.49 | 0.21 |
| 9 | 4.52 | 4.56 | −0.03 | 4.57 | −0.05 |
| 10 | 4.89 | 4.91 | −0.03 | 4.87 | 0.01 |
| 11 | 4.33 | 4.29 | 0.04 | 4.26 | 0.07 |
| 12 | 4.89 | 4.95 | −0.06 | 4.71 | 0.18 |
| 13 | 4.07 | 4.06 | 0.01 | 4.09 | −0.02 |
| 14 | 3.40 | 3.41 | −0.01 | 3.38 | 0.02 |
| 15 | 3.40 | 3.42 | −0.02 | 3.26 | 0.14 |
| 16 | 3.40 | 3.42 | −0.02 | 3.55 | −0.15 |
| 17 | 3.40 | 3.46 | −0.06 | 3.07 | 0.33 |
| 18 | 3.40 | 3.40 | −0.01 | 3.26 | 0.14 |
| 19 | 6.52 | 6.59 | −0.07 | 6.34 | 0.18 |
| 20 | 6.30 | 6.27 | 0.03 | 6.36 | −0.06 |
| 21 | 6.30 | 6.31 | −0.01 | 6.24 | 0.06 |
| 22 | 5.40 | 5.45 | −0.05 | 5.45 | −0.05 |
| 23 | 6.52 | 6.51 | 0.01 | 6.35 | 0.17 |
| 24 | 6.40 | 6.40 | 0.00 | 6.47 | −0.07 |
| 25 | 5.59 | 5.59 | 0.00 | 5.58 | 0.01 |
| 26 | 4.74 | 4.70 | 0.04 | 4.86 | −0.12 |
| 27 | 3.97 | 3.97 | 0.00 | 3.98 | 0.00 |
| 28 | 5.15 | 5.11 | 0.04 | 5.27 | −0.11 |
| 29 | 4.62 | 4.41 | 0.21 | 4.33 | 0.29 |
| 30 | 4.28 | 4.24 | 0.04 | 4.28 | 0.01 |
| 31 | 4.24 | 4.19 | 0.05 | 5.57 | −1.33 |
| 32 | 3.52 | 3.51 | 0.02 | 3.58 | −0.05 |
| 33 | 6.15 | 6.13 | 0.02 | 5.98 | 0.17 |
| 34 | 6.52 | 6.52 | 0.00 | 6.55 | −0.02 |
| 35 | 7.10 | 7.13 | −0.03 | 6.94 | 0.16 |
| 36 | 4.22 | 4.42 | −0.20 | 4.33 | −0.11 |
| 37 | 4.60 | 4.43 | 0.17 | 4.49 | 0.11 |
| 38 | 4.19 | 4.45 | −0.26 | 4.50 | −0.31 |
| 39 | 3.80 | 3.78 | 0.02 | 3.81 | −0.02 |
| 40 | 6.82 | 6.85 | −0.03 | 6.87 | −0.05 |
| 41 | 6.96 | 7.00 | −0.04 | 6.92 | 0.04 |
| 42 | 4.28 | 4.21 | 0.07 | 4.17 | 0.10 |
| 43 | 3.91 | 3.98 | −0.07 | 5.35 | −1.44 |
| 44 | 3.18 | 3.20 | −0.03 | 3.12 | 0.06 |
| 45 | 8.52 | 8.49 | 0.03 | 8.30 | 0.22 |
| 46 | 3.00 | 3.01 | −0.01 | 3.25 | −0.25 |
| 47 | 3.00 | 2.90 | 0.10 | 2.99 | 0.01 |
| 48 | 3.31 | 3.365 | −0.06 | 3.31 | 0.00 |
| 49 | 3.00 | 3.01 | −0.01 | 3.12 | −0.12 |
| 50 | 3.26 | 3.27 | −0.01 | 3.37 | −0.11 |
| 51 | 3.00 | 2.79 | 0.21 | 3.11 | −0.11 |
| 52 | 4.60 | 4.50 | 0.10 | 3.84 | 0.77 |
| 53 | 3.26 | 3.22 | 0.04 | 2.97 | 0.29 |
| 54 | 4.80 | 4.89 | −0.10 | 4.15 | 0.64 |
| 55 | 3.00 | 3.05 | −0.04 | 3.65 | −0.65 |
| 56 | 4.11 | 4.09 | 0.03 | 3.48 | 0.63 |
| 57 | 4.70 | 4.79 | −0.09 | 5.28 | −0.58 |
| 58 | 3.00 | 3.14 | −0.14 | 2.96 | 0.04 |
| 59 | 3.00 | 2.96 | 0.04 | 2.86 | 0.14 |
| 60 | 3.00 | 3.00 | 0.00 | 3.34 | −0.34 |
| 61 | 6.26 | 6.26 | 0.00 | 5.37 | 0.89 |
| 62 | 3.00 | 3.03 | −0.02 | 3.39 | −0.39 |
| 63 | 3.00 | 3.12 | −0.12 | 3.28 | −0.28 |
| 64 | 6.41 | 6.36 | 0.05 | 6.11 | 0.30 |
| 65 | 5.59 | 5.58 | 0.01 | 5.64 | −0.05 |
| 66 | 3.72 | 3.72 | 0.00 | 3.62 | 0.09 |
| 67 | 3.00 | 2.98 | 0.02 | 3.18 | −0.18 |
| 68 | 5.10 | 5.08 | 0.02 | 5.00 | 0.10 |
| 69 | 6.66 | 6.72 | −0.06 | 6.59 | 0.07 |
| 70 | 6.82 | 6.78 | 0.05 | 6.98 | −0.15 |
| 71 | 8.00 | 8.04 | −0.04 | 8.00 | 0.00 |
| 72 | 8.00 | 7.98 | 0.02 | 8.05 | −0.05 |
| Test Set | |||||
| 73 | 6.70 | 6.69 | 0.01 | 6.47 | 0.23 |
| 74 | 4.96 | 4.48 | 0.48 | 4.83 | 0.13 |
| 75 | 3.40 | 4.83 | −1.43 | 6.52 | −3.12 |
| 76 | 3.40 | 3.88 | −0.49 | 2.80 | 0.60 |
| 77 | 6.52 | 6.53 | −0.01 | 6.53 | −0.01 |
| 78 | 6.00 | 6.27 | −0.27 | 6.23 | −0.23 |
| 79 | 5.51 | 4.31 | 1.20 | 3.45 | 2.06 |
| 80 | 3.97 | 5.20 | −1.23 | 5.36 | −1.39 |
| 81 | 6.52 | 6.64 | −0.12 | 5.63 | 0.89 |
| 82 | 6.40 | 8.84 | −2.45 | 11.19 | −4.79 |
| 83 | 4.32 | 3.85 | 0.47 | 4.49 | −0.17 |
| 84 | 3.70 | 4.96 | −1.26 | 3.81 | −0.11 |
| 85 | 6.77 | 5.48 | 1.29 | 6.26 | 0.51 |
| 86 | 3.00 | 3.38 | −0.38 | 3.21 | −0.21 |
| 87 | 3.00 | 4.21 | −1.21 | 5.48 | −2.48 |
| 88 | 3.00 | 2.05 | 0.95 | 5.24 | −2.24 |
| 89 | 5.80 | 5.72 | 0.08 | 6.38 | −0.58 |
| 90 | 3.82 | 4.21 | −0.39 | 3.83 | −0.01 |
| 91 | 3.00 | 4.95 | −1.95 | 6.38 | −3.38 |
| 92 | 6.82 | 7.54 | −0.72 | 7.54 | −0.71 |
| 93 | 8.00 | 7.70 | 0.30 | 7.62 | 0.38 |
| 94 | 7.10 | 7.63 | −0.54 | 7.56 | −0.46 |
| 95 | 8.54 | 9.44 | −0.90 | 8.12 | 0.42 |
| 96 | 4.30 | 3.83 | 0.48 | 3.38 | 0.92 |
| 97 | 4.30 | 3.98 | 0.32 | 3.34 | 0.96 |
| Compounds | Qcv2 | Rncv2 | ONC | SEE | F value |
|---|---|---|---|---|---|
| 1 | −0.181 | 0.069 | 1 | 1.445 | 5.212 |
| 2 | 0.038 | 0.101 | 1 | 1.42 | 7.849 |
| 3 | −0.181 | 0.055 | 1 | 1.456 | 4.109 |
| 4 | −0.11 | 0.04 | 1 | 1.468 | 2.928 |
| 5 | 0.207 | 0.722 | 5 | 0.813 | 34.288 |
| 6 | −0.225 | 0.135 | 1 | 1.393 | 10.968 |
| 7 | −0.061 | 0.055 | 1 | 1.456 | 4.061 |
| 8 | −0.046 | 0.063 | 1 | 1.45 | 4.685 |
| 9 | −0.411 | 0.088 | 1 | 1.43 | 6.781 |
| 10 | −0.019 | 0.069 | 1 | 1.446 | 5.156 |
| 11 | −0.121 | 0.246 | 2 | 1.311 | 11.228 |
| 12 | −0.023 | 0.058 | 1 | 1.454 | 4.272 |
| 13 | −0.028 | 0.242 | 2 | 1.314 | 11.005 |
| 14 | −0.164 | 0.059 | 1 | 1.453 | 4.389 |
| 15 | 0.012 | 0.087 | 1 | 1.431 | 6.681 |
| Compounds | Fitness score | Orientation 1 | Compound | Fitness score | Orientation 1 |
|---|---|---|---|---|---|
| Training Set | |||||
| 1 | 46.7447 | Yes | 37 | 67.605 | Yes |
| 2 | 65.4864 | Yes | 38 | 57.7153 | No |
| 3 | 69.988 | Yes | 39 | 50.9253 | Yes |
| 4 | 68.7748 | Yes | 40 | 69.5187 | Yes |
| 5 | 75.6942 | Yes | 41 | 73.0713 | Yes |
| 6 | 65.2663 | Yes | 42 | 61.2473 | No |
| 7 | 77.8612 | Yes | 43 | 57.4067 | No |
| 8 | 56.0559 | Yes | 44 | 68.8039 | No |
| 9 | 85.5617 | Yes | 45 | 79.0696 | Yes |
| 10 | 77.7628 | Yes | 46 | 51.0306 | Yes |
| 11 | 70.8855 | Yes | 47 | 55.4352 | Yes |
| 12 | 82.8049 | Yes | 48 | 57.8608 | Yes |
| 13 | 63.7736 | Yes | 49 | 59.4957 | Yes |
| 14 | 70.8527 | No | 50 | 65.029 | Yes |
| 15 | 56.3686 | Yes | 51 | 65.035 | Yes |
| 16 | 72.0652 | No | 52 | 56.6576 | Yes |
| 17 | 54.8263 | Yes | 53 | 64.2358 | Yes |
| 18 | 71.7425 | Yes | 54 | 60.8265 | Yes |
| 19 | 69.7473 | Yes | 55 | 51.5799 | Yes |
| 20 | 68.1959 | Yes | 56 | 58.7258 | Yes |
| 21 | 77.6519 | Yes | 57 | 56.3411 | Yes |
| 22 | 78.6929 | Yes | 58 | 66.6992 | Yes |
| 23 | 79.6385 | Yes | 59 | 57.0687 | No |
| 24 | 74.5361 | Yes | 60 | 42.5776 | No |
| 25 | 74.043 | Yes | 61 | 63.0642 | Yes |
| 26 | 79.7058 | Yes | 62 | 60.9584 | Yes |
| 27 | 90.9285 | Yes | 63 | 63.0642 | Yes |
| 28 | 88.8448 | Yes | 64 | 62.6583 | Yes |
| 29 | 59.6149 | No | 65 | 75.5489 | Yes |
| 30 | 69.544 | No | 66 | 52.7054 | Yes |
| 31 | 63.492 | Yes | 67 | 57.6862 | Yes |
| 32 | 45.5814 | Yes | 68 | 88.0785 | Yes |
| 33 | 60.9885 | Yes | 69 | 89.4423 | Yes |
| 34 | 67.8952 | Yes | 70 | 91.0785 | Yes |
| 35 | 75.3107 | Yes | 71 | 100.3275 | Yes |
| 36 | 67.5022 | Yes | 72 | 102.9909 | Yes |
| Test Set | |||||
| 73 | 70.0894 | Yes | 85 | 67.2837 | Yes |
| 74 | 71.9324 | Yes | 86 | 58.983 | Yes |
| 75 | 46.0479 | No | 87 | 55.1520 | Yes |
| 76 | 64.2422 | Yes | 88 | 47.7195 | Yes |
| 77 | 70.7896 | Yes | 89 | 65.82375 | Yes |
| 78 | 66.3318 | Yes | 90 | 56.9968 | Yes |
| 79 | 81.0177 | Yes | 91 | 57.2802 | Yes |
| 80 | 98.5388 | Yes | 92 | 93.4818 | Yes |
| 81 | 58.0293 | Yes | 93 | 94.082 | Yes |
| 82 | 64.0589 | Yes | 94 | 87.3304 | Yes |
| 83 | 61.4473 | Yes | 95 | 70.0355 | Yes |
| 84 | 58.8713 | No | 96 | 70.7513 | No |
| 97 | 81.0516 | No | |||
| Comp. | R1 | R2 | R3 | R4 | pIC50 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Training Set | ||||||||||
| 1 | -OCH3 | -NH2 | H | H | 3.46 | |||||
| 2 |  | -NH2 | H | H | 6.70 | |||||
| 3 |  | -NH2 | H | H | 6.70 | |||||
| 4 |  | -NH2 | H | H | 6.70 | |||||
| 5 |  | -NH2 |  | H | 5.70 | |||||
| 6 |  | -NH2 |  | H | 5.00 | |||||
| 7 |  | -NH2 |  | H | 5.05 | |||||
| 8 | -OCH2CH=CH2 | -NH2 | H | H | 4.70 | |||||
| 9 |  | -NH2 | -CH2COOCH2CH3 | H | 4.52 | |||||
| 10 |  | -NH2 | -CH2C≡N | H | 4.89 | |||||
| 11 |  | -NH2 | -CH2CONH2 | H | 4.33 | |||||
| 12 (R/S) 1 |  | -NH2 | -CH2CH(OH)CH2CH3 (R/S) | H | 4.89 | |||||
| 13 |  | H | H | H | 4.07 | |||||
| 14 |  | -NH2 | -CH2COOCH2CH3 | - | 3.40 | |||||
| 15 |  | -NH2 | -CH2CONH2 | - | 3.40 | |||||
| 16 (R/S) |  | -NH2 | -CH2CH(OH)CH2CH3 (R/S) | - | 3.40 | |||||
| 17 |  | -NH2 | H | H | 3.40 | |||||
| 18 |  | -NH2 | H | H | 3.40 | |||||
| 19 |  | -NH2 | H | H | 6.52 | |||||
| 20 |  | -NH2 | H | H | 6.30 | |||||
| 21 |  | -NH2 | H | H | 6.30 | |||||
| 22 |  | -NH2 | H | H | 5.40 | |||||
| 23 |  | -NH2 | H | H | 6.52 | |||||
| 24 |  | -NH2 | -CHO | H | 6.40 | |||||
| 25 |  | -NH2 | -CH3 | H | 5.59 | |||||
| 26 (R/S) |  | -NH2 | -CH2CH(OH)CH2Cl (R/S) | H | 4.74 | |||||
| 27 (R/S) |  | -NH2 | -CH2CH(OH)CH2NHCH-(CH3)2 (R/S) | H | 3.97 | |||||
| 28 (R/S) |  | -NH2 | -CH2CH(OH)CH2OCH-(CH3)2 (R/S) | H | 5.15 | |||||
| 29 |  | -NHCOCH3 | H | H | 4.62 | |||||
| 30 |  | -NH2 | -CH3 | - | 4.28 | |||||
| 31 |  | -NH2 | H | H | 4.24 | |||||
| 32 |  | -NH2 | H | H | 3.52 | |||||
| 33 |  | -NH2 | H | -NH2 | 6.15 | |||||
| 34 |  | -NH2 | H | -OH | 6.52 | |||||
| 35 |  | -NH2 | H | -Br | 7.10 | |||||
| 36 |  | -OH | H | H | 4.22 | |||||
| 37 |  | -OH | H | -OH | 4.60 | |||||
| 38 |  | -NHCOCH3 | H | -OH | 4.19 | |||||
| 39 |  | -NHCH3 | H | H | 3.80 | |||||
| 40 |  | -NH2 | H | H | 6.82 | |||||
| 41 |  | -NH2 | H | H | 6.96 | |||||
| 42 |  | -NH2 | H | H | 4.28 | |||||
| 43 |  | -NH2 | H | H | 3.91 | |||||
| 44 |  | -NH2 | H | H | 3.81 | |||||
| 45 |  | -NH2 | H | H | 8.52 | |||||
| 46 | -OCH2CH3 | -NH2 | H | H | 3.00 | |||||
| 47 | -O(CH2)2CH3 | -NH2 | H | H | 3.00 | |||||
| 48 | -O(CH2)3CH3 | -NH2 | H | H | 3.31 | |||||
| 49 | -O(CH2)2CH(CH3)2 | -NH2 | H | H | 3.00 | |||||
| 50 | -O(CH2)5CH3 | -NH2 | H | H | 3.26 | |||||
| 51 |  | -NH2 | H | H | 3.00 | |||||
| 52 | -OCH2CH(=CH2)CH3 | -NH2 | H | H | 4.60 | |||||
| 53 |  | -NH2 | H | H | 3.26 | |||||
| 54 | -OCH2CH(=CH2)CH2CH3 | -NH2 | H | H | 4.80 | |||||
| 55 | -OCH2CH(=CH2)CH2(CH3)2 | -NH2 | H | H | 3.00 | |||||
| 56 |  | -NH2 | H | H | 4.11 | |||||
| 57 | -OCH2C≡CH | -NH2 | H | H | 4.70 | |||||
| 58 |  | -NH2 | H | H | 3.00 | |||||
| 59 |  | -NH2 | H | H | 3.00 | |||||
| 60 |  | -NH2 | H | H | 3.00 | |||||
| 61 |  | -NH2 | H | H | 6.26 | |||||
| 62 |  | -NH2 | H | H | 3.00 | |||||
| 63 |  | -NH2 | H | H | 3.00 | |||||
| 64 |  | -NH2 | H | H | 6.41 | |||||
| 65 |  | -NH2 | H | H | 5.59 | |||||
| 66 | -OCH2COCH3 | -NH2 | H | H | 3.72 | |||||
| 67 | -OCH2COCH(CH3)2 | -NH2 | H | H | 3.00 | |||||
| 68 |  | -NH2 |  | H | 5.10 | |||||
| 69 |  | -NH2 |  | H | 6.66 | |||||
| 70 |  | -NH2 |  | H | 6.82 | |||||
| 71 |  | -NH2 |  | H | 8.00 | |||||
| 72 |  | -NH2 |  | H | 8.00 | |||||
| Test Set | ||||||||||
| 73 |  | -NH2 | H | H | 6.70 | |||||
| 74 |  | -NH2 |  | H | 4.96 | |||||
| 75 |  | -NH2 | -CH2C≡CH | - | 3.40 | |||||
| 76 |  | -NH2 | H | H | 3.40 | |||||
| 77 |  | -NH2 | H | H | 6.52 | |||||
| 78 |  | -NH2 | H | H | 6.00 | |||||
| 79 |  | -NH2 | -CH2OCOC(CH3)3 | H | 5.51 | |||||
| 80 (R/S) |  | -NH2 | -CH2CH(OH)CH2NH-C(CH3)3 (R/S) | H | 3.97 | |||||
| 81 |  | -NH2 | H | -CH3 | 6.52 | |||||
| 82 |  | -NH2 | H | -CF3 | 6.40 | |||||
| 83 |  | -F | H | H | 4.32 | |||||
| 84 |  | -N(CH3)2 | H | H | 3.70 | |||||
| 85 |  | -NH2 | H | H | 6.77 | |||||
| 86 | -O(CH2)4CH3 | -NH2 | H | H | 3.00 | |||||
| 87 (R/S) |  (R/S) (R/S) | -NH2 | H | H | 3.00 | |||||
| 88 |  | -NH2 | H | H | 3.00 | |||||
| 89 |  | -NH2 | H | H | 5.80 | |||||
| 90 | -OCH2COCH2CH3 | -NH2 | H | H | 3.82 | |||||
| 91 |  | -NH2 | H | H | 3.00 | |||||
| 92 |  | -NH2 |  | H | 6.82 | |||||
| 93 |  | -NH2 |  | H | 8.00 | |||||
| 94 |  | -NH2 |  | H | 7.10 | |||||
| 95 |  | -NH2 | H | H | 8.54 | |||||
| 96 |  |  | H | H | 4.30 | |||||
| 97 |  |  |  | H | 4.30 | |||||
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, G.; Fan, T.; Zhang, N.; Ren, T.; Zhao, L.; Zhong, R. Identification of the Structural Features of Guanine Derivatives as MGMT Inhibitors Using 3D-QSAR Modeling Combined with Molecular Docking. Molecules 2016, 21, 823. https://doi.org/10.3390/molecules21070823
Sun G, Fan T, Zhang N, Ren T, Zhao L, Zhong R. Identification of the Structural Features of Guanine Derivatives as MGMT Inhibitors Using 3D-QSAR Modeling Combined with Molecular Docking. Molecules. 2016; 21(7):823. https://doi.org/10.3390/molecules21070823
Chicago/Turabian StyleSun, Guohui, Tengjiao Fan, Na Zhang, Ting Ren, Lijiao Zhao, and Rugang Zhong. 2016. "Identification of the Structural Features of Guanine Derivatives as MGMT Inhibitors Using 3D-QSAR Modeling Combined with Molecular Docking" Molecules 21, no. 7: 823. https://doi.org/10.3390/molecules21070823