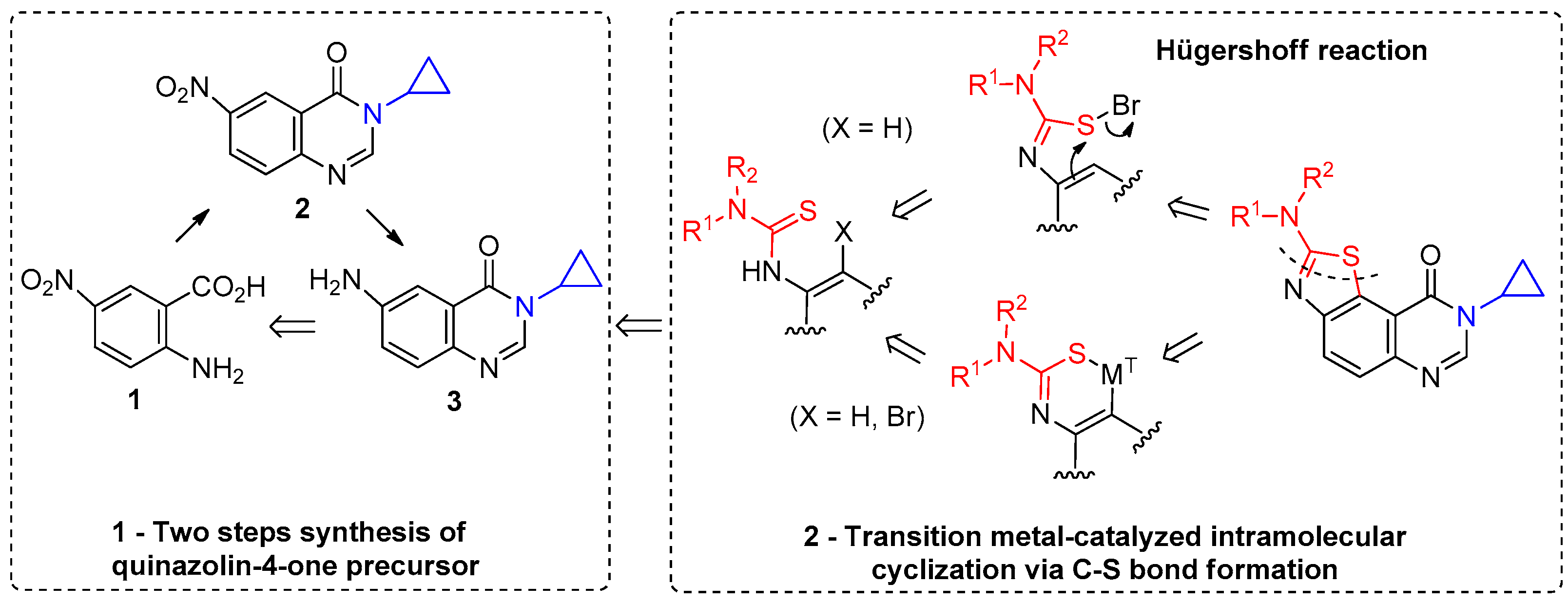
3.2. Chemistry
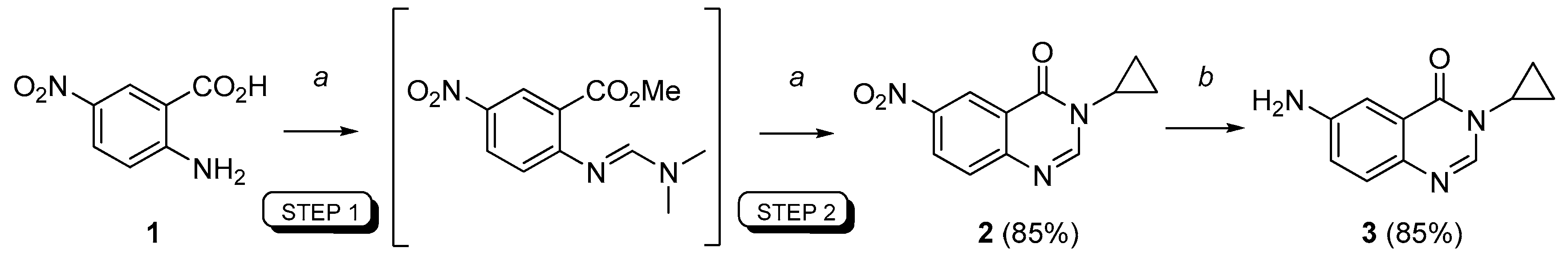
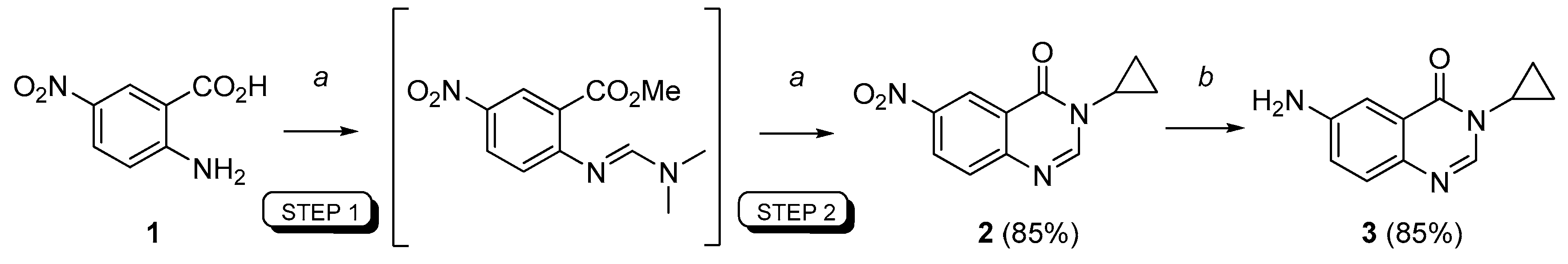
3-Cyclopropyl-6-nitroquinazolin-4(3H)-one (2). A stirred suspension of 5-nitroanthranilic acid 1 (1.5 mmol, 1 equiv.) in DMFDMA (500 µL, 2.5 equiv.) and DMF (1.5 mL) was heated at 100 °C for 15 min under microwave (after a ramp period of 2 min at 900 W). Solvents were removed in vacuo and cyclopropylamine (1.65 mmol, 1.1 equiv.) was added followed with AcOH (1.5 mL). The mixture was irradiated at 100 °C (900 W) for 15 min (ramp: 2 min). Evaporation of the solvent gave a crude product which was purified by column chromatography using petroleum ether/methylene chloride (100:0 to 0:100, v/v) as eluent. Compound 2 was isolated as a light yellow solid (295 mg, 85%), mp. 174–176 °C; 1H-NMR (DMSO-d6) δ 8.82 (d, J = 2.7 Hz, 1H, H5), 8.54 (dd, J = 9.0, 2.7 Hz, 1H, H7), 8.45 (s, 1H, H2), 7.86 (d, J = 9.0 Hz, 1H, H8), 3.34–3.25 (m, 1H, NCH), 1.08–0.99 (m, 4H, CH);13C-NMR (DMSO-d6) δ 160.6, 151.6, 151.3, 145.1, 128.9, 128.1, 121.9, 121.4, 29.5, 5.9; νmax 3356, 3102, 1675, 1600, 1512, 1300, 1275, 936, 855, 752 cm−1; HRMS calcd for C11H10N3O3 [M + H]+ 232.0722 found 232.0719.
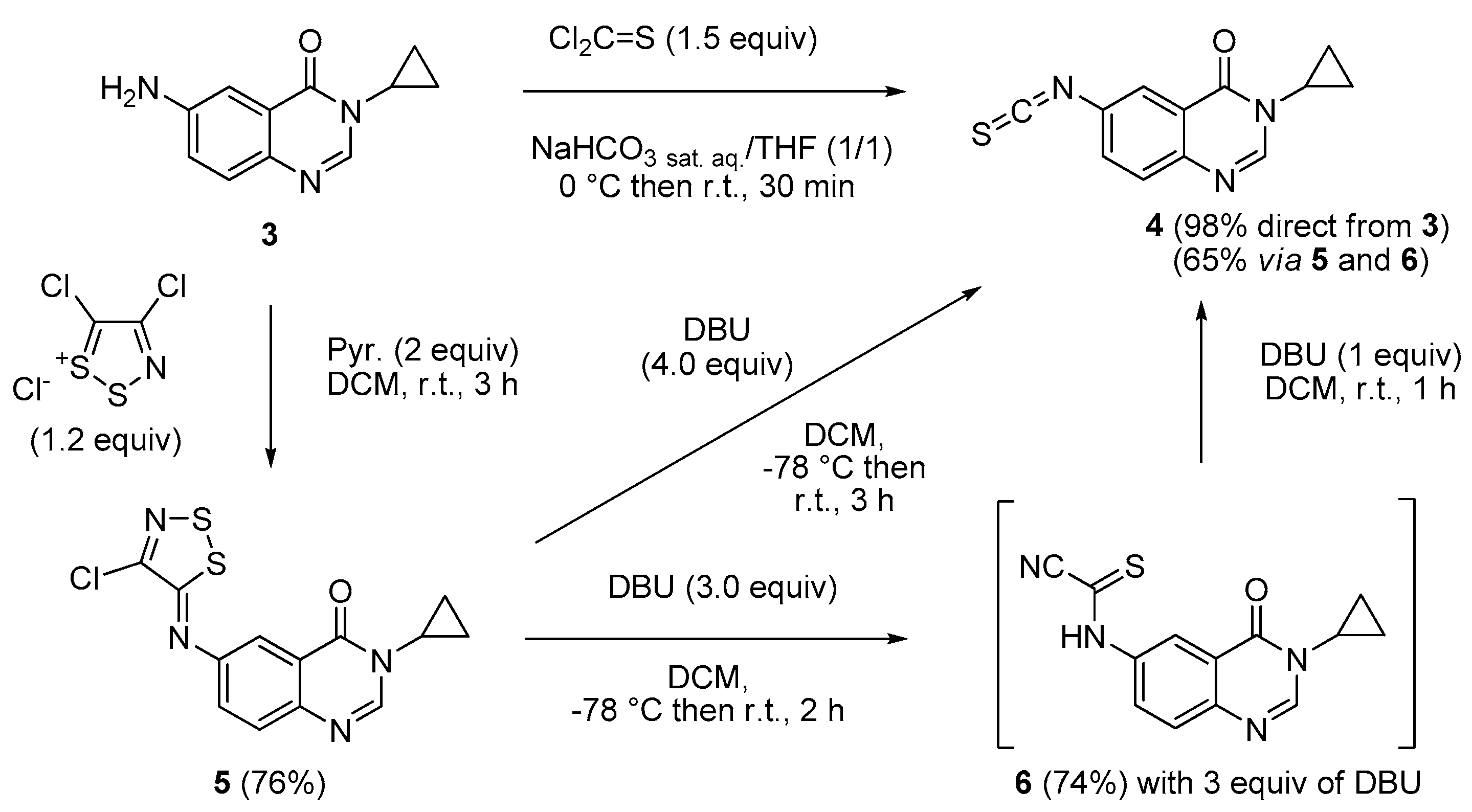
6-Amino-3-cyclopropylquinazolin-4(3H)-one (3). A stirred mixture of 6-nitro-3-cyclopropyl-quinazolin-4(3H)-one 2 (15 mmol, 1.0 equiv.), ammonium formate (5.0 equiv.) and a catalytic (10 mol %) amount of 10% palladium charcoal in ethanol (0.2 M solution) was heated under microwaves for 20 min (300 W, 85 °C). The reaction mixture was filtered through Celite® and washed with hot ethanol. The solvent was removed in vacuo to give the crude product which was dissolved in ethyl acetate, washed with water, dried over MgSO4 and concentrated under reduced pressure to give the reduced compound as a pale yellow solid (2.6 g, 85%), mp. 172–174 °C; 1H-NMR (DMSO-d6) δ 7.94 (s, 1H, H2), 7.37 (d, J = 8.7 Hz, 1H, H8), 7.22 (d, J = 2.7 Hz, 1H, H5), 7.07 (dd, J = 8.7, 2.7 Hz, 1H, H7), 5.66 (br s, 2H, NH2), 3.20–3.16 (m, 1H, NCH), 1.03–1.01 (m, 2H, CH), 0.90 (m, 2H, CH); 13C-NMR (DMSO-d6) δ 161.3, 148.1, 142.7, 138.3, 127.9, 122.3, 122.0, 106.1, 28.9, 6.0; νmax 3420, 3336, 3222, 1653, 1627, 1600, 1491, 1325, 830, 791 cm−1; HRMS calcd for C11H12N3O [M + H]+ 202.0980 found 202.0974.
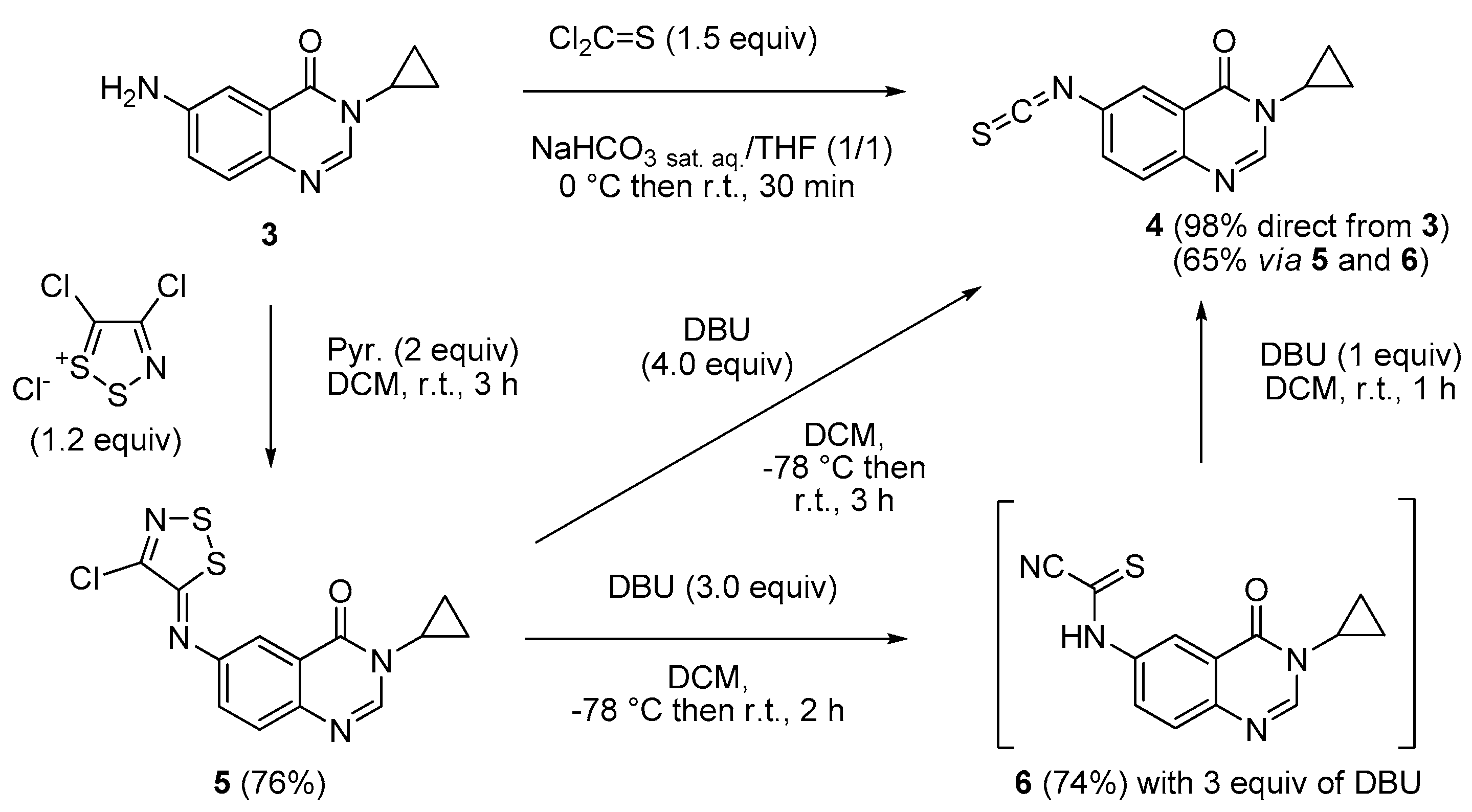
3-Cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (4). Method A: A solution of DBU (542 mg, 3.57 mmol, 3.0 equiv.) in methylene chloride (1.0 mL) was added drop wise to a stirred solution of 6-aminoquinazolin-4(3H)-one 5 (400 mg, 1.19 mmol) in methylene chloride (7 mL) maintained at −78 °C. The resulting mixture was warmed to room temperature and stirred for 2 h. One equivalent of DBU (181 mg, 1.19 mmol) was added and the black solution was stirred for 1 h. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (0/100 to 50/50, v/v) as eluent to furnish 4 as a white solid (188 mg, 65%). Method B: Thiophosgene (0.57 mL, 7.47 mmol, 1.5 equiv.) was added drop wise to a stirred solution of 6-aminoquinazolin-4(3H)-one 3 (1.00 g, 4.97 mmol) in a mixture of THF (5.0 mL) and a saturated solution of NaHCO3 (5.0 mL) maintained at 0 °C. The resulting mixture was warmed to room temperature and stirred for 30 min. On completion, the reaction mixture was diluted with water (25 mL) and ethyl acetate (25 mL). The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed with water and brine. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (0/100 to 50/50, v/v) as eluent to furnish 4 as a colorless solid (1.18 g, 98%); mp. 146–148 °C; 1H-NMR (DMSO-d6) δ 8.31 (s, 1H, H2), 8.04 (d, J = 2.2 Hz, 1H, H5), 7.82 (dd, J = 8.6, 2.2 Hz, 1H, H7), 7.70 (d, J = 8.6 Hz, 1H, H8), 3.28–3.21 (m, 1H, NCH), 1.08–0.97 (m, 4H, CH); 13C-NMR (CDCl3) δ 161.2, 147.4, 146.2, 137.8, 131.7, 130.6, 129.2, 123.3, 122.9, 29.6, 6.7; νmax 2118, 1669, 1602, 833, 535 cm−1; HRMS calcd for C12H10N3OS [M + H]+ 244.0545 found 244.0537.
6-[(4-Chloro-5H-1,2,3-dithiazol-5-yl)amino]-3-cyclopropylquinazolin-4(3H)-one (5). A suspension of 6-amino-3-cyclopropylquinazolin-4(3H)-one (3, 300 mg, 1.49 mmol) and 4,5-dichloro-1,2,3-dithiazolium chloride (1.2 equiv.) in DCM (0.1 M solution) was stirred for 1 h at room temperature under an argon atmosphere. Pyridine (2.0 equiv.) was added and the mixture was stirred again for 2 h at room temperature. The resulting solution was concentrated under vacuo to give a crude residue which was purified by chromatography on silica gel with EtOAc/DCM (5/95 then 50/50, v/v) to give the expected iminodithiazole as a yellow solid (382 mg, 76%), mp. 190–192 °C; 1H-NMR (DMSO-d6) δ 8.29 (s, 1H, H2), 7.97 (d, J = 2.4 Hz, 1H, H5), 7.78 (d, J = 8.7 Hz, 1H, H8), 7.65 (dd, J = 8.7, 2.4 Hz, 1H, H7), 3.20–3.16 (m, 1H, NCH), 1.09–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 161.0, 160.4, 149.0, 147.7, 147.0, 145.4, 129.1, 128.2, 122.3, 114.2, 29.3, 5.9; νmax 3073, 1659, 1592, 1530, 1454, 1305, 951, 842, 581 cm−1; HRMS calcd for C13H10N4OS2Cl2 [M + H]+ 336.9985 found 336.9974.
(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)carbamothioyl cyanide (6). A solution of DBU (542 mg, 3.57 mmol, 3.0 equiv.) in methylene chloride (1.0 mL) was added drop wise to a stirred solution of iminodithiazole 5 (400 mg, 1.19 mmol) in methylene chloride (7.0 mL) maintained at −78 °C. The resulting mixture was warmed to room temperature and stirred for 2 h. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate/methylene chloride (0/100 to 50/50, v/v) as eluent to furnish 6 as an orange solid (250 mg, 74%), mp. 196–198 °C; 1H-NMR (DMSO-d6) δ 13.72 (br s, 1H, NH), 8.91 (d, J = 2.4 Hz, 1H, H5), 8.32 (s, 1H, H2), 8.16 (dd, J = 9.0, 2.7 Hz, 1H, H7), 7.76 (d, J = 9.0 Hz, H8), 3.20–3.16 (m, 1H, NCH), 1.09–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 161.1, 160.8, 148.4, 146.0, 136.9, 128.7, 128.0, 121.4, 118.7, 114.0, 29.3, 5.9; νmax 3519, 3430, 2944, 2055, 1641, 1601, 1487, 1389, 1256, 1193, 1021, 847, 739 cm−1; HRMS calcd for C13H11N4OS [M + H]+ 271.0654 found 271.0655.
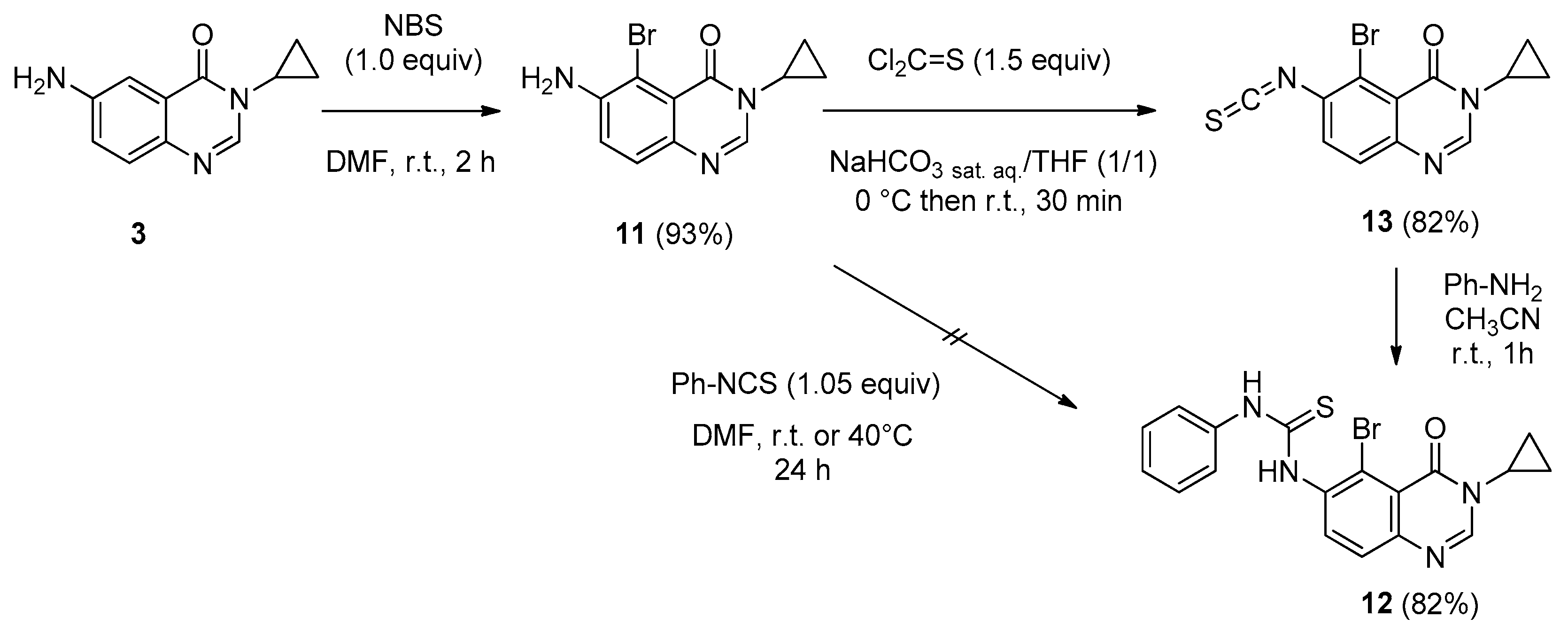
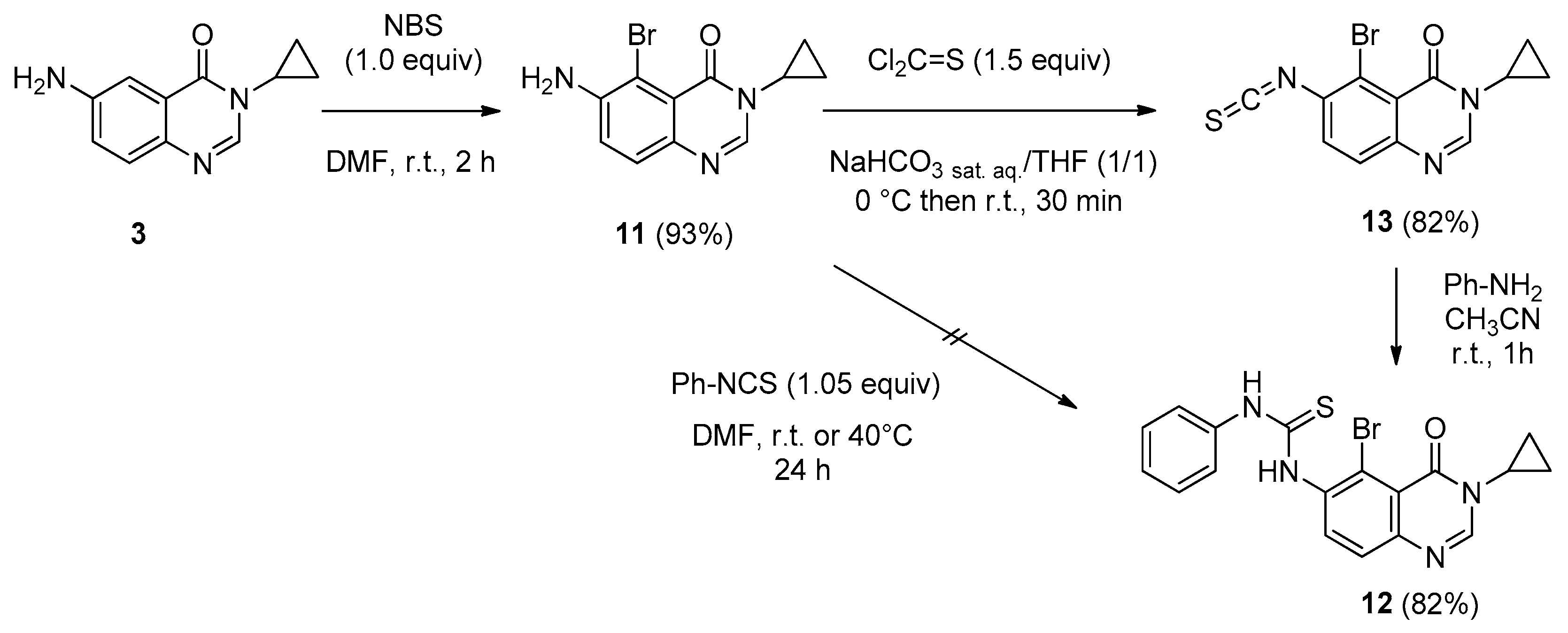
6-Amino-5-bromo-3-cyclopropylquinazolin-4(3H)-one (11). To a stirred solution of 6-amino-3-cyclo-propylquinazolinone 3 (5.5 g, 27.3 mmol) was added NBS (4.9 g, 27.3 mmol, 1.0 equiv.) at room temperature. After 2 h of stirring at room temperature, the solvent was removed under vacuum and the crude residue was purified by chromatography on silica gel using EtOAc/DCM (20/80 to 50/50, v/v) as eluent to give the compound 11 as a colorless solid (7.1 g, 93%), mp. 172–174 °C; 1H-NMR (DMSO-d6) δ 8.03 (s, 1H, H2), 7.40 (d, J = 8.7 Hz, 1H, H8), 7.28 (d, J = 8.7 Hz, 1H, H7), 5.86 (br s, 2H, NH2), 3.18–3.13 (m, 1H, NCH), 1.03–0.99 (m, 2H, CH), 0.91–0.85 (m, 2H, CH); 13C-NMR (DMSO-d6) δ 159.6, 146.1, 144.0, 140.4, 127.4, 121.7, 119.5, 100.5, 29.4, 6.0; νmax 3088, 3017, 2926, 2848, 1675, 1508, 1335, 1141, 855, 752, 699 cm−1; HRMS calcd for C11H11N3O79Br [M + H]+ 280.0085 found 280.0095 (100%), calcd for C11H11N3O81Br [M + H]+ 282.0065 found 282.0072 (93%).
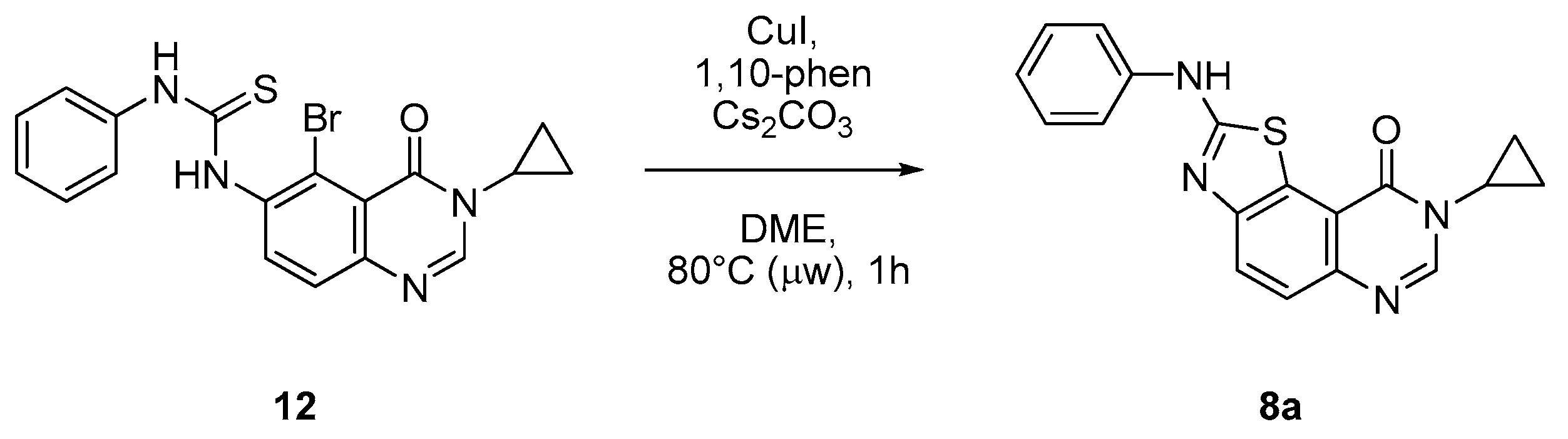
1-(5-Bromo-3-cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-3-phenylthiourea (12). A solution of isothiocyanate 13 (0.200 g, 0.62 mmol) and aniline (61 µL, 0.65 mmol, 1.05 equiv.) in acetonitrile (2.0 mL) was stirred at room temperature overnight. the solvent was removed under vacuum und the crude residue was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (0/1 to 5/5, v/v) to furnish the expected thiourea 12 as a colorless solid (0.211 g, 82%), mp. 212–214 °C; 1H-NMR (DMSO-d6) δ 10.11 (s, 1H, NH), 9.47 (s, 1H, NH), 8.31 (s, 1H, H2), 7.92 (d, J = 8.7 Hz, 1H, H8), 7.62 (d, J = 8.7 Hz, 1H, H7), 7.57–7.55 (m, 2H, Ph), 7.39–7.34 (m, 2H, Ph), 7.19–7.14 (m, 1H, Ph), 3.22–3.17 (m, 1H, NCH), 1.04–0.92 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.2, 159.6, 148.2, 147.8, 139.0, 137.9, 135.1, 128.6, 126.4, 124.8, 123.7, 119.6, 119.1, 29.2, 6.0; νmax 3315, 2930, 1677, 1619, 1515, 1324, 1297, 897, 696, 501 cm−1; HRMS calcd for C18H16N4OS79Br [M + H]+ 415.0228 found 415.0233 (93%), for C18H16N4OS81Br [M + H]+ 417.0208 found 417.0197 (100%).
5-Bromo-3-cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (13). According to the previous procedure described for compound 4, 6-amino-5-bromoquinazolin-4(3H)-one (11, 1.00 g, 3.57 mmol) was stirred with thiophosgene (0.410 g, 5.35 mmol, 1.5 equiv.) in a mixture of THF (3.5 mL) and a saturated solution of NaHCO3 (3.5 mL) to furnish 13 as a colorless solid (0.940 g, 82%), mp. 210–212 °C; 1H-NMR (DMSO-d6) δ 8.36 (s, 1H, H2), 7.90 (d, J = 8.7 Hz, 1H, H8), 7.67 (d, J = 8.7 Hz, 1H, H7), 3.23–3.16 (m, 1H, NCH), 1.05–0.93 (m, 4H, CH); 13C-NMR (CDCl3) δ 159.9, 148.0, 147.6, 138.7, 132.1, 131.3, 128.3, 121.1, 120.0, 30.1, 6.7 (2C); νmax 3061, 2921, 2122, 1688, 1588, 1454, 1314, 1259, 838 cm−1; HRMS calcd for C12H9N3OS79Br [M + H]+ 321.9650 found 321.9647, for C12H9N3OS81Br [M + H]+ 323.9629 found 323.9630.
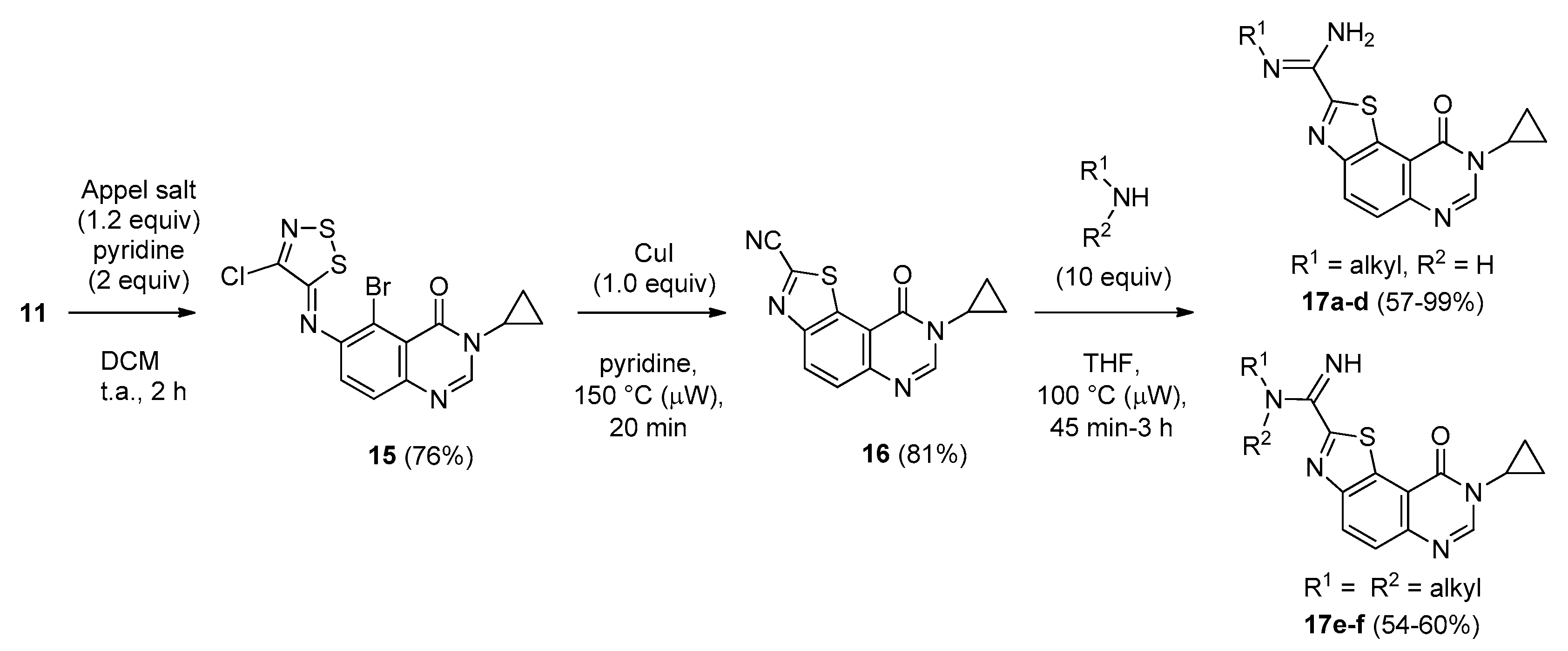
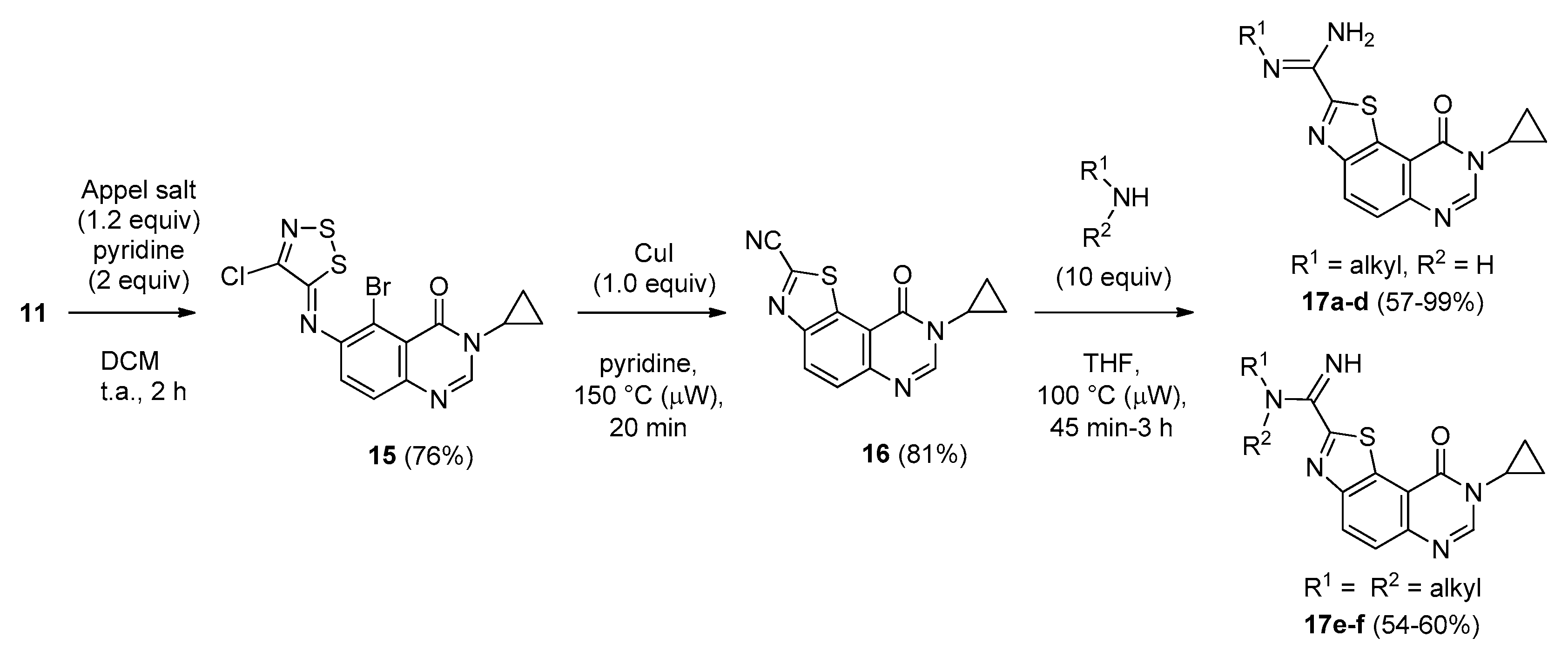
(Z)-5-Bromo-6-[(4-chloro-5H-1,2,3-dithiazol-5-ylidene)amino]-3-cyclopropylquinazolin-4(3H)-one (15). A suspension of 6-amino-5-bromo-3-cyclopropylquinazolin-4(3H)-one (11, 2.0 g, 1.0 equiv.) and 4,5-dichloro-1,2,3-dithiazolium chloride (1.2 equiv.) in DCM (0.1 M solution) was stirred for 1 h at room temperature under an argon atmosphere. Pyridine (2.0 equiv.) was added and the mixture was stirred again for 2 h at room temperature. The resulting solution was concentrated under vacuo to give a crude residue which was purified by chromatography on silica gel with EtOAc/DCM (5/95 then 50/50, v/v) to give the desired iminodithiazole as an orange solid (2.3 g, 76%), mp. 204–206 °C; 1H-NMR (DMSO-d6) δ 8.33 (s, 1H, H2), 7.75 (d, J = 8.7 Hz, 1H, H7), 7.58 (d, J = 8.7 Hz, 1H, H8), 3.24–3.17 (m, 1H, NCH), 1.06–1.00 (m, 2H, CH), 0.97–0.94 (m, 2H, CH); 13C-NMR (DMSO-d6) δ 163.2, 159.6, 150.5, 147.9, 146.8, 145.8, 129.0, 124.6, 120.3, 111.3, 29.7, 6.0 (2C); νmax 3003, 1862, 1675, 1593, 1452, 1325, 1293, 1148, 826, 759 cm−1; HRMS calcd for C13H9N4OS279BrCl [M + H]+ 414.9090 found 414.9088.
8-Cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carbonitrile (16). A suspension of imine 15 (2.00 g, 1.0 equiv.), copper iodide (CuI, 2.0 equiv.) in pyridine (0.33 M solution) was irradiated under microwaves at 115 °C (power input: 300 W) for 20 min. After cooling, the mixture was diluted in EtOAc, washed with a saturated aqueous solution of sodium thiosulfate. The organic layer was dried over MgSO4 and the solvent was removed in vacuo. The crude residue was purified by column chromatography on silica gel with EtOAc/methylene chloride (0/100 to 20/80, v/v) as eluent to give the expected compound as a colorless solid (1.05 g, 81%), mp. 248–250 °C; 1H-NMR (DMSO-d6) δ 8.63 (d, J = 8.7 Hz, 1H, H4), 8.61 (s, 1H, H2), 7.99 (d, J = 9.0 Hz, 1H, H5), 3.42–3.36 (m, 1H, NCH), 1.13–1.09 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 160.2, 150.3, 149.5, 148.2, 139.2, 131.6, 129.9, 127.8, 115.4, 113.5, 29.7, 5.9 (2C); νmax 3067, 2233 (C≡N), 1664, 1579, 1441, 1353, 1303, 1222, 1038, 839, 692 cm−1; HRMS calcd for C13H9N4OS [M + H]+ 269.0497 found 269.0487.
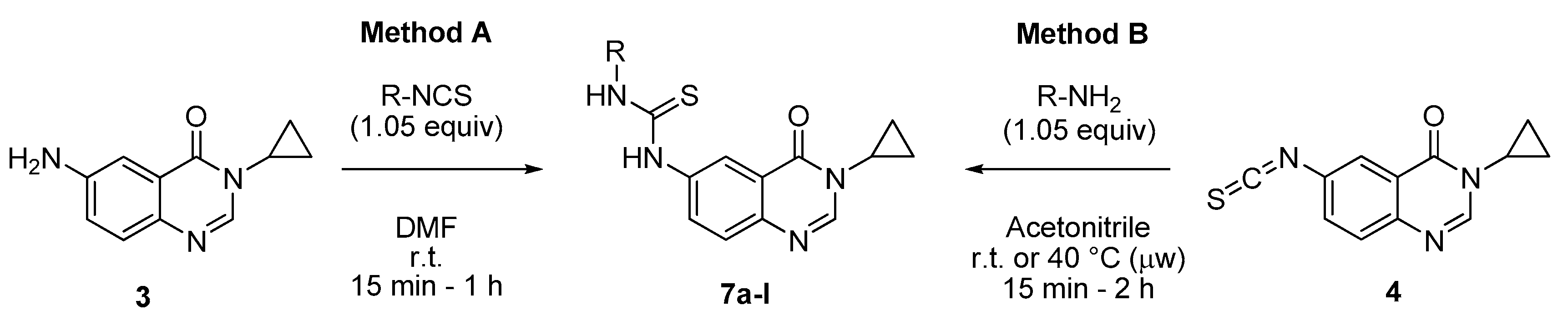
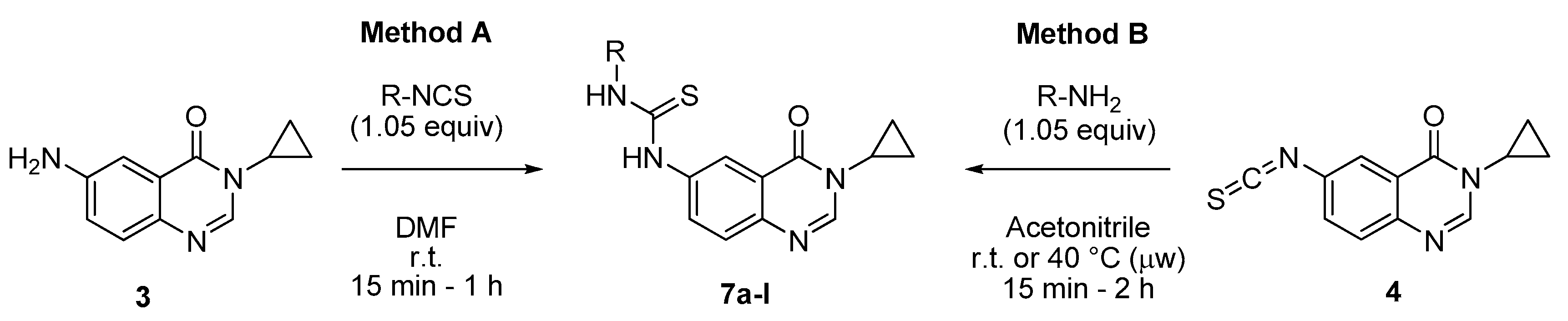
3.2.1. General Procedures for the Synthesis of N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-aryl or Alkylthioureas 7a–l from 3-Cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (4) or 6-Amino-3-cyclopropylquinazolin-4(3H)-one (3)
Method A: To a stirred solution of isothiocyanate 4 (250 mg, 1.03 mmol) in acetonitrile (3 mL) was added the appropriate amine (1.08 mmol, 1.05 equiv.) at room temperature. The resulting mixture was stirred at to room temperature for nucleophilic amines for 15 min to 2 h or under microwave at 40 °C for 1 h. On completion, the solvent was evaporated under vacuum. Trituration of crude mixture in diethyl ether (6 mL) followed by filtration furnished the expected thiourea derivatives 7a, c–d, g, j–l.
Method B: To a stirred solution of 6-amino-3-cyclopropylquinazolinone (3, 250 mg, 1.24 mmol) in DMF (4 mL) was added the appropriate arylisothiocyanate (1.05 equiv.) at room temperature. The resulting mixture was stirred at room temperature for 15 min to 4 h. On completion, the solvent was evaporated under vacuum. Trituration of crude mixture in diethyl ether (8 mL) followed by filtration furnished the expected thiourea derivatives 7a–b; e–f; h, i.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-phenylthiourea (7a): According to the method A (40 °C, 1 h) or the method B (r.t., 2 h), product 7a was obtained as a colorless solid (291 mg, 84%) or (333 mg, 80%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.09 (s, 1H, NH), 10.00 (s, 1H, NH), 8.25 (d, J = 1.8 Hz, 1H, H5), 8.21 (s, 1H, H2), 7.92 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.60 (d, J = 8.7 Hz, 1H, H8), 7.46 (d, J = 8.0 Hz, 2H, Ph), 7.34 (t, J = 8.0 Hz, 2H, Ph), 7.14 (t, J = 8.0 Hz, 1H, Ph), 3.26–3.19 (m, 1H, NCH), 1.03–0.92 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.0, 146.8, 144.1, 139.1, 138.3, 130.1, 128.5, 127.0, 124.5, 123.6, 121.1, 118.8, 29.1, 5.9; νmax 3292, 3069, 2990, 1649, 1592, 1511, 1492, 1303, 1246, 1163, 1037, 821, 610, 580 cm−1; HRMS calcd for C18H17N4OS [M + H]+ 337.1123 found 337.1109.
N-(4-Chlorophenyl)-N′-(3-cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)thiourea (7b): According to the method B (r.t., 1 h), compound 7b was obtained as a colorless solid (418 mg, 91%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.17 (s, 1H, NH), 10.06 (s, 1H, NH), 8.26 (d, J = 1.8 Hz, 1H, H5), 8.23 (s, 1H, H2), 7.93 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.63 (d, J = 8.7 Hz, 1H, H8), 7.53 (d, J = 8.7 Hz, 2H, ArH), 7.42 (d, J = 8.7 Hz, 2H, ArH), 3.28–3.18 (m, 1H, NCH), 1.08–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.8, 161.1, 147.0, 144.2, 138.2, 130.2, 128.5, 128.4, 127.2, 125.3, 121.3, 119.0, 29.2, 6.0; νmax 3290, 1645, 1506, 1488, 1305, 1245, 1087, 787, 654, 525 cm−1; HRMS calcd for C18H16N4OS Cl [M + H]+ 371.0733 found 371.0721.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-fluorophenyl)thiourea (7c): According to the method A (r.t., 2 h), compound 7c was isolated as a colorless solid (331 mg, 90%), mp. 194–196 °C; 1H-NMR (DMSO-d6) δ 10.07 (s, 1H, NH), 9.94 (s, 1H, NH), 8.26 (d, J = 2.4 Hz, 1H, H5), 8.22 (s, 1H, H2), 7.93 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.62 (d, J = 8.7 Hz, 1H, H8), 7.50–7.46 (m, 2H, ArH), 7.22–7.16 (m, 2H, ArH), 3.25–3.20 (m, 1H, NCH), 1.08–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.1, 161.1, 159.3 (d, J = 240 Hz, C4′-F), 146.9, 144.1, 138.3, 135.5, 130.2, 127.1, 126.2 (d, J = 8.25 Hz, 2C, C2′-F), 121.2, 119.0, 115.2 (d, J = 22.5 Hz, 2C, C3′-F), 29.2, 6.0; νmax 3324, 3195, 3055, 1651, 1633, 1606, 1510, 1300, 1221, 836, 717, 545 cm−1; HRMS calcd for C18H16N4OFS [M + H]+ 355.1029 found 355.1014.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-[4-(trifluoromethyl)phenyl)]-thiourea (7d): According to the method A (40 °C, 1 h), compound 7d was isolated as a light yellow solid (351 mg, 84%), mp. 198–200 °C; 1H-NMR (DMSO-d6) δ 10.33 (s, 1H, NH), 10.29 (s, 1H, NH), 8.28 (d, J = 2.7 Hz, 1H, H5), 8.23 (s, 1H, H2), 7.94 (dd, J = 8.7, 2.7 Hz, 1H, H7), 7.76 (d, J = 8.7 Hz, 2H, ArH), 7.67 (d, J = 8.7 Hz,2H, ArH), 7.64 (d, J = 8.7 Hz, 1H, H8), 3.25–3.21 (m, 1H, NCH), 1.08–0.94 (m, 4H, CH); 19F NMR (282 MHz, DMSO-d6) δ −60.4 (3F, CF3); 13C-NMR (DMSO-d6) δ 179.8, 161.1, 147.1, 144.3, 143.1, 138.0, 130.2, 127.2, 125.7 (q, J = 37.5 Hz, 2C, C3′-F), 124.1 (q, J = 32.0 Hz, C4′-F), 122.9, 121.2, 119.0, 29.2, 6.0; νmax 3167, 2988, 1684, 1604, 1487, 1323, 1289, 1190, 1068, 835, 613 cm−1; HRMS calcd for C19H16N4OF3S [M + H]+ 405.0997 found 405.0996.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-nitrophenyl)thiourea (7e): According to the method B (r.t., 30 min.), compound 7e was isolated as a yellow solid (351 mg, 74%), mp. 198–200 °C; 1H-NMR (DMSO-d6) δ 10.56 (s, 1H, NH), 10.55 (s, 1H, NH), 8.31 (d, J = 2.3 Hz, 1H, H5), 8.24 (s, 1H, H2), 8.22 (d, J = 9.1 Hz, 2H, ArH), 7.95 (dd, J = 8.7, 2.3 Hz, 1H, H7), 7.85 (d, J = 8.7 Hz, 2H, ArH), 7.65 (d, J = 8.7 Hz, 1H, H8), 3.28–3.22 (m, 1H, NCH), 1.06–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.5, 161.0, 147.2, 146.0, 144.4, 142.4, 137.8, 130.1, 127.3, 122.4, 121.3, 119.1, 29.2, 6.0; νmax 3331, 3215, 3094, 3067, 1650, 1626, 1490, 1322, 1246, 1106, 847, 705, 527 cm−1; HRMS calcd for C18H16N5O3S [M + H]+ 382.0974 found 382.0959.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(2,4-dichlorophenyl)thiourea (7f): According to the method B (r.t., 1 h), compound 7f was isolated as a colorless solid (446 mg, 89%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.28 (s, 1H, NH), 9.70 (s, 1H, NH), 8.32 (d, J = 2.4 Hz, 1H, H5), 8.24 (s, 1H, H2), 7.95 (dd, J = 2.4, 8.7 Hz, 1H, H7), 7.72 (d, J = 2.4 Hz, 1H, H3′); 7.64 (d, J = 8.7 Hz, 1H, H8), 7.61 (d, J = 8.4 Hz, 1H, H6′), 7.46 (dd, J = 2.4, 8.4 Hz, 1H, H5′), 3.28–3.20 (m, 1H, NCH), 1.08–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.5, 161.1, 147.1, 144.3, 138.0, 135.4, 131.4, 131.2, 131.0, 130.2, 129.0, 127.5, 127.2, 121.3, 119.3, 29.2, 6.0; νmax 3234, 1650, 1518, 1445, 1346, 1209, 840, 808, 665, 499 cm−1; HRMS calcd for C18H15N4OSCl2 [M + H]+ 405.0344 found 405.0344.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(pyridin-3-yl)thiourea (7g): According to the method A (40 °C (µW), 1 h), compound 7g was isolated as a colorless solid (309 mg, 89%), mp. 188–190 °C; 1H-NMR (DMSO-d6) δ 10.29 (s, 1H, NH), 10.05 (s, 1H, NH), 8.62 (d, J = 2.4 Hz, 1H, H2′), 8.35 (d, J = 4.5 Hz, 1H, H6′), 8.27 (d, J = 2.1 Hz, 1H, H5), 8.24 (s, 1H, H2), 7.96–7.92 (m, 2H, H7 + H4′), 7.64 (d, J = 8.7 Hz, 1H, H8), 7.39 (dd, J = 2.4, 4.5 Hz, 1H, H5′), 3.27–3.22 (m, 1H, NCH), 1.06–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.4, 161.1, 147.0, 145.44, 145.37, 144.3, 138.0, 136.1, 131.4, 130.3, 127.2, 123.2, 121.3, 119.1, 29.2, 6.0; νmax 3217, 3011, 1656, 1539, 1485, 1363, 1257, 839, 707, 558 cm−1; HRMS calcd for C17H16N5OS [M + H]+ 338.1076 found 338.1071.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(p-tolyl)thiourea (7h): According to the method B (r.t., 2 h), compound 7h was obtained as a colorless solid (386 mg, 89%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 9.99 (s, 1H, NH), 9.90 (s, 1H, NH), 8.27 (d, J = 1.8 Hz, 1H, H5), 8.22 (s, 1H, H2), 7.94 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.61 (d, J = 8.7 Hz, 1H, H8), 7.35 (d, J = 8.1 Hz, 2H, ArH), 7.16 (d, J = 8.1 Hz, 2H, ArH), 3.26–3.19 (m, 1H, NCH), 2.30 (s, 3H, CH3), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.1, 146.8, 144.0, 138.5, 136.5, 133.9, 130.2, 129.0, 127.0, 123.9, 121.2, 118.8, 29.2, 20.5, 6.0; νmax 3290, 3072, 2993, 1648, 1596, 1513, 1492, 1303, 1246, 1163, 1037, 827, 654, 608, 580 cm−1; HRMS calcd for C19H19N4OS [M + H]+ 351.1280 found 351.1276.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-methoxyphenyl)thiourea (7i): According to the method B (r.t., 2 h), compound 7i was isolated as a colorless solid (395 g, 87%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 9.91 (s, 1H, NH), 9.81 (s, 1H, NH), 8.27 (d, J = 1.8 Hz, 1H, H5), 8.22 (s, 1H, H2), 7.94 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.61 (d, J = 8.7 Hz, 1H, H8), 7.34 (d, J = 8.1 Hz, 2H, ArH), 6.93 (d, J = 8.1 Hz, 2H, ArH), 3.76 (s, 3H, OCH3), 3.26–3.19 (m, 1H, NCH), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.0, 161.1, 156.7, 146.8 144.0, 138.54 131.8, 130.2, 127.0, 126.0, 121.2, 118.9, 113.8, 55.2, 29.2, 6.0; νmax 3296, 1648, 1565, 1514, 1306, 817, 719, 574 cm−1; HRMS calcd for C19H19N4O2S [M + H]+ 367.1229 found 367.1222.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-methoxyphenyl)thiourea (7j): According to the method A (r.t., 10 min.), compound 7j was isolated as a grey solid (340 mg, 87%), mp. 210–212 °C; 1H-NMR (DMSO-d6) δ 9.77 (s, 1H, NH), 9.70 (s, 1H, NH), 8.27 (d, J = 2.4 Hz, 1H, H5), 8.20 (s, 1H, H2), 7.93 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.59 (d, J = 8.7 Hz, 1H, H8), 7.22 (d, J = 9.0 Hz, 2H, ArH), 6.72 (d, J = 9.0 Hz, 2H, ArH), 2.89 (s, 6H, NCH3), 3.33–3.21 (m, 1H, NCH), 1.05–0.93 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.1, 148.3, 146.7, 143.9, 138.7, 130.2, 127.9, 126.9, 125.7, 121.1, 118.8, 112.3, 40.3, 29.1, 6.0; νmax 3290, 3245, 1649, 1524, 1311, 1243, 704 cm−1; HRMS calcd for C20H22N5OS [M + H]+ 380.1545 found 380.1530.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(2,4-dimethoxyphenyl)thiourea (7k): According to the method A (r.t., 30 min.), product 7k was isolated as a black solid (396 mg, 97%), mp. 166–168 °C; 1H-NMR (DMSO-d6) δ 9.90 (s, 1H, NH), 9.22 (s, 1H, NH), 8.33 (d, J = 2.4 Hz, 1H, H5), 8.21 (s, 1H, H2), 7.95 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.60 (d, J = 8.7 Hz, 1H, H8), 7.47 (d, J = 8.4 Hz, 1H, H6′), 6.64 (d, J = 2.7 Hz, 1H, H3′), 6.53 (dd, J = 8.4, 2.7 Hz, 1H, H5′), 3.83 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.25–3.20 (m, 1H, NCH), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.1, 161.1, 159.5, 154.2, 146.8, 143.9, 138.6, 130.1, 128.2, 126.9, 121.1, 120.1, 118.8, 104.3, 99.0, 55.7, 55.3, 29.1, 6.0; νmax 3156, 2969, 1674, 1601, 1510, 1487, 1282, 1236, 1206, 1031, 834, 686, 540 cm−1; HRMS calcd for C20H21N4O3S [M + H]+ 397.1334 found 397.1319.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(3,4-dimethoxyphenyl)thiourea (7l): According to the method A (r.t., 5 min.), compound 7l was isolated as a black solid (359 mg, 88%), mp. 196–198 °C; 1H-NMR (DMSO-d6) δ 9.87 (s, 1H, NH), 9.83 (s, 1H, NH), 8.24 (d, J = 2.4 Hz, 1H, H5), 8.21 (s, 1H, H2), 7.93 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.60 (d, J = 8.7 Hz, 1H, H8), 7.10 (s, 1H, ArH), 6.93 (s, 2H, ArH), 3.75 (s. 3H, OCH3), 3.74 (s, 3H, OCH3), 3.26–3.16 (m, 1H, NCH), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.1, 148.4, 146.8, 146.4, 144.1, 138.5, 131.9, 130.4, 126.9, 121.2, 119.1, 116.5, 111.7, 109.3, 55.7, 55.5, 29.1, 6.0; νmax 3271, 1670, 1602, 1511, 1233, 1130, 1024, 835, 566 cm−1; HRMS calcd for C20H21N4O3S [M + H]+ 397.1334 found 397.1342.
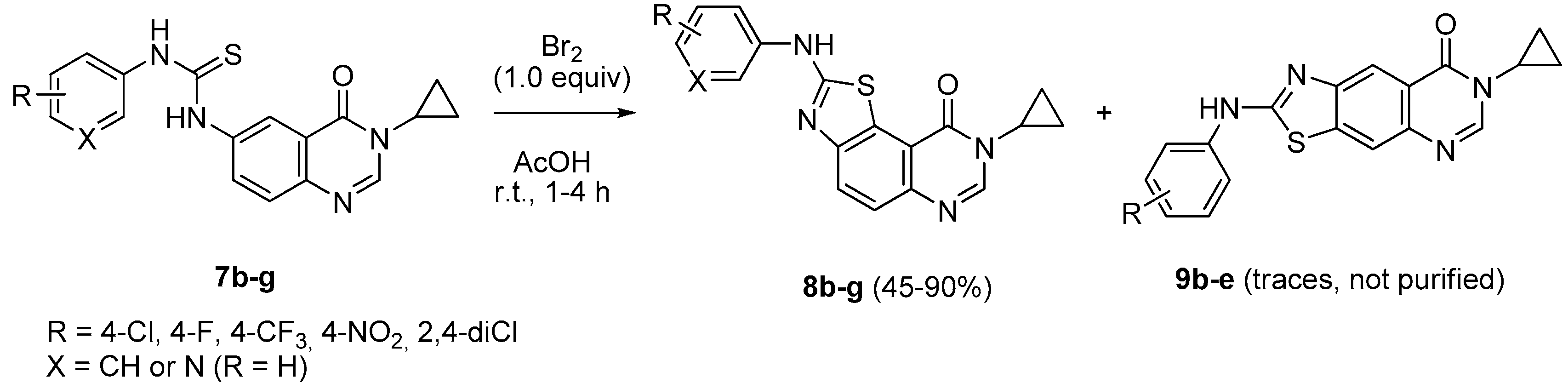
3.2.2. General Procedure for the Synthesis of 8-Cyclopropyl-2-arylaminothiazolo[5,4-f]quinazolin-9(8H)-ones 8d–g and 3-Cyclopropyl-6-[(benzo[d]thiazol-2-yl)amino]quinazolin-4(3H)-ones 10a; 10l via the Hügershoff Reaction
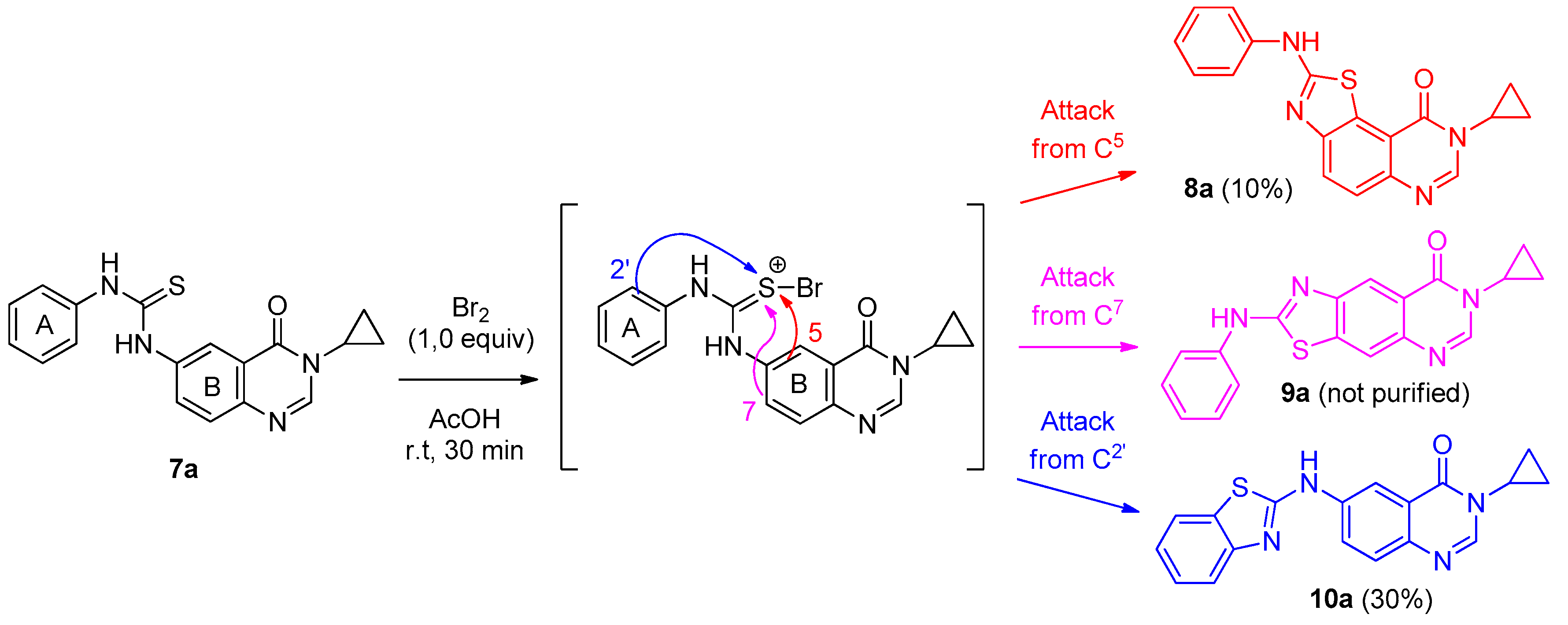
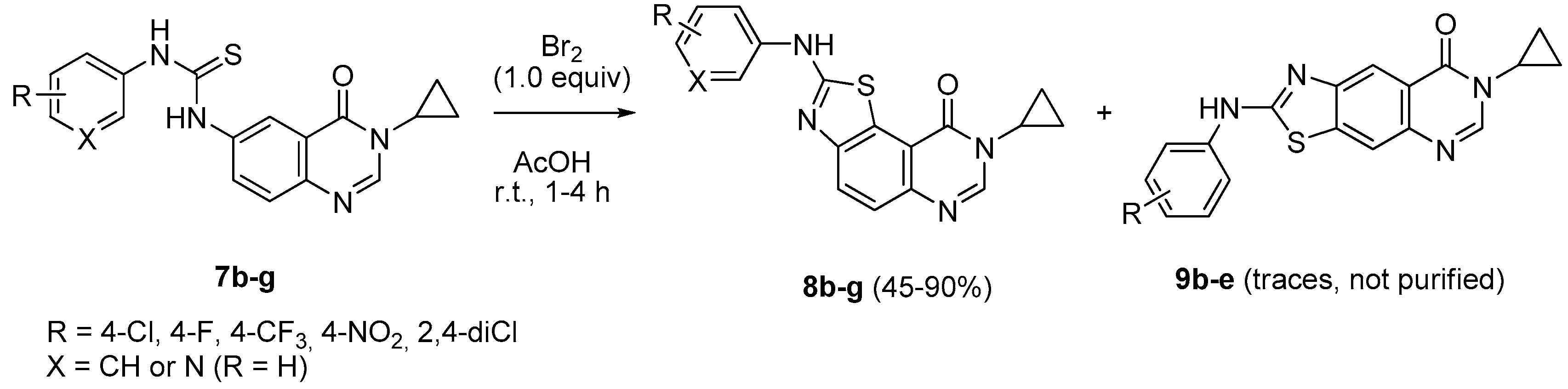
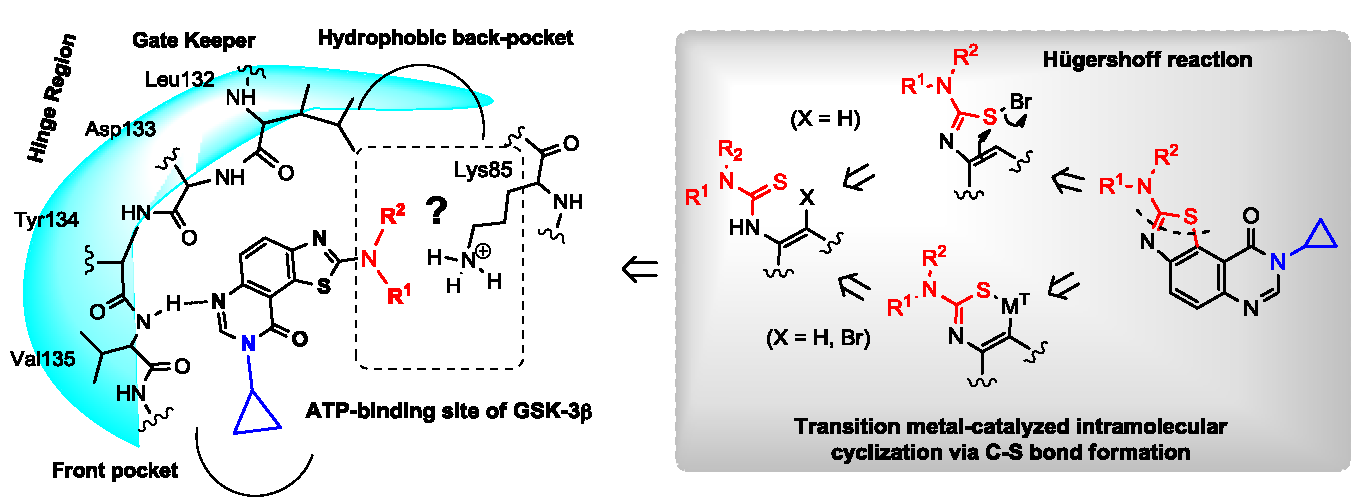
To a solution of appropriate thiourea derivative 7 (125 mg, 1 equiv.) in acetic acid (0.2 M solution) maintained at 0 °C was added dropwise bromine (Br2, 1.0 equiv.). The resulting mixture was warmed to room temperature and stirred for 30 min. to 4 h. On completion, the solvent was removed under vacuum. The crude residue was dissolved in dichloromethane (25 mL). The organic layer was washed with a saturated solution of NaHCO3, then with water and brine. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (100:0 to 0:100%, v/v) as eluent to furnish the expected fused 2-arylaminothiazole 8 or 10.
8-Cyclopropyl-2-([4-(trifluoromethyl)phenyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (8d): from 7d (125 mg, 0.31 mmol) in presence of bromine (16.0 µL, 0.31 mmol, 1.0 equiv.), 1 h at room temperature. After purification, 8d was isolated as a colorless solid (98 mg, 79%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 11.06 (s, 1H, NH), 8.31 (s, 1H, H2), 8.09 (d, J = 8.2 Hz, 1H, H4), 8.03 (d, J = 9.0 Hz, 2H, Ph), 7.75 (d, J = 9.0 Hz, 2H, Ph), 7.67 (d, J = 8.2 Hz, 1H, H5), 3.36–3.28 (m, 1H, NCH), 1.10–1.03 (m, 4H, CH); 19F NMR (282 MHz, DMSO-d6) δ −60.4 (3F, CF3); 13C-NMR (DMSO-d6) δ 164.5, 160.5, 150.7, 146.2, 143.8, 144.3, 143.1, 126.3, 125.9, 125.6, 124.8 (q, J = 37.5 Hz, 2C, C3′-F), 124.1 (q, J = 32.0 Hz, C4′-F), 122.9 (2C), 121.2, 117.5, 115.4, 29.3, 5.9; νmax 3263, 3061, 3018, 1661, 1532, 1318, 1066, 1108, 831 cm−1; HRMS calcd for C19H14N4OSF3 [M + H]+ 403.0842 found 403.0840.
8-Cyclopropyl-2-[(4-nitrophenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8e): from 7e (125 mg, 0.33 mmol) in presence of bromine (16.8 µL, 0.33 mmol, 1.0 equiv.), 4 h at room temperature. After purification, 8e was isolated as a yellow solid (108 mg, 86%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 11.41 (s, 1H, NH), 8.34 (s, 1H, H7), 8.31 (d, J = 9.3 Hz, 2H, Ph), 8.15 (d, J = 8.6 Hz, 1H, H4), 8.07 (d, J = 9.3 Hz, 2H, Ph), 7.72 (d, J = 8.6 Hz, 1H, H5), 3.38–3.24 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 164.0, 160.5, 150.4, 146.4, 146.2, 144.0, 140.8, 125.9, 125.3, 125.2, 117.1, 115.4, 29.3, 5.9; νmax 3291, 3111, 1643, 1560, 1500, 1327, 1307, 1254, 1110, 826 cm−1; HRMS calcd for C18H14N5O3S [M + H]+ 380.0817 found 380.0824.
8-Cyclopropyl-2-(2,4-dichlorophenylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8f): from 7f (125 mg, 0.31 mmol) in presence of bromine (15.8 µL, 0.31 mmol, 1.0 equiv.), 1 h 30 at room temperature. After purification, 8f was isolated as a colorless solid (112 mg, 90%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.25 (s, 1H, NH), 8.47 (d, J = 8.9 Hz, 1H, H6′), 8.31 (s, 1H, H7), 8.03 (d, J = 8.6 Hz,1H, H4), 7.71 (d, J = 2.4 Hz, 1H, H3′), 7.65 (d, J = 8.4 Hz, 1H, H5), 7.51 (dd, J = 2.4, 8.6 Hz, 1H, H5′), 3.33–3.28 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 165.9, 160.5, 150.4, 146.1, 143.6, 136.0, 129.1, 127.8, 127.5, 125.4, 124.9, 124.7, 124.0, 115.4, 29.3, 6.0; νmax 3217, 3094, 1662, 1584, 1521, 1456, 1298, 901, 831, 622, 536 cm−1; HRMS calcd for C18H13N4OSCl2 [M + H]+ 403.0187 found 403.0180.
8-Cyclopropyl-2-(pyridin-3-ylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8g): from 7g (125 mg, 0.37 mmol) and bromine (19.0 µL, 0.37 mmol, 1.0 equiv.), 2 h at room temperature. After purification, 8g was isolated as a colorless solid (100 mg, 80%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.89 (s, 1H, NH), 8.94 (d, J = 2.4 Hz, 1H, H2′), 8.39 (dd, J = 2.4, 8.2 Hz, 1H, H6′), 8.31 (s, 1H, H7), 8.26 (dd, J = 2.4, 4.5 Hz, 2H, H4′), 8.08 (d, J = 8.6 Hz, 1H, H4), 7.67 (d, J = 8.6 Hz, 1H, H5), 7.43 (dd, J = 4.5, 8.2 Hz, 1H, H5′), 3.38–3.24 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 164.8, 160.5, 150.8, 146.1, 143.6, 142.9, 139.5, 137.2, 125.6, 125.5, 125.0, 124.3, 123.8, 115.4, 29.3, 5.9; νmax 3659, 3256, 2973, 1662, 1521, 1426, 1280, 826, 799 cm−1; HRMS calcd for C17H14N5OS [M + H]+ 336.0919 found 336.0910.
3-Cyclopropyl-6-[(benzo[d]thiazol-2-yl)amino]quinazolin-4(3H)-one (10a): from 7a (125 mg, 0.37 mmol) in presence of bromine (18.9 µL, 0.37 mmol, 1.0 equiv.), after 1 h at room temperature. After purification, 10a was isolated as a colorless solid (37 mg, 30%), mp. 258–260 °C; 1H-NMR (DMSO-d6) δ 10.93 (s, 1H, NH), 8.72 (d, J = 2.3 Hz, 1H, H5), 8.18 (s, 1H, H2), 8.10 (dd, J = 2.3, 8.7 Hz, 1H, H7), 7.85 (d, J = 8.7 Hz, 1H, H8), 7.69–7.64 (m, 2H, H5′+H6′), 7.40–7.35 (m, 1H, H4′), 7.22–7.17 (m, 1H, H7′), 3.31–3.21 (m, 1H, NCH), 1.09–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 161.3, 161.2, 151.9, 146.0, 142.5, 139.4, 130.1, 128.0, 126.1, 124.7, 122.7, 122.0, 121.3, 119.5, 112.4, 29.2, 6.0; νmax 3401, 1657, 1623, 1599, 1537, 1490, 1315 cm−1; HRMS calcd for C18H15N4OS [M + H]+ 335.0967 found 335.0966.
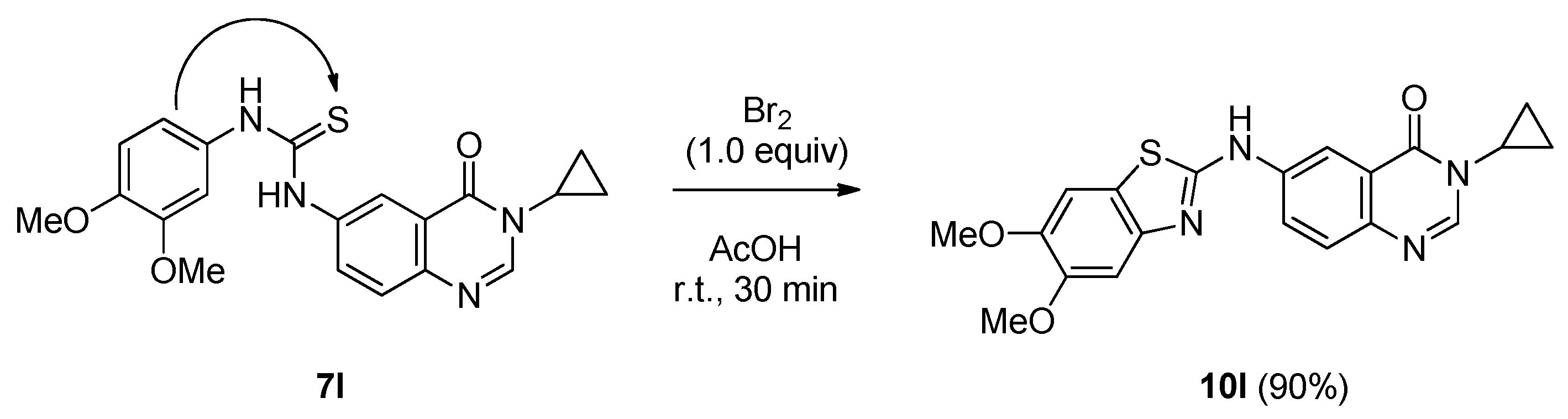
3-Cyclopropyl-6-((5,6-dimethoxybenzo[d]thiazol-2-yl)amino)quinazolin-4(3H)-one (10l): from 7l (125 mg, 0.32 mmol) and bromine (16.4 µL, 0.32 mmol, 1.0 equiv.), 30 min at room temperature. After purification, 10l was isolated as a colorless solid (113 mg, 90%), mp. 200 °C; 1H-NMR (DMSO-d6) δ 10.73 (s, 1H, NH), 8.71 (d, J = 2.5 Hz, 1H, H5), 8.35 (s, 1H, H2), 8.07 (dd, J = 2.5, 8.8 Hz, 1H, H7), 7.68 (d, J = 8.8 Hz, 1H, H8), 7.48 (s, 1H, H4′), 7.28 (s, 1H, H7′), 3.85 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.33–3.28 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 160.3, 160.1, 150.4, 148.6, 147.3, 146.1, 145.3, 140.3, 137.5, 125.0, 121.4, 120.7, 104.3, 103.3, 56.0, 55.7, 30.0, 6.0; νmax 3380, 3240, 2999, 1693, 1552, 1482, 1219, 625, 527 cm−1; HRMS calcd for C20H19N4O3S [M + H]+ 395.1178 found 395.1173.
3.2.3. General Procedure for the Synthesis of 8-Cyclopropyl-2-arylaminothiazolo[5,4-f]quinazolin-9(8H)-ones 8a; c; h–n and 8-Cyclopropyl-2-alkylaminothiazolo[5,4-f]quinazolin-9(8H)-ones 14a–f from 5-Bromo-3-cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (13) via a One-Pot Regio-Controlled Copper(I) Mediated C-S Coupling of Intermediates 7
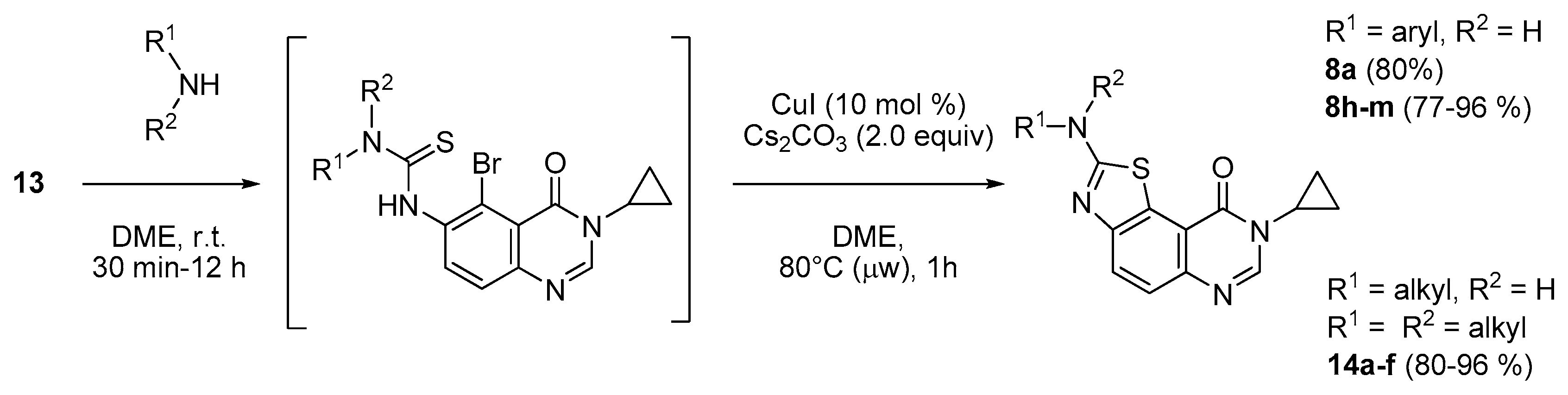
To a solution of 5-bromo-3-cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (13, 100 mg, 0.31 mmol, 1.0 equiv.) in DME (3 mL) was added the appropriate amine (0.33 mmol, 1.05 equiv.) at room temperature for 30 min. to 12 h. After completion, copper(I) iodide (5.7 mg, 0.03 mmol, 10 mol %) and cesium carbonate (208 mg, 0.64 mmol, 2.0 equiv.) were added and the resulting suspension was heated under microwave at 80 °C for 1 h. The resulting mixture was adsorbed on Celite® and purified by flash chromatography using ethyl acetate/methylene chloride (0/100 to 50/50, v/v) as eluent to furnish the expected fused 2-arylaminothiazoloquinazolinones 8a; c; h–n and 14a; f or methanol/methylene chloride (0:100 to 5:95, v/v) as eluent to furnish the 2-alkylaminothiazoloquinazolinones 14b–e.
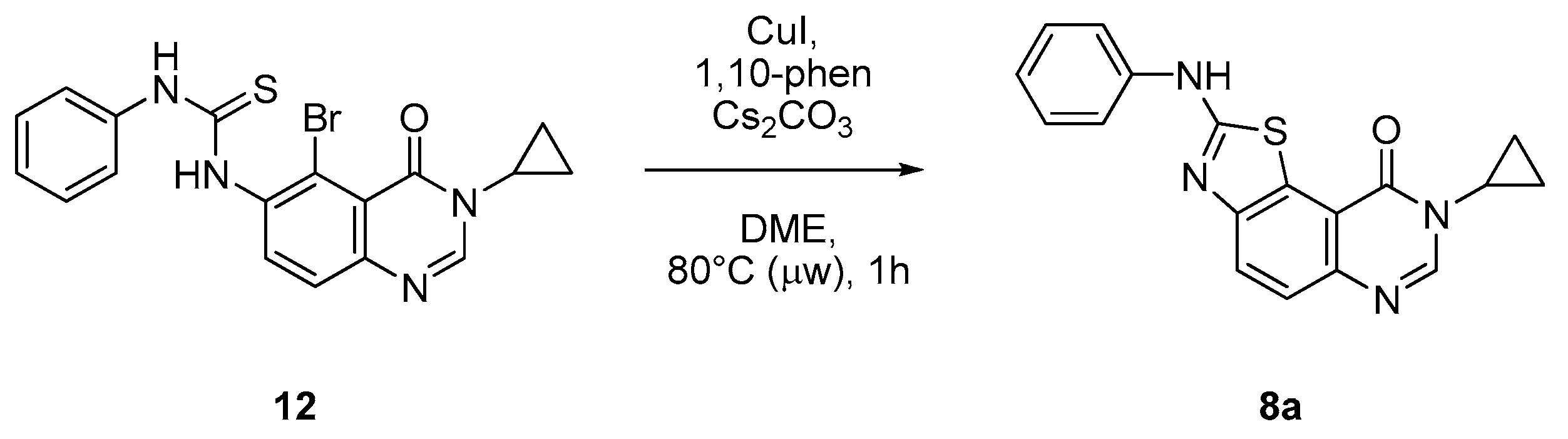
8-Cyclopropyl-2-(phenylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8a): According to the general procedure, thiourea intermediate was obtained from 13 and aniline (30.7 mg) after 12 h at room temperature. After purification, 8a was isolated as a colorless solid (82.9 mg, 80%), mp. 204–206 °C; 1H-NMR (DMSO-d6) δ 10.69 (s, 1H, NH), 8.30 (s, 1H, H7), 8.04 (d, J = 8.7 Hz, 1H, H4), 7.85–7.83 (m, 2H, H2 + H6′), 7.64 (d, J = 8.7 Hz, 1H, H5), 7.41–7.37 (m, 2H, Ph), 7.07–7.03 (m, 1H, Ph), 3.38–3.24 (m, 1H, NCH), 1.09–1.02 (m, 4H, CH); 13C-NMR (75 MHz, -d6) δ 165.1, 160.6, 151.2, 145.9, 143.3, 140.5, 129.0 (2C), 125.5, 125.2, 124.8, 122.2, 117.8, 115.4, 20.3, 6.0; νmax 3257, 3078, 1674, 1516, 1317, 1243, 828, 751, 696 cm−1; HRMS calcd for C18H15N4OS [M + H]+ 335.0967 found 335.0951.
8-Cyclopropyl-2-[(4-fluorophenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8c): According to the general procedure, thiourea intermediate was obtained from 13 and 4-fluoroaniline (56.8 mg) after 2 h at room temperature. After purification, 8c was isolated as a white solid (104.9 mg, 96%), mp > 265 °C; 1H-NMR (300 MHz,DMSO-d6) δ 10.69 (s, 1H, NH), 8.29 (s, 1H, H7), 8.05 (d, J = 8.7 Hz, 1H, H4), 7.88–7.86 (m, 1H, Ph), 7.66 (d, J = 8.7 Hz, 1H, H5), 7.24 (m, 2H, Ph), 3.38–3.24 (m, 1H, NCH), 1.09–1.02 (m, 4H, CH); 19F NMR (282 MHz, DMSO-d6) δ −120.8; 13C-NMR (DMSO-d6) δ 165.1, 160.6, 157.4 (d, J = 237 Hz, C4′-F), 151.1, 145.9, 143.4, 137.0, 125.4, 125.2, 124.9, 119.5 (d, J = 7.50 Hz, 2C, C2′-F), 115.7, 115.4 (d, J = 22.5 Hz, 2C, C3′-F), 29.2, 5.9 (2C); νmax 3291, 3111, 1643, 1560, 1500, 1327, 1307, 1254, 1110, 826 cm−1; HRMS calcd for C18H14N4OSF [M + H]+ 353.0872 found 353.0861.
8-Cyclopropyl-2-(p-tolylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8h): According to the general procedure, thiourea intermediate was obtained from 13 and p-toluidine (35.4 mg,) after 2 h at room temperature. After purification, 8h was isolated as a colorless solid (96 mg, 89%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.56 (s, 1H, NH), 8.28 (s, 1H, H7), 8.02 (d, J = 8.7 Hz, 1H, H4), 7.71 (d, J = 8.1 Hz, 2H, Ph), 7.63 (d, J = 8.7 Hz, 1H, H5), 7.20 (d, J = 8.1 Hz, 2H, Ph), 3.38–3.24 (m, 1H, NCH), 2.29 (s, 3H, CH3), 1.10–1.01 (m, 4H, CH); 13C-NMR (75 MHz,DMSO-d6) δ 165.2, 160.6, 151.3, 145.8, 143.3, 138.0, 131.1, 129.4, 125.4, 125.1, 124.8, 118.0, 115.4, 29.3, 20.4, 5.9; νmax 3191, 3041, 1640, 1564, 1501, 1327, 1317, 1254, 1140, 840 cm−1; HRMS calcd for C19H17N4OS [M + H]+ 349.1118 found 349.1123.
8-Cyclopropyl-2-[(4-methoxyphenyl]amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8i): According to the general procedure, thiourea intermediate was obtained from 13 and p-anisidine (40.6 mg) after 2 h at room temperature. After purification, 8i was isolated as a colorless solid (99.4 mg, 88%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.50 (s, 1H, NH), 8.26 (s, 1H, H7), 7.78 (d, J = 8.7 Hz, 1H, H4), 7.72 (d, J = 9.0 Hz, 2H, Ph), 7.62 (d, J = 8.7 Hz, 1H, H5), 6.98 (d, J = 9.0 Hz, 2H, Ph), 3.78 (s, 3H, OCH3), 3.38–3.26 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 165.6, 160.6, 154.8, 151.4, 145.7, 143.1, 133.8, 126.5, 125.3, 124.9, 124.8, 119.8, 115.4, 114.2, 115.4, 55.2, 29.1, 5.9; νmax 3267, 3195, 2994, 1679, 1565, 1505, 1295, 1228, 1029, 826, 522 cm−1; HRMS calcd for C19H17N4O2S [M + H]+ 365.1072 found 365.1066.
8-Cyclopropyl-2-([4-(dimethylamino)phenyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (8j): According to the general procedure, thiourea intermediate was obtained from 13 and 4-(dimethylamino)aniline (44.9 mg) after 1 h at room temperature. After purification, 8j was isolated as a pale yellow solid (101.7 mg, 87%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.27 (s, 1H, NH), 8.24 (s, 1H, H7), 7.94 (d, J = 8.7 Hz, 1H, H4), 7.61–7.56 (m, 3H, Ph + H5), 7.78 (d, J = 9.1 Hz, 1H, Ph), 3.38–3.24 (m, 1H, NCH), 2.88 (s, 6H, NCH3), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 166.2, 160.6, 151.7, 146.9, 145.5, 142.9, 130.4, 125.3, 124.7, 120.3, 115.4, 113.1, 40.3, 29.2, 5.9; νmax 3184, 2992, 1644, 1574, 1501, 1427, 1347, 1244, 840 cm−1; HRMS calcd for C20H20N5OS [M + H]+ 378.1389 found 378.1376.
8-Cyclopropyl-2-[(2,4-dimethoxyphenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8k): According to the general procedure, thiourea intermediate was obtained from 13 and 2,4-dimethoxyaniline (50.5 mg) after 30 min. at room temperature. After purification, 8k was isolated as a brown solid (105.1 mg, 86%), mp. 240–242 °C; 1H-NMR (DMSO-d6) δ 9.75 (s, 1H, NH), 8.24 (s, 1H, H7), 8.03 (d, J = 8.7 Hz, 1H, H6′), 7.92 (d, J = 8.6 Hz, 1H, H4), 7.59 (d, 1H, J = 8.6 Hz, H5), 6.69 (d, J = 2.4 Hz, 1H, H3′), 6.60 (dd, J = 2.4, 8.7 Hz, 1H, H5′), 3.85 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.38–3.24 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 167.9, 160.6, 151.8, 151.4, 145.5, 142.9, 125.9, 124.6, 124.5, 123.4, 122.3, 115.4, 104.4, 99.1, 55.7, 55.3, 29.2, 5.9; νmax 3402, 3061, 2932, 2826, 1669, 1563, 1532, 1451, 1178, 1023, 822, 514 cm−1; HRMS calcd for C20H19N4O3S [M + H]+ 395.1178 found 395.1178.
8-Cyclopropyl-2-[(3,4-dimethoxyphenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8l): According to the general procedure, thiourea intermediate was obtained from 13 and 4-aminoveratrole (50.5 mg) after 30 min. at room temperature. After purification, 8l was isolated as a colorless solid (110 mg, 90%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.47 (s, 1H, NH), 8.27 (s, 1H, H7), 7.99 (d, J = 8.6 Hz, 1H, H4), 7.63 (d, J = 8.6 Hz, 1H, H5), 7.46 (d, J = 2.4 Hz, 1H, H2′), 7.34 (d, J = 2.4, 8.8 Hz, 1H, H6′), 6.98 (d, J = 8.8 Hz, 1H, H5′), 3.81 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.38–3.24 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 165.5, 160.6, 151.4, 148.9, 145.7, 144.4, 143.2, 134.2, 125.3, 125.0, 124.8, 115.4, 112.5, 110.1, 103.8, 55.8, 55.4, 29.2, 5.9; νmax 3273, 3201, 2937, 2832, 1659, 1564, 1516, 1455, 1226, 1025, 826, 537 cm−1; HRMS calcd for C20H19N4O3S [M + H]+ 395.1178 found 395.1166.
4-[(8-Cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazolin-2-yl)amino]benzenesulfonamide (8m): According to the general procedure, thiourea intermediate was obtained from 13 and 4-aminobenzenesulfonamide (56.8 mg) after 12 h at room temperature. After purification, 8l was isolated as a beige solid (128.1 mg, 77%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 11.10 (s, 1H, NH), 8.30 (s, 1H, H7), 8.10 (d, J = 8.6 Hz, 1H, H4), 8.00 (d, J = 8.8, 2H, Ph), 7.83 (d, J = 8.8 Hz, Ph), 7.67 (d, J = 8.6 Hz, 1H, H5), 7.28 (br, 2H, NH2), 3.39–3.28 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 164.5, 160.5, 150.7, 146.2, 143.7, 143.2, 136.9, 127.0, 125.6, 125.0, 117.2, 115.4, 29.3, 6.0; νmax 3486, 3296, 3067, 1655, 1591, 1556, 1518, 1153, 832, 534 cm−1; HRMS calcd for C18H16N5O3S2 [M + H]+ 414.0695 found 414.0692.
2-(Benzylamino)-8-cyclopropylthiazolo[5,4-f]quinazolin-9(8H)-one (14a): According to the general procedure, thiourea intermediate was obtained from 13 and benzylamine (35.4 mg) after 30 min. at room temperature. After purification, 14a was isolated as a colorless solid (91.8 mg, 85%), mp. 262–264 °C; 1H-NMR (DMSO-d6) δ 8.73 (t, J =5.7 Hz, 1H, NH), 8.21 (s, 1H, H7), 7.84 (d, J = 8.7 Hz, 1H, H4), 7.55 (d, J = 8.7 Hz, 1H, H5), 7.40–7.27 (m, 5H Ph), 4.64 (d, J =5.7 Hz, 2H, CH2), 3.33–3.28 (m, 1H, NCH), 1.07–0.99 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 169.8, 160.5, 151.6, 145.2, 142.4, 138.7, 128.4 (2C), 127.4 (2C), 127.1, 125.7, 124.5, 124.2, 115.4, 47.2, 29.1, 5.9; νmax 3223, 3067, 2907, 1658, 1589, 1455, 1311, 826, 703, 686 cm−1; HRMS calcd for C19H17N4OS [M + H]+ 349.1117 found 349.1123.
8-Cyclopropyl-2-([2-(dimethylamino)ethyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (14b): According to the general procedure, thiourea intermediate was obtained from 13 and N,N-dimethylethylenediamine (29.1 mg) after 30 min. at room temperature. After purification, 14b was isolated as a colorless solid (98.0 mg, 96%), mp. 210–212 °C; 1H-NMR (DMSO-d6/CDCl3) δ 8.12 (s, 1H, H7), 7.90 (d, J = 8.6 Hz, 1H, H4), 7.63 (d, 1H, J = 8.6 Hz, H5), 7.37 (s, 1H, NH), 3.70 (m, 2H, CH2NH), 3.35–3.30 (m, 1H, NCH), 2.89–2.85 (m, 2H, NCH2), 2.50 (s, 6H, NCH3), 1.28 (m, 2H, CH), 1.04 (m, 2H, CH); 13C-NMR (DMSO-d6/CDCl3) δ 170.1, 160.8, 151.6, 143.9, 142.2, 126.0, 124.2, 124.1, 115.3, 56.9, 44.3, 41.1, 25.7, 6.0; νmax 3201, 2942, 2776, 1659, 1592, 1571, 1456, 1340, 1314, 1164, 825 cm−1; HRMS calcd for C16H20N5OS [M + H]+ 330.1389 found 330.1392.
8-Cyclopropyl-2-([2-(piperidin-1-yl)ethyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (14c): According to the general procedure, thiourea intermediate was obtained from 13 and 2-piperiridinylethylamine (43.0 mg) after 30 min. at room temperature. After purification, 14c was isolated as a colorless solid (104 mg, 91%), mp. 218–220 °C; 1H-NMR (DMSO-d6) δ 8.25–8.20 (m, 2H, H7 + NH), 7.82 (d, J = 8.4 Hz, 1H, H4), 7.54 (d, 1H, J = 8.4 Hz, H5), 3.56–3.54 (m, 2H, NCH2), 3.41–3.31 (m, 2H, CH2NH), 3.30–3.24 (m, 1H, NCH), 2.65–2.59 (m, 2H, CH2N), 1.55–1.54 (m, 4H, CH), 1.41–1.39 (m, 4H, CH), 1.10–1.01 (m, 4H,CH); 13C-NMR (DMSO-d6) δ 169.6, 160.5, 151.7, 145.2, 142.3, 125.6, 124.4, 124.0, 115.4, 64.8, 54.1, 53.8, 40.1, 29.1, 15.1, 5.9; νmax 3279, 2960, 2860, 1660, 1566, 1529, 1453, 1297, 1113, 833 cm−1; HRMS calcd for C18H22N5O2S [M + H]+ 372.1494 found 372.1487.
8-Cyclopropyl-2-[(2-morpholinoethyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (14d): According to the general procedure, thiourea intermediate was obtained from 13 and 2-morpholinoethylamine (43.0 mg) after 30 min. at room temperature. After purification, 14d was isolated as a colorless solid (102 mg, 89%), mp. 222–224 °C; 1H-NMR (DMSO-d6) δ 8.27 (s, 1H, H7), 8.25 (br, 1H, NH), 7.89 (d, J = 8.4 Hz, 1H, H4), 7.61 (d, 1H, J = 8.4 Hz, H5), 3.67–3.63 (m, 4H, OCH2), 3.60–3.57 (m, 2H, CH2NH), 3.30–3.24 (m, 1H, NCH), 2.57–2.53 (m, 2H, CH2N), 2.44–2.42 (m, 4H, NCH2), 1.09–1.00 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 169.6, 160.6, 151.8, 145.1, 142.3, 125.6, 124.4, 124.0, 115.4, 66.1, 57.0, 53.3, 41.1, 29.1, 5.9; νmax 3279, 2960, 1665, 1566, 1529, 1454, 1111, 835 cm−1.
8-Cyclopropyl-2-(piperidin-1-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (14e): According to the general procedure, thiourea intermediate was obtained from 13 and piperidine (26.3 mg) after 30 min. at room temperature. After purification, 14e was isolated as a colorless solid (90.1 mg, 89%), mp. 244–246 °C; 1H-NMR (CDCl3) δ 8.05 (s, 1H, H7), 7.88 (d, J = 8.4 Hz, 1H, H4), 7.63 (d, 1H, J = 8.4 Hz, H5), 3.68 (m, 4H, NCH2), 3.30–3.25 (m, 1H, NCH), 1.71 (m, 6H, CH2), 1.21 (m, 2H, CH), 0.97 (m, 2H, CH); 13C-NMR (CDCl3) δ 172.3, 161.6, 152.8, 144.0, 142.0, 126.1, 125.1, 124.9, 49.5, 29.3, 25.3, 24.2, 6.5; νmax 2949, 2860, 1659, 1522, 1432, 1254, 1120, 824 cm−1; HRMS calcd for C17H19N4OS [M + H]+ 327.1280 found 327.1283.
8-Cyclopropyl-2-(piperidin-1-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (14f): According to the general procedure, thiourea intermediate was obtained from 13 and morpholine (28.7 mg) after 30 min. at room temperature. After purification, 14f was isolated as a colorless solid (93.6 mg, 92%), mp. 230–232 °C; 1H-NMR (DMSO-d6) δ 8.26 (s, 1H, H7), 7.92 (d, J = 8.7 Hz, 1H, H4), 7.62 (d, J = 8.7 Hz, 1H, H5), 3.79–3.75 (m, 4H, OCH2), 3.65–3.60 (m, 4H, NCH2), 3.33–3.28 (m, 1H, NCH), 1.08–1.00 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 171.7, 160.5, 151.4, 145.6, 142.7, 125.4, 125.0, 124.8, 115.5, 65.5, 47.9, 29.2, 6.0; νmax 3061, 2971, 2860, 1655, 1561, 1456, 1432, 1340, 1112, 823 cm−1; HRMS calcd for C16H17N4O2S [M + H]+ 329.1072 found 329.1060.
3.2.4. General Procedure for the Synthesis of N-Alkyl-8-cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamides 17a–f from 16
In a sealed tube, a solution of carbonitrile derivative 16 (75 mg, 0.28 mmol) and the appropriate amine (2.8 mmol, 10 equiv.) in THF (1 mL) was heated under microwaves at 100 °C for 45 min under argon atmosphere. The solvent was removed under vacuum and the crude residue was adsorbed on Celite® and purified by flash chromatography on silica gel with methanol/methylene chloride (0/100 to 2/98, v/v) as eluent to furnish the desired amidine compound.
N-Benzyl-8-cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17a): beige powder (105 mg, 93%), mp. 148–150 °C; 1H-NMR (DMSO-d6) δ 8.48 (s, 1H, H7), 8.43 (d, J = 9.0 Hz, 1H, H4), 7.84 (d, J = 9.0 Hz, 1H, H5), 7.49–7.23 (m, 5H, Ph), 6.99 (br s, 2H, NH2), 4.48 (s, 2H, CH2Ph), 3.39–3.33 (m, 1H, NCH), 1.14–1.01 (m, 4H, CH); 13C-NMR (CDCl3) δ 161.0, 152.1, 146.9, 146.8, 129.4, 129.0, 128.6, 127.5, 127.1, 126.1, 29.7, 6.6; νmax 3456, 3328, 3187, 1699, 1653, 1604, 1586, 1448, 1342, 1311, 1091, 826, 738 cm−1; HRMS calcd for C20H18N5OS [M + H]+ 376.1232 found 376.1240.
(Z)-8-Cyclopropyl-N′-[2-(dimethylamino)ethyl]-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17b): colorless solid (99 mg, 99%), mp. 168–170 °C; 1H-NMR (DMSO-d6) δ 8.46 (s, 1H, H7), 8.40 (d, J = 8.7 Hz, 1H, H4), 7.82 (d, J = 8.7 Hz, 1H, H5), 6.75 (br s, 2H, NH2), 3.38–3.33 (m, 1H, NCH), 3.31 (m, 2H, NCH2), 2.56 (m, 2H, NCH2), 2.23 (s, 6H, NCH3), 1.11–1.04 (m, 4H, CH); νmax 3398, 3329, 3203, 2829, 1653, 1589, 1507, 1453, 1350, 1314, 829 cm−1; HRMS calcd for C17H21N6OS [M + H]+ 357.1498 found 357.1500.
(Z)-8-Cyclopropyl-N′-(2-morpholinoethyl)-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17c): colorless solid (63 mg, 57%), mp. 154–156 °C; 1H-NMR (CDCl3) δ 8.32 (d, J = 8. 7 Hz, 1H, H4), 8.22 (s, 1H, H7), 7.80 (d, J = 8.7 Hz, 1H, H5), 3.76–3.74 (m, 4H, OCH2), 3.48 (t, J = 6.0 Hz, H1′), 3.37–3.32 (m, 1H, NCH), 2.76 (t, J = 6.0 Hz, H2′), 2.63–2.60 (m, 4H, NCH2), 1.30–1.23 (m, 2H, CH), 1.22–1.03 (m, 2H, CH), 13C-NMR (CDCl3) δ 169.2, 161.0, 153.0, 152.2, 146.9, 133.4, 129.5, 126.3, 116.4, 67.0, 58.3, 53.9, 42.6, 29.7, 6.7; νmax 3480, 3374, 2966, 1642, 1316, 1110, 832, 521 cm−1; HRMS calcd for C19H23N6O2S [M + H]+ 399.1603 found 399.1604.
(Z)-8-Cyclopropyl-9-oxo-N′-[2-(piperidin-1-yl)ethyl]-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17d): colorless solid (96 mg, 87%), mp. 180–182 °C; 1H-NMR (CDCl3) δ 8.33 (d, J = 8. 7 Hz, 1H, H4), 8.22 (s, 1H, H7), 7.80 (d, J = 8.7 Hz, 1H, H5), 3.52–3.48 (m, 2H, H1′), 3.37–3.33 (m, 1H, NCH), 2.70–2.66 (m, 1H, H2′), 2.52–2.50 (m, 4H, NCH), 1.65–1.59 (m, 4H, CH), 1.49–1.30 (m, 4H, CH), 1.30–1.26 (m, 2H, CH), 1.22–1.03 (m, 2H, CH), 13C-NMR (CDCl3) δ 169.8, 161.0, 152.3, 146.8, 133.5, 129.4, 126.1, 116.5, 54.9, 29.7, 26.1, 24.4, 6.7; νmax 3463, 3325, 2933, 1650, 1588, 1345, 1040, 825, 568 cm−1; HRMS calcd for C20H25N6OS [M + H]+ 397.1811 found 397.1811.
8-Cyclopropyl-2-[imino(morpholino)methyl]thiazolo[5,4-f]quinazolin-9(8H)-one (17e): colorless solid (53.7 mg, 54%), mp. 184–186 °C ; 1H-NMR (DMSO-d6) δ 8.48 (s, 1H, H7), 8.45 (d, J = 9.0 Hz, 1H, H4), 8.83 (d, J = 9.0 Hz, 1H, H5), 3.71–3.68 (m, 4H, OCH2), 3.48–3.45 (m, 4H, NCH2), 3.39–3.33 (m, 1H, NCH), 1.12–1.07 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 162.0, 160.8, 159.5, 151.6, 148.7, 147.1, 131.4, 129.7, 126.7, 116.0, 66.3, 46.9, 30.0, 6.4; νmax 3279, 2955, 2854, 1660, 1586, 1496, 1436, 1347, 1109, 830 cm−1; HRMS calcd for C17H25N5O2S. [M + H]+ 356.1188 found 356.1190.
8-Cyclopropyl-2-[imino(piperidin-1-yl)methyl]thiazolo[5,4-f]quinazolin-9(8H)-one (17f): colorless solid (54.0 mg, 59%), mp. 204–206 °C ; 1H-NMR (DMSO-d6) δ 8.50 (s, 1H, H7), 8.48 (d, J = 8.7 Hz, 1H, H4), 7.18 (br s, 1H, NH), 7.87 (d, J = 8.7 Hz, 1H, H5), 3.43 (m, 4H, NCH2), 3.39–3.33 (m, 1H, NCH), 1.70–1.56 (m, 6H, CH2), 1.11–1.05 (m, 4H, CH); νmax 3162, 2936, 2848, 1678, 1587, 1345, 829 cm−1; HRMS calcd for C17H25N5O2S. [M + H]+ 354.1389 found 354.1389.


 ,
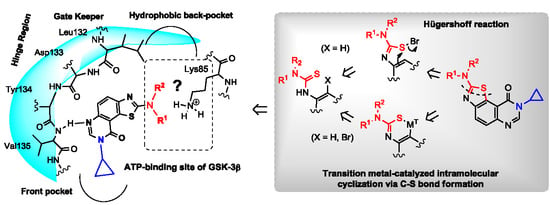
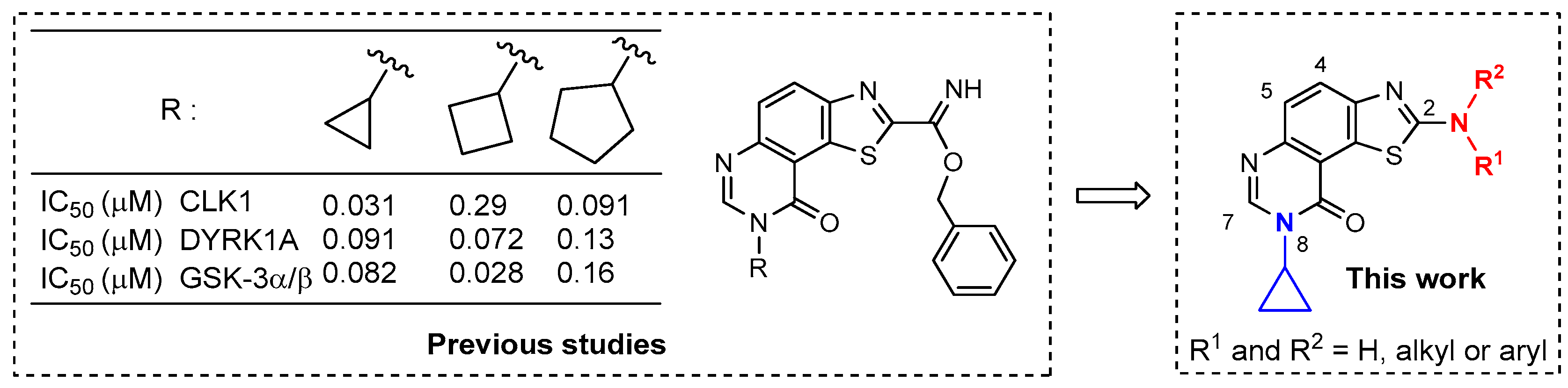
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



























