
Total Synthesis and Antifungal Activity of Palmarumycin CP17 and Its Methoxy Analogues


Abstract
:
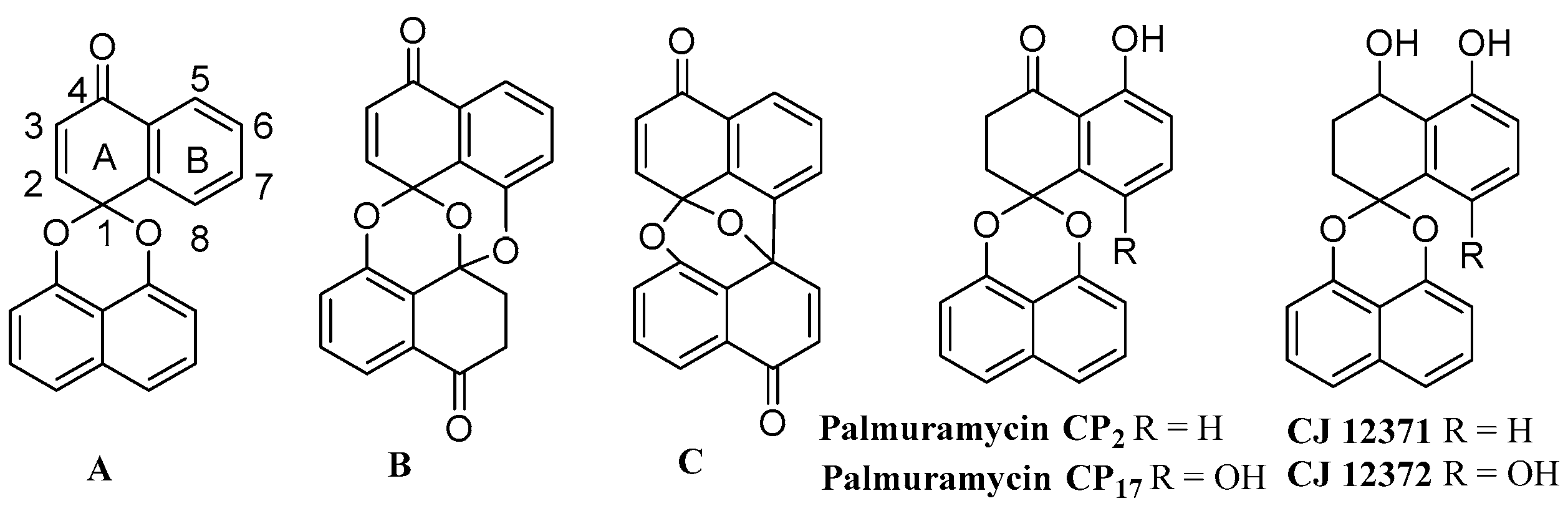
1. Introduction
2. Results and Discussion
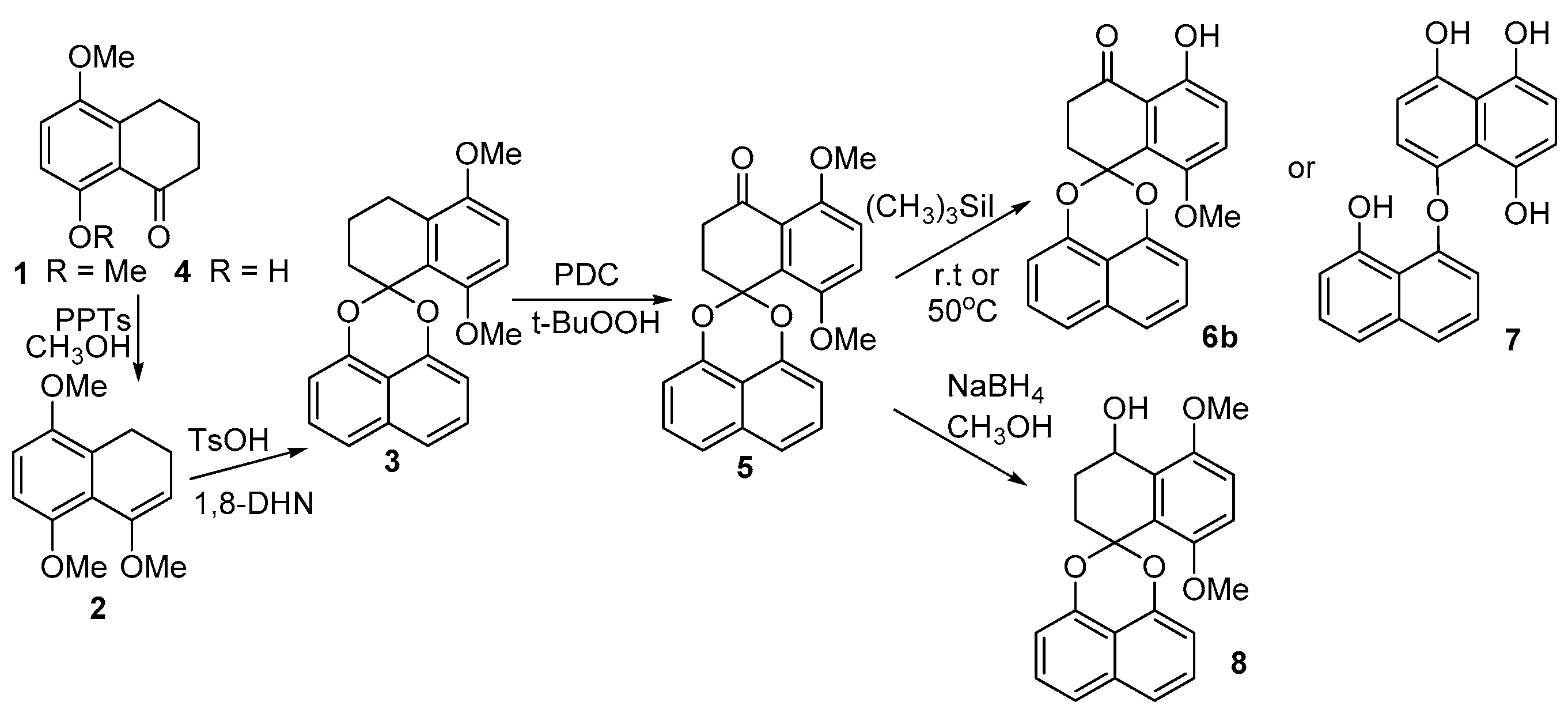
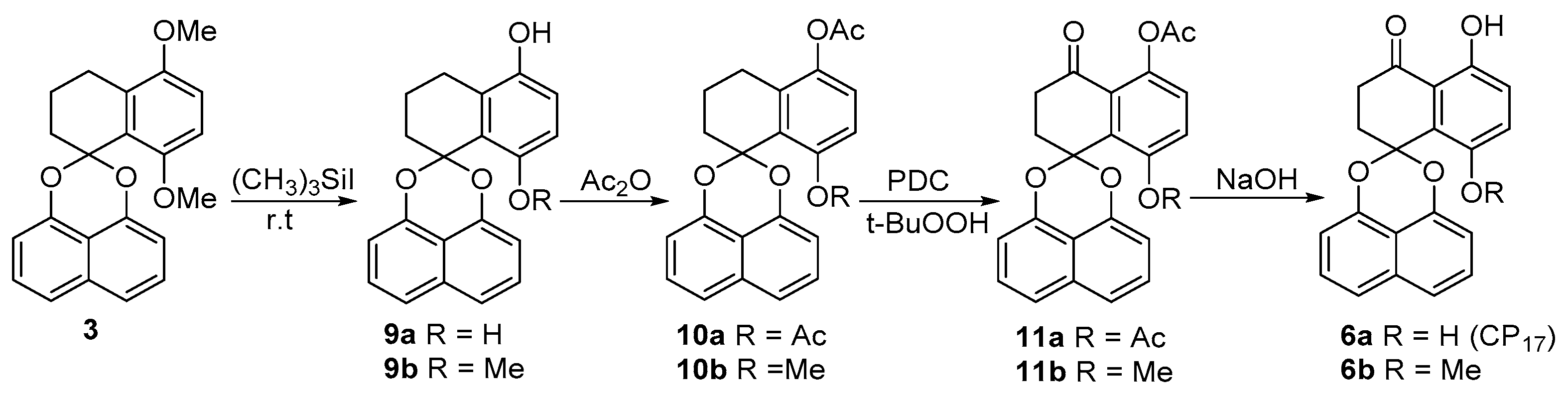
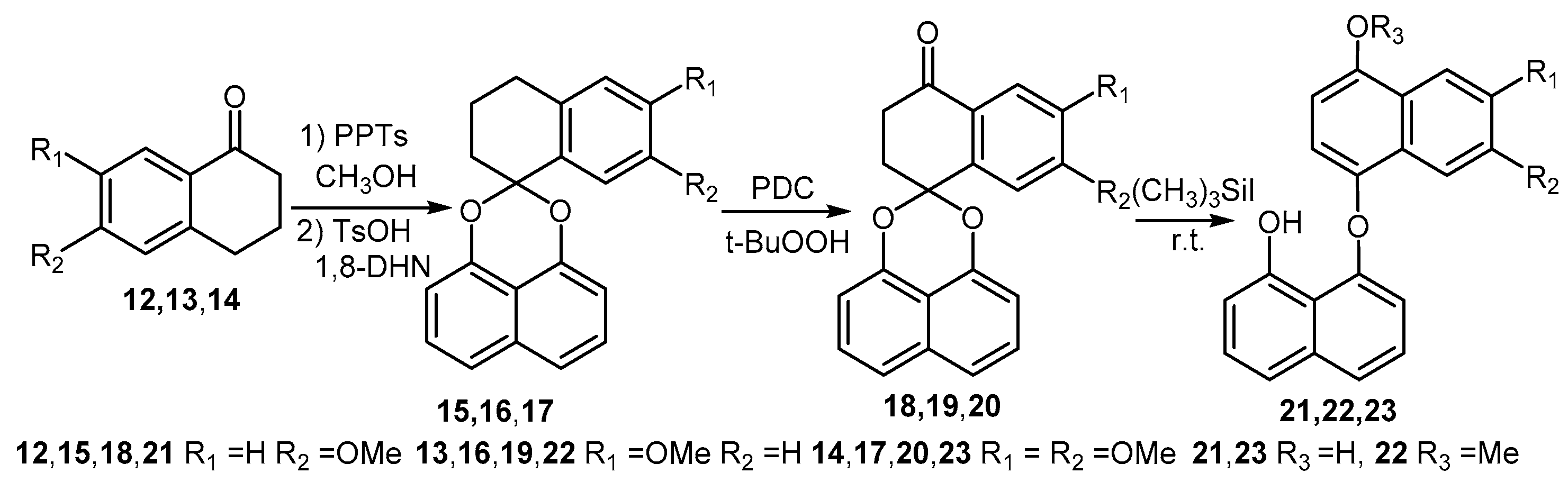
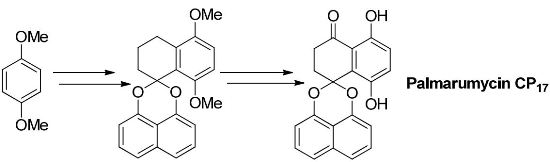
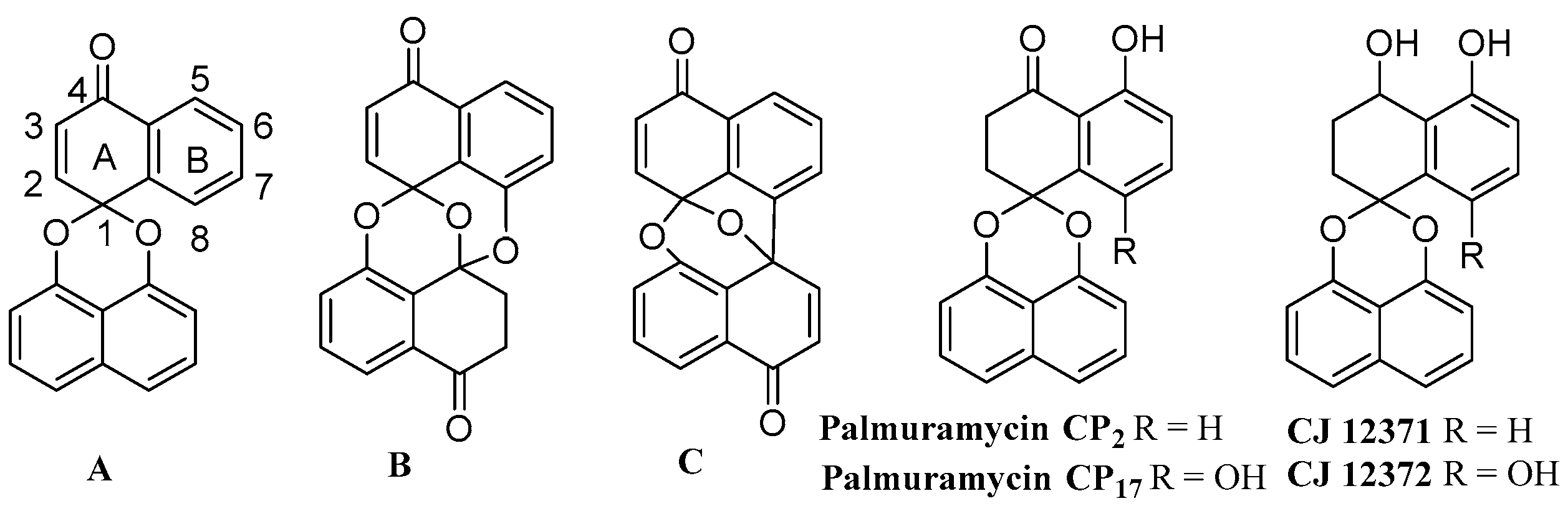
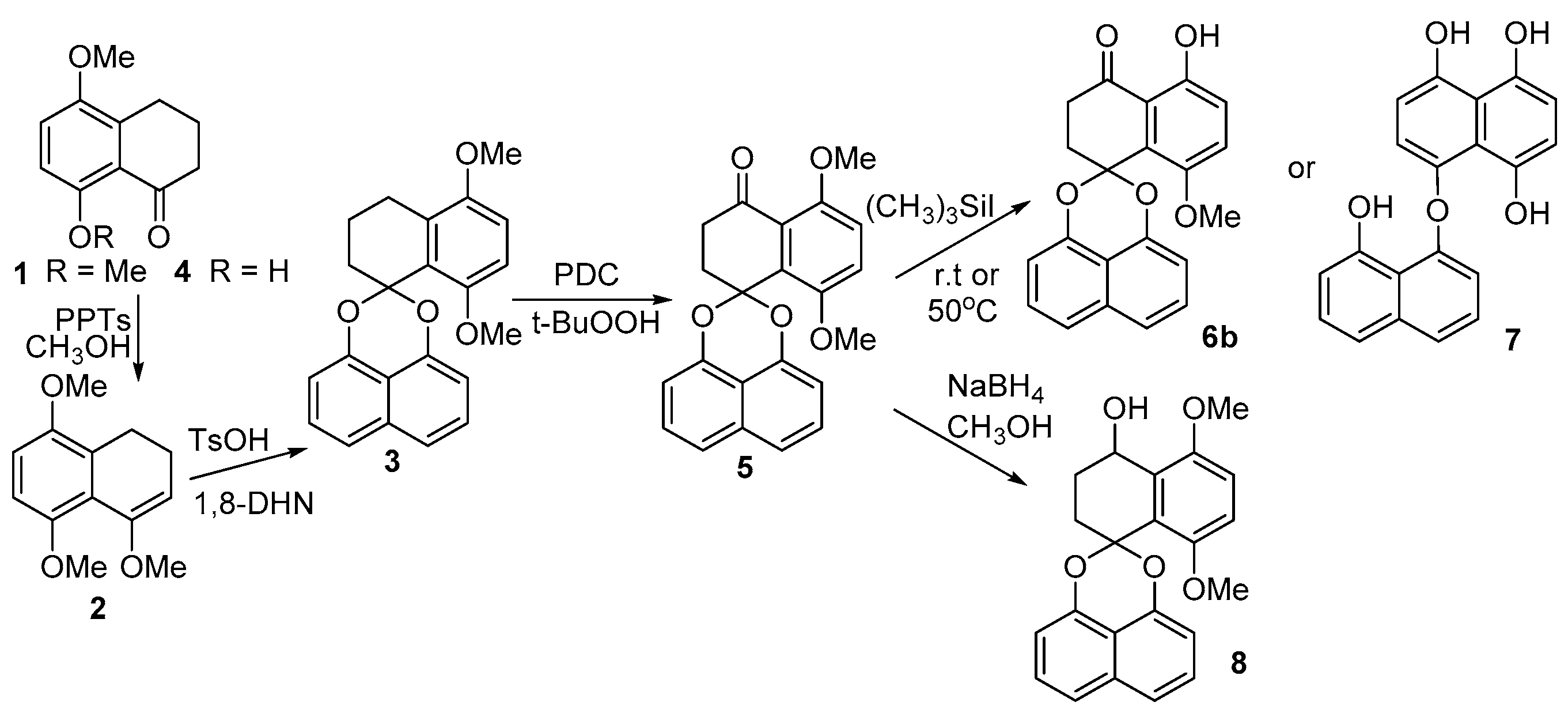
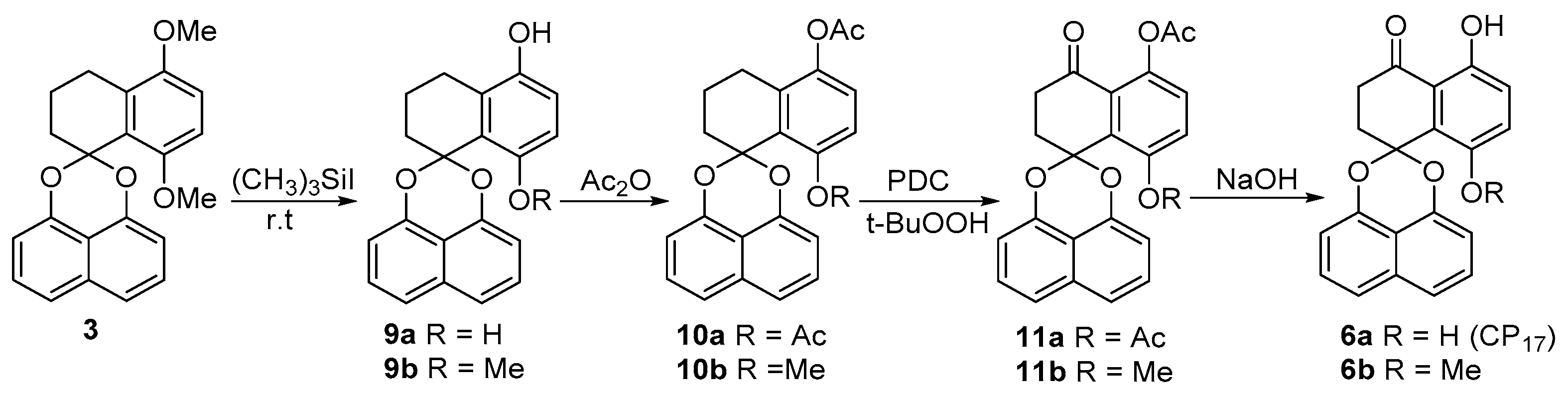
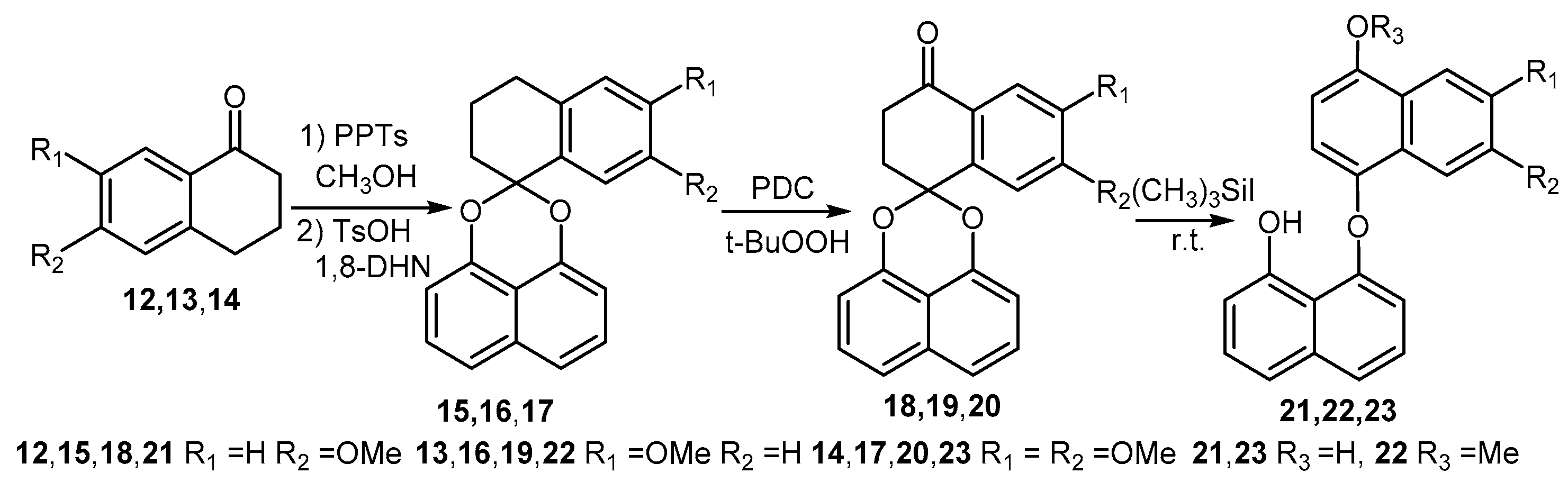
2.1. Synthesis of Palmarumycin CP17 and Its Methoxy Analogues
2.2. Antifungal Activity of Spirobisnaphthalene Palmarumycin CP17 and Its Methoxy Analogues
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Spirobisnaphthalene Palmarumycin CP17 and Their Methoxy Analogues
5,8-Dimethoxytetralone (1)
5,8-Dimethoxytetralone methyl enol ether (2)
5,8-Dimethoxy-1,2,3,4-tetrahydrospiro[naphtha-ene-1,2′-naphtho-[1,8-de][1,3]dioxine (3)
5,8-Dimethoxy-2,3-dihydrospiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine-4-one (5)
5-Hydroxy-8-methoxy-2,3-dihydrospiro[naphthalene-1,2′-naphtho-[1,8-de][1,3]dioxine-4-one (6b)
8-Hydroxynaphthalen-1-yl-4,5,8-trihydroxynaphthalene ether (7)
8-((8-Hydroxynaphthalen-1-yl)oxy)naphthalene-1,4,5-triol (8)
5,8-Dihydroxy-1,2,3,4-tetrahydrospiro[naphthalene-1,2′-naphtho-[1,8-de][1,3]dioxine (9a) and 5-Hydroxy-8-methoxy-1,2,3,4-tetrahydrospiro[naphthalene-1,2′-naphtho-[1,8-de][1,3]dioxine (9b)
5,8-Diacetoxy-1,2,3,4-tetrahydrospiro[naphthalene-1,2’-naphtho-[1,8-de][1,3]dioxine (10a)
5-Acetoxy-8-methoxy-1,2,3,4-tetrahydrospiro[naphthalene-1,2′-naphtho-[1,8-de][1,3]dioxine (10b)
5,8-Diacetoxy-2,3-dihydrospiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine-4-one (11a) and 5-Acetoxy-8-methoxy-2,3-dihydrospiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine-4-one (11b)
5,8-Dihydroxy-2,3-dihydrospiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine-4-one (Palmarumycin CP17, 6a) and 5-Hydroxy-8-methoxy-2,3-di-hydrospiro[naphthalene-1,2′-naphtho[1,8-de][1,3]-dioxine-4-one (6b)
7-Methoxytetralone (13) and 6,7-Dimethoxytetralone (14)
6-Methoxy-3,4-dihydro-2H-spironaphthalene-1,2′-naphtho[1,8-de][1,3]dioxine (15), 7-Methoxy-3,4-dihydro-2H-spironaphthalene-1,2′-naphtho[1,8-de][1,3]dioxine (16) and 6,7-Dimethoxy-3,4-dihydro-2H-spironaphthaene-1,2′-naphtho[1,8-de][1,3]dioxine (17)
6-Methoxy-2,3-dihydrospiro[naphthalene-1,2′-naphtho[1,8-de][1,3]dioxine-4-one (18), 7-Methoxy-2,3-dihydrospiro[naphthalene-1,2′-naphtho-[1,8-de][1,3]dioxine-4-one (19) and 6,7-Dimethoxy-2,3-di-hydrospiro[naphthalene-1,2′-naphtho-[1,8-de][1,3]dioxine-4-one (20)
8-Hydroxynaphthalen-1-yl-4-hydroxy-6-methoxynaphthalene ether (21), 8-Hydroxynaphthalen-1-yl-4-hydroxy-7-methoxynaphthalene ether (22) and 8-Hydroxy-naphthalen-1-yl-4,7-dimethoxynaphthalene ether (23)
3.3. Bioassay of Spirobisnaphthalene Palmarumycin CP17 and Its Methoxy Analogues
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miyashita, K.; Imanishi, T. Syntheses of natural products having an epoxyquinone structure. Chem. Rev. 2005, 105, 4515–4536. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Schwan, W.R.; Volk, T.J.; Rott, M.; Liu, M.; Huang, P.; Liu, Z.; Wang, Y.; Zitomer, N.C.; Sleger, C.; et al. Antibacterial spirobisnaphthalenes from the North American cup fungus Urnula craterium. J. Nat. Prod. 2012, 75, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Jung, J.K. Long-range electrostatic effects in synthesis: Dipole-controlled nucleophilic addition to a naphthoquinone acetal in model studies toward diepoxin sigma. Angew. Chem. Int. Ed. 1997, 36, 764–767. [Google Scholar] [CrossRef]
- Jiao, P.; Swenson, D.; Gloer, J.; Campbell, J.; Shearer, C. Decaspirones A–E, new bioactive spirodioxynaphthalenes from the freshwater aquatic fungus decaisnella thyridioides. J. Nat. Prod. 2006, 69, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Guo, H.; Li, E.; Liu, X.; Zhou, Y.; Che, Y. Decaspirones F–I, bioactive secondary metabolites from the saprophytic fungus helicoma viridis. J. Nat. Prod. 2006, 69, 1672–1675. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Krohn, K.; Elsasser, B.; Florke, U.; Draeger, S.; Schulz, B. Metabolic products of the endophytic fungus Microsphaeropsis sp. from Larix decidua. Eur. J. Org. Chem. 2007, 29, 4845–4854. [Google Scholar] [CrossRef]
- Kanoh, K.; Okada, A.; Adachi, K.; Imagawa, H.; Nishizawa, M.; Matsuda, S.; Shizuri, Y.; Utsumi, R. Ascochytatin, a novel bioactive spirodioxynaphthalene metabolite producted by the marine-derived fungus Ascochyta sp. NGB4. J. Antibiot. 2008, 61, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, Q.; Lin, G.; Guo, S.; Yang, J. Spirobisnaphthalene analogues from the endophytic fungus Preussia sp. J. Nat. Prod. 2009, 72, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Kurtan, T.; Miao, Z.; Mandi, A.; Istvan, K.; Liu, H.; Ding, J.; Guo, Y. Palmarumycins BG1–BG7 and Preussomerin BG1: Establishment of their absolute configurations using theoretical calculations of electronic circular dichroism spectra. J. Org. Chem. 2011, 76, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Macías-Rubalcava, M.; Sobrino, M.; Hernández-Bautista, C.; Hernández-Ortega, S. Naphthoquinone spiroketals and organic extracts from the endophytic fungus Edenia gomezpompae as potential herbicides. J. Agric. Food Chem. 2014, 62, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Macías-Rubalcava, M.; Hernández-Bautista, B.; Jiménez-Estrada, M.; González, M.; Glenn, A.; Hanlin, R. Naphthoquinone spiroketal with allelochemical activity from the newly discovered endophytic fungus Edenia gomezpompae. Phytochemistry 2008, 69, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Song, H.; Li, J.; Tang, Y.; Sun, R.; Wang, L. Ymf 1029A–E, preussomerin analogues from the fresh-water-derived fungus YMF 1.01029. J. Nat. Prod. 2008, 71, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Dersar, S.A.; Blunt, J.W.; Munro, M.H.G. Spiro-mamakone A: A unique relative of the spirobisnaphthalene class of compounds. Org. Lett. 2006, 8, 2059–2061. [Google Scholar]
- Pudhom, K.; Teerawatananond, T.; Chookpaiboon, S. Spirobisnaphthalenes from the mangrove-derived fungus Rhytidhysteron sp. AS21B. Mar. Drugs 2014, 12, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-M.; Mafezoli, J.; Oliveira, M.C.F.; U’Ren, J.M.; Arnold, A.E.; Gunatilaka, A.A.L. Anteaglonialides A−F and Palmarumycins CE1−CE3 from Anteaglonium sp. FL0768, a fungal endophyte of the spikemoss Selaginella arenicola. J. Nat. Prod. 2015, 78, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Krohn, K.; Wang, S.; Ahmed, I.; Altun, S.; Aslan, A.; Florke, U.; Kock, I.; Schlummer, S. Flexible route to Palmarumycin CP1 and CP2 and CJ-12.371 methyl ether. Eur. J. Org. Chem. 2010, 4476–4481. [Google Scholar] [CrossRef]
- Wipf, P.; Lynch, S.; Powis, G.; Birmingham, A.; Englund, E. Synthesis and biological activity of prodrug inhibitors of the thioredoxin–thioredoxin reductase system. Org. Biomol. Chem. 2005, 3, 3880–3882. [Google Scholar] [CrossRef] [PubMed]
- Aslan, A.; Altun, S.; Ahmed, I.; Florke, U.; Schulz, B.; Krohn, K. Synthesis of biologically active nonnatural palmarumycin derivatives. Eur. J. Org. Chem. 2011, 1176–1188. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, J.; Shan, X.; Cai, X.; Peng, Y. Spirobisnaphthalenes from fungi and their biological activities. Mini-Rev. Med. Chem. 2010, 10, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.S.; Guo, Y.W.; Krohn, K. Structure, bioactivities, biosynthetic relationships and chemical synthesis of the spirodioxynaphthalenes. Nat. Prod. Rep. 2010, 27, 1840–1870. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Tian, J.; Wang, X.; Mou, Y.; Mao, Z.; Lai, D.; Dai, J.; Peng, Y.; Zhou, L.; Wang, M. Bioactive spirobisnaphthalenes from the endophytic fungus Berkleasmium sp. J. Nat. Prod. 2014, 77, 2151–2160. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Luo, H.; Mao, Z.; Shan, T.; Sun, W.; Zhou, K.; Zhou, L. Enhancement of palmarumycins C12 and C13 production in liquid culture of endophytic fungus Berkleasmium sp. Dzf12 after treatments with metal ions. Int. J. Mol. Sci. 2013, 14, 979–998. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Lu, S.; Luo, C.; Luo, R.; Mou, Y.; Wang, M.; Peng, Y.; Zhou, L. Preparative separation of spirobisnaphthalenes from endophytic fungus Berkleasmium sp. Dzf12 by high-speed counter-current chromatography. Molecules 2013, 18, 12896–12908. [Google Scholar] [CrossRef] [PubMed]
- Nabatame, K.; Hirama, M.; Inoue, M. A simple desymmetrization approach to the spiroxin framework. Heterocycles 2008, 76, 1011–1016. [Google Scholar]
- Kwan, A.; Stein, J.; Carrico-Moniz, D. A catalytic asymmetric entry to enantioenriched tertiary naphthoquinols via a facile tandem oxidation/ring-opening sequence. Tetrahedron Lett. 2011, 52, 3426–3428. [Google Scholar] [CrossRef]
- Martinez-Luis, S.; Della-Togna, G.; Coley, P.; Kursar, T.; Gerwick, W.; Cubilla-Rios, L. Antileishmanial constituents of the Panamanian endophytic fungus Edenia sp. J. Nat. Prod. 2008, 71, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Luis, S.; Cherigo, L.; Spadafora, C.; Gerwick, W.H.; Cubilla-Rios, L. Additional antileishmanial constituents of the Panamanian endophytic fungus Edenia sp. Rev. Latinoamer. Quim. 2009, 37, 104–114. [Google Scholar]
- Cai, X.; Shan, T.; Li, P.; Huang, Y.; Xu, L.; Zhou, L.; Wang, M.; Jiang, W. Spirobisnaphthalenes from the endophytic fungus Dzf12 of Dioscorea zingiberensis and their antimicrobial activities. Nat. Prod. Commun. 2009, 4, 1469–1472. [Google Scholar] [PubMed]
- Wipf, P.; Jung, J.K. Total synthesis of palmarumycin CP1 and (±)-deoxypreussomerin A. J. Org. Chem. 1998, 63, 3530–3531. [Google Scholar] [CrossRef]
- Karsten, K.; Karsten, B.; Hans-Jiirgen, A.; Siegfried, D.; Barbara, S.; Stefan, B.; Gerhard, B. Generation of the palmarumycin spiroacetal framework by oxidative cyclization of an open chain metabolite from coniothyrium palmarum. Liebigs Ann. Recueil 1997, 2531–2534. [Google Scholar]
- Ragot, J.; Alcaraz, M.; Taylor, R. Syntheses of palmarumycin CP1 and CP2, CJ-12,371 and novel analogues. Tetrahedron Lett. 1998, 39, 4921–4924. [Google Scholar] [CrossRef]
- Ragot, J.; Steeneck, C.; Alcaraz, M.; Taylor, R. The synthesis of 1,8-dihydroxynaphthalene-derived natural products: Palmarumycin CP1. palmarumycin CP2, palmarumycin C11, CJ-12,371, deoxypreussomerin A and novel analogues. J. Chem. Soc. Perkin Trans. 1 1999, 1073–1082. [Google Scholar] [CrossRef]
- Inoue, M.; Nabatame, K.; Hirama, M. Communication-a novel route to 1,8-dihydroxynaphthalene- derived natural products. Synthesis of (±)-CJ-12,372. Heterocycles 2003, 59, 87–92. [Google Scholar] [CrossRef]
- Barrett, A.G.M.; Hamprecht, D.; Meyer, T. Total syntheses of palmarumycins CP1 and CP2 and CJ-12,371: novel spiro-ketal fungal metabolites. Chem. Commun. 1998, 809–810. [Google Scholar] [CrossRef]
- Barrett, A.G.M.; Blaney, F.; Campbell, A.D.; Hamprecht, D.; Meyer, T.; White, A.J.P.; Witty, D.; Williams, D.J. Unified route to the palmarumycin and preussomerin natural products. Enantioselective Synthesis of (−)-Preussomerin G. J. Org. Chem. 2002, 67, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.; Heathcock, C.H. Total syntheses of (±)-preussomerins G and I. Org. Lett. 1999, 1, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Jung, J. Formal total synthesis of (+)-diepoxin σ. J. Org. Chem. 2000, 65, 6319–6337. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.C. The Bioassay Technologies for Pesticides; Beijing Agricultural University Press: Beijing, China, 1991; pp. 161–162. [Google Scholar]
- Sample Availability: Samples of the compounds 1–23 are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | R.S | P.P | B.C | F.O | P.A | F.G | C.A |
|---|---|---|---|---|---|---|---|
| 3 | 44.5 | 33.6 | 50.5 | 10.3 | 21.7 | 18.6 | 49.7 |
| 5 | 54.5 | 73.7 | 61.2 | 13.0 | 33.3 | 18.6 | 49.7 |
| 8 | 48.9 | 43.5 | 60.5 | 16.0 | 38.3 | 23.0 | 51.4 |
| 9a | 52.1 | 57.0 | 68.4 | 16.3 | 23.0 | 13.9 | 44.7 |
| 9b | 51.1 | 75.0 | 57.9 | 14.0 | 29.8 | 11.6 | 54.8 |
| 10a | 55.3 | 63.5 | 69.0 | 21.7 | 36.7 | 18.6 | 58.0 |
| 10b | 42.3 | 50.6 | 67.2 | 14.9 | 26.5 | 18.6 | 53.1 |
| 11b | 71.3 | 0 | 67.1 | 10.0 | 2.7 | 23.0 | 59.0 |
| 6a | 48.9 | 43.5 | 60.5 | 16.0 | 38.3 | 23.0 | 51.4 |
| 6b | 52.4 | 64.7 | 60.5 | 21.3 | 39.0 | 41.9 | 50.6 |
| Compd. | Fungi | Regression Eq. | EC50 (µg/mL) | Correlation efficient (R2) |
|---|---|---|---|---|
| 5 | P. piricola | Y = −1.278 + 1.282X | 9.34 | 0.9604 |
| 9b | P. piricola | Y = −3.014 + 2.760X | 12.35 | 0.9525 |
| 11b | R. solani | Y = −2.076 + 1.980X | 11.18 | 0.9645 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Liu, G.; Yang, M.; Wang, M.; Zhou, L. Total Synthesis and Antifungal Activity of Palmarumycin CP17 and Its Methoxy Analogues. Molecules 2016, 21, 600. https://doi.org/10.3390/molecules21050600
Wang R, Liu G, Yang M, Wang M, Zhou L. Total Synthesis and Antifungal Activity of Palmarumycin CP17 and Its Methoxy Analogues. Molecules. 2016; 21(5):600. https://doi.org/10.3390/molecules21050600
Chicago/Turabian StyleWang, Ruina, Guoyue Liu, Mingyan Yang, Mingan Wang, and Ligang Zhou. 2016. "Total Synthesis and Antifungal Activity of Palmarumycin CP17 and Its Methoxy Analogues" Molecules 21, no. 5: 600. https://doi.org/10.3390/molecules21050600