
Adapting Document Similarity Measures for Ligand-Based Virtual Screening

 and
and
Abstract
:1. Introduction
2. Related Work
- The presence or absence of a feature is more essential than the difference between the two values associated with a present feature.
- The similarity degree should increase when the difference between two non-zero values of a specific feature decreases.
- The similarity degree should decrease when the number of presence-absence features increases.
- Two documents are less similar to each other if none of the features have non-zero values in both documents.
- The similarity measure should be symmetric.
- The value distribution of a feature is considered, i.e., the standard deviation of the feature is taken into account, for its contribution to the similarity between two documents. A feature with a larger spread offers more contribution to the similarity between d1 and d2.
- .
- .
- J: number of features (vectors).
- σ: standard deviation of all non-zero values in a vector.
- λ : small value of λ, e.g., 0.01–0.0001
3. Similarity Searching
4. Methods
4.1. Tanimoto Similarity Method
4.2. The Adapted Similarity Measure of Text Processing (ASMTP)
- (a)
- The feature appears in both documents.
- (b)
- The feature appears in only one document.
- (c)
- The feature appears in none of the documents.
- .
- : Molecule1 (query).
- : Molecule 2(reference).
- J = feature index (vector).
- μ: is the average of all non-zero values of vector (feature) j.
- λ: small value λ, e.g., 0.01–0.0001.
- j: total number of non-zero values in the features index.
5. Experimental Design
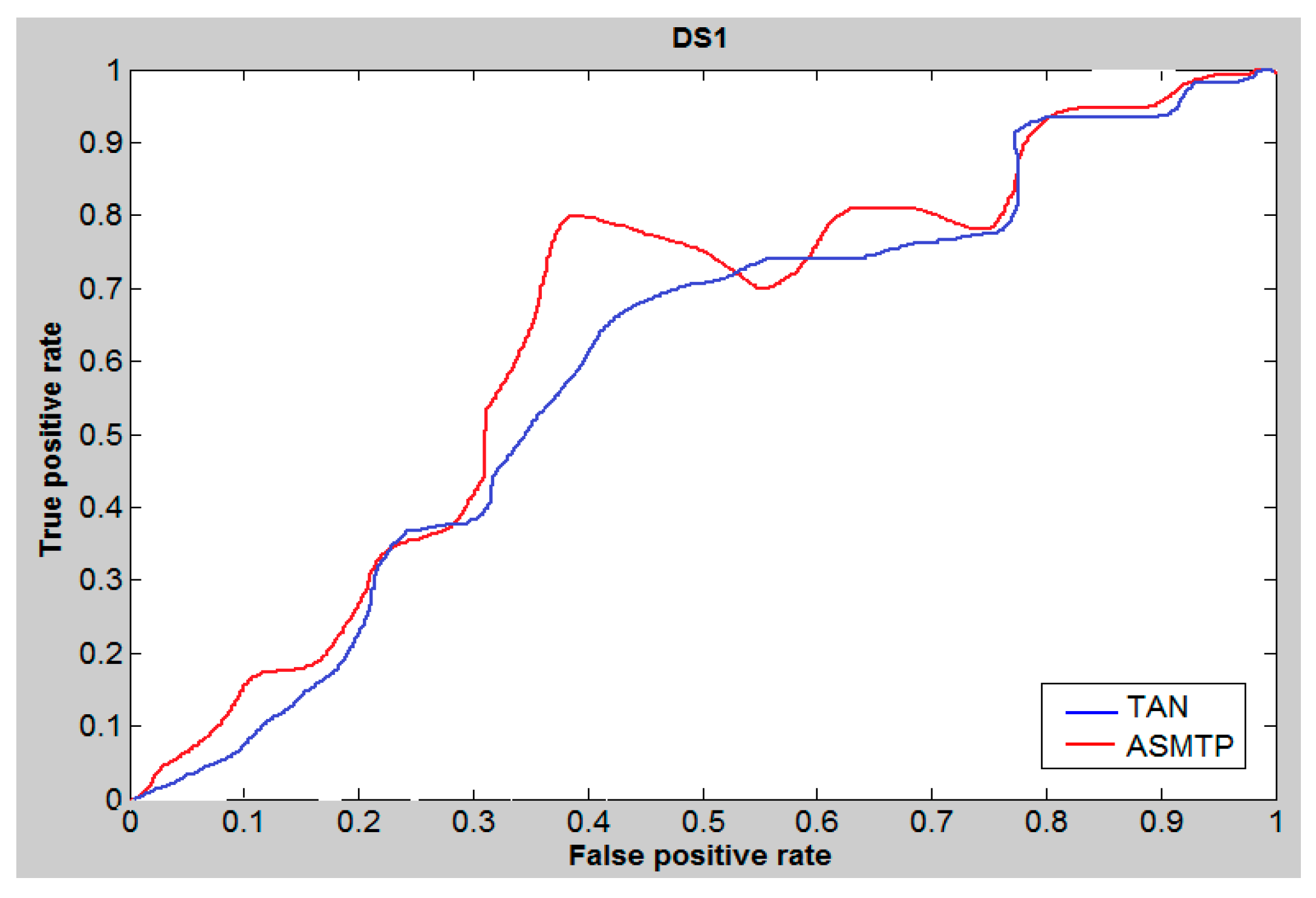
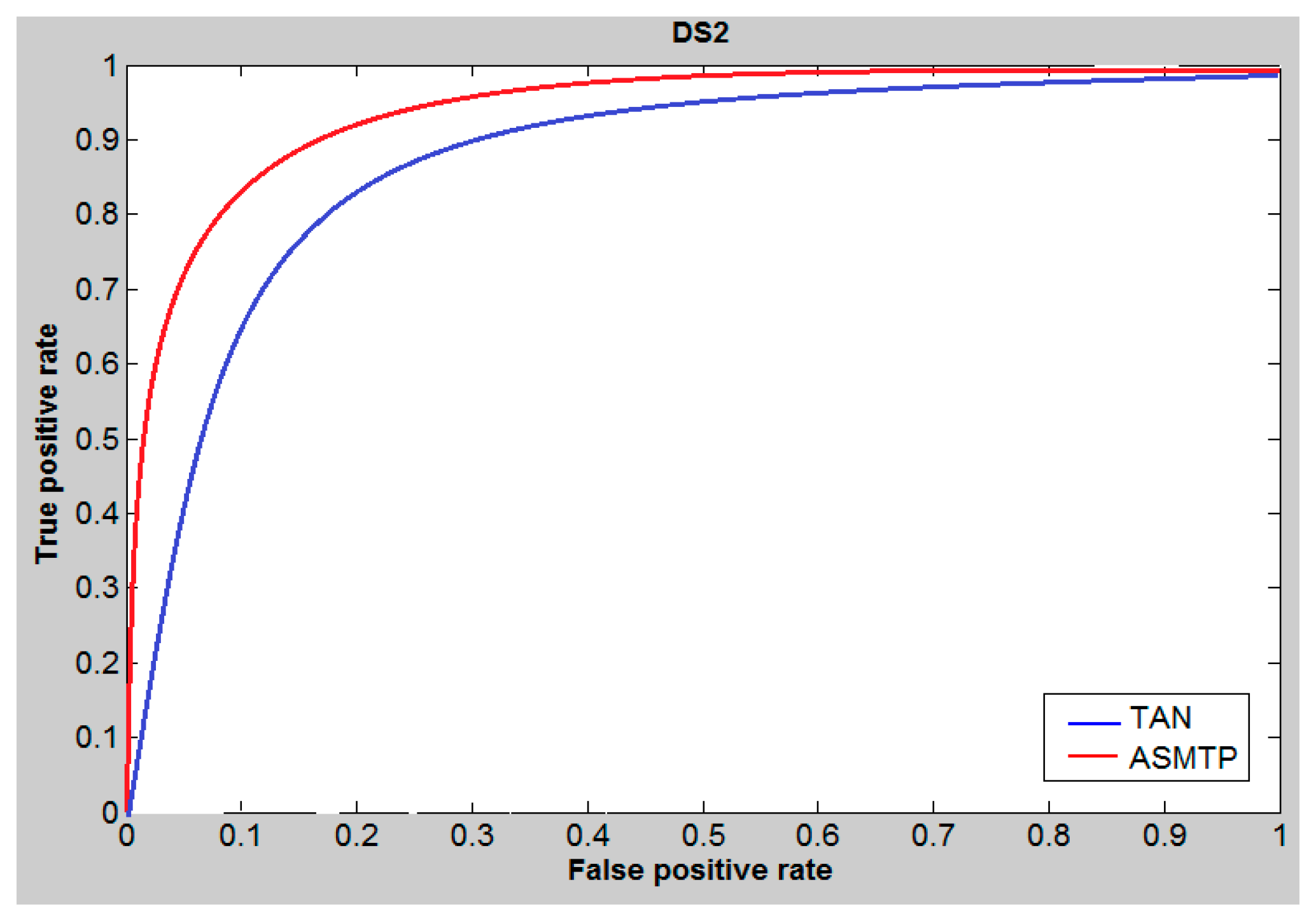
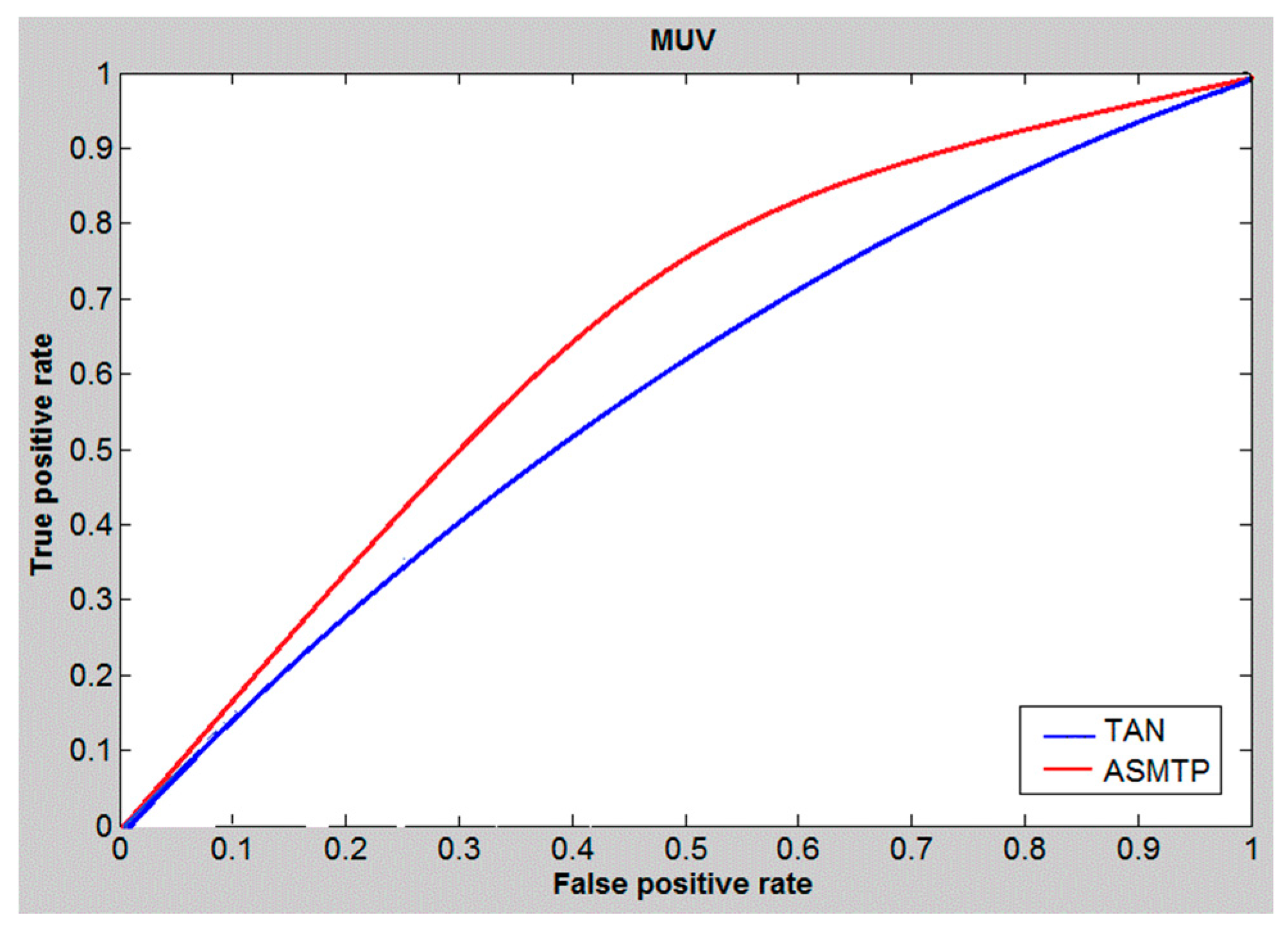
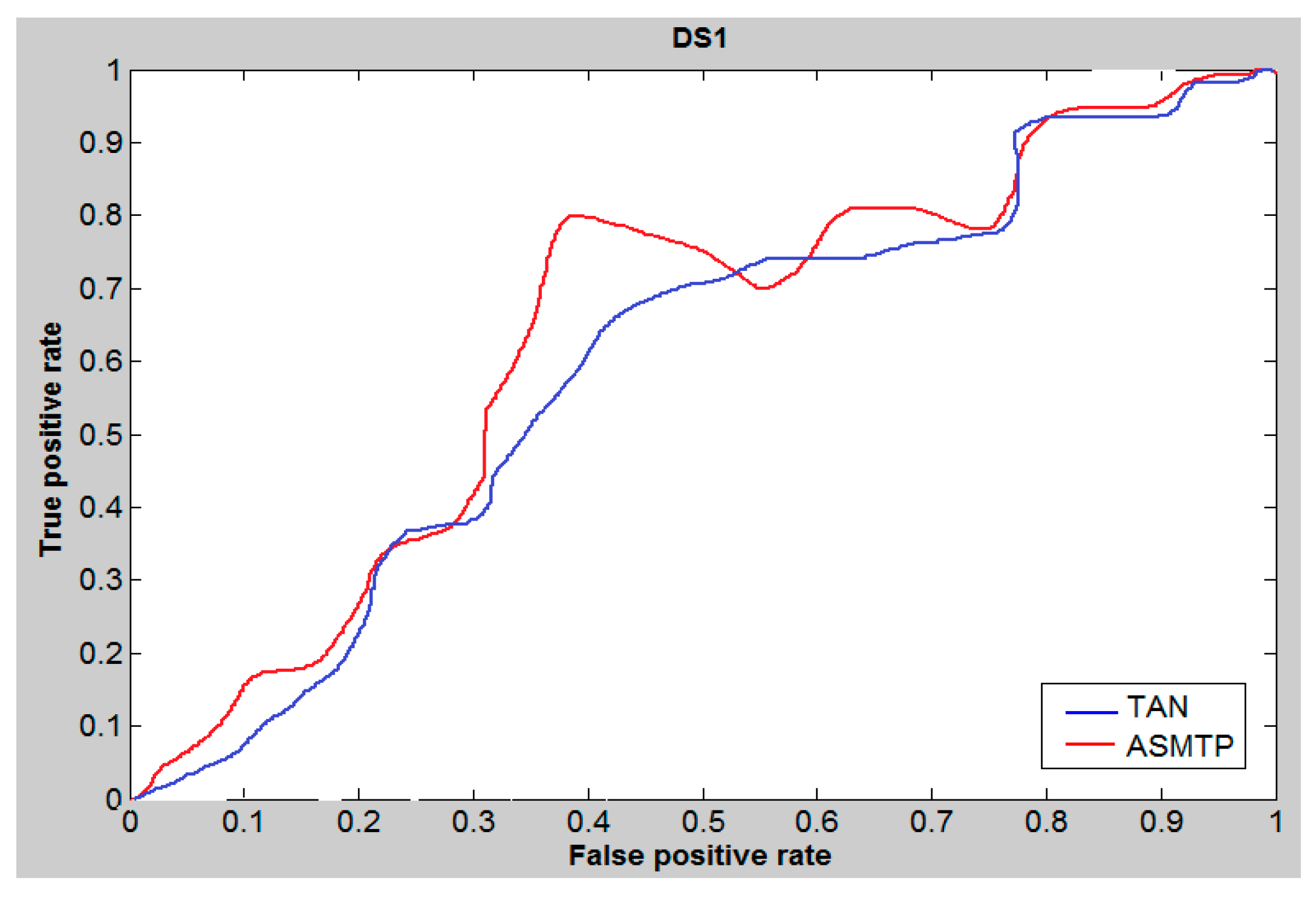
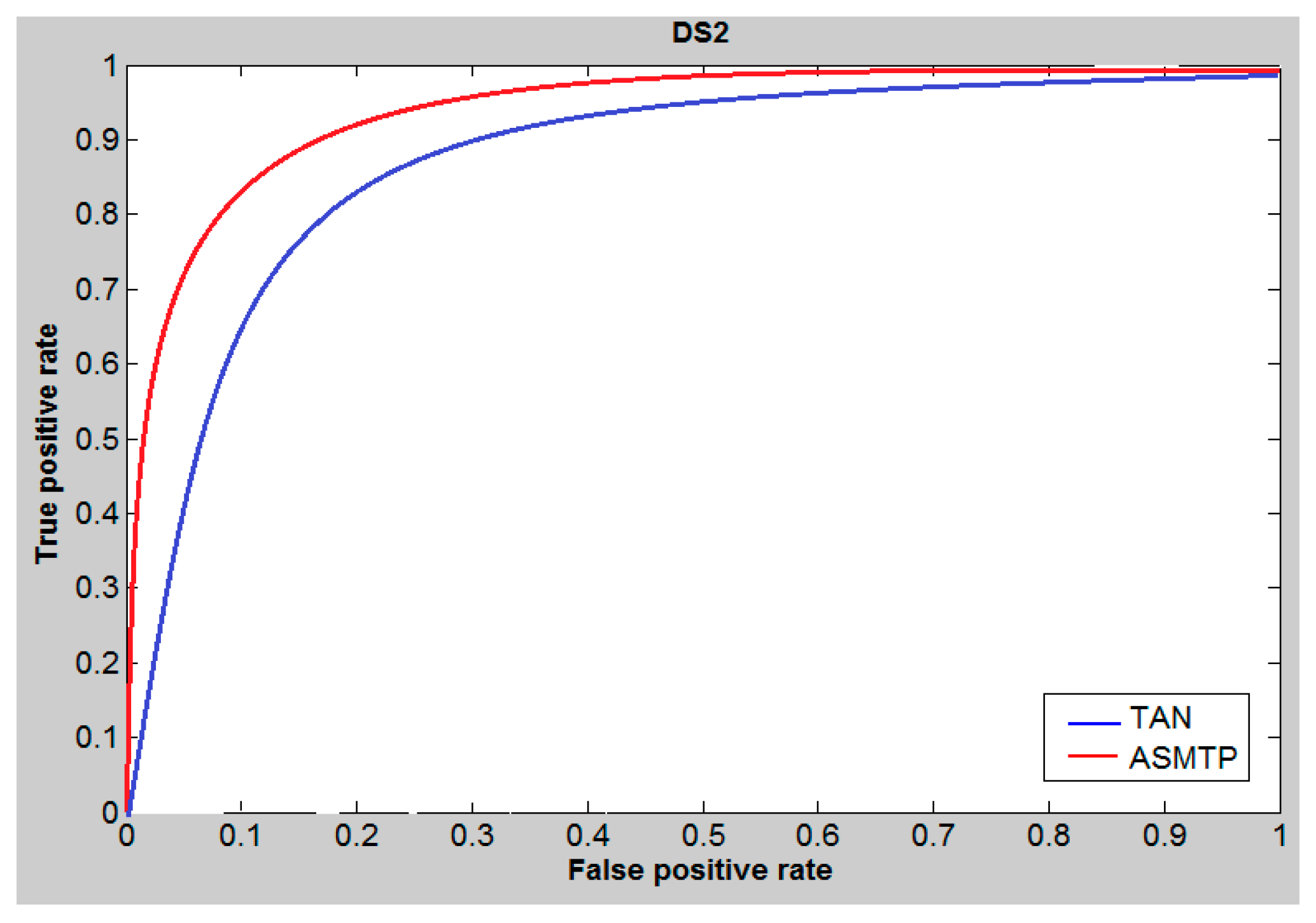
6. Results and Discussion
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Willett, P. Similarity methods in chemoinformatics. Annu. Rev. Inf. Sci. Technol. 2009, 43, 1–117. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Discov. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Cano, G.; García-Rodríguez, J.; Pérez-Sánchez, H. Improvement of Virtual Screening Predictions using Computational Intelligence Methods. Lett. Drug Des. Discov. 2014, 11, 33–39. [Google Scholar] [CrossRef]
- Jain, A. Virtual screening in lead discovery and optimization. Curr. Opin. Drug Discov. Dev. 2004, 7, 396–403. [Google Scholar]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics, Volume 41 (2 Volume Set); John Wiley & Sons: New York, NY, USA, 2009; Volume 41. [Google Scholar]
- Sheridan, R.P. Alternative global goodness metrics and sensitivity analysis: Heuristics to check the robustness of conclusions from studies comparing virtual screening methods. J. Chem. Inf. Model. 2008, 48, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Von Korff, M.; Freyss, J.; Sander, T. Comparison of ligand-and structure-based virtual screening on the DUD data set. J. Chem. Inf. Model. 2009, 49, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: England, UK, 1990. [Google Scholar]
- Ahmed, A.; Abdo, A.; Salim, N. Ligand-based virtual screening using Bayesian inference network and reweighted fragments. Sci. World J. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Saeed, F.; Salim, N.; Abdo, A. Condorcet and borda count fusion method for ligand-based virtual screening. J. Cheminform. 2014, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Liu, Z.; Yan, X.; Ding, Q.; Gu, Q.; Xu, J. LBVS: An online platform for ligand-based virtual screening using publicly accessible databases. Mol. Divers. 2014, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Fusing similarity rankings in ligand-based virtual screening. Comput. Struct. Biotechnol. J. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Textual and Chemical Information Processing: Different Domains but Similar Algorithms. Inf. Res. 2000, 5. Available online: http://www.informationr.net/ir/5-2/paper69.html (accessed on 20 February 2015). [Google Scholar]
- Lin, Y.-S.; Jiang, J.-Y.; Lee, S.-J. A similarity measure for text classification and clustering. IEEE Trans. Knowl. Data Eng. 2014, 26, 1575–1590. [Google Scholar] [CrossRef]
- Downs, G.M.; Willett, P.; Fisanick, W. Similarity searching and clustering of chemical-structure databases using molecular property data. J. Chem. Inf. Comput. Sci. 1994, 34, 1094–1102. [Google Scholar] [CrossRef]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; K Vassilatis, D.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.M. Substructure searching methods: Old and new. J. Chem. Inf. Comput. Sci. 1993, 33, 532–538. [Google Scholar] [CrossRef]
- Willett, P.; Winterman, V.; Bawden, D. Implementation of nonhierarchic cluster analysis methods in chemical information systems: Selection of compounds for biological testing and clustering of substructure search output. J. Chem. Inf. Comput. Sci. 1986, 26, 109–118. [Google Scholar] [CrossRef]
- Willett, P. Similarity-based approaches to virtual screening. Biochem. Soc. Trans. 2003, 31, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Whittle, M.; Gillet, V.J.; Willett, P.; Alex, A.; Loesel, J. Enhancing the effectiveness of virtual screening by fusing nearest neighbor lists: A comparison of similarity coefficients. J. Chem. Inf. Comput. Sci. 2004, 44, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity searching using 2D structural fingerprints. In Chemoinformatics and Computational Chemical Biology; Springer: New York, NY, USA, 2011; pp. 133–158. [Google Scholar]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2014, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Jenkins, J.L.; Scheiber, J.; Sukuru, S.C.K.; Glick, M.; Davies, J.W. How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Downs, G.M.; Willett, P. Similarity searching in databases of chemical structures. Rev. Comput. Chem. 1996, 7, 1–66. [Google Scholar]
- Drwal, M.N.; Griffith, R. Combination of ligand-and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V.; Xiang, H.; Holliday, J.; Buscema, M.; Willett, P. Similarity coefficients for binary chemoinformatics data: Overview and extended comparison using simulated and real data sets. J. Chem. Inf. Model. 2012, 52, 2884–2901. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): Evaluation of performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ekins, S. Computer Applications in Pharmaceutical Research and Development; John Wiley & Sons: New York, NY, USA, 2006; Volume 2. [Google Scholar]
- Han, L.; Ma, X.; Lin, H.; Jia, J.; Zhu, F.; Xue, Y.; Li, Z.; Cao, Z.; Ji, Z.; Chen, Y. A support vector machines approach for virtual screening of active compounds of single and multiple mechanisms from large libraries at an improved hit-rate and enrichment factor. J. Mol. Gr. Model. 2008, 26, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Gilson, M.K. Virtual screening of molecular databases using a support vector machine. J. Chem. Inf. Model. 2005, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. New methods for ligand-based virtual screening: Use of data fusion and machine learning to enhance the effectiveness of similarity searching. J. Chem. Inf. Model. 2006, 46, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Holliday, J.; Bradshaw, J. A machine learning approach to weighting schemes in the data fusion of similarity coefficients. J. Chem. Inf. Model. 2009, 49, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Symyx technologies. Mdl drug data report: Sci Tegic Accelrys Inc., the MDL Drug Data Report (MDDR). Available online: http://www.accelrys.com/ (accessed on 12 March 2016).
- Rohrer, S.G.; Baumann, K. Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar]
- Al-Dabbagh, M.M.; Salim, N.; Himmat, M.; Ahmed, A.; Saeed, F. A quantum-based similarity method in virtual screening. Molecules 2015, 20, 18107–18127. [Google Scholar] [CrossRef] [PubMed]
- Pipeline Pilot Software; Scitegic Accelrys Inc.: San Diego, CA, USA, 2008.
- Truchon, J.-F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminform. 2013, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nagwani, N.K. A comment on “a similarity measure for text classification and clustering”. IEEE Trans. Knowl. Data Eng. 2015, 27, 2589–2590. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.

{kind=link}
{kind=link}
{kind=link}
| Similarity Measure | Formula |
|---|---|
| Extended Jaccard coefficient | S= |
| Dice | = |
| Euclidean distance | |
| Cosine similarity | |
| Pairwise-adaptive | = |
| IT-Sim |
| Activity Index | Activity Class | Active Molecules |
|---|---|---|
| 31420 | Renin inhibitors | 1130 |
| 71523 | HIV protease | 750 |
| 37110 | Thrombin inhibitors | 803 |
| 31432 | Angiotensin II AT1antagonists | 943 |
| 42731 | Substance P antagonists | 1246 |
| 06233 | Substance P antagonists | 752 |
| 06245 | 5HT reuptake inhibitors | 359 |
| 07701 | D2 antagonists | 395 |
| 06235 | 5HT1A agonists | 827 |
| 78374 | Protein kinase C inhibitors | 453 |
| Activity Index | Activity Class | Active Molecules |
|---|---|---|
| 07707 | Adenosine (AI) agonists | 207 |
| 07708 | Adenosine (A2) agonists | 156 |
| 31420 | Rennin inhibitors 1 | 1300 |
| 42710 | CCK agonists | 111 |
| 64100 | Monocycle_ lactams | 1346 |
| 64200 | Cephalosporin’s | 113 |
| 64220 | Carbacephems | 1051 |
| 64500 | Carbapenems | 126 |
| 64350 | Tribactams | 388 |
| 75755 | Vitamin D analogues | 455 |
| Activity Index | Activity Class |
|---|---|
| 466 | S1P1 rec. (agonists) |
| 548 | PKA (inhibitors) |
| 600 | SF1 (inhibitors) |
| 644 | Rho-Kinase2 (inhibitors) |
| 652 | HIV RT-RNase (inhibitors) |
| 689 | Eph rec. A4 (inhibitors) |
| 692 | SF1 (agonists) |
| 712 | HSP 90 (inhibitors) 30 |
| 713 | ER-a-Coact. Bind. (inhibitors) |
| 733 | ER-b-Coact. Bind. (inhibitors) |
| 737 | ER-a-Coact. Bind. (potentiators) |
| 810 | FAK (inhibitors |
| 832 | Cathepsin G (inhibitors) |
| 846 | FXIa (inhibitors) |
| 852 | FXIIa (inhibitors) |
| 858 | D1 rec. (allosteric modulators) |
| 859 | M1 rec. (allosteric inhibitors) |
| Activity Classes | TAN | SQB | ASMTP | |||
|---|---|---|---|---|---|---|
| 1% | 5% | 1% | 5% | 1% | 5% | |
| 31420 | 57.3 | 85.85 | 73.73 | 87.22 | 78.83 | 96.81 |
| 71523 | 29.96 | 58.09 | 26.84 | 48.7 | 12.82 | 51.94 |
| 37110 | 14.38 | 29.98 | 24.73 | 45.62 | 39.53 | 63.84 |
| 31432 | 36.37 | 76.85 | 36.66 | 70.44 | 45.22 | 97.45 |
| 42731 | 16.89 | 27.74 | 21.17 | 19.35 | 13.95 | 20.88 |
| 6233 | 22.72 | 37.78 | 12.49 | 21.04 | 22.77 | 36.75 |
| 6245 | 5.03 | 14.83 | 6.03 | 13.63 | 11.73 | 26.26 |
| 7701 | 8.45 | 23.07 | 11.35 | 21.85 | 8.95 | 17.26 |
| 6235 | 9.03 | 21 | 10.15 | 19.13 | 21.91 | 37.17 |
| 78374 | 12.08 | 17.81 | 13.08 | 20.55 | 1.77 | 2.65 |
| 78331 | 8.77 | 16.71 | 5.92 | 13.1 | 3.31 | 10.24 |
| Mean | 20.08909 | 37.24636 | 22.01364 | 34.05 | 23.65 | 41.93182 |
| Shaded cells | 3 | 4 | 0 | 0 | 8 | 7 |
| Activity Classes | TAN | SQB | Proposed Method | |||
|---|---|---|---|---|---|---|
| 1% | 5% | 1% | 5% | 1% | 5% | |
| 09249 | 61.84 | 70.39 | 58.5 | 74.22 | 72.82 | 73.3 |
| 12455 | 47.03 | 56.58 | 55.61 | 100 | 99.35 | 100 |
| 12464 | 65.1 | 88.19 | 62.22 | 95.24 | 81.66 | 96.46 |
| 31281 | 81.82 | 86.64 | 83 | 93 | 92.73 | 99.09 |
| 43210 | 80.31 | 93.75 | 80.73 | 98.94 | 88.2 | 99.85 |
| 71522 | 53.84 | 77.68 | 53.13 | 98.93 | 81.25 | 99.11 |
| 75721 | 46.8 | 63.94 | 34.61 | 90.9 | 77.27 | 98.67 |
| 78331 | 30.56 | 44.8 | 29.04 | 92.72 | 80 | 96.8 |
| 78348 | 80.18 | 91.71 | 81.86 | 93.75 | 82.17 | 99.74 |
| 78351 | 87.56 | 94.82 | 85.4 | 95.39 | 96.48 | 96.92 |
| Mean | 63.504 | 76.85 | 62.41 | 93.31 | 85.193 | 95.994 |
| Shaded cells | 0 | 0 | 0 | 0 | 10 | 10 |
| Activity Index | Tanimoto | SQB | Proposed Method | |||
|---|---|---|---|---|---|---|
| 1% | 5% | 1% | 5% | 1% | 5% | |
| 466 | 3.1 | 5.86 | 1.38 | 8.62 | 5.86 | 9.66 |
| 548 | 8.62 | 22.76 | 11.38 | 24.14 | 10.34 | 17.93 |
| 600 | 3.79 | 11.38 | 5.52 | 16.21 | 6.21 | 13.45 |
| 644 | 7.59 | 17.59 | 8.97 | 17.93 | 7.24 | 12.41 |
| 652 | 2.76 | 7.93 | 3.79 | 9.66 | 5.86 | 11.38 |
| 689 | 3.79 | 9.66 | 4.48 | 11.72 | 5.86 | 9.71 |
| 692 | 0.69 | 4.83 | 1.38 | 4.83 | 3.79 | 6.55 |
| 712 | 4.14 | 10.34 | 5.17 | 11.03 | 6.21 | 8.97 |
| 713 | 3.1 | 7.24 | 2.76 | 5.86 | 6.21 | 9.31 |
| 733 | 3.45 | 8.97 | 4.14 | 8.62 | 5.86 | 9.31 |
| 737 | 2.41 | 8.28 | 1.72 | 8.28 | 7.59 | 14.14 |
| 810 | 2.07 | 6.9 | 1.72 | 11.03 | 7.24 | 13.1 |
| 832 | 6.55 | 13.1 | 8.28 | 14.83 | 13.1 | 20 |
| 846 | 9.66 | 28.62 | 12.41 | 26.9 | 10.69 | 25.52 |
| 852 | 12.41 | 21.38 | 9.66 | 20 | 13.45 | 21.03 |
| 858 | 1.72 | 5.86 | 1.38 | 6.21 | 6.21 | 7.93 |
| 859 | 1.38 | 8.97 | 2.41 | 8.62 | 5.86 | 10.69 |
| Avg | 4.54 | 11.70 | 5.09 | 12.61 | 7.50 | 12.991 |
| Shaded cells | 0 | 2 | 3 | 5 | 14 | 11 |
| Methods | DS1 | DS2 | MUV | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BEDROC( α 20) | EF (1%) | BEDROC( α 20) | EF (1%) | BEDROC( α 20) | EF (1%) | |||||||
| Mean | Median | Mean | Median | Mean | Median | Mean | Median | Mean | Median | Mean | Median | |
| Tan | 0.48 | 0.46 | 80.01 | 86.01 | 0.33 | 0.34 | 23.01 | 23.01 | 0.37 | 0.37 | 16.69 | 17.92 |
| SQB | 0.53 | 0.57 | 90.01 | 89.31 | 0.44 | 0.39 | 29.01 | 22.01 | 0.41 | 0.39 | 18.01 | 19.74 |
| ASMTP | 0.61 | 0.64 | 92.9 | 90.23 | 0.46 | 0.50 | 28.27 | 25.32 | 0.44 | 0.42 | 18.93 | 20.14 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Himmat, M.; Salim, N.; Al-Dabbagh, M.M.; Saeed, F.; Ahmed, A. Adapting Document Similarity Measures for Ligand-Based Virtual Screening. Molecules 2016, 21, 476. https://doi.org/10.3390/molecules21040476
Himmat M, Salim N, Al-Dabbagh MM, Saeed F, Ahmed A. Adapting Document Similarity Measures for Ligand-Based Virtual Screening. Molecules. 2016; 21(4):476. https://doi.org/10.3390/molecules21040476
Chicago/Turabian StyleHimmat, Mubarak, Naomie Salim, Mohammed Mumtaz Al-Dabbagh, Faisal Saeed, and Ali Ahmed. 2016. "Adapting Document Similarity Measures for Ligand-Based Virtual Screening" Molecules 21, no. 4: 476. https://doi.org/10.3390/molecules21040476