
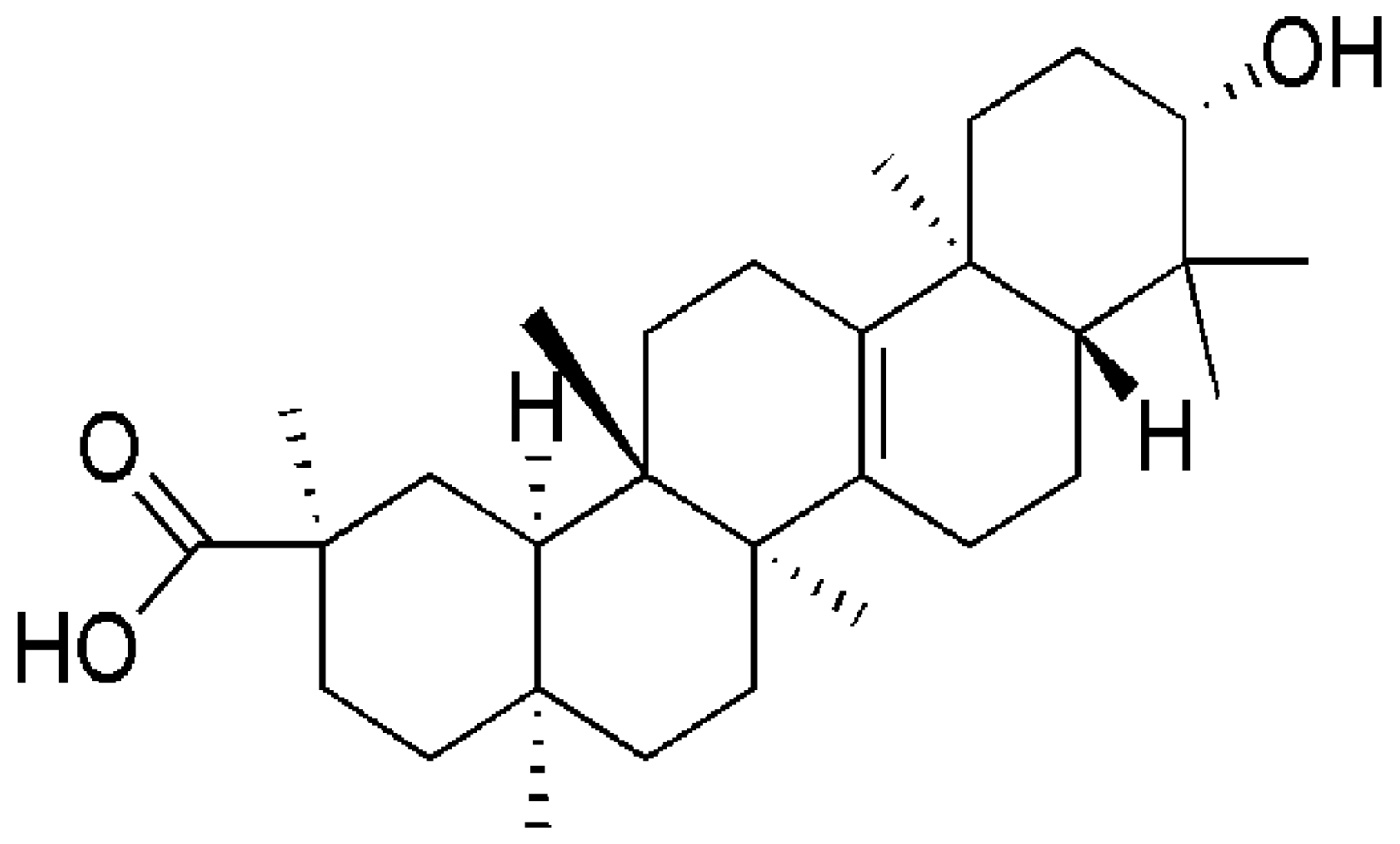
Bryonolic Acid, a Triterpenoid, Protect Against N-methyl-d-Aspartate-Induced Neurotoxicity in PC12 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
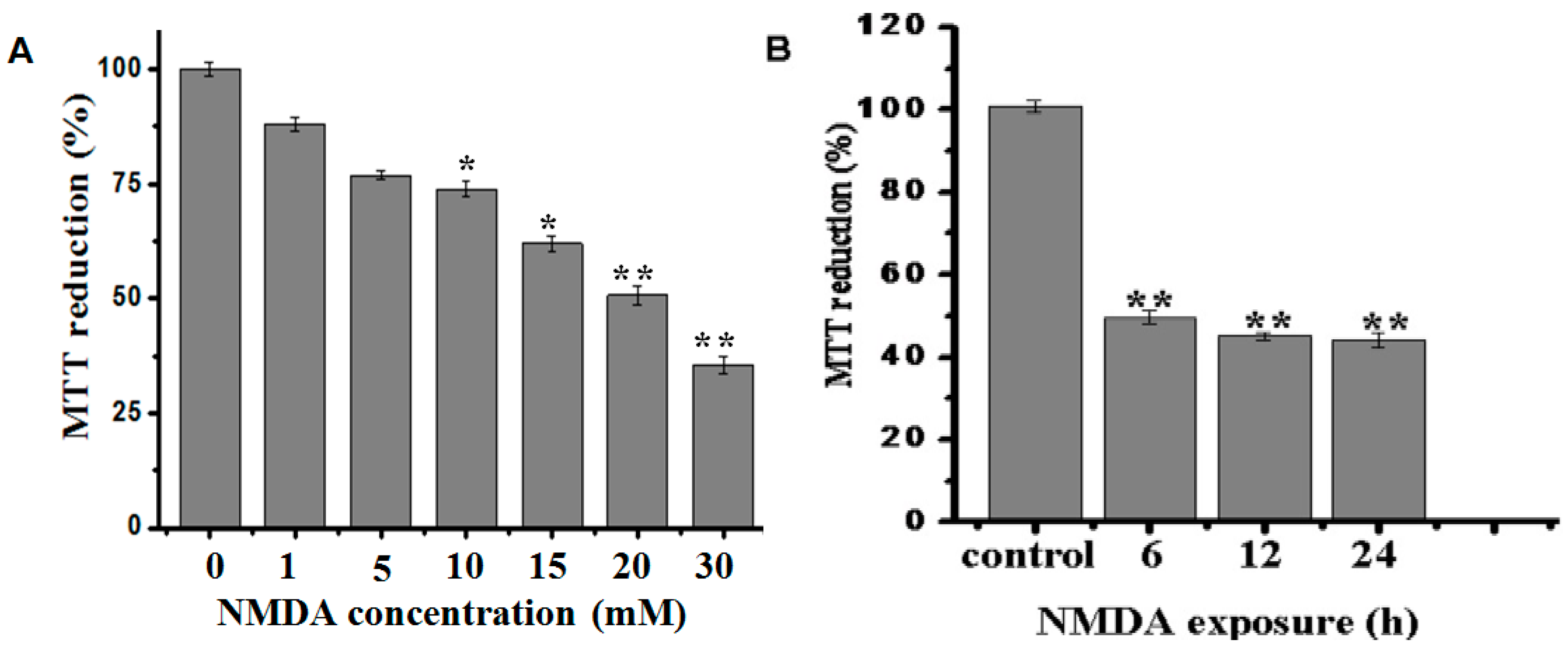
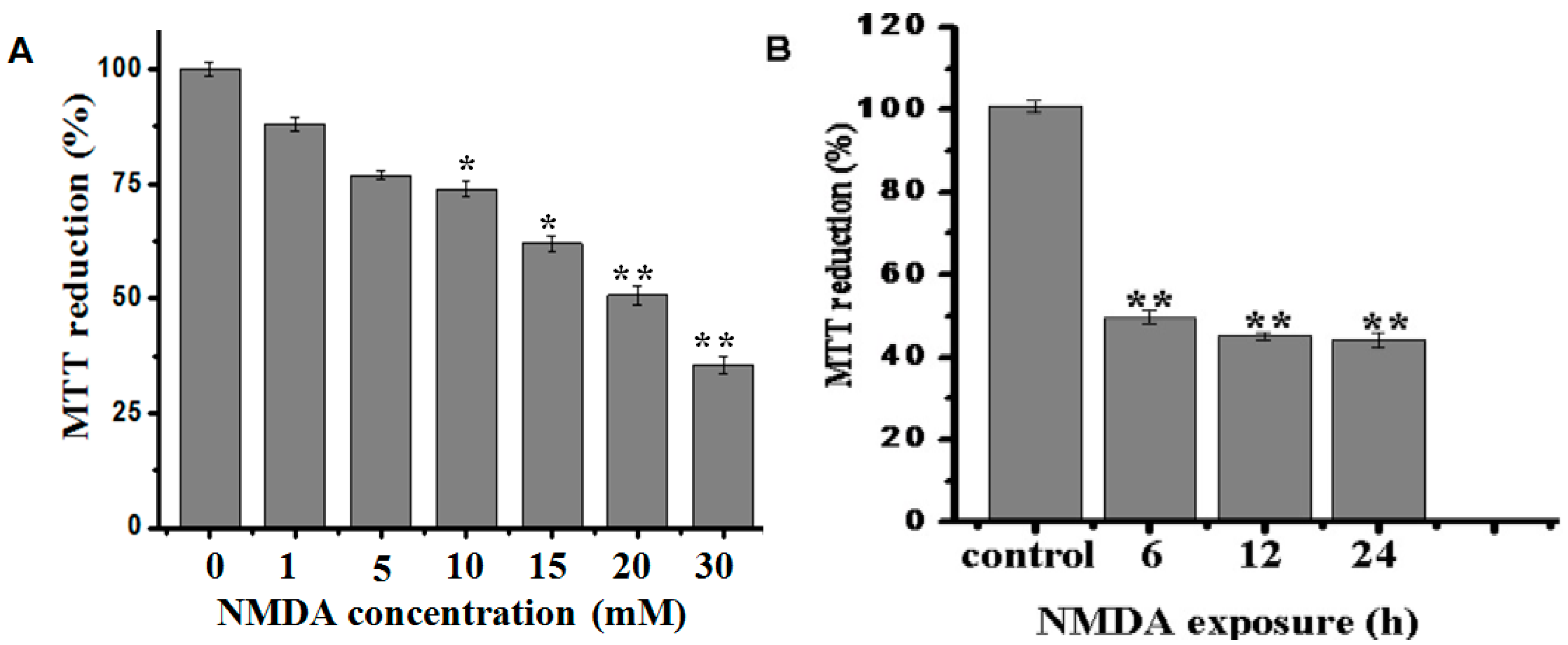
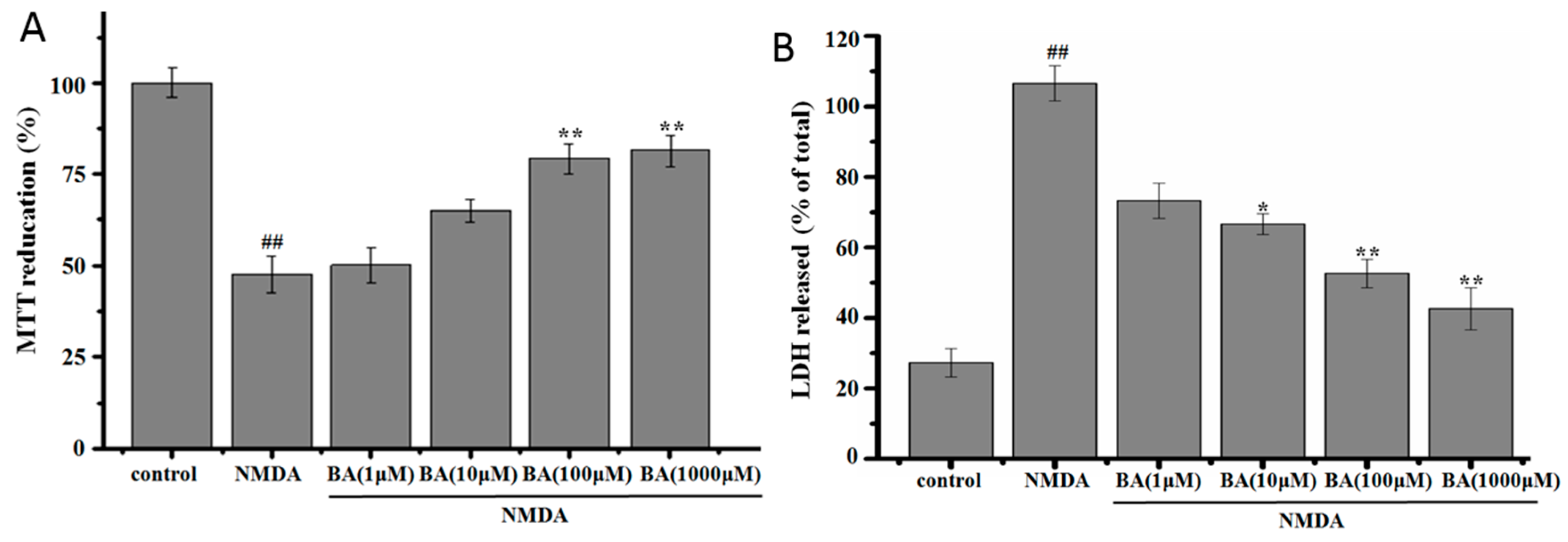
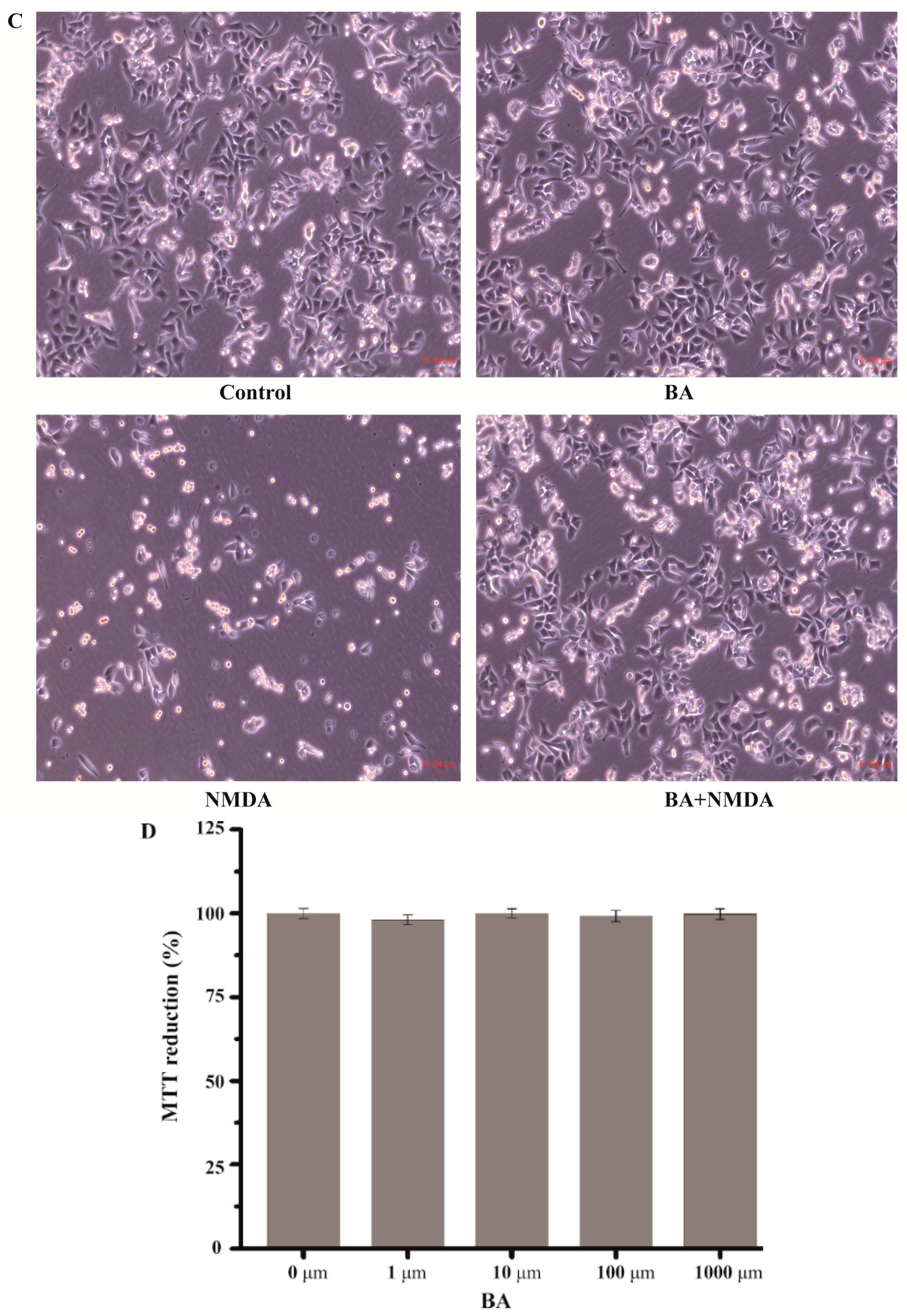
2.1. Effect of BA on the Cell Viability in NMDA-Induced Excitotoxicity
2.2. Effect of BA on LDH Release
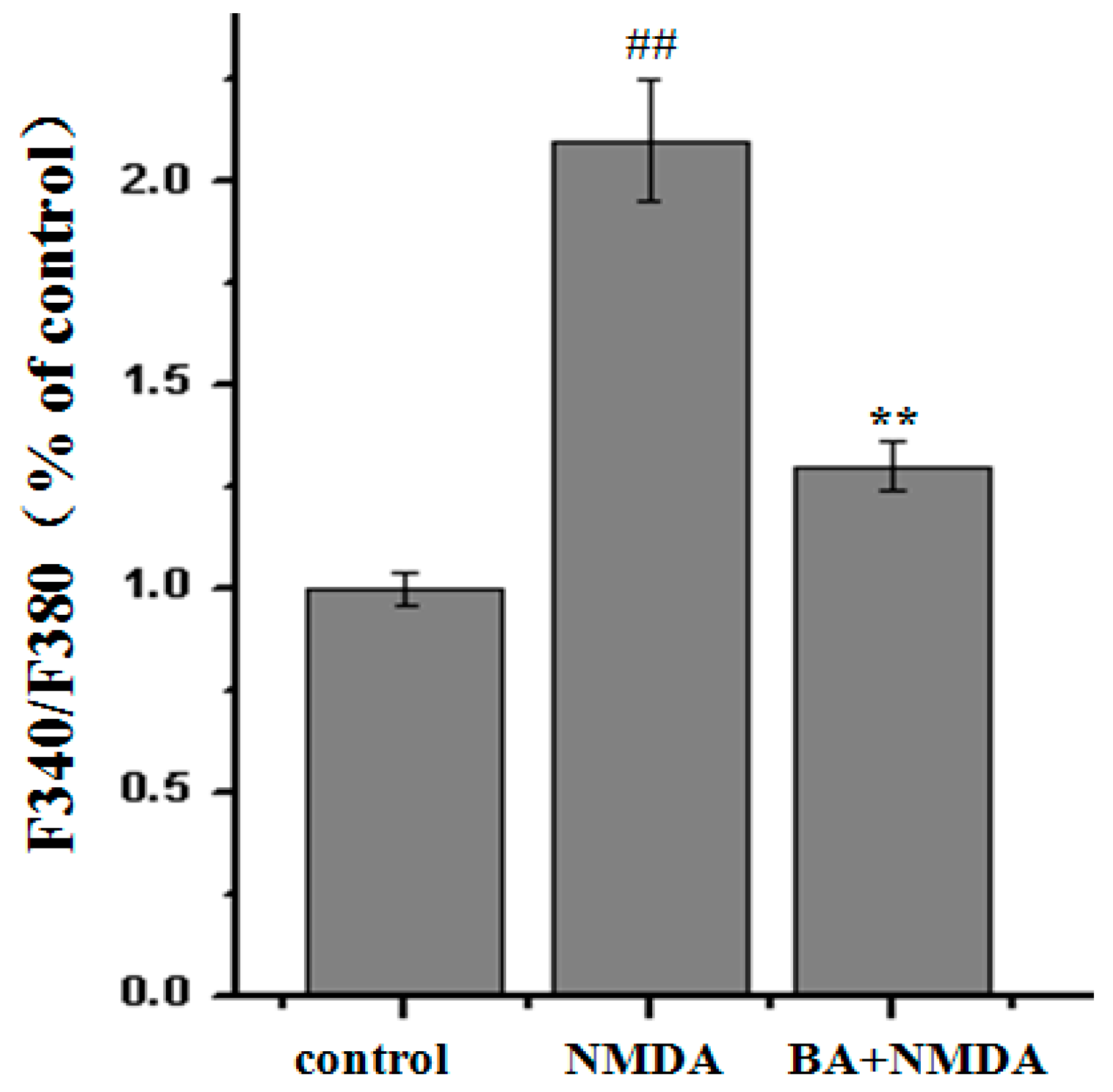
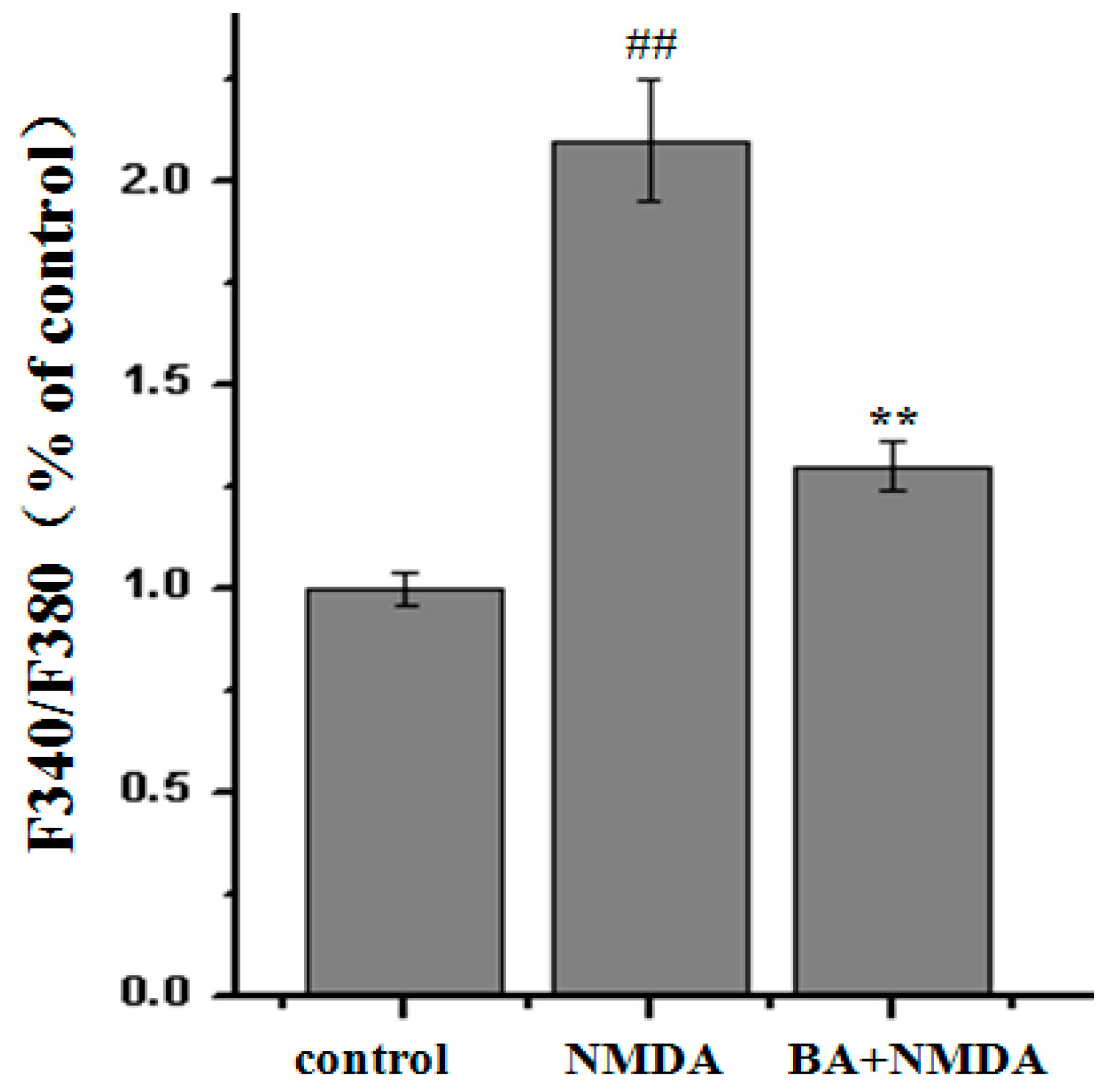
2.3. Effect of BA on Intracellular Ca2+ Concentration
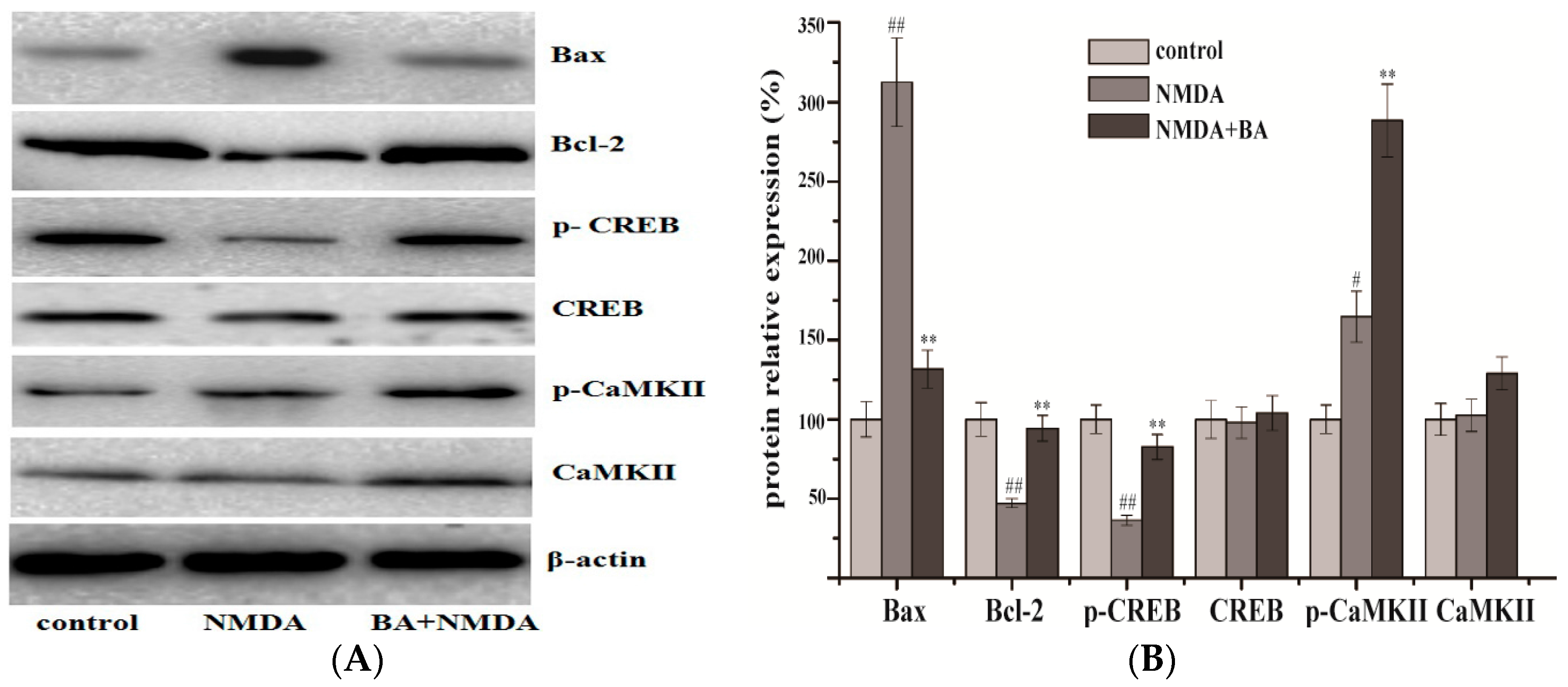
2.4. Effects of BA on the Protein Expression of CaMKII, p-CaMKII, CREB, p-CREB, Bax, Bcl-2
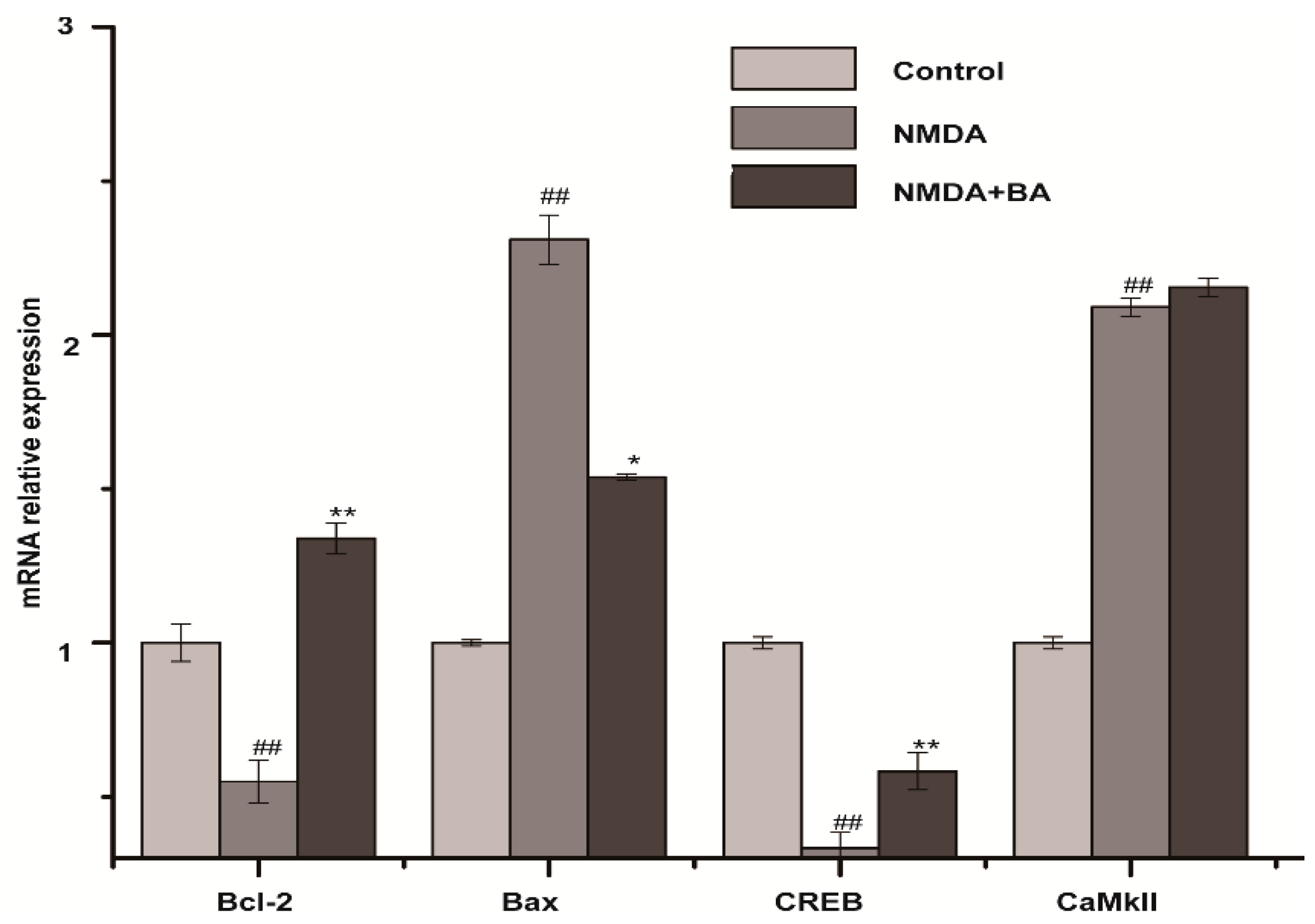
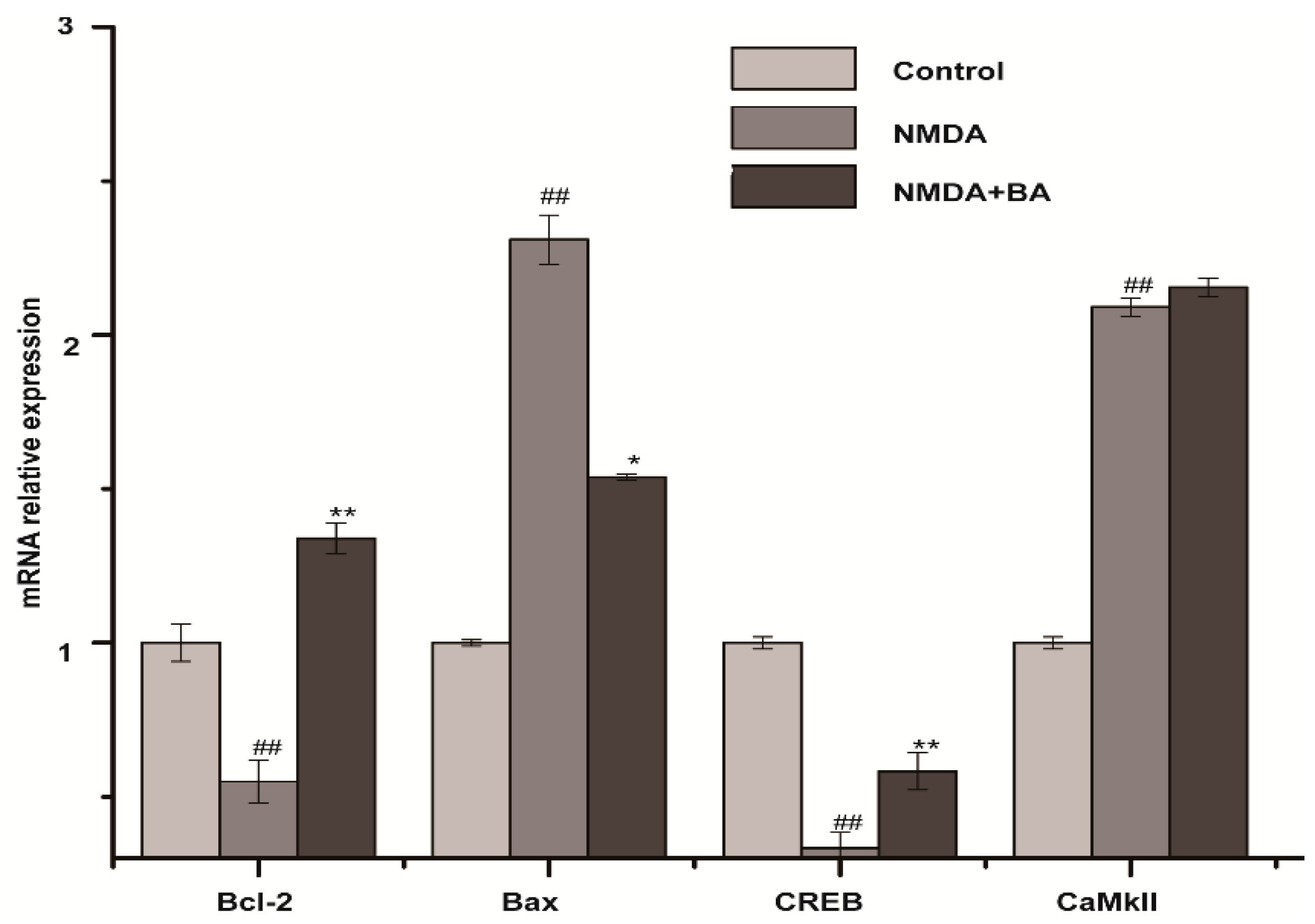
2.5. Effects of BA on the mRNA Expression of CaMKII, CREB, Bax, Bcl-2
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Treatment
4.3. Cell Viability Assay
4.4. Lactate Dehydrogenase (LDH) Assay
4.5. Measurement of Intracellular Ca2+ Concentration
4.6. Western Blot Analysis
4.7. Real-Time PCR Analysis
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CREB | cAMP response element-binding protein |
| NMDA | N-Methyl-d-aspartate |
| PC12 | pheochromocytoma cell line |
| BA | bryonolic acid |
| MTT | methyl thiazolyl tetrazolium |
References
- Simerabet, M.; Robin, E.; Aristi, I.; Adamczyk, S.; Tavernier, B.; Vallet, B.; Bordet, R.; Lebuffe, G. Preconditioning by an in situ administration of hydrogen peroxide: Involvement of reactive oxygenspecies and mitochondrial ATP-dependent potassium channel in a cerebral Ischemia-Reperfusion model. Brain Res. 2008, 1240, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Nappi, G.; Bernardi, G.; Bagetta, G.; Corasaniti, M.T. Post-ischemic Brain Damage: Pathophysiology and Role of Inflammatory Mediators. FEBS J. 2009, 276, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E. Injury and Repair Mechanisms in Ischemic Stroke: Considerations for the Development of Novel Neurotherapeutics. Curr. Opin. Investig. Drug 2009, 10, 644–654. [Google Scholar]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of Ischemic Brain Damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, V.; Qureshi, A.I. Mechanisms of Ischemic Brain Injury. Curr. Cardiol. Rep. 2004, 6, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.W.; Liu, H.X.; Jiao, H.Y.; Wang, L.Y.; Chen, L.Y.; Liang, J.; Zhao, M.; Zhang, X.T. Neuroprotective Effect of Ginkgolide K on Glutamate-induced Cytotoxicity in PC 12 Cells via Inhibition of ROS Generation and Ca2+ Influx. Neurotoxicology 2012, 33, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Minamisawa, H.; Katayama, Y.; Ueda, M.; Terashi, A.; Nakamura, K.; Kudo, Y. Increased Intracellular Ca2+ Concentration in the Hippocampal CA1 Area During Global Ischemia and Reperfusion in the Rat: A Possible Cause of Delayed Neuronal Death. Neurosci. 1999, 88, 57–67. [Google Scholar] [CrossRef]
- Bano, D.; Nicotera, P. Ca2+ Signals and Neuronal Death in Brain Ischemia. Stroke 2007, 38 (Suppl. 2), S674–S676. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular Targets in Cerebral Ischemia for Developing Novel Therapeutics. Brain Res. Rev. 2007, 54, 34–66. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.S. Pathophysiology of Focal Cerebral Ischemia: A Therapeutic Perspective. J. Vasc. Interv. Radiol. 2004, 15, Pt 2. S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Erondu, N.E.; Kennedy, M.B. Regional Distribution of Type II Ca2+/Calmodulin-dependent Protein Kinase in Rat Brain. J. Neurosci. 1985, 5, 3270–3277. [Google Scholar] [PubMed]
- Takeda, H.; Kitaoka, Y.; Hayashi, Y.; Kumai, T.; Munemasa, Y.; Fujino, H.; Kobayashi, S.; Ueno, S. Calcium/Calmodulin-dependent Protein Kinase II Regulatesthe Phosphorylation of CREB in NMDA-induced Retinal Neurotoxicity. Brain Res. 2007, 1184, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.S.; Lim, I.A.; Hemsworth, D.E.; Horne, M.C. Calcium/Calmodulin-dependent Protein Kinase II is Associated with the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Barria, A.; Muller, D.; Derkach, V.; Griffith, L.C.; Soderling, T.R. Regulatory Phosphorylation of AMPA-type Glutamate Receptors by CaM-KII During Long-term Potentiation. Science 1997, 276, 2042–2045. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.; Edin, S.; Antonsson, A.; Grundström, T. Calmodulin-dependent Kinase II Mediates T Cell Receptor/CD3- and Phorbol Ester-induced Activation of IkappaB Kinase. J. Biol. Chem. 2001, 276, 36008–36013. [Google Scholar] [CrossRef] [PubMed]
- Barkai, U.; Prigent-Tessier, A.; Tessier, C.; Gibori, G.B.; Gibori, G. Involvement of SOCS-1, the Suppressor of Cytokine Signaling, in the Prevention of Prolactin-Responsive Gene Expression in Decidual Cells. Mol. Endocrinol. 2000, 14, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Seo, Y.J.; Kwon, M.S.; Shim, E.J.; Lee, J.Y.; Ham, Y.O.; Lee, H.K.; Suh, H.W. Increase of Phosphorylation of Calcium/Calmodulin-dependent Protein Kinase-II in Several Brain Regions by Substance P Administered Intrathecally in Mice. Brain Res. Bull. 2005, 65, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Fukushi, H.; Morishima, Y.; Shirasaki, Y. Calmodulin and Calmodulin-Dependent Kinase II Mediate Neuronal Cell Death Induced by Depolarization. Brain Res. 2003, 962, 89241–89247. [Google Scholar] [CrossRef]
- Meng, F.; Guo, J.; Zhang, Q.; Song, B.; Zhang, G. Autophosphorylated Calcium/Calmodulin-dependent Protein Kinase II Alpha (CaMKII alpha) Reversibly Targets to and Phosphorylates N-methyl-d-Aspartate Receptor Subunit 2B (NR2B) in Cerebral Ischemia and Reperfusion in Hippocampus of Rats. Brain Res. 2003, 967, 161–169. [Google Scholar] [CrossRef]
- Tang, K.; Liu, C.; Kuluz, J.; Hu, B. Alterations of CaMKII after Hypoxia–ischemia During Brain Development. J. Neurochem. 2004, 91, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Y.; Chang, Y.C.; Lee, H.T.; Huang, C.C. CREB Activation in the Rapid, Intermediate, and Delayed Ischemic Preconditioning Against Hypoxic-Ischemia in Neonatal Rat. J. Neurochem. 2009, 108, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.N.; Waxham, M.N.; Dash, P.K. Neuronal Activity Increases the Phosphorylation of the Transcription Factor cAMP Response Element-Binding Protein (CREB) in Rat Hippocampus and Cortex. J. Biol. Chem. 1996, 271, 14214–14220. [Google Scholar] [PubMed]
- Lee, M.M.; Badache, A.; DeVries, G.H. Phosphorylation of CREB in Axon-induced Schwann Cell Proliferation. J. Neurosci. Res. 1999, 55, 702–712. [Google Scholar] [CrossRef]
- Lonze, B.E.; Riccio, A.; Cohen, S.; Ginty, D.D. Apoptosis, Axonal Growth Defects, and Degeneration of Peripheral Neurons in Mice Lacking CREB. Neuron 2002, 34, 371–385. [Google Scholar] [CrossRef]
- Redmond, L.; Kashani, A.H.; Ghosh, A. Calcium Regulation of Dendritic Growth via CaM Kinase IV and CREB-mediated Transcription. Neuron 2002, 34, 999–1010. [Google Scholar] [CrossRef]
- Wong, K.; Zhang, J.; Awasthi, S.; Sharma, A.; Rogers, L.; Matlock, E.F.; Van Lint, C.; Karpova, T.; McNally, J.; Harrod, R. Nerve Growth Factor Receptor Signaling Induces Histone Acetyltransferase Domain-dependent Nuclear Translocation of p300/CREB-binding Protein-associated Factor and hGCN5 Acetyltransferases. J. Biol. Chem. 2004, 279, 55667–55674. [Google Scholar] [CrossRef] [PubMed]
- Meller, R.; Minami, M.; Cameron, J.A.; Impey, S.; Chen, D.; Lan, J.Q.; Henshall, D.C.; Simon, R.P. CREB-mediated Bcl-2 Protein Expression after Ischemic Preconditioning. J. Cereb. Blood Flow Metab. 2005, 25, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, H.; Zhang, S.W. Effect of Trichosanthin on High Plasm a Homocystein in Rats with Cerebral Ischemia-reperfasion. JETCM 2011, 20, 275–277. [Google Scholar]
- Chen, W.; Zhang, S.W.; Wang, H. Study of Radices Trichosanthis on Neuronal Apoptosis in Rats with Local Cerebral Ischemia Reperfusion. Mod. J. Integr. Tradit. Chin West. Med. 2011, 20, 1844–1845. [Google Scholar]
- Tanaka, S.; Uno, C.; Akimoto, M.; Tabata, M.; Honda, C.; Kamisako, W. Anti Allergic Effect of Bryonolic Acid from Luffa Cylindrica Cell Suspension Cultures. Planta Med. 1991, 57, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Akihisa, T.; Tokuda, H.; Ichiishi, E.; Mukainaka, T.; Toriumi, M.; Ukiya, M.; Yasukawa, K.; Nishino, H. Anti-tumor Promoting Effects of Multiflorane-type Triterpenoids and Cytotoxic Activity of Karounidiol Against Humancancer cell Lines. Cancer Lett. 2001, 173, 9–14. [Google Scholar] [CrossRef]
- Kondo, T.; Inoue, M.; Mizukami, H.; Ogihara, Y. Cytotoxic Activity of Bryonolic Acid Isolated from Transformed Hairy Roots of Trichosanthes Kirilowii Var. Japonica. Biol. Pharm. Bull. 1995, 18, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Kondo, T.; Mizukami, H.; Ogihara, Y. Bryonolic Acid Production in Hairy Roots of Trichosanthes Kirilowii Max. Var Japonica Kitam. Transformed with Agrobacterium Rhizogenes and Its Cytotoxic activity. Chem. Pharm. Bull. 1994, 42, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Barker, E.C.; Gatbonton-Schwager, T.N.; Han, Y.; Clay, J.E.; Letterio, J.J.; Tochtrop, G.P. Bryonolic Acid: A Large-scale Isolation and Evaluation of Heme Oxygenase 1 Expression in Activated Macrophages. J. Nat. Prod. 2012, 73, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.Q.; Zhong, X.M.; Li, Z.Y.; Huang, Z. Paeoniflorin Protects Against NMDA-induced Neurotoxicity in PC12 Cells via Ca2+ Antagonism. Phytother. Res. 2011, 25, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Jeong, H.Y.; Lee, H.K.; Kim, S.; Hwang, B.Y.; Bae, K.; Seong, Y.H. Neuroprotection of the Leaf and Stem of Vitis Amurensis and Their Active Compounds Against Ischemic Brain Damage in Rats and Excitotoxicity in Cultured Neurons. Phytomedicine 2012, 19, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Negis, Y.; Unal, A.Y.; Korulu, S.; Karabay, A. Cell Cycle Markers Have Different Expression and Localization Patterns in Neuron-like PC12 Cells and Primary Hippocampal Neurons. Neurosci. Lett. 2011, 496, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Dzubak, P.; Hajduch, M.; Vydra, D.; Hustova, A.; Kvasnica, M.; Biedermann, D.; Markova, L.; Urban, M.; Sarek, J. Pharmacological Activities of Natural Triterpenoids and Their Therapeutic Implications. Nat. Prod. Rep. 2006, 23, 394–411. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Que, J.; Ye, M.; Zhang, Y.; Xu, W.; Li, H.; Xu, W.; Chu, K. Bryonolic Acid, a Triterpenoid, Protect Against N-methyl-d-Aspartate-Induced Neurotoxicity in PC12 Cells. Molecules 2016, 21, 418. https://doi.org/10.3390/molecules21040418
Que J, Ye M, Zhang Y, Xu W, Li H, Xu W, Chu K. Bryonolic Acid, a Triterpenoid, Protect Against N-methyl-d-Aspartate-Induced Neurotoxicity in PC12 Cells. Molecules. 2016; 21(4):418. https://doi.org/10.3390/molecules21040418
Chicago/Turabian StyleQue, Jinhua, Miao Ye, Yuqin Zhang, Wen Xu, Huang Li, Wei Xu, and Kedan Chu. 2016. "Bryonolic Acid, a Triterpenoid, Protect Against N-methyl-d-Aspartate-Induced Neurotoxicity in PC12 Cells" Molecules 21, no. 4: 418. https://doi.org/10.3390/molecules21040418