
Synthesis of Novel Quaternary Ammonium Salts and Their in Vitro Antileishmanial Activity and U-937 Cell Cytotoxicity

 ,
,
Abstract
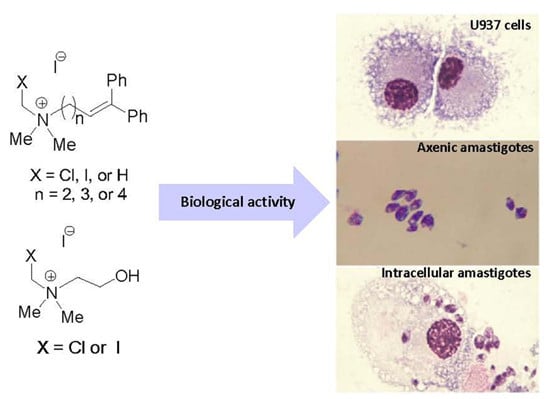
:
1. Introduction
2. Results
2.1. Synthesis
2.2. In Vitro Cytotoxicity of Quaternary N-(Halomethylated) Ammonium Salts 1 and 2, Non N-(Halomethylated) Ammonium Salts 3 and Halogenated Cholines 4
3. Discussion
4. Materials and Methods
4.1. Structural Characterization
4.2. Synthesis
4.2.1. General Procedure for the Preparation of ω-Bromo-α,α-diphenyl Alcohols 9
4.2.2. General Procedure for the Preparation of ω-Bromo-α,α-diphenyl Olefins 10
4.2.3. General Procedure for the Preparation of ω-(N,N-Dimethylamino)-α,α-diphenyl Olefins 11
4.2.4. General Procedure for the Preparation of Quaternary Ammonium Salts 1–3 (Series I and II)
4.2.5. General Procedure for the Preparation of Choline 4c and Choline Analogs 4a and 4b (Series III)
4.3. Biological Activity
4.3.1. Cells and Culture Conditions
4.3.2. Leishmania Strain and Cultivation of Parasites
4.3.3. In Vitro Cytotoxicity Using U-937 Cells
4.3.4. In Vitro Leishmanicidal Activity on Intracellular Amastigotes
4.3.5. In Vitro Leishmanicidal Activity with Axenic Amastigotes
4.3.6. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| VL | Visceral leishmaniasis |
| ML | Mucocutaneous leishmaniasis |
| CL | Cutaneous leishmaniasis |
| MA | Meglumine antimoniate |
| AmB | Amphotericin B |
| EC50 | Effective Concentration 50 |
| LC50 | Lethal Concentration 50 |
| FBS | Fetal Bovine Serum |
| NNN | Novy-MacNeal-Nicholle |
| PMA | Phorbol 12-myristate 13-acetate |
| RPMI | Roswell Park Memorial Institute |
| O.D. | Optical Density |
| THF | Tetrahydrofuran |
| DMSO | Dimethylsulfoxide |
References
- World Health Organization. Investing to Overcome the Global Impact of Neglected Tropical Diseases: Third WHO Report on Neglected Diseases; WHO/HTM/NTD/2015.1; World Health Organization: Geneva, Swistzerland, 2015. [Google Scholar]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar]
- Alvar, J.; Yactayo, S.; Bern, C. Leishmaniasis and poverty. Trends Parasitol. 2006, 22, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, L. Leishmaniasis vaccine: Where are we today? J. Glob. Infect. Dis. 2010, 2, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Den Boer, M.; Argaw, D.; Jannin, J.; Alvar, J. Leishmaniasis impact and treatment access. Clin. Microbiol. Infect. 2011, 17, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Yamey, G.; Torreele, E. The world’s most neglected diseases. BMJ 2002, 325, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Obando, D.; Widmer, F.; Wright, L.C.; Sorrell, T.C.; Jolliffe, C.K. Correlation of Antifungal Activity with Fungal Phospholipase Inhibition Using a Series of Bisquaternary Ammonium Salts. J. Med. Chem. 2006, 49, 811–816. [Google Scholar]
- Calvani, M.; Critelli, L.; Gallo, G.; Giorgi, F.; Gramiccioli, G.; Santaniello, M.; Scafetta, N.; Tinti, M.; de Angelis, F. L-Carnitine Esters as Soft, Broad-Spectrum Antimicrobial Amphiphiles. J. Med. Chem. 1998, 41, 2227–2233. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, M.; Aramak, Y.; Seto, M.; Imoto, H.; Nishikawa, Y.; Kanzaki, N.; Okamoto, M.; Sawada, H.; Nishimura, O.; Baba, M.; et al. Discovery of Novel, Potent, and Selective Small-Molecule CCR5 Antagonists as Anti-HIV-1 Agents: Synthesis and Biological Evaluation of Anilide Derivatives with a Quaternary Ammonium. J. Med. Chem. 2000, 43, 2049–2063. [Google Scholar] [PubMed]
- Calas, M.; Cordina, G.; Bompart, J.; Ben Bari, M.; Jei, T.; Ancelin, M.L.; Vial, H. Antimalarial Activity of Molecules Interfering with Plasmodium falciparum Phospholipid Metabolism. Structure-Activity Relationship Analysis. J. Med. Chem. 1997, 40, 3557–3566. [Google Scholar] [PubMed]
- Avlonitis, N.; Lekka, E.; Detsi, A.; Koufaki, M.; Calogeropoulou, T.; Scoulica, E.; Siapi, E.; Kyrikou, I.; Mavromoustakos, T.; Tsotinis, A.; et al. Antileishmanial Ring-Substituted Ether Phospholipids. J. Med. Chem. 2003, 46, 755–767. [Google Scholar]
- Pérez, J.; Castanys, S.; Gamorro, F. Leishmania donovani Resistance to Miltefosine Involves a Defective Inward Translocation of the Drug. Antimicrob. Agents Chemother. 2003, 47, 2397–2403. [Google Scholar] [CrossRef]
- Unger, C.; Sindermann, H.; Peukert, M.; Hilgard, P.; Engel, J.; Eibl, H. Hexadecylphosphocholine in the topical treatment of skin metastases in breast cancer patients. Prog. Exp. Tumor Res. 1992, 34, 153–159. [Google Scholar] [CrossRef]
- Croft, S.L.; Yardley, V. Chemotherapy of leishmaniasis. Curr. Pharm. Des. 2002, 8, 319–342. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Mamoun, C.B. Choline transport in Leishmania major promastigotes and its inhibition by choline and phosphocholine analogs. Mol. Biochem. Parasitol. 2002, 125, 127–134. [Google Scholar] [CrossRef]
- Boumann, H.A.; Damen, M.J.A.; Versluis, C.; Heck, A.J.R.; de Kruijff, B.; de Kroon, A.I. The two biosynthetic routes leading to phosphatidylcholine in yeast produce different sets of molecular species. Evidence for lipid remodeling. Biochemistry 2003, 42, 3054–3059. [Google Scholar] [PubMed]
- Azzou, S.; Maache, M.; García, R.G.; Osuna, A. Leishmanicidal activity of edelfosine, miltefosine, and ilmofosine. Basic Clin. Pharmacol. Toxicol. 2005, 96, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.M.; Muñoz, D.L.; Cedeño, D.L.; Vélez, I.D.; Jones, M.A.; Robledo, S.M. Leishmania tarentolae: Utility as an in vitro model for screening of antileishmanial agents. Exp. Parasitol. 2010, 126, 471–475. [Google Scholar]
- Ancelin, M.L.; Vial, H.J. Quaternary ammonium compounds efficiently inhibit Plasmodium falciparum growth in vitro by impairment of choline transport. Antimicrob. Agents Chemother. 1986, 29, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Múnera-Orozco, C.; Ocampo-Cardona., R.; Cedeño, D.L.; Toscano, R.A.; Ríos-Vásquez, L.A. Crystal structures of three new N-halomethylated quaternary ammonium salts. Acta Cryst. 2015, E71, 1230–1235. [Google Scholar]
- Ríos, L.A.; Ocampo, R.; Duque, S.M.; Robledo, S.M.; Vélez, I.D.; Cedeño, D.L.; Jones, M.A. Quaternary N-(Halomethyl) Ammonium Salts as Therapeutic Agents. Patent US 9145352 B2, 29 September 2015. [Google Scholar]
- Sangshetti, J.N.; Khan, F.A.K.; Kulkarni, A.A.; Arote, R.; Patil, R.H. Antileishmanial drug discovery: Comprehensive review of the last 10 years. RSC Adv. 2015, 5, 32376–32415. [Google Scholar] [CrossRef]
- Horner, J.; Martínez, F.; Musa, O.; Newcomb, M.; Shahin, H.E. Kinetics of Dialkylaminium Cation Radical Reactions: Radical Clocks, Solvent Effects, Acidity Constants, and Rate Constants for Reactions with Hydrogen Atom Donors. J. Am. Chem. Soc. 1995, 117, 11124–11133. [Google Scholar] [CrossRef]
- Ríos, L.A.; Dolbier, W.R.; Paredes, R.; Lusztyk, J.; Ingold, K.U.; Jonsson, M. Generation and Study of the Reactivity of α-Ammonium Distonic Radical Cations in Solution. J. Am. Chem. Soc. 1996, 118, 11313–11314. [Google Scholar] [CrossRef]
- Mistry, J.S.; Abraham, D.J.; Kozikowski, A.P.; Hanin, I. Neurochemistry of Aging. 2. Design, Synthesis, and Biological Evaluation of Halomethyl Analogues of Choline with High Affinity Choline Transport Inhibitory Activity. J. Med. Chem. 1991, 34, 2031–2036. [Google Scholar] [PubMed]
- Taylor, V.M.; Cedeño, D.L.; Muñoz, D.L.; Jones, M.A.; Lash, T.D.; Young, A.M.; Constantin, M.H.; Esposito, N.; Vélez, I.D.; Robledo, S.M. In Vitro and in Vivo Studies of the Utility of Dimethyl and Diethyl Carbaporphyrin Ketals in Treatment of Cutaneous Leishmaniasis. Antimicrob. Agents Chemother. 2011, 55, 4755–4764. [Google Scholar] [CrossRef] [PubMed]
- Pulido, S.A.; Muñoz, D.L.; Restrepo, A.M.; Mesa, C.V.; Alzate, J.F.; Vélez, I.D.; Robledo, S.M. Improvement of the green fluorescent protein reporter system in Leishmania spp. for the in vitro and in vivo screening of antileishmanial drugs. Acta Trop. 2012, 122, 36–45. [Google Scholar] [PubMed]
- Finney, J.D. Statistical Method in Biological Assay, 3rd ed.; Charles Griffin & Co.: London, UK, 1978; p. 508. [Google Scholar]
- Sample Availability: Samples of compounds 1a, 1b, 1c, 2a, 2b, 2c, 3a, 3b and 3c are available from the authors.


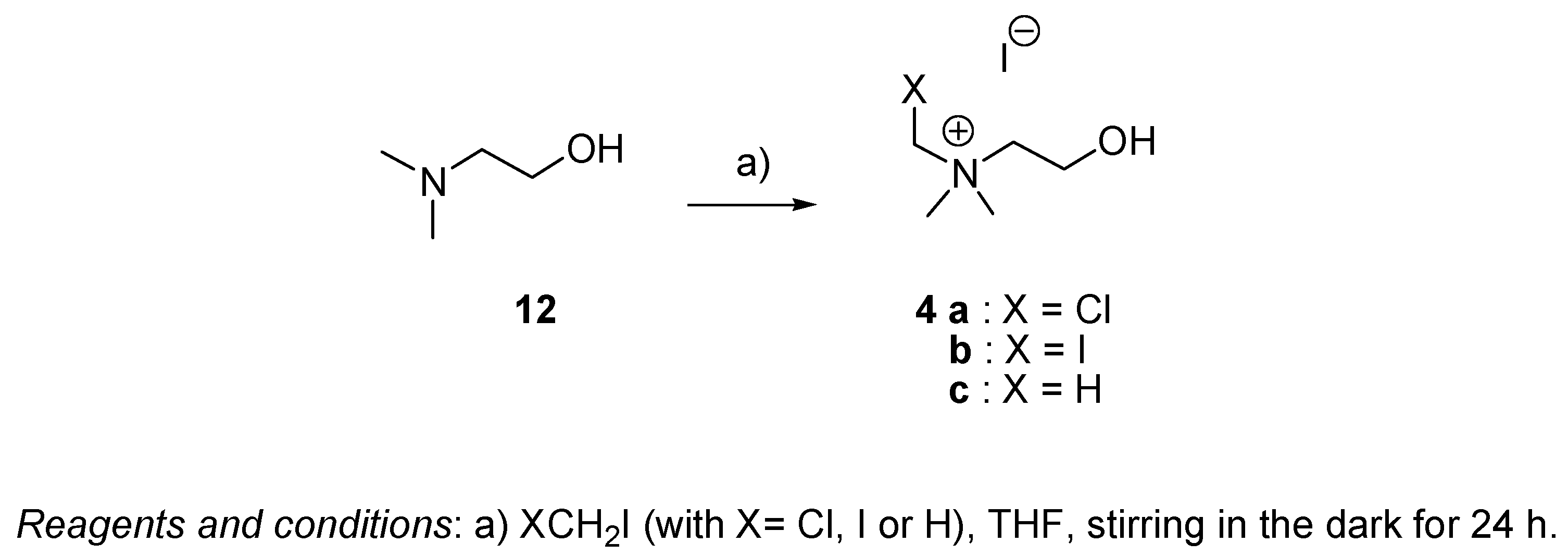


{kind=link}
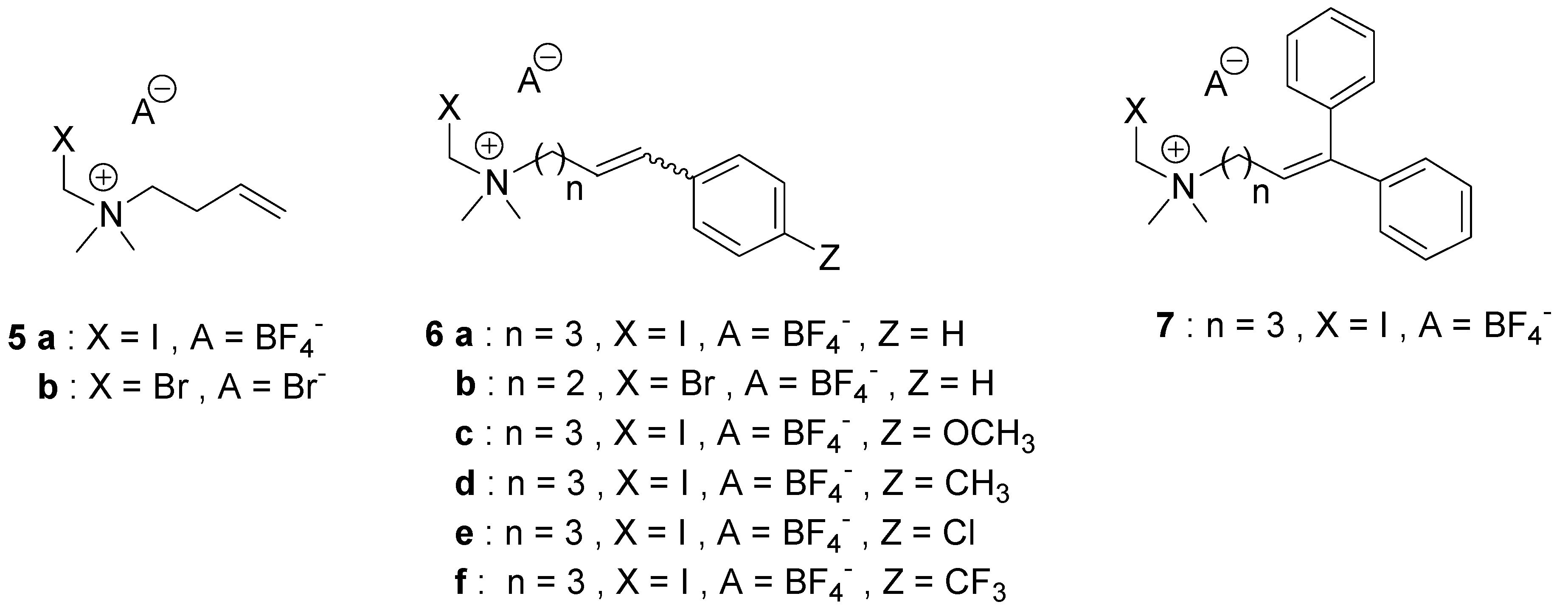{kind=link}
{kind=link}
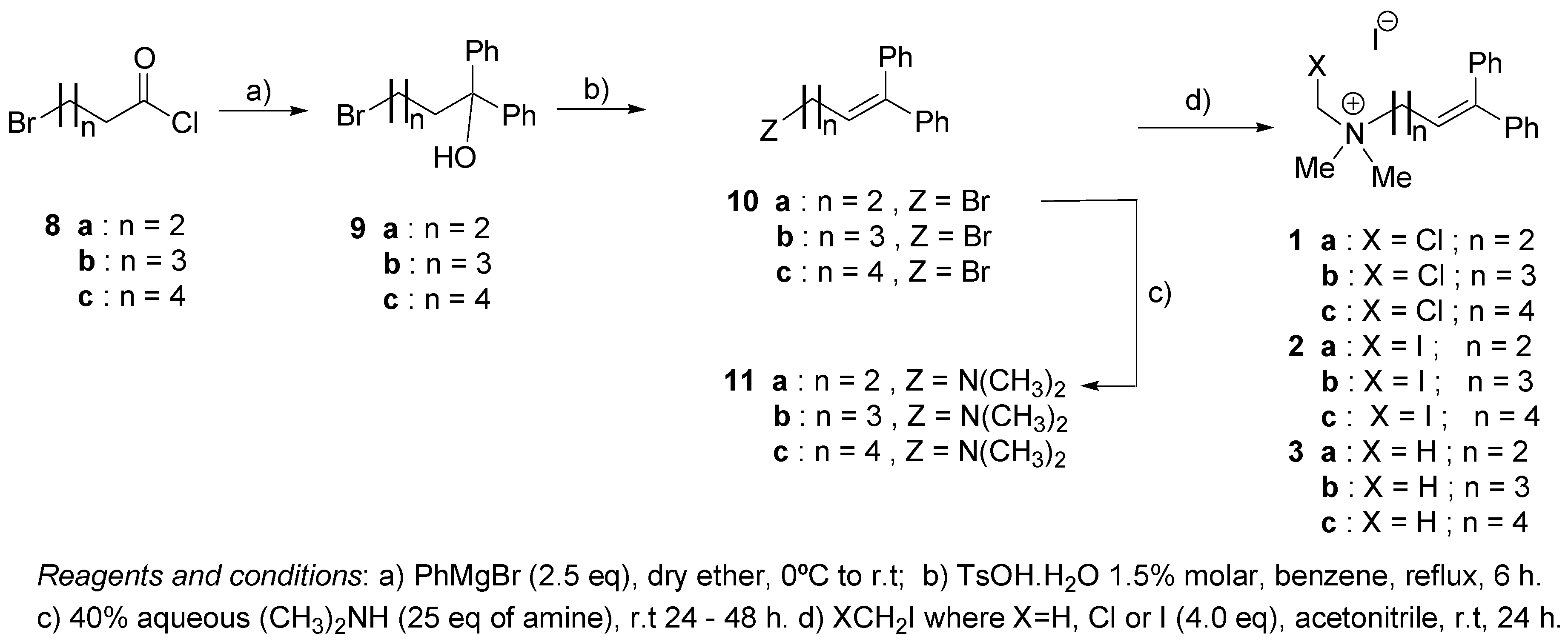{kind=link}
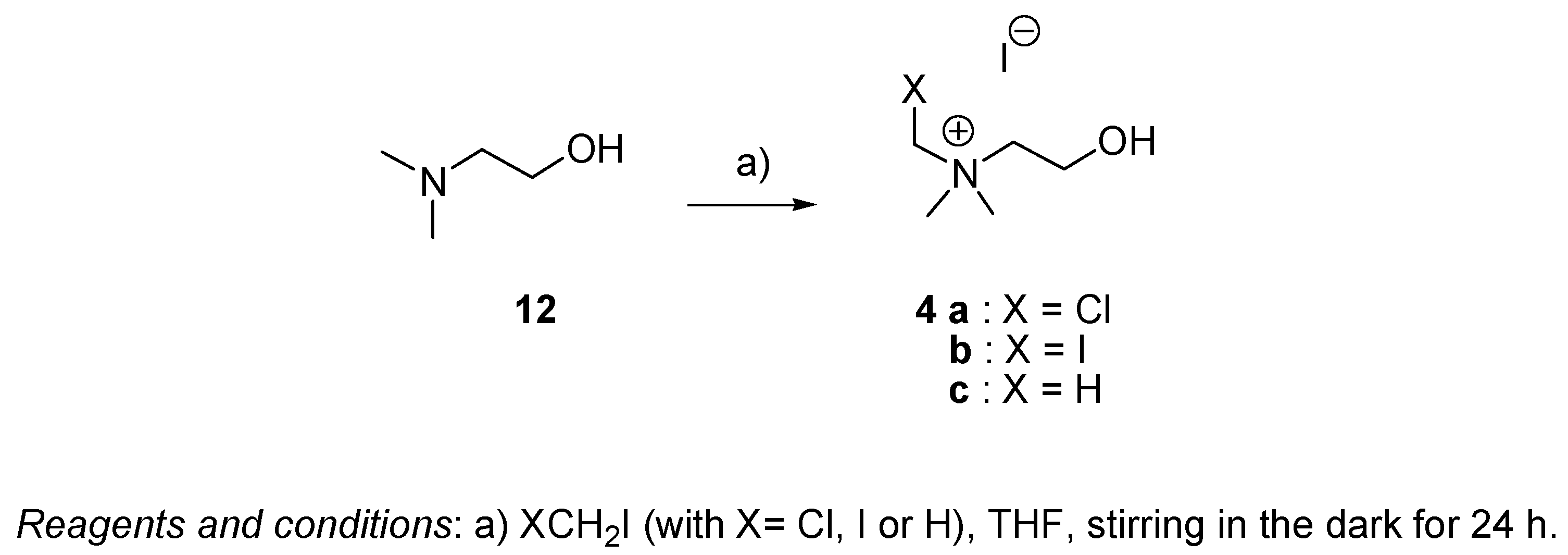{kind=link}
| Compound | Cytotoxicity | Intracellular Amastigotes | Axenic Amastigotes | ||
|---|---|---|---|---|---|
| LC50 | EC50 | SI | EC50 | SI | |
| 1a | 44.4 ± 4.9 | 38.4 ± 0.2 | 1.2 | 35.0 ± 5.7 | 1.3 |
| 1b | 21.0 ± 2.0 | 46.8 ± 1.2 | 0.5 | 56.3 ± 3.7 | 0.4 |
| 1c | 21.0 ± 0.5 | 49.4 ± 3.1 | 0.4 | 40.5 ± 3.3 | 0.5 |
| 2a | 46.0 ± 5.4 | 27.8 ± 3.9 | 1.7 | 36.2 ± 0.1 | 1.3 |
| 2b | 10.1 ± 0.2 | 24.0 ± 0.7 | 0.4 | 19.7 ± 1.5 | 0.5 |
| 2c | 9.5 ± 0.3 | 17.6 ± 1.0 | 0.5 | 14.0 ± 0.9 | 0.7 |
| 3a | 39.2 ± 4.8 | 30.0 ± 1.4 | 1.3 | 25.0 ± 0.7 | 1.5 |
| 3b | 27.2 ± 0.7 | 24.7 ± 1.9 | 1.2 | 25.0 ± 0.5 | 1.1 |
| 3c | 12.5 ± 0.8 | 41.6 ± 2.8 | 0.3 | 34.1 ± 5.1 | 0.4 |
| 4a | 17.9 ± 0.02 | 29.6 ± 2.8 | 0.6 | 24.3 ± 1.3 | 0.7 |
| 4b | 17.8 ± 0.2 | 23.9 ± 1.0 | 0.7 | 19.6 ± 1.8 | 0.9 |
| 4c | 372.0 ± 43.1 | 149.5 ± 17.9 | 2.5 | 47.2 ± 7.1 | 7.9 |
| AmB | 28.2 ± 0.5 | 0.05 ± 0.001 | 560 | 0.041 ± 0.001 | 690 |
| MA | 358.2 ± 1.7 | 6.33 ± 0.86 | 56.6 | 312.1 ± 18.6 | 1.4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duque-Benítez, S.M.; Ríos-Vásquez, L.A.; Ocampo-Cardona, R.; Cedeño, D.L.; Jones, M.A.; Vélez, I.D.; Robledo, S.M. Synthesis of Novel Quaternary Ammonium Salts and Their in Vitro Antileishmanial Activity and U-937 Cell Cytotoxicity. Molecules 2016, 21, 381. https://doi.org/10.3390/molecules21040381
Duque-Benítez SM, Ríos-Vásquez LA, Ocampo-Cardona R, Cedeño DL, Jones MA, Vélez ID, Robledo SM. Synthesis of Novel Quaternary Ammonium Salts and Their in Vitro Antileishmanial Activity and U-937 Cell Cytotoxicity. Molecules. 2016; 21(4):381. https://doi.org/10.3390/molecules21040381
Chicago/Turabian StyleDuque-Benítez, Sandra M., Luz Amalia Ríos-Vásquez, Rogelio Ocampo-Cardona, David L. Cedeño, Marjorie A. Jones, Iván D. Vélez, and Sara M. Robledo. 2016. "Synthesis of Novel Quaternary Ammonium Salts and Their in Vitro Antileishmanial Activity and U-937 Cell Cytotoxicity" Molecules 21, no. 4: 381. https://doi.org/10.3390/molecules21040381