
Structure, Solubility and Stability of Orbifloxacin Crystal Forms: Hemihydrate versus Anhydrate



Abstract
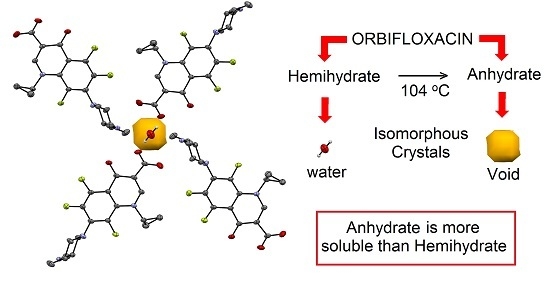
:
1. Introduction
2. Results and Discussions
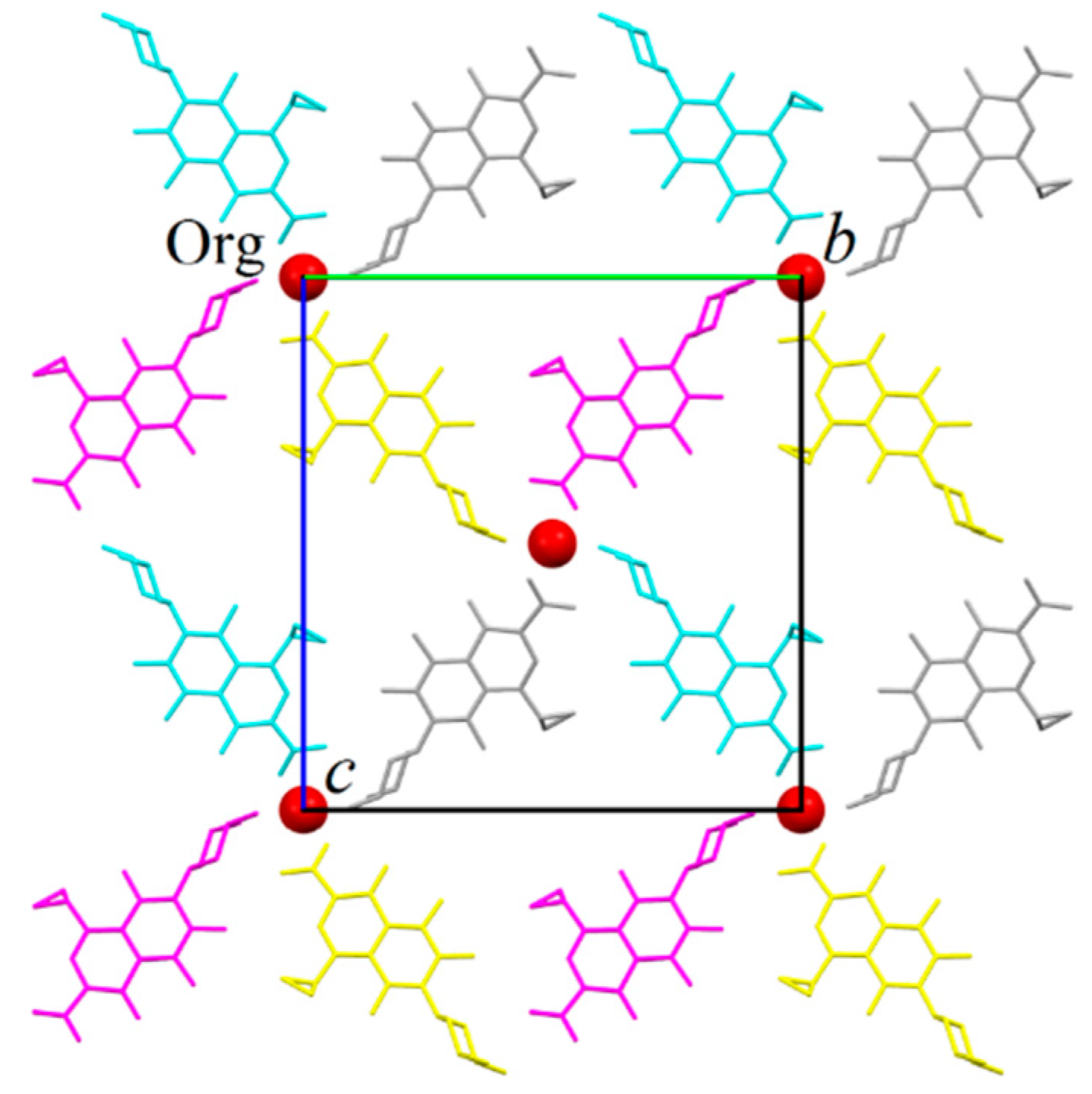
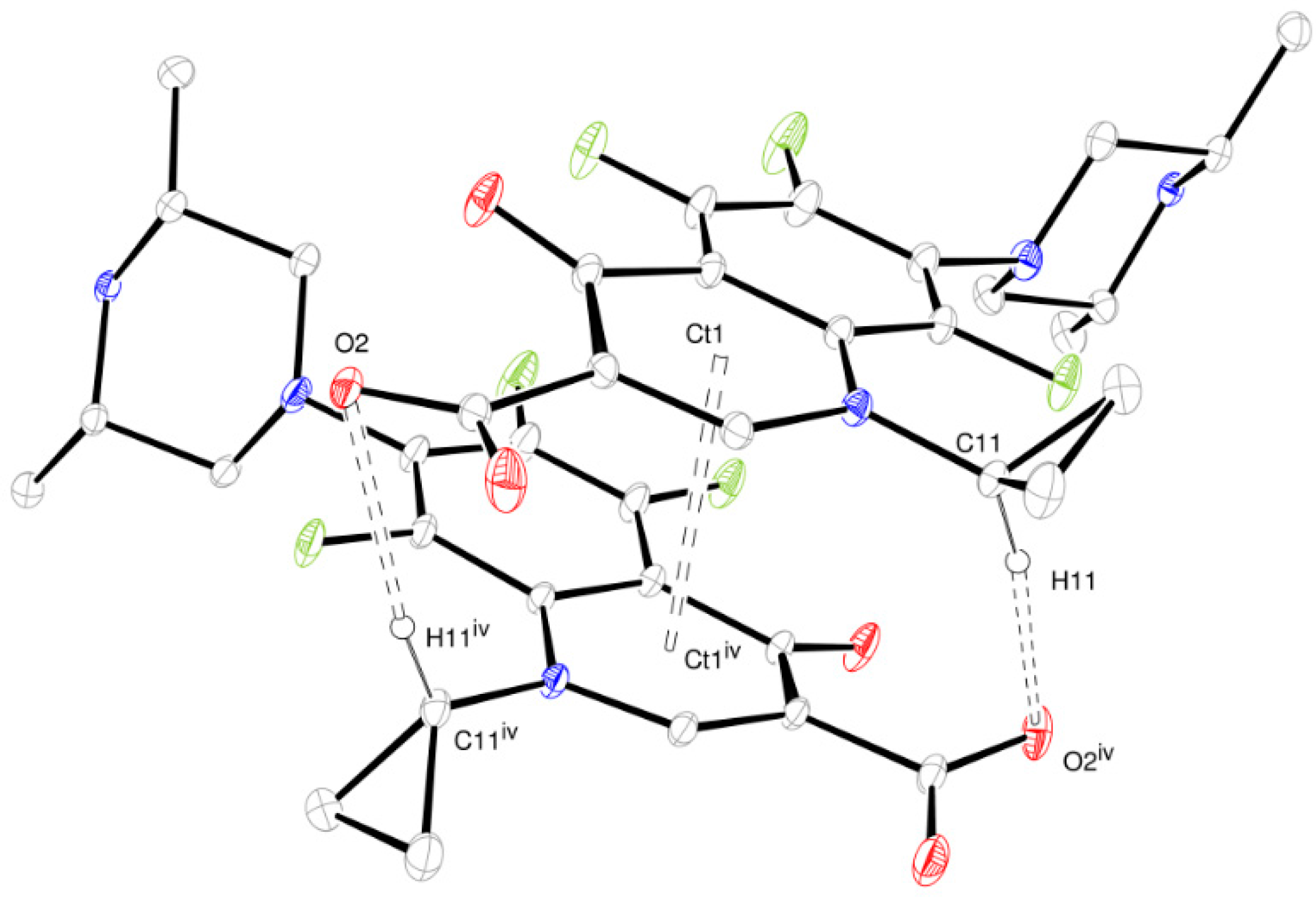
2.1. Structure Determination by Single-Crystal X-ray Diffraction
- (a)
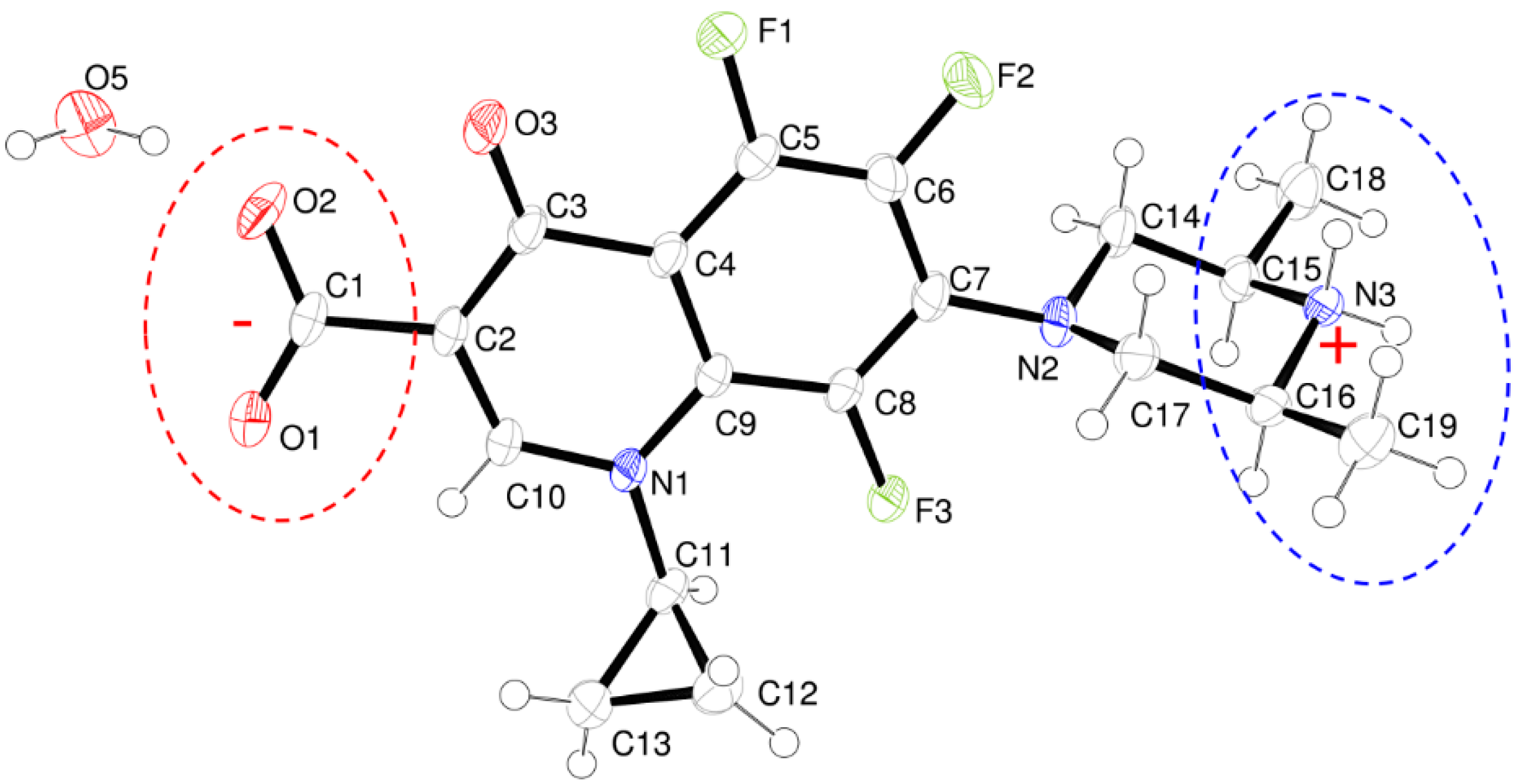
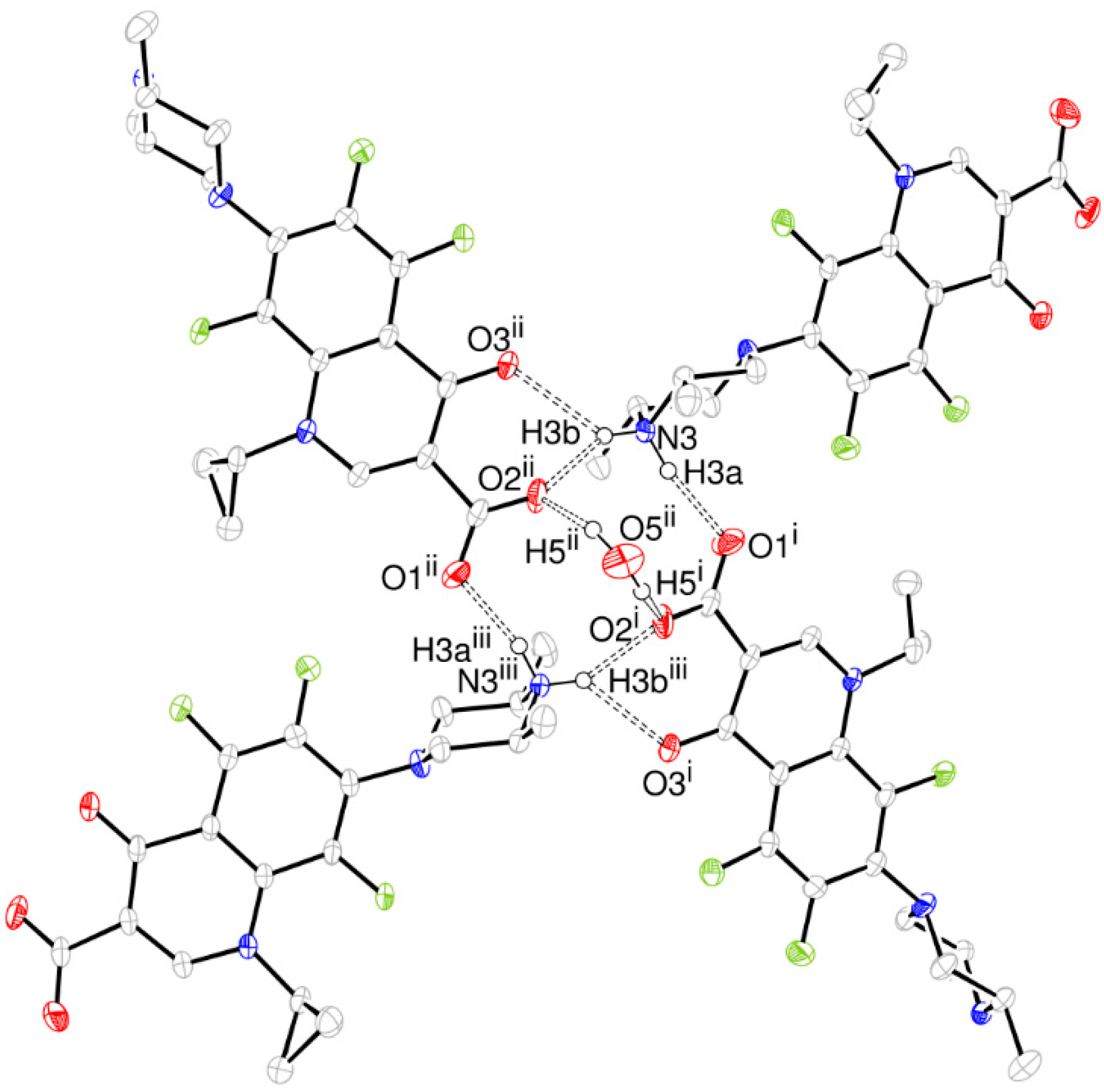
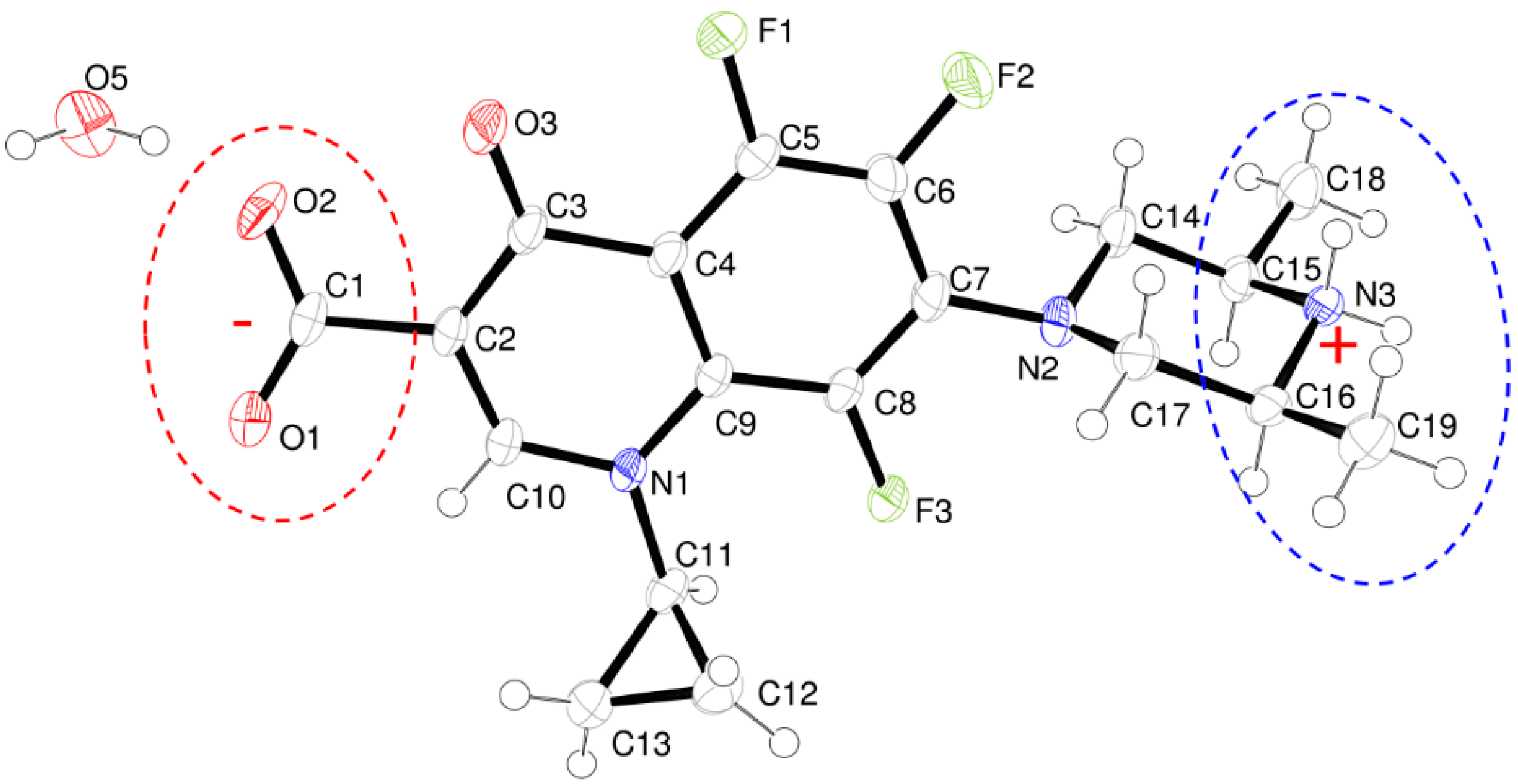
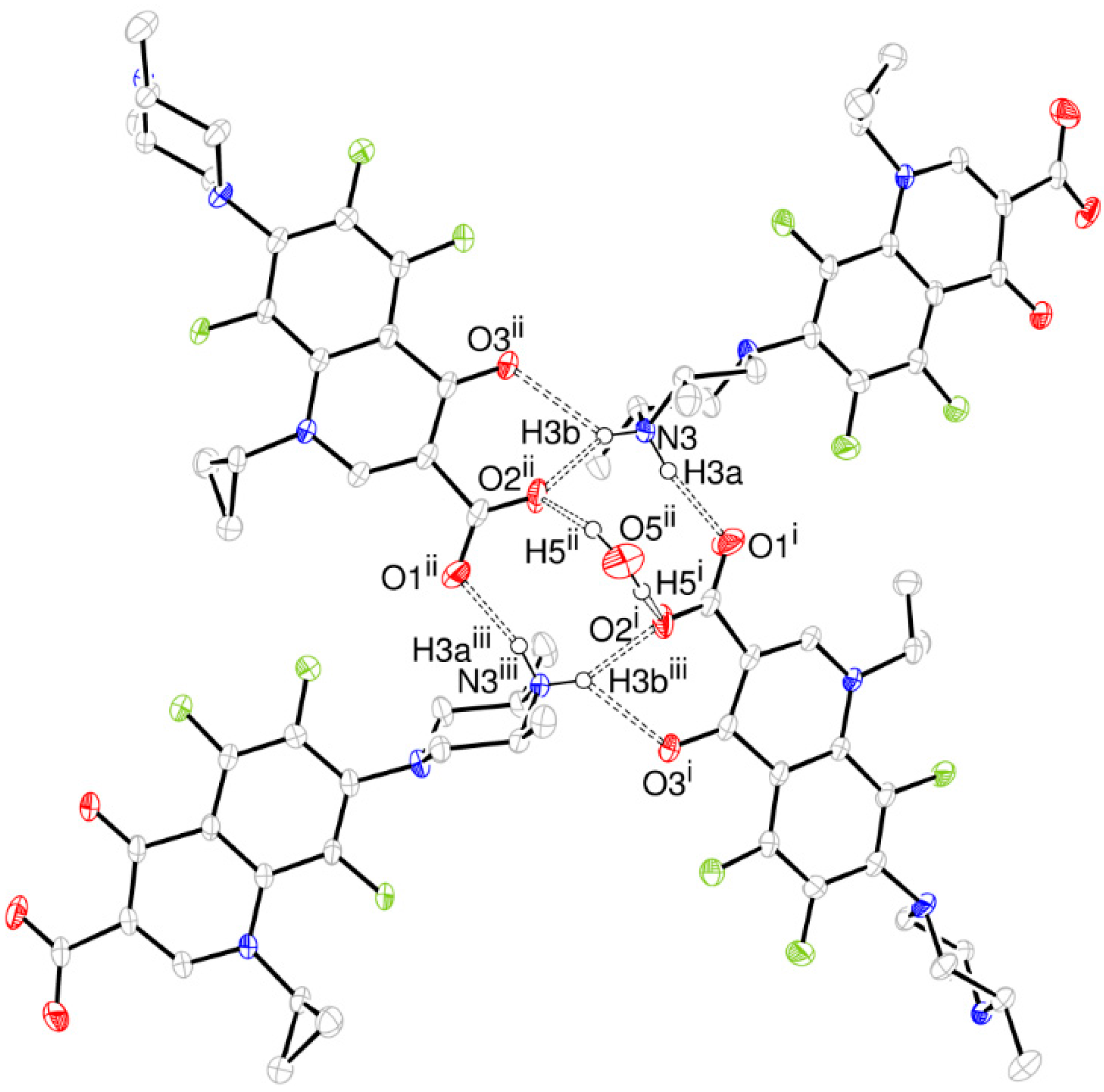
- The structure is a hemihydrate (one half water molecule for each ORBI molecule), and the water oxygen atom lies in a special position (lies in the 2-fold axis parallel to the unit cell a axis) of the C2221 space group. Therefore, the two water hydrogen atoms shown in Figure 2 are crystallographic dependent by symmetry.
- (b)
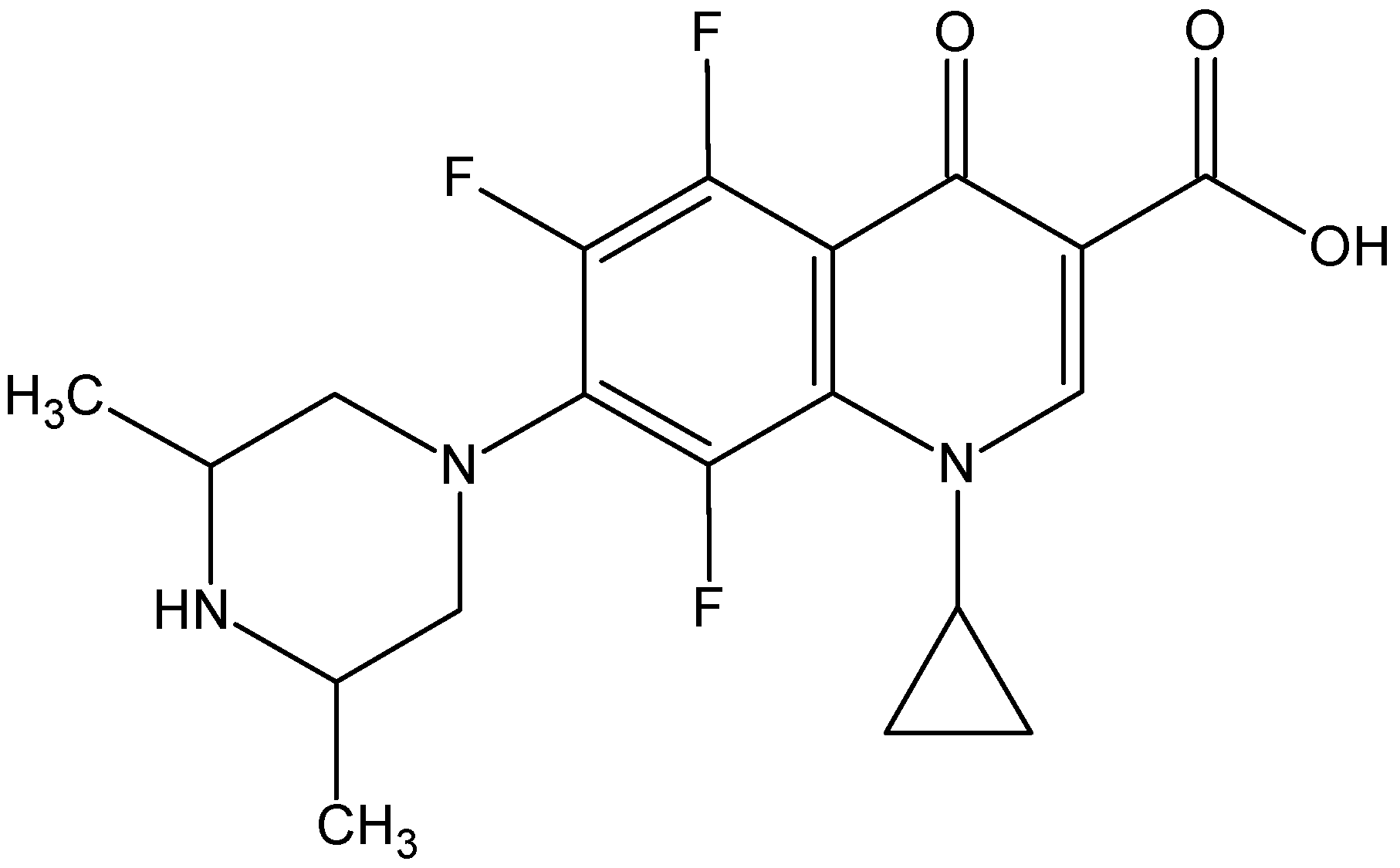
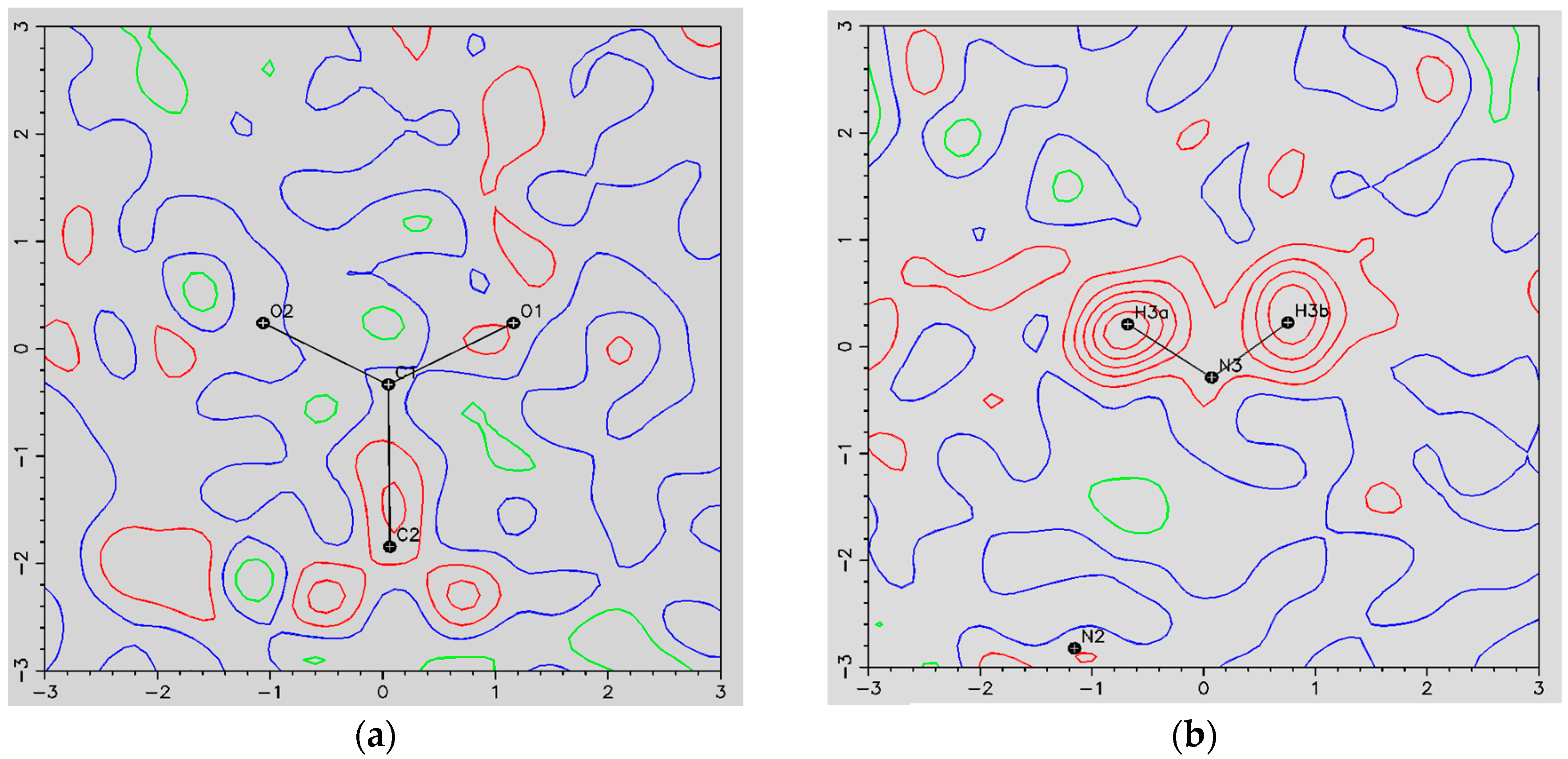
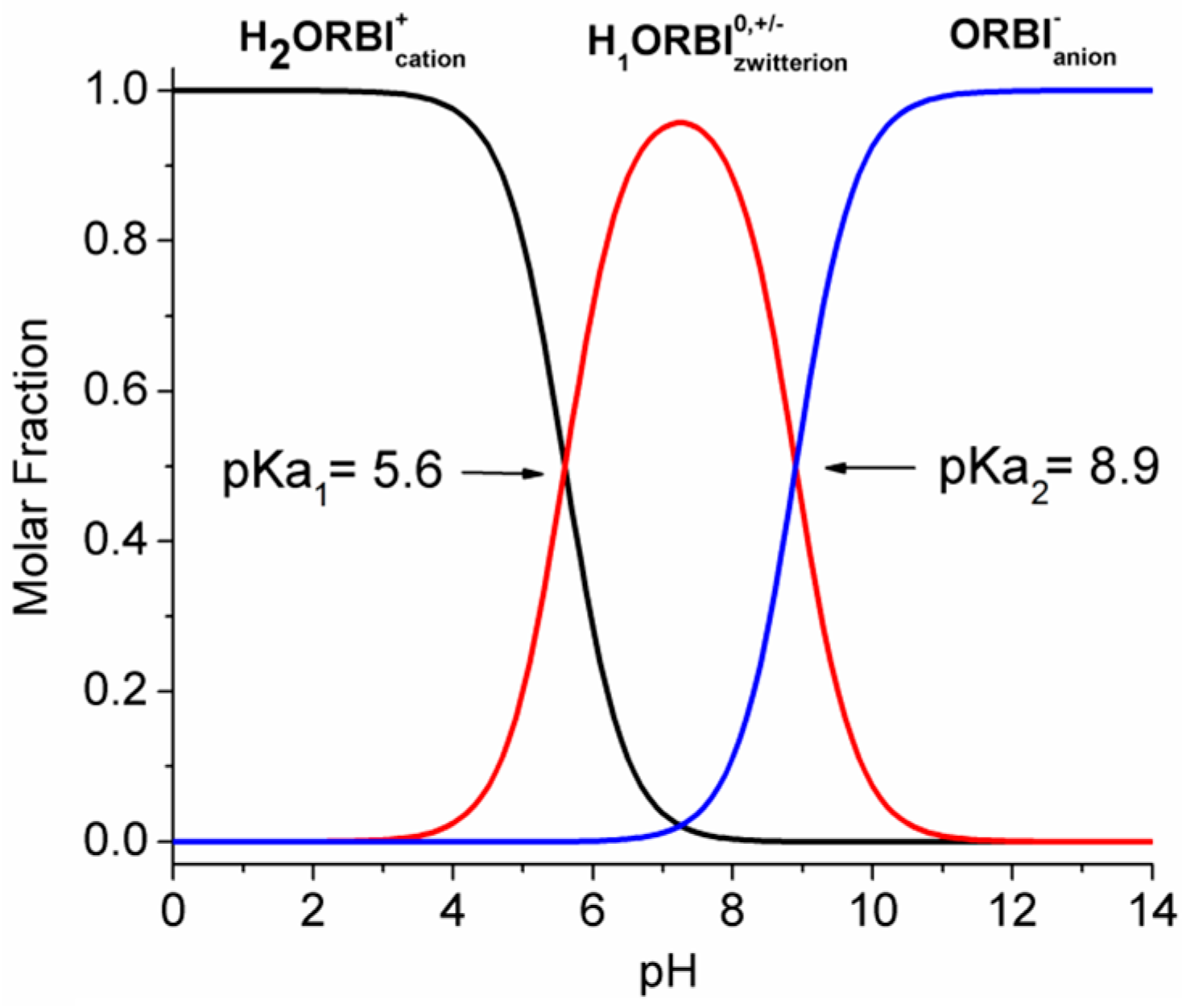
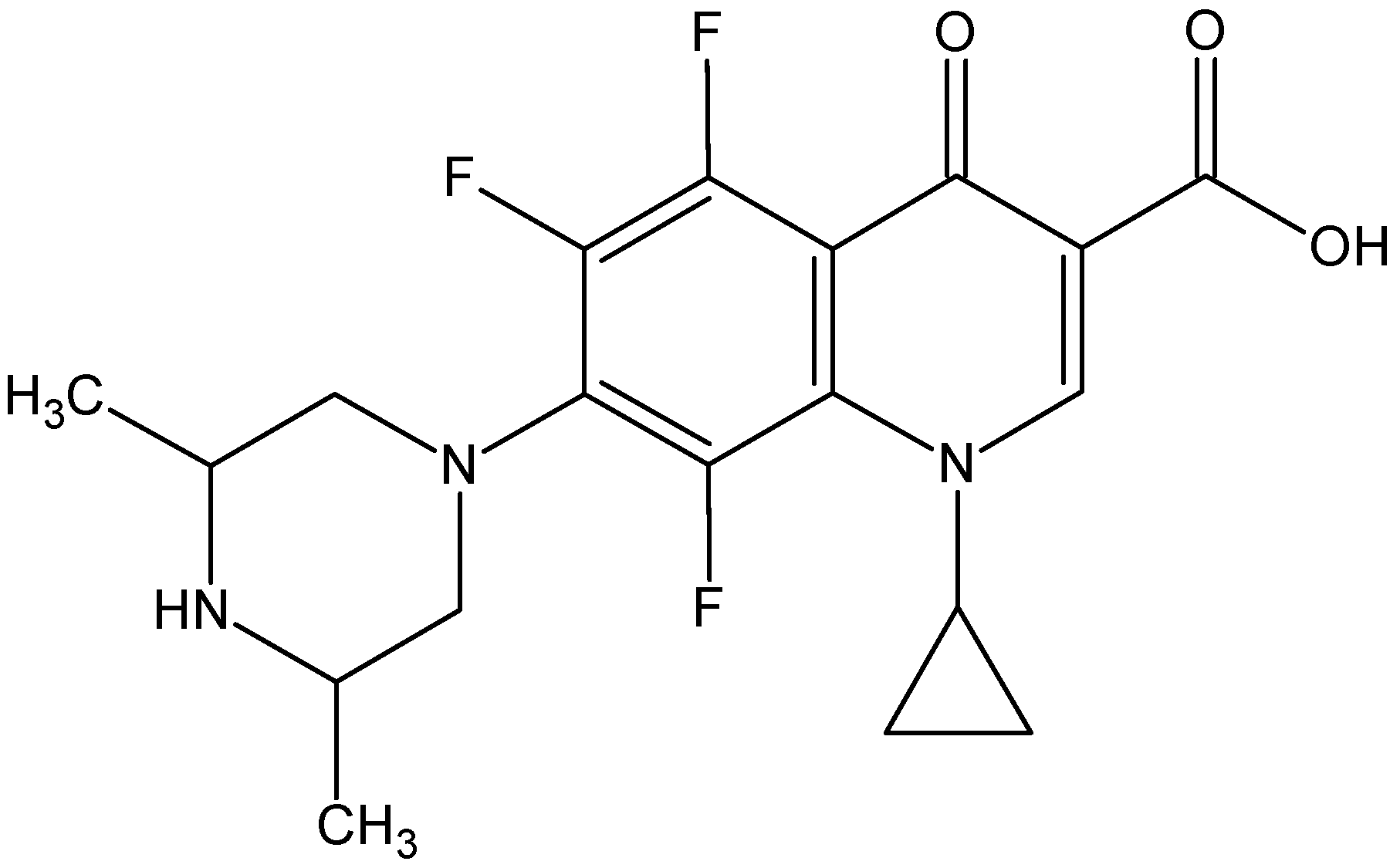
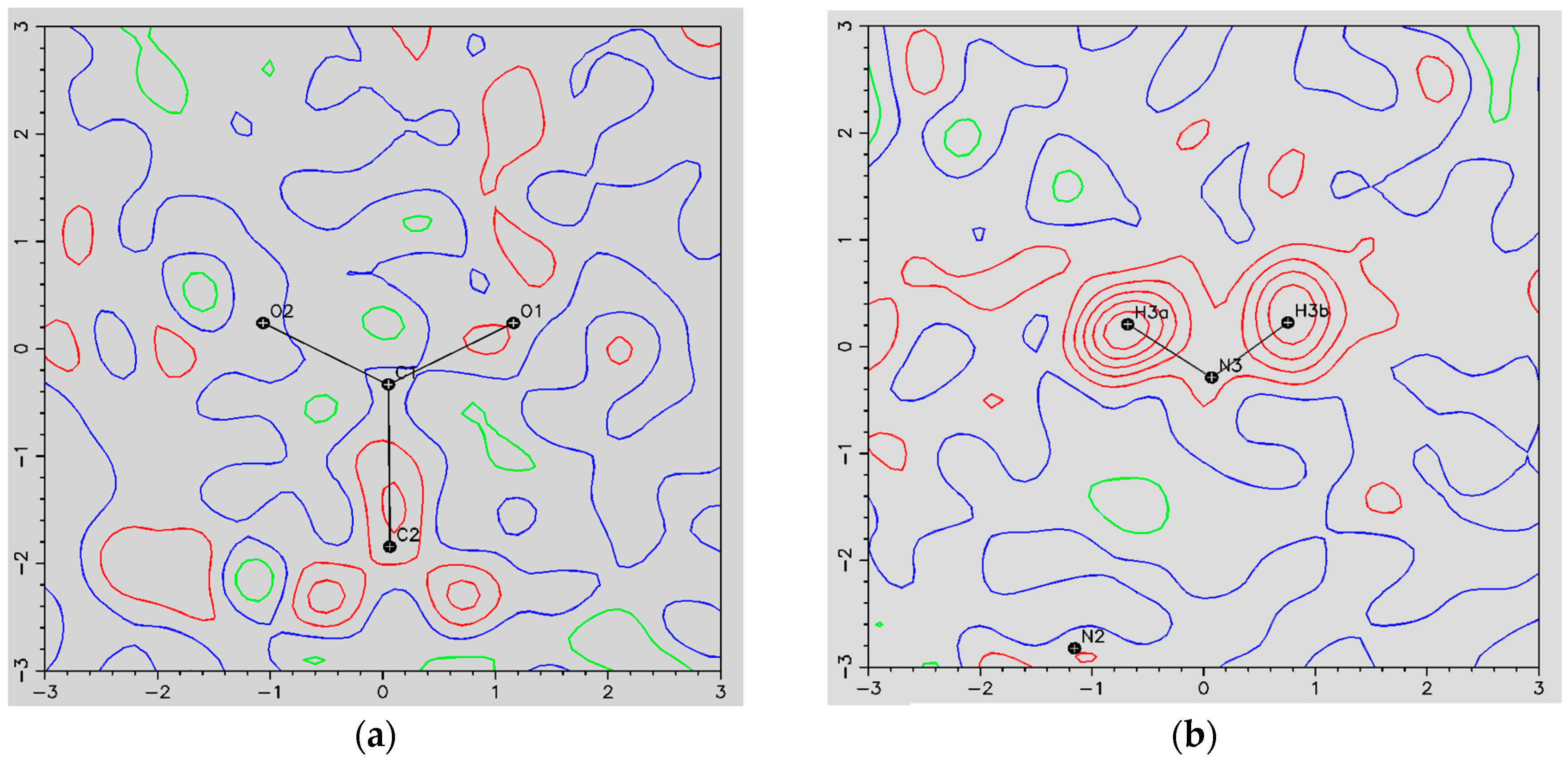
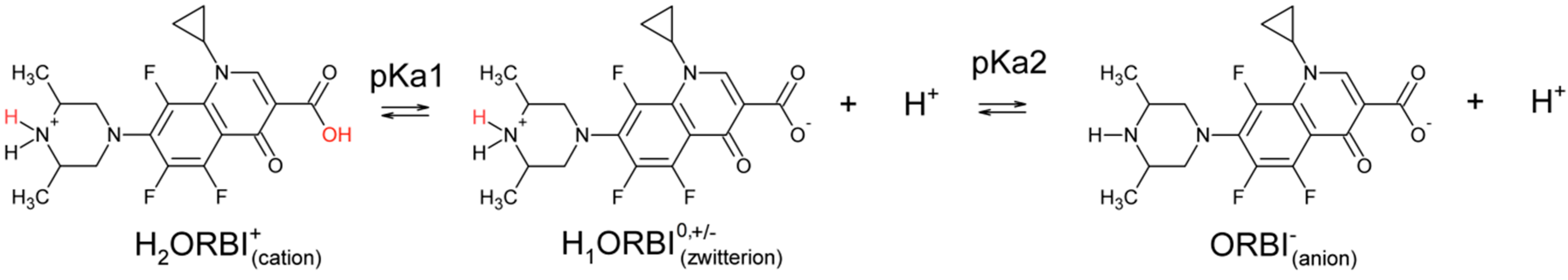
- The ORBI molecule is in its zwitterion form with the carboxylic hydrogen transferred to pyperazine nitrogen. The proton transfer was confirmed by Fourier difference maps, which show the absence of residual electron density close to the oxygen atoms of the carboxylic group and the presence of two large peaks of electronic density close to the pyperazine nitrogen, which clearly reveals the presence of two hydrogens linked to it (Figure 3). Moreover, identical bond lengths are observed for C1-O1 (1.250(3) Å) and C1-O2 (1.251(2) Å), indicating the formation of a carboxylate ion (negative charge resulting from deprotonation is delocalized between the two oxygen atoms).
- (c)
- Since ORBI molecule has two stereocenters (at C15 and C16) and it is crystallized in a non-centrosymmetric space group (Table 1) containing just one molecule in the asymmetric unit, its crystal structure is expected to contain a pure enantiomer [31]. However, this is not the case because the piperazine ring containing the two stereocenters has as internal mirror plane (through N2, N3, and the hydrogens linked to N3) that bisects this moiety. The symmetrical nature arising from the presence of two stereogenic centres with identical substitution but opposite configuration makes ORBI a meso-compound. As highlighted by Hoffmann, meso-compounds can reach the same conformation with a plane of symmetry or a centre of inversion among the continuum of freely accessible conformations [32]. The ORBI C15(R),C16(S) and C15(S),C16(R) isomers are equivalent (superimposable on its internal mirror image) considering the pyperazine moiety. Additionally, because the quinolone and pyperazine rings can freely rotate around the C7-N2 bond, the whole molecules containing the two isomers are also equivalent. ORBI is therefore optically inactive; the contributions of the two stereogenic centres to the Cotton effect compensate for each other [32]. Therefore, the C15(R),C16(S) isomer shown in Figure 2 was arbitrarily chosen.
2.2. ORBI Characterization
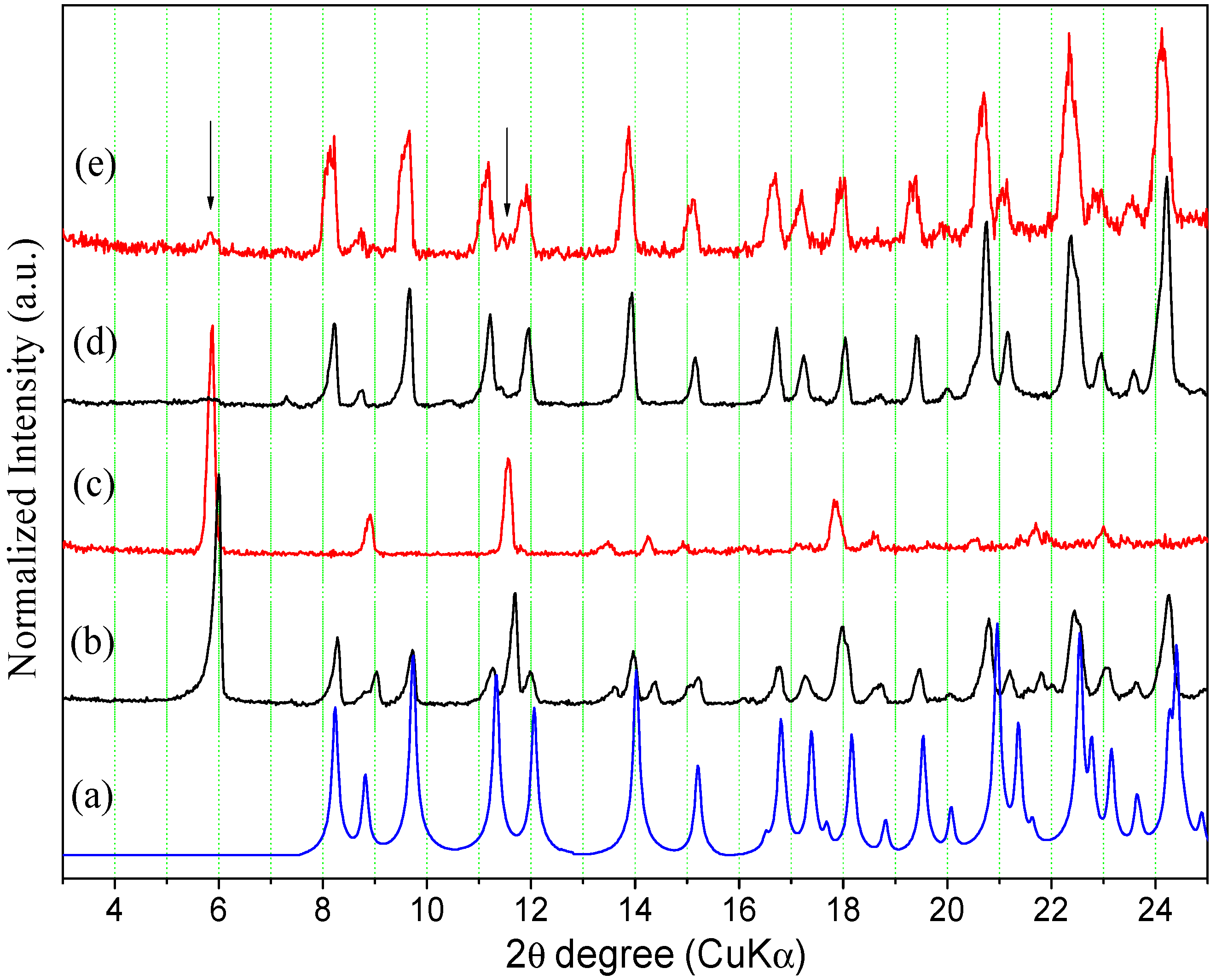
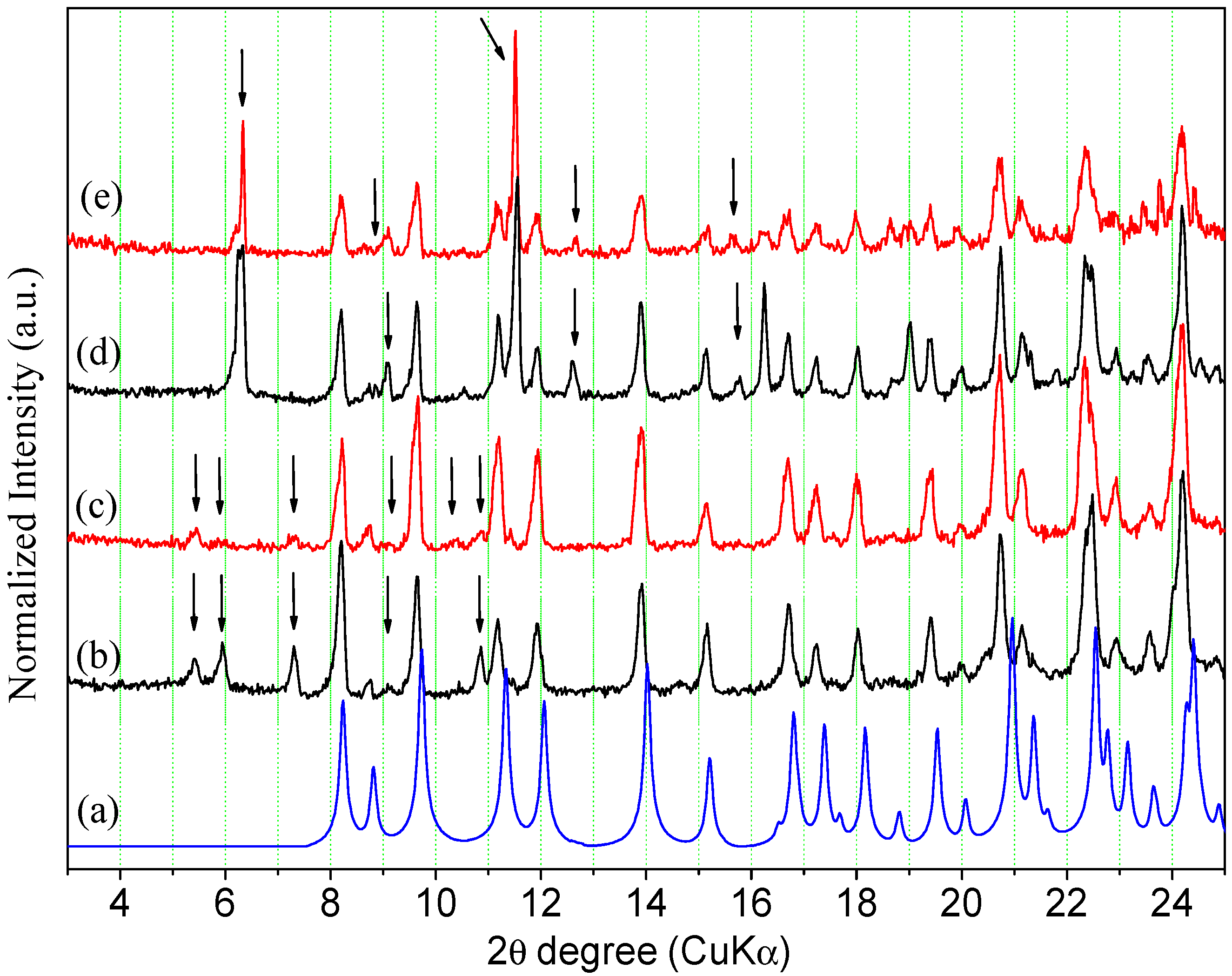
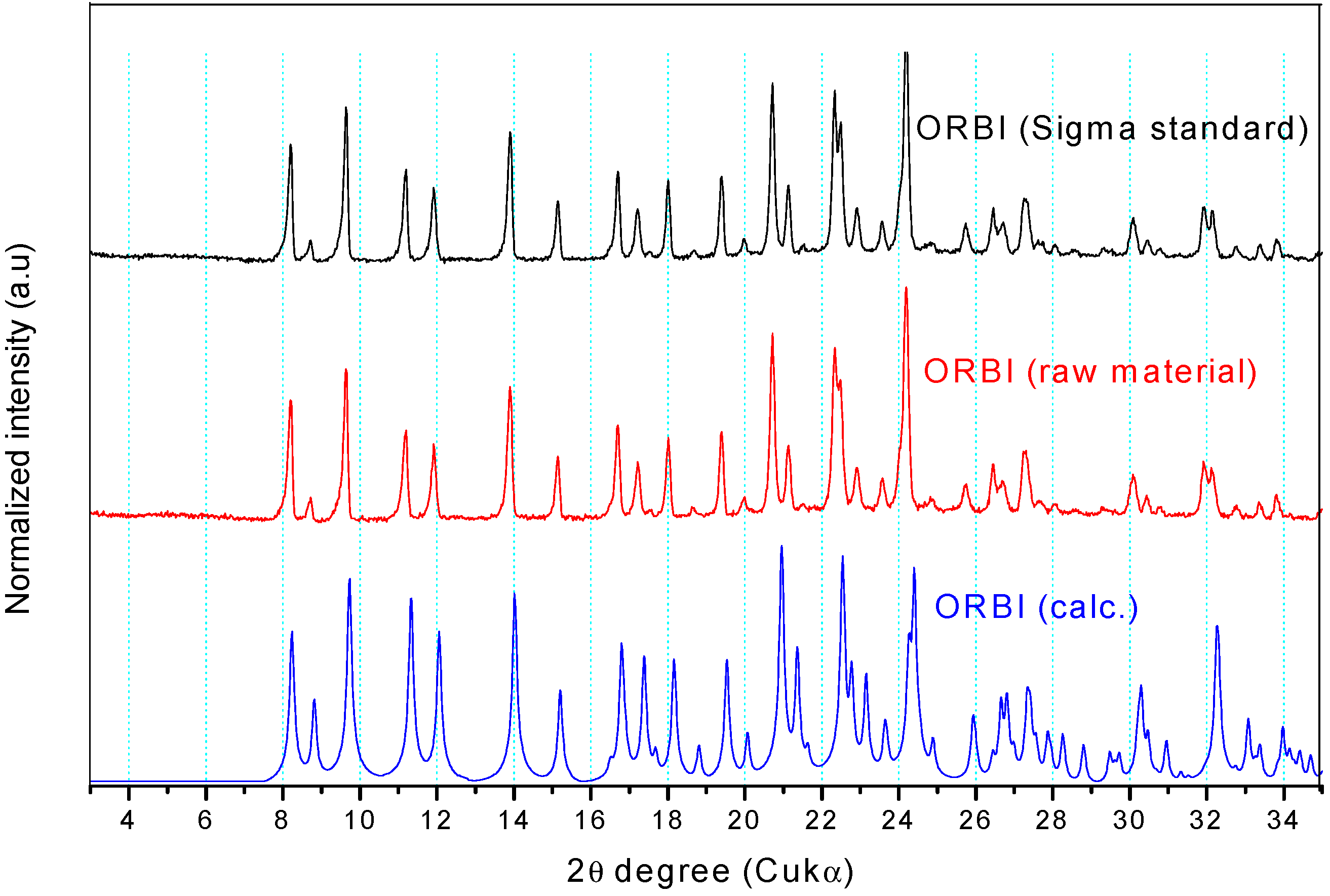
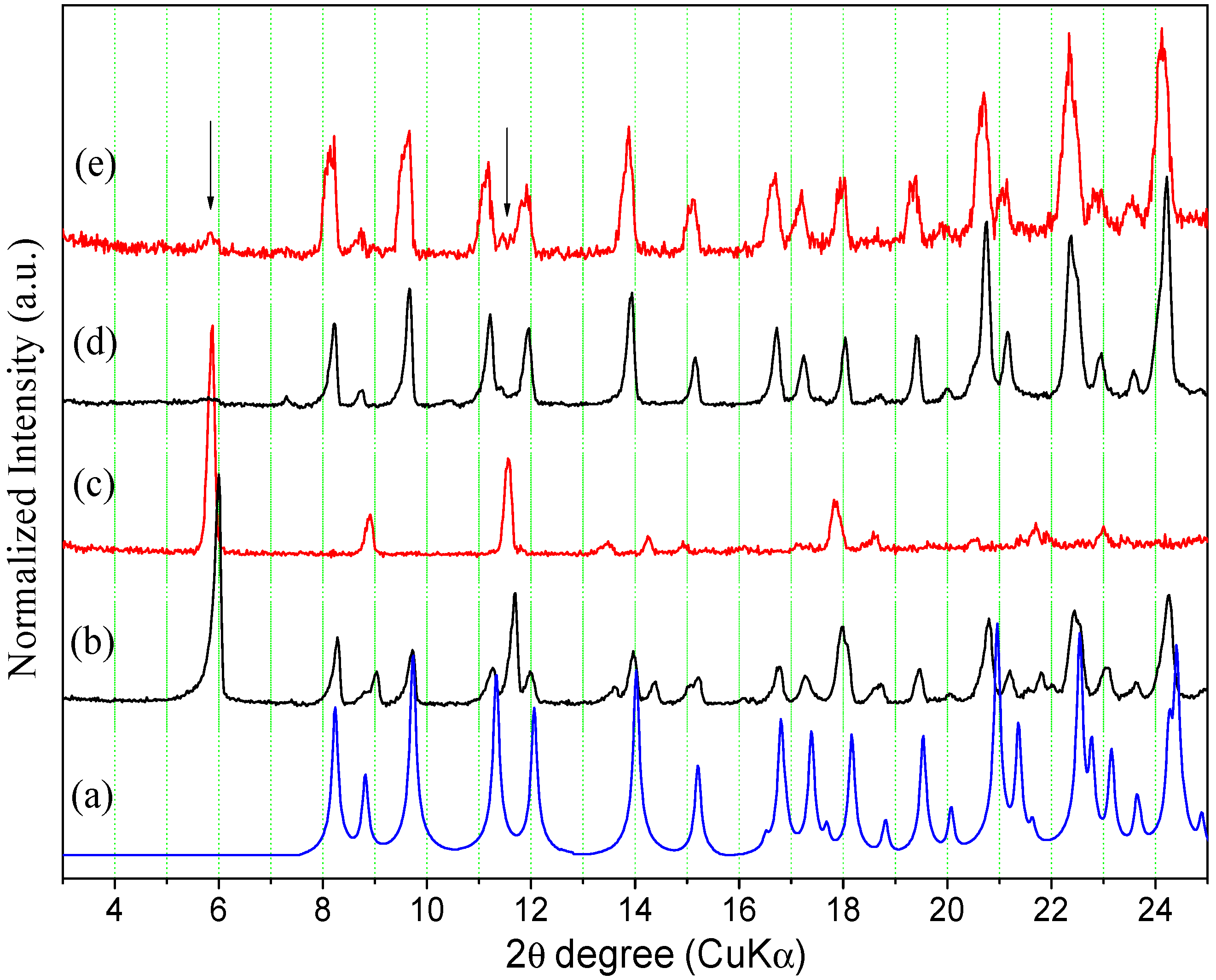
2.2.1. Powder X-ray Diffraction
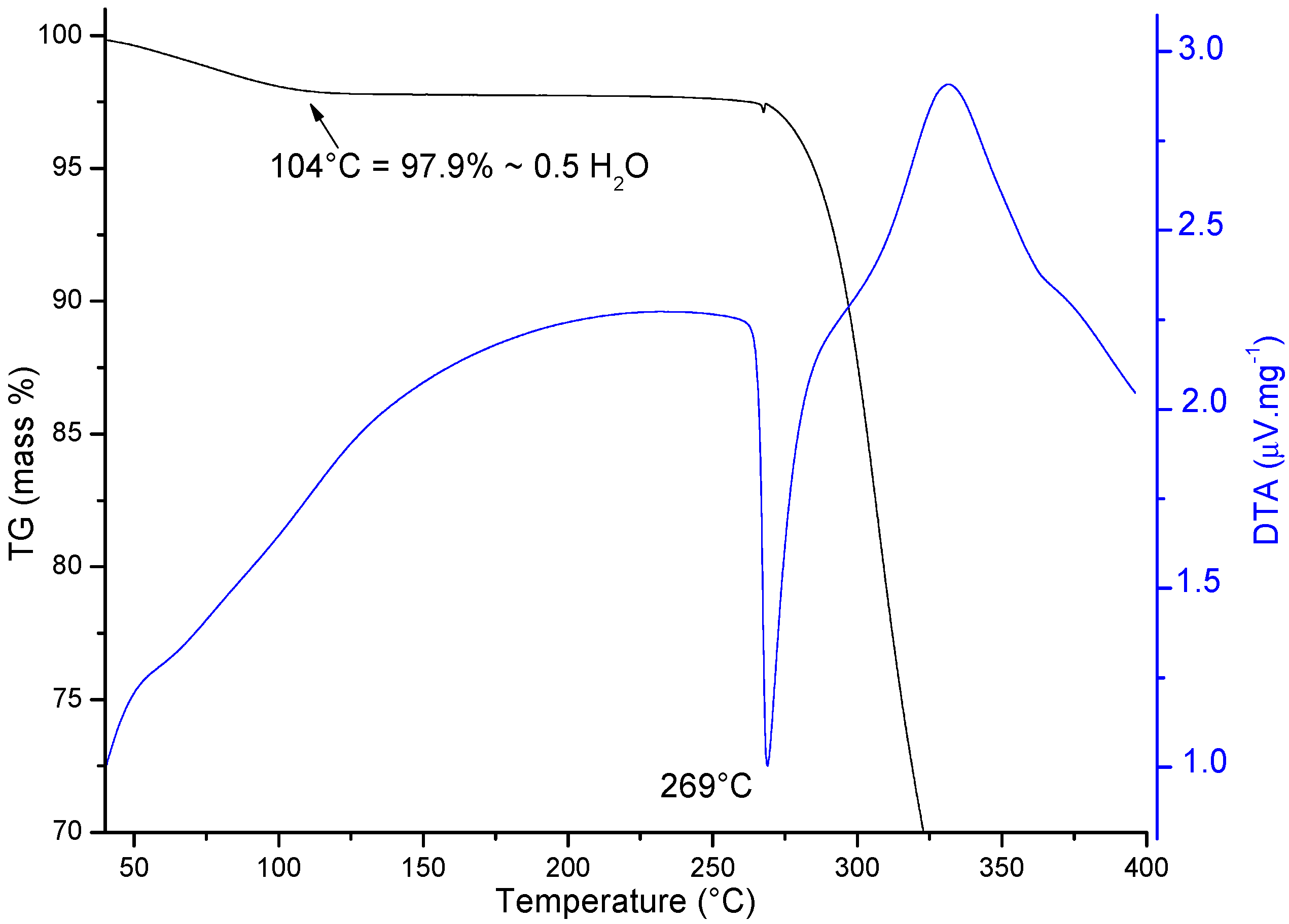
2.2.2. Thermal Analysis
2.2.3. Infrared Spectroscopy with Attenuated Total Reflectance by Fourier Transform (FTIR-ATR)
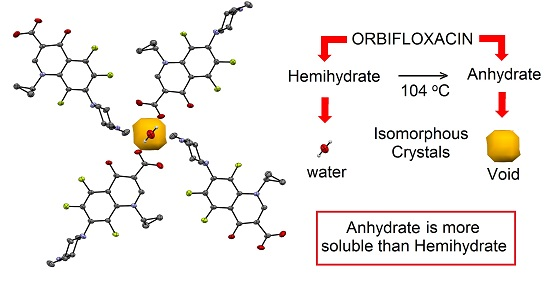
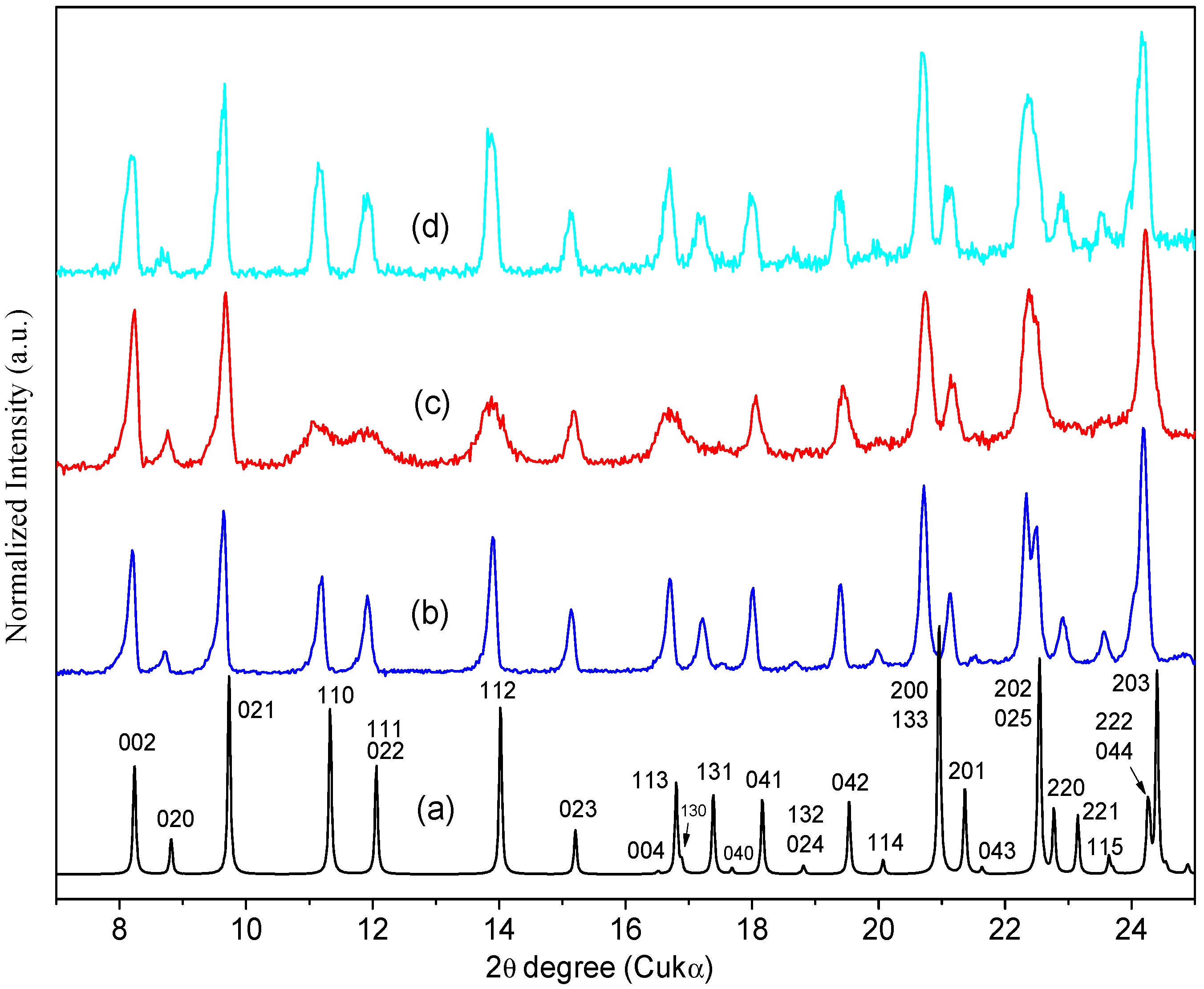
2.3. Interconversion Study between ORBI Anhydrous and Hemihydrate Forms
2.4. Characterization of ORBI Tablets
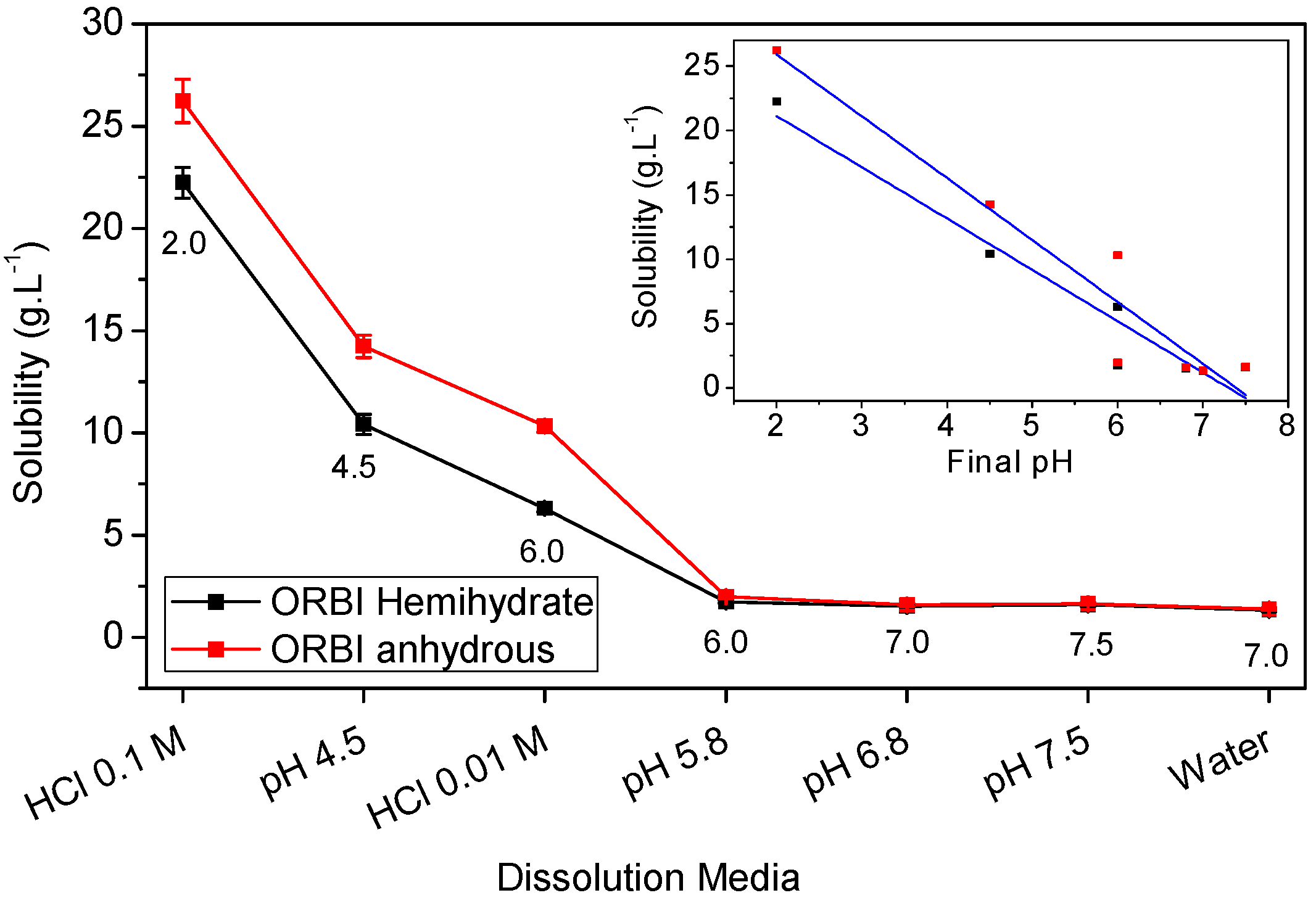
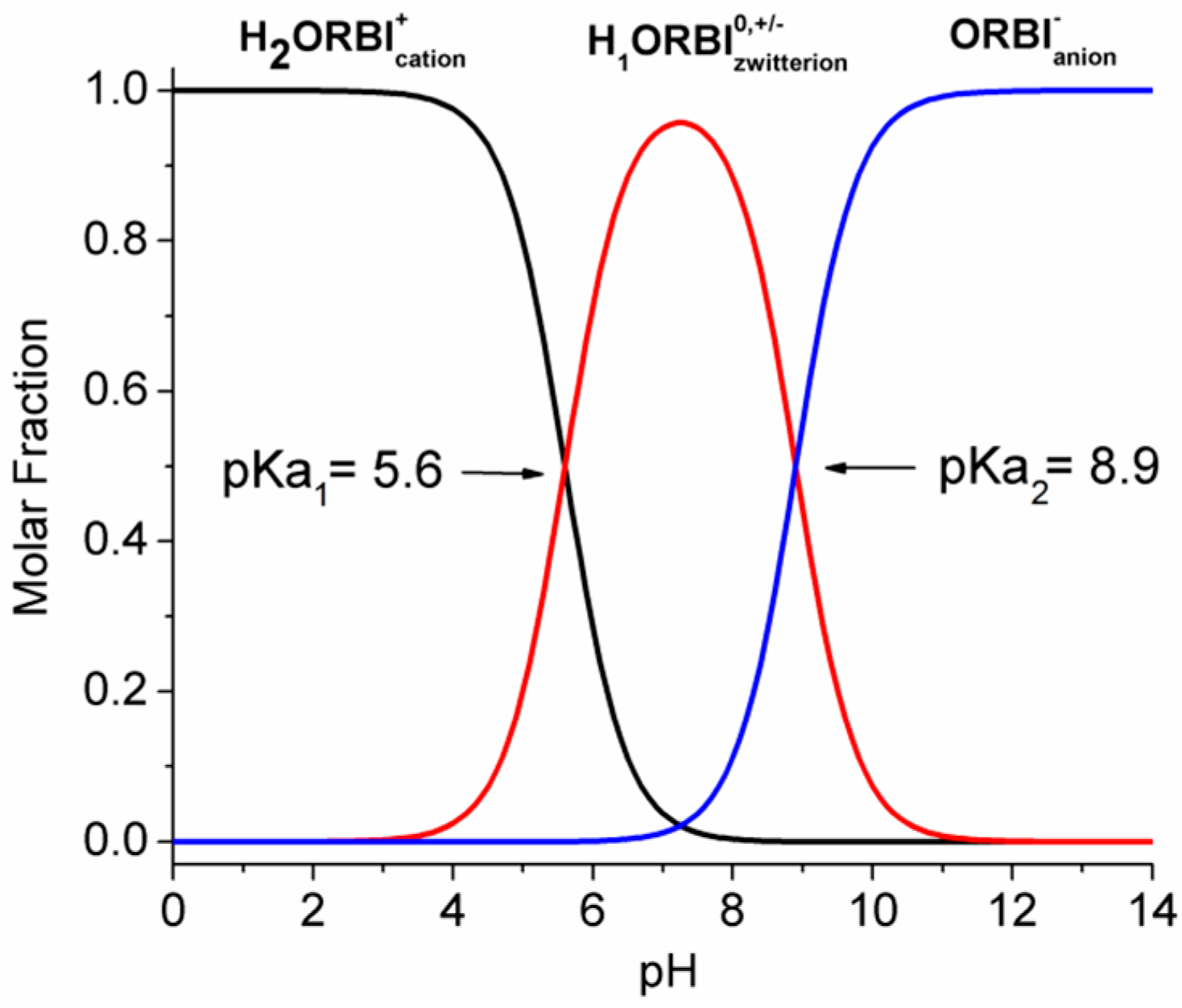
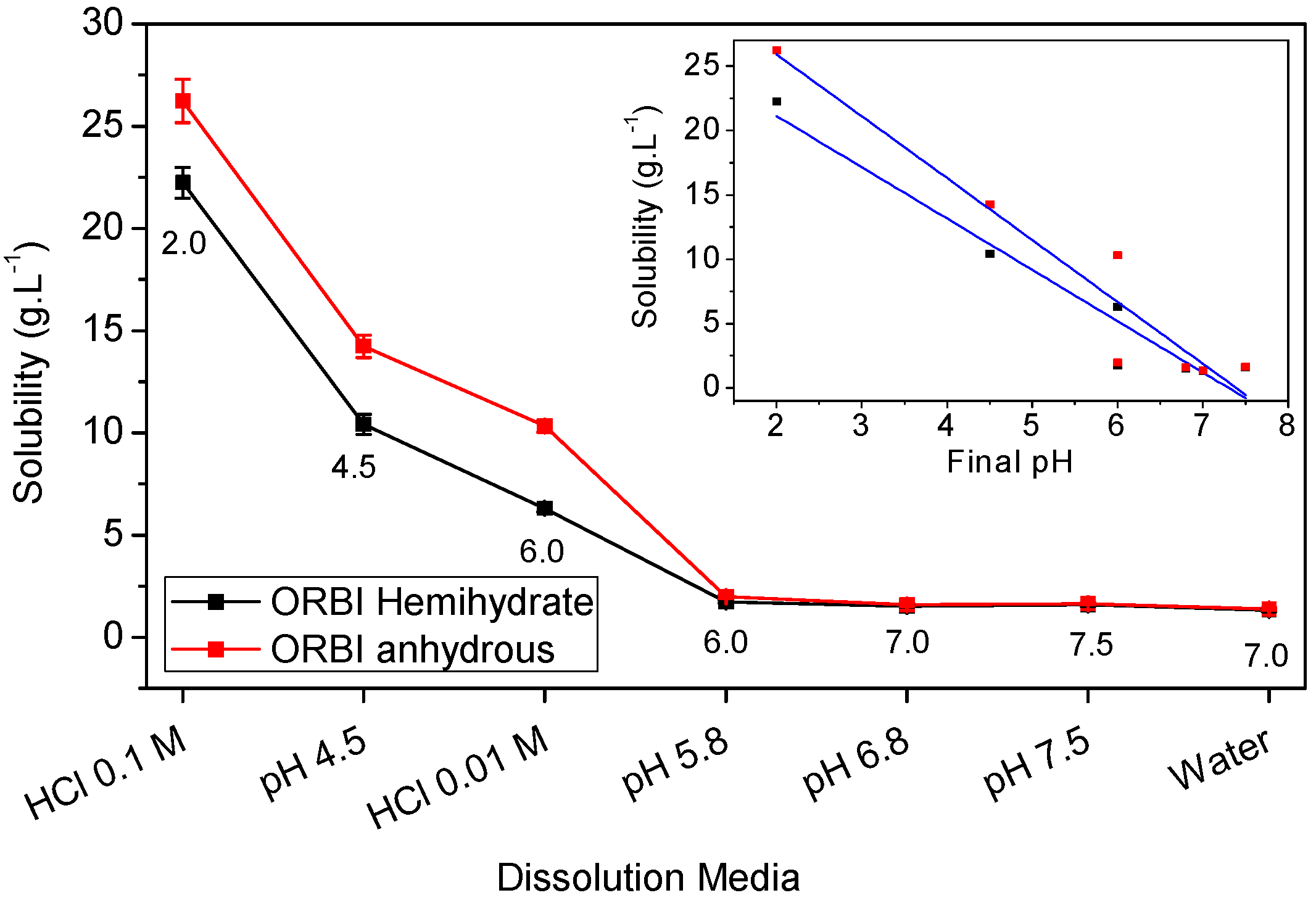
2.5. Solubility Studies of Hemihydrated and Anhydrous Forms of ORBI
3. Materials and Methods
3.1. Chemicals
3.2. Crystal Preparation
3.3. Structural Determination by X-ray Diffraction for Single Crystals
3.4. Powder X-ray Diffraction Analysis
3.5. Infrared Spectroscopy with Attenuated Total Reflectance by Fourier Transform (FTIR-ATR)
3.6. Thermal Analysis
3.7. Interconversion Study between ORBI Anhydrous and Hemihydrate Forms
3.8. ORBI Quantification
3.9. Solubility Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ORBI | orbifloxacin |
| API | Active Pharmaceutical Ingredient |
| SCXRD | Single-Crystal X-ray Diffraction |
| PXRD | Powder X-ray Diffraction |
| TG/DTA | Thermogravimetric/Differential Thermal Analysis |
| FTIR-ATR | Fourier transform infrared spectroscopy-Attenuated Total Rectance |
References
- Ihrke, P.J.; Papich, M.G.; Demanuelle, T.C. The use of fluoroquinolones in veterinary dermatology. Vet. Dermatol. 1999, 10, 193–204. [Google Scholar] [CrossRef]
- Dudhatra, G.B.; Kumar, A.; Awale, M.M.; Kamani, D.R.; Chauhan, B.N.; Patel, H.B.; Mody, S.K. Determination of orbifloxacin in sheep plasma by high performance liquid chromatography with ultraviolet detection after intravenous and intramuscular administration. J. Pharmacol. Toxicol. Methods 2013, 67, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Dong, X.; Wang, Y.; Peng, D.; Wang, X.; Hao, H.; Xie, S.; Qu, W.; Liu, Z.; Yuan, Z. Development of a novel genetically modified bioluminescent-bacteria-based assay for detection of fluoroquinolones in animal-derived foods. Anal. Bioanal. Chem. 2014, 406, 7899–7910. [Google Scholar] [CrossRef] [PubMed]
- Albarellos, G.A.; Landoni, M.F. Current concepts on the use of antimicrobials in cats. Vet. J. 2009, 180, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.E.F. Martindale the Extra Pharmacopoeia, 35th ed.; Pharmaceutical Press: London, UK, 2007; p. 278. [Google Scholar]
- Goudah, A.; Abo-El-Sooud, K. Pharmacokinetics and milk penetration of orbifloxacin after intravenous and intramuscular injections to dromedary lactating camels (Camelus dromedaries). J. Vet. Pharmacol. Ther. 2008, 31, 276–280. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, M.J. The Merck Index: An Encyclopedia of Chemicals, Drugs and Biologicals, 14th ed.; Merck & Co.: Whitehouse Station, NJ, USA, 2006; p. 1182. [Google Scholar]
- British Pharmacopoeia; Her Majesty’s Stationery Office: London, UK, 2011; Volume 1, pp. 88–90.
- United States Pharmacopeia National Formulary—USP38 NF33; U.S. Pharmacopeial Convention: Rockville, MD, USA, 2015; p. 3000.
- Brog, J.P.; Chanez, C.L.; Crochet, A.; Fromm, K.M. Polymorphism, what it is and how to identify it: A systematic review. RSC Adv. 2013, 3, 16905–16931. [Google Scholar] [CrossRef]
- Láng, P.; Kiss, V.; Ambrus, R.; Farkas, G.; Szabó-Révész, P.; Aigner, Z.; Várkonyi, E. Polymorph screening of an active material. J. Pharm. Biomed. Anal. 2013, 84, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Bonfilio, R.; Leal, J.S.; Santos, O.M.; Pereira, G.R.; Doriguetto, A.C.; Araújo, M.B. Analysis of chlorthalidone polymorphs in raw materials and tablets and the effect of forms I and II on the dissolution properties of drug products. J. Pharm. Biomed. Anal. 2014, 88, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, J.; Allesø, M.; Mirza, S.; Koradia, V.; Gordon, K.C.; Rantanen, J. Solid form screening—A review. Eur. J. Pharm. Biopharm. 2009, 71, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, J.A.; Krc, J., Jr.; Kinkel, A.W.; Samyn, J.C. Effect of polymorphism on the absorption of chloramphenicol from chloramphenicol palmitate. J. Pharm. Sci. 1967, 56, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Erdemir, D.; Myerson, A.S. Crystal polymorphism in chemical process development. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs, 2nd ed.; SSCI: West Lafayette, IN, USA, 1999. [Google Scholar]
- Purohit, R.; Venugopalan, P. Polymorphism: An overview. Resonance 2009, 14, 882–893. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Guidance for Industry—ANDAs: Pharmaceutical Solid Polymorphism Chemistry, Manufacturing, and Controls Information, 2007. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072866.pdf (accessed on 18 December 2015).
- International Conference on Harmonisation (ICH). Q6A: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances; ICH: Genebra, Switzerland, 1999; Available online: http://www.ich.org/products/guidelines/quality/article/qualityguidelines.html (accessed on 18 December 2015).
- Censi, R.; Di Martino, P. Polymorph impact on the bioavailability and stability of poorly soluble drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed]
- Santos, O.M.M.; Silva, D.M.; Martins, F.T.; Legendre, A.O.; Azarias, L.C.; Rosa, I.M.L.; Neves, P.P.; Araujo, M.B.; Doriguetto, A.C. Protonation pattern, tautomerism, conformerism, and physicochemical analysis in new crystal forms of the antibiotic doxycycline. Cryst. Growth Des. 2014, 14, 3711–3726. [Google Scholar] [CrossRef]
- Martins, F.T.; Bocelli, M.D.; Bonfilio, R.; Araújo, M.B.; Lima, P.V.; Neves, P.P.; Veloso, M.P.; Ellena, J.; Doriguetto, A.C. Conformational polymorphism in racemic crystals of the diuretic drug chlortalidone. Cryst. Growth Des. 2009, 9, 3235–3244. [Google Scholar] [CrossRef]
- Martins, F.T.; Paparidis, N.; Doriguetto, A.C.; Ellena, J. Crystal engineering of an anti-HIV drug based on the recognition of assembling molecular frameworks. Cryst. Growth Des. 2009, 9, 5283–5292. [Google Scholar] [CrossRef]
- Martins, F.T.; Doriguetto, A.C.; Ellena, J. From rational design of drug crystals to understanding of nucleic acid structures: Lamivudine duplex. Cryst. Growth Des. 2010, 10, 676–684. [Google Scholar] [CrossRef]
- Martins, F.T.; Legendre, A.O.; Honorato, S.B.; Ayala, A.P.; Doriguetto, A.C.; Ellena, J. Solvothermal preparation of drug crystals: Didanosine. Cryst. Growth Des. 2010, 10, 1885–1891. [Google Scholar]
- Martins, F.T.; Lima, P.V.; Azarias, L.C.; Abreu, P.J.; Neves, P.P.; Legendre, A.O.; Andrade, F.M.; Oliveira, G.R.; Ellena, J.; Doriguetto, A.C. Increasing the symmetry of drug crystals: A monoclinic conformational polymorph of the platelet antiaggregating agent ticlopidine hydrochloride. CrystEngComm 2011, 13, 5737–5743. [Google Scholar] [CrossRef]
- Legendre, A.O.; Silva, L.R.R.; Silva, D.M.; Rosa, I.M.L.; Azarias, L.C.; Abreu, P.J.; Araújo, M.B.; Neves, P.P.; Torres, C.; Martins, F.T.; et al. Solid state chemistry of the antibiotic doxycycline: Structure of the neutral monohydrate and insights into its poor water solubility. CrystEngComm 2012, 14, 2532–2540. [Google Scholar] [CrossRef]
- Martins, F.T.; Abreu, P.J.; Azarias, L.C.; Villis, P.C.M.; Campos Melo, A.C.; Ellena, J.; Doriguetto, A.C. Form III-like conformation and Form I-like packing in a chloroform channel solvate of the diuretic drug chlortalidone. CrystEngComm 2012, 14, 6173–6177. [Google Scholar] [CrossRef] [Green Version]
- Ellena, J.; Bocelli, M.D.; Honorato, S.B.; Ayala, A.P.; Doriguetto, A.C.; Martins, F.T. Base-paired and base-stacked structures of the anti-HIV drug lamivudine: A nucleoside DNA-mimicry with unprecedented topology. Cryst. Growth Des. 2012, 12, 5138–5147. [Google Scholar] [CrossRef]
- Martins, F.T; Bonfilio, R.; Rosa, I.M.L.; Santos, L.M.; Santos, O.M.M.; Araújo, M.B.; Doriguetto, A.C. The form II of the antihypertensive drug chlorthalidone. CrystEngComm 2013, 15, 3767–3771. [Google Scholar] [CrossRef]
- Flack, H.D. Chiral and achiral crystal structures. Helv. Chim. Acta 2003, 86, 905–921. [Google Scholar] [CrossRef]
- Hoffmann, R.W. Meso compounds: Stepchildren or favored children of stereoselective synthesis? Angew. Chem. Int. Ed. 2003, 42, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Boeyens, J.C.A.; Evans, D.G. Conformational analysis of ring pucker. Acta Crystallogr. B 1989, B45, 581–590. [Google Scholar]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H. The Cambridge structural database: A quarter of a million crystal structures and rising. Acta Crystallogr. B 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Chakraborti, C.K.; Mishra, S.C.; Nanda, U.N.; Naik, S. FTIR and XRD investigations of some fluoroquinolones. Int. J. Pharm. Pharm. Sci. 2011, 3, 165–170. [Google Scholar]
- Herbstein, F.H. IUCr Monographs on Crystallography—18: Crystalline Molecular Complexes and Compounds: Structures and Principles; Oxford University Press Inc.: New York, NY, USA, 2005. [Google Scholar]
- Nesterenko, V.F.; Bondar, M.P.; Ershov, V. Instability of plastic flow at dynamic pore collapse. In Proceedings of the AIP Conference Proceedings, Colorado Springs, CO, USA, 28−2 July 1994; Volume 309, pp. 1173–1176.
- Simonot-Grange, M.H. Thermodynamic and structural features of water sorption in zeolites. Clays Clay Miner. 1979, 27, 423–428. [Google Scholar] [CrossRef]
- Byrn, S.; Pfeiffer, R.; Ganey, M.; Hoiberg, C.; Poochikian, G. Pharmaceutical solids: A strategic approach to regulatory considerations. Pharm. Res. 1995, 12, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Borgadus, J.B.; Blackwood, R.B., Jr. Solubility of doxycycline in aqueous solution. J. Pharm. Sci. 1979, 68, 188–194. [Google Scholar]
- Aulton, M.E. Delineamento de Formas Farmacêuticas, 2nd ed.; Artmed: Rio de Janeiro, Brazil, 2005; p. 675. [Google Scholar]
- CrysAlis Pro 1.7.; Oxford Diffraction Ltd.: Abingdon, UK, 2006.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 568. [Google Scholar] [CrossRef]
- Cazedey, E.C.L.; Othman, A.; Garg, S.; Salgado, H.R.N. A validated stability-indicating lc method for orbifloxacin in the presence of degradation products. Curr. Pharm. Anal. 2011, 7, 176–181. [Google Scholar] [CrossRef]
- Baka, E.; Comer, J.E.; Takács-Novák, K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J. Pharm. Biomed. Anal. 2008, 46, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Empirical Formula | C19H20F3N3O3. 1/2H2O |
| Formula Weight (g∙mol−1) | 404.4 |
| Temperature (K) | 150 (2) |
| Wavelength (Å) | 0.71073 |
| Crystal System | Orthorhombic |
| Space group | C2221 |
| Cell parameters (Å, °) | a = 8.4713(4); b = 20.0456(7); c = 21.4550(7); α = β = γ = 90 |
| Cell Volume (Å3) | 3643.3(2) |
| Z; Calc. density (g∙cm−3) | 8; 1.474 |
| Absorption coefficient (mm−1) | 0.123 |
| F(000) | 1688 |
| Crystal size (mm3) | 0.25 × 0.15 × 0.10 |
| θ-range (°) | 1.90 a 29.49 |
| Index ranges | −11≤ h ≤ 11; −27 ≤ k ≤ 25; −29 ≤ l ≤28 |
| Reflexions collected/unique | 22,842/4627 [R(int) = 0.0281] |
| Completeness to θmax = 29.49 (%) | 100 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4627/0/267 |
| Goodness-of-fit on F2 | 1.041 |
| R1/wR2 for I > 2σ(I) | R1 = 0.0357, wR2 = 0.0883 |
| R1/wR2 for all data | R1 = 0.0428, wR2 = 0.0938 |
| Largest diff. peak/hole (e∙ų) | 0.287 and −0.202 |
| D-H···A | d(D-H) | d(H···A) | d(D···A) | <(D-H-A) |
|---|---|---|---|---|
| O5ii-H5ii···O2ii | 0.92(8) | 1.98(8) | 2.854(3) | 159(7) |
| N3-H3a···O1i | 0.88(3) | 1.86(3) | 2.720(2) | 164(2) |
| N3-H3b···O2ii | 0.86(3) | 1.94(3) | 2.714(2) | 150(2) |
| N3-H3b···O3ii | 0.86(3) | 2.35(3) | 2.955(2) | 128(2) |
| C11-H11···O2iv | 0.98 | 2.42 | 3.382(3) | 168.6 |
| Media | Hemihydrate (g·L−1) | SD (g·L−1) | Solubility * | Anhydrous (g·L−1) | SD (g·L−1) | Classification * | Anhydrous/ Hemihydrate (%) | Final pH † |
|---|---|---|---|---|---|---|---|---|
| Water | 1.32 | 0.0678 | SS | 1.38 | 0.06634 | SS | 4.38 | 7.0 |
| pH 7.5 | 1.58 | 0.0644 | SS | 1.64 | 0.05327 | SS | 3.80 | 7.5 |
| pH 6.8 | 1.52 | 0.039 | SS | 1.59 | 0.07758 | SS | 4.60 | 7.0 |
| pH 5.8 | 1.73 | 0.0601 | SS | 1.98 | 0.07915 | SS | 14.45 | 6.0 |
| HCl 0.01 mol·L−1 | 6.30 | 0.1739 | SS | 10.33 | 0.29854 | SP | 63.97 | 6.0 |
| pH 4.5 | 10.41 | 0.5007 | SP | 14.24 | 0.5539 | SP | 36.79 | 4.5 |
| HCl 0.1 mol·L−1 | 22.24 | 0.7506 | SP | 26.23 | 1.0623 | SP | 17.94 | 2.0 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, O.M.M.; Freitas, J.T.J.; Cazedey, E.C.L.; Araújo, M.B.d.; Doriguetto, A.C. Structure, Solubility and Stability of Orbifloxacin Crystal Forms: Hemihydrate versus Anhydrate. Molecules 2016, 21, 328. https://doi.org/10.3390/molecules21030328
Santos OMM, Freitas JTJ, Cazedey ECL, Araújo MBd, Doriguetto AC. Structure, Solubility and Stability of Orbifloxacin Crystal Forms: Hemihydrate versus Anhydrate. Molecules. 2016; 21(3):328. https://doi.org/10.3390/molecules21030328
Chicago/Turabian StyleSantos, Olimpia Maria Martins, Jennifer Tavares Jacon Freitas, Edith Cristina Laignier Cazedey, Magali Benjamim de Araújo, and Antonio Carlos Doriguetto. 2016. "Structure, Solubility and Stability of Orbifloxacin Crystal Forms: Hemihydrate versus Anhydrate" Molecules 21, no. 3: 328. https://doi.org/10.3390/molecules21030328