
Photochemistry of the α-Al2O3-PETN Interface

 and
and
Abstract
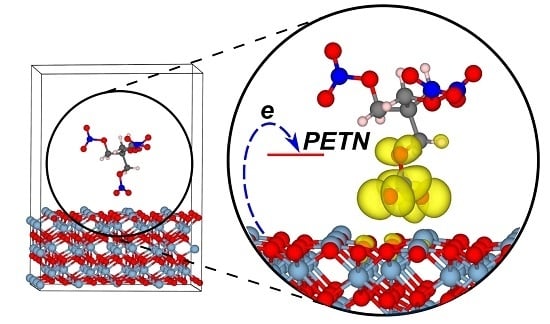
:
1. Introduction
2. Results and Discussion
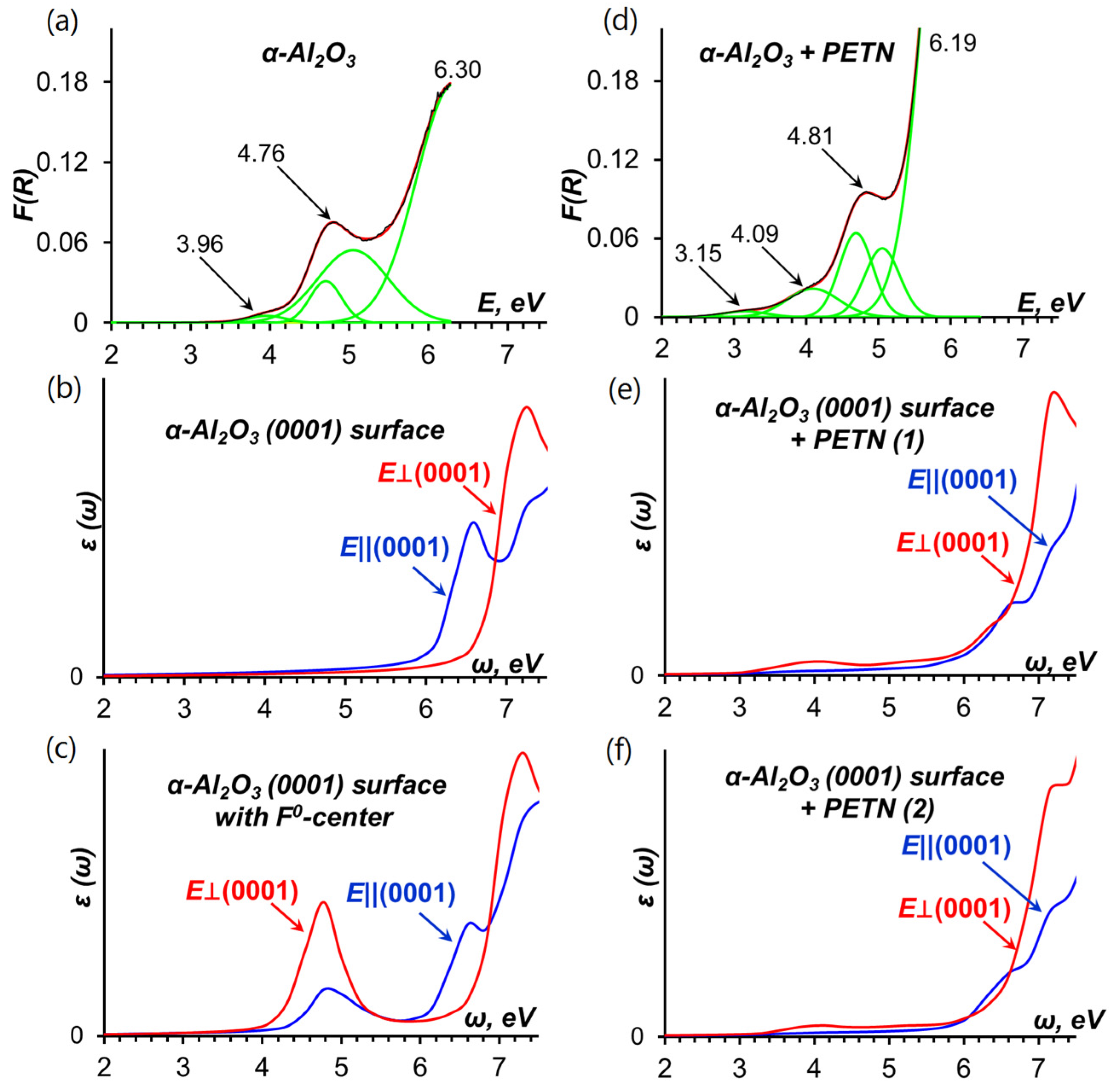
2.1. Optical Absorption of α-Al2O3-PETN Composites
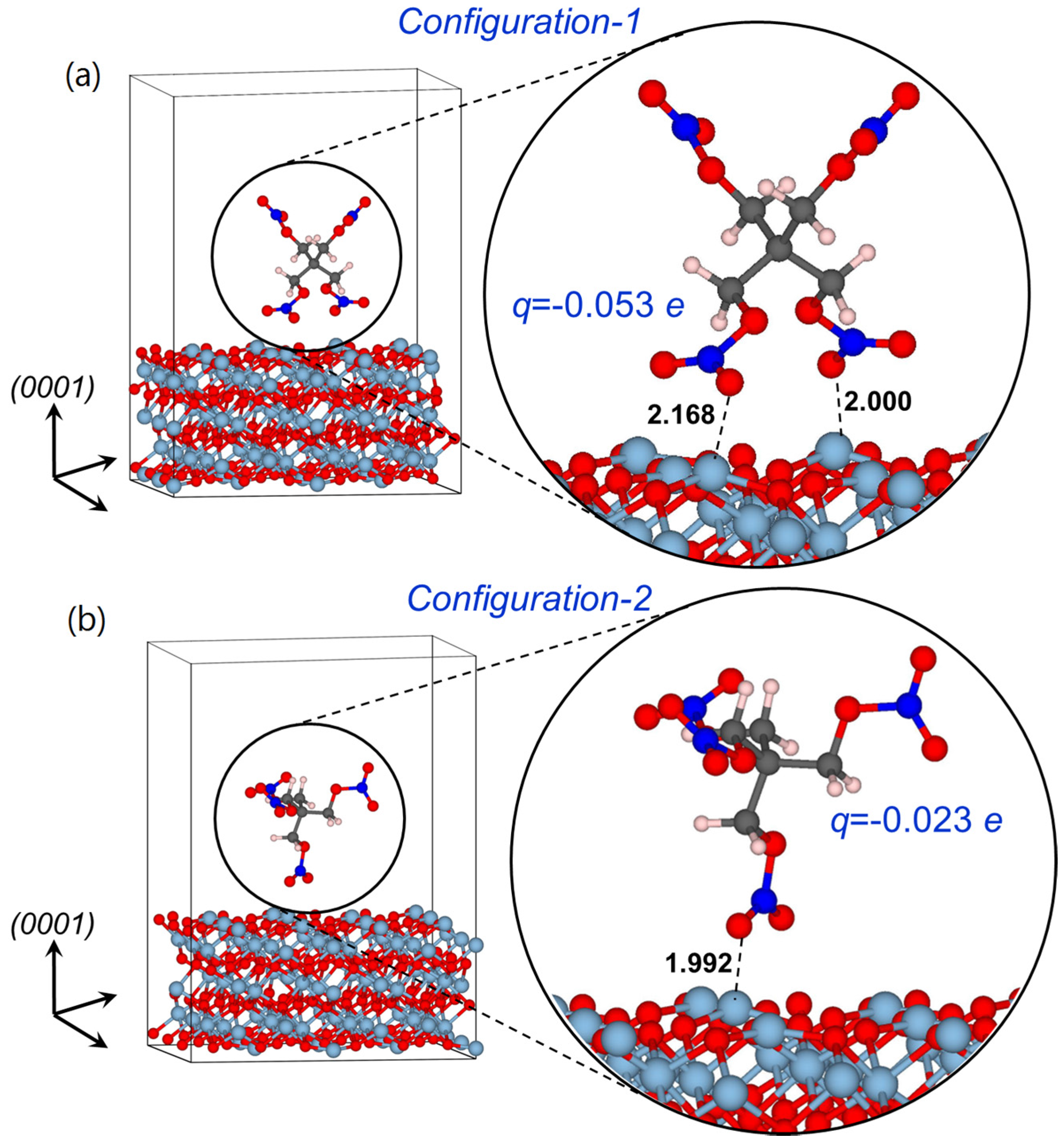
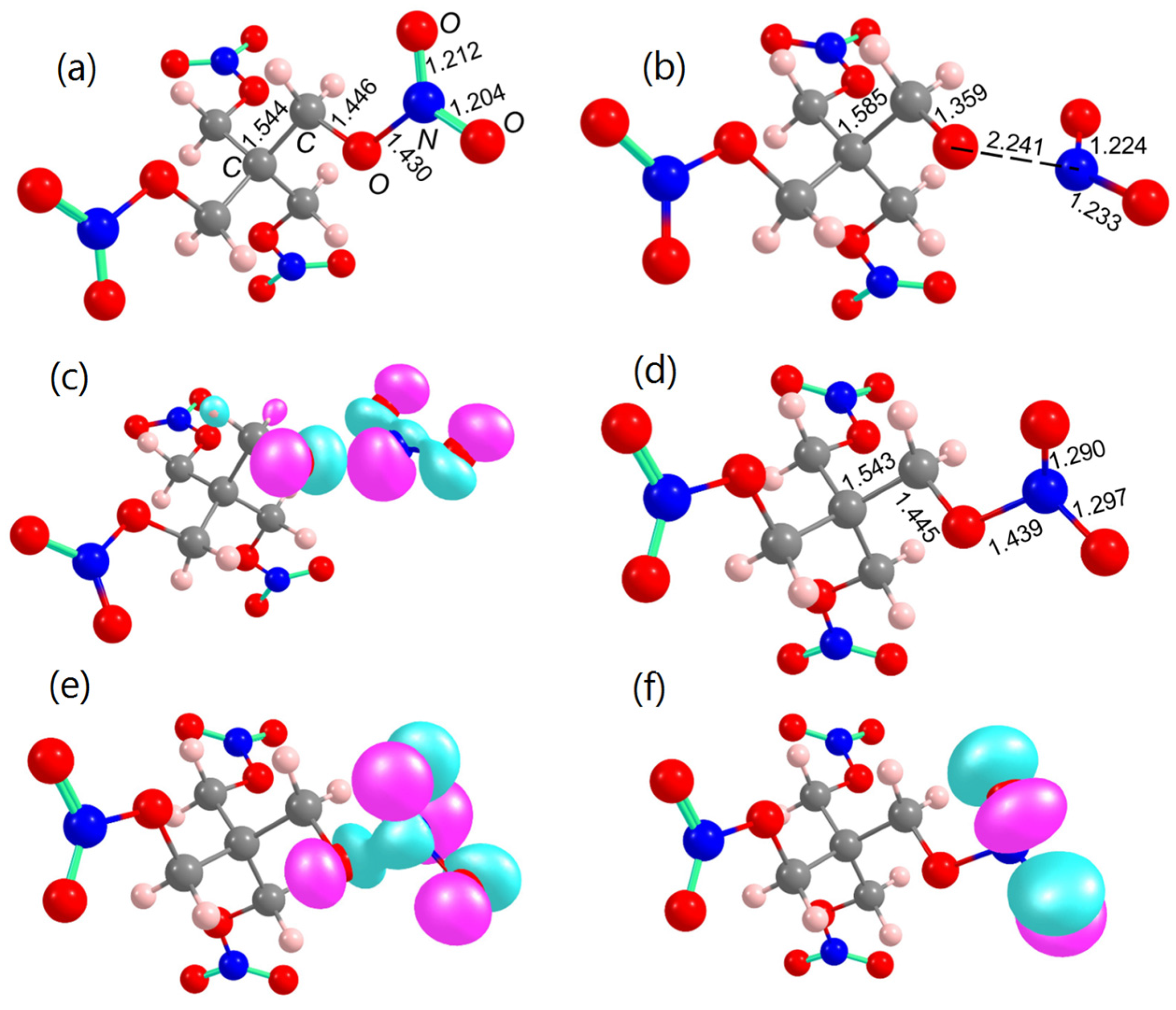
2.1.1. Structures of Model Supercells
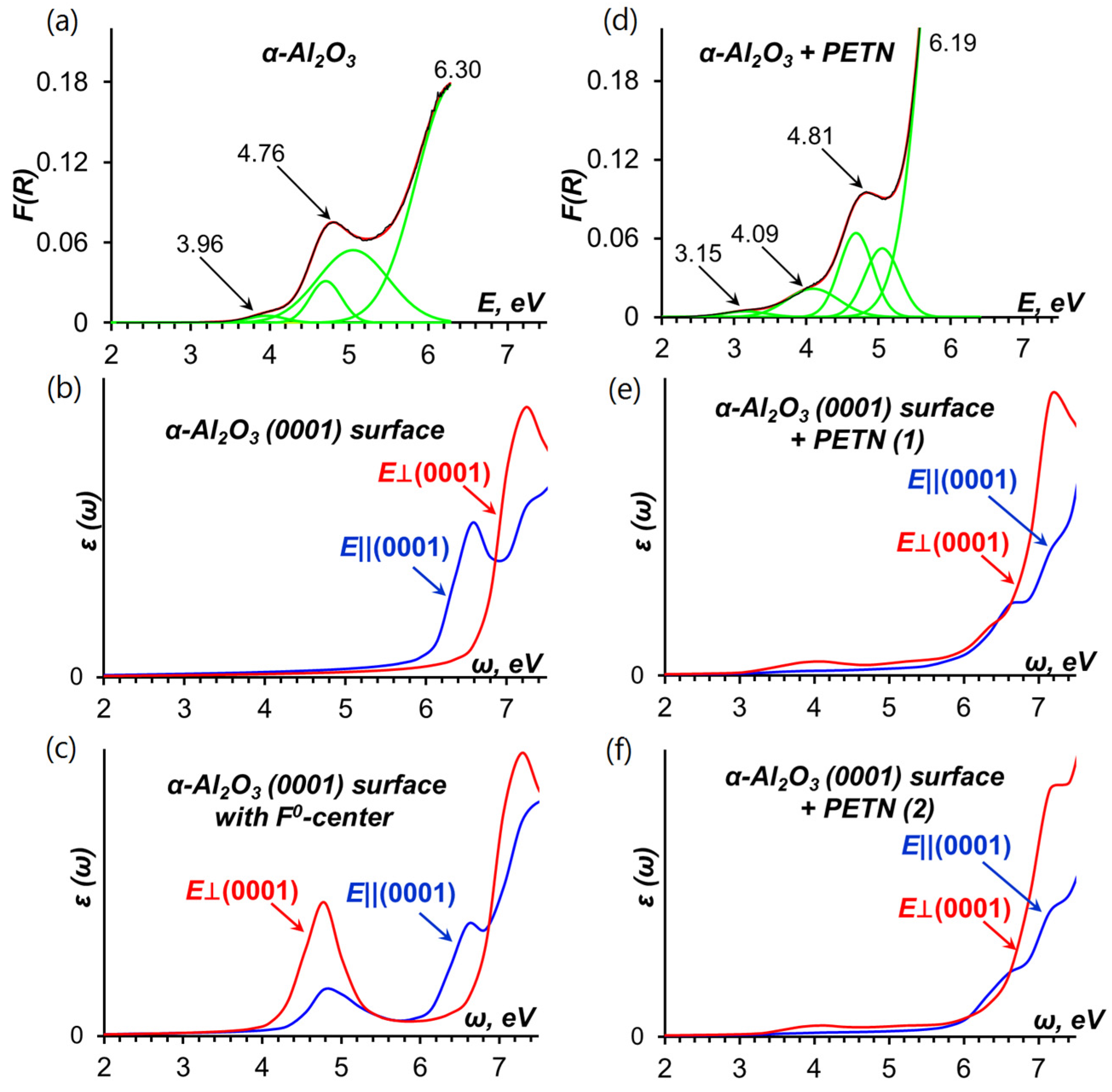
2.1.2. Optical Absorption of the Pristine α-Al2O3 (0001) Surface
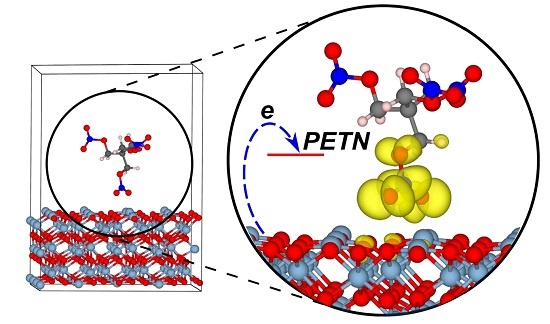
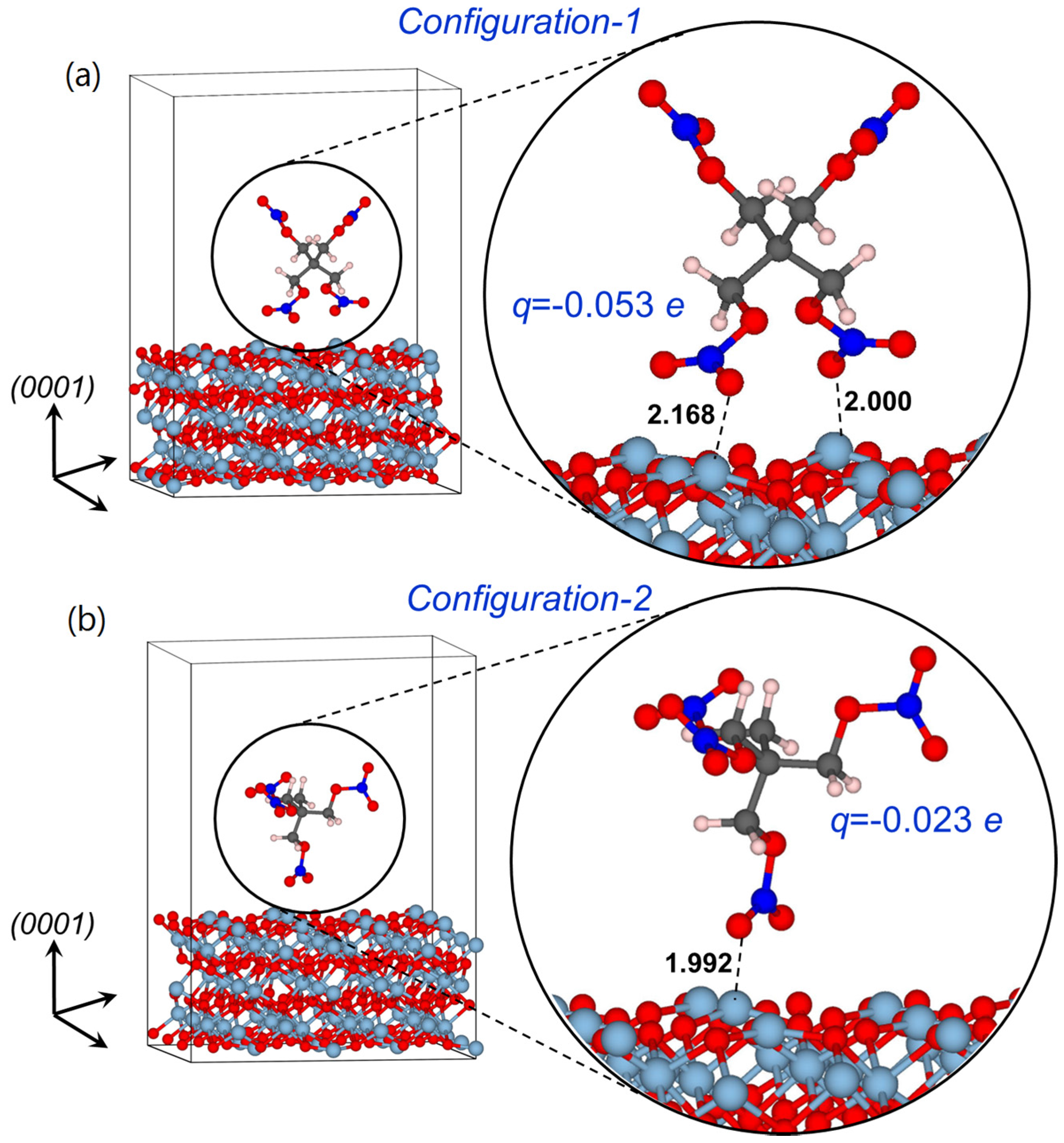
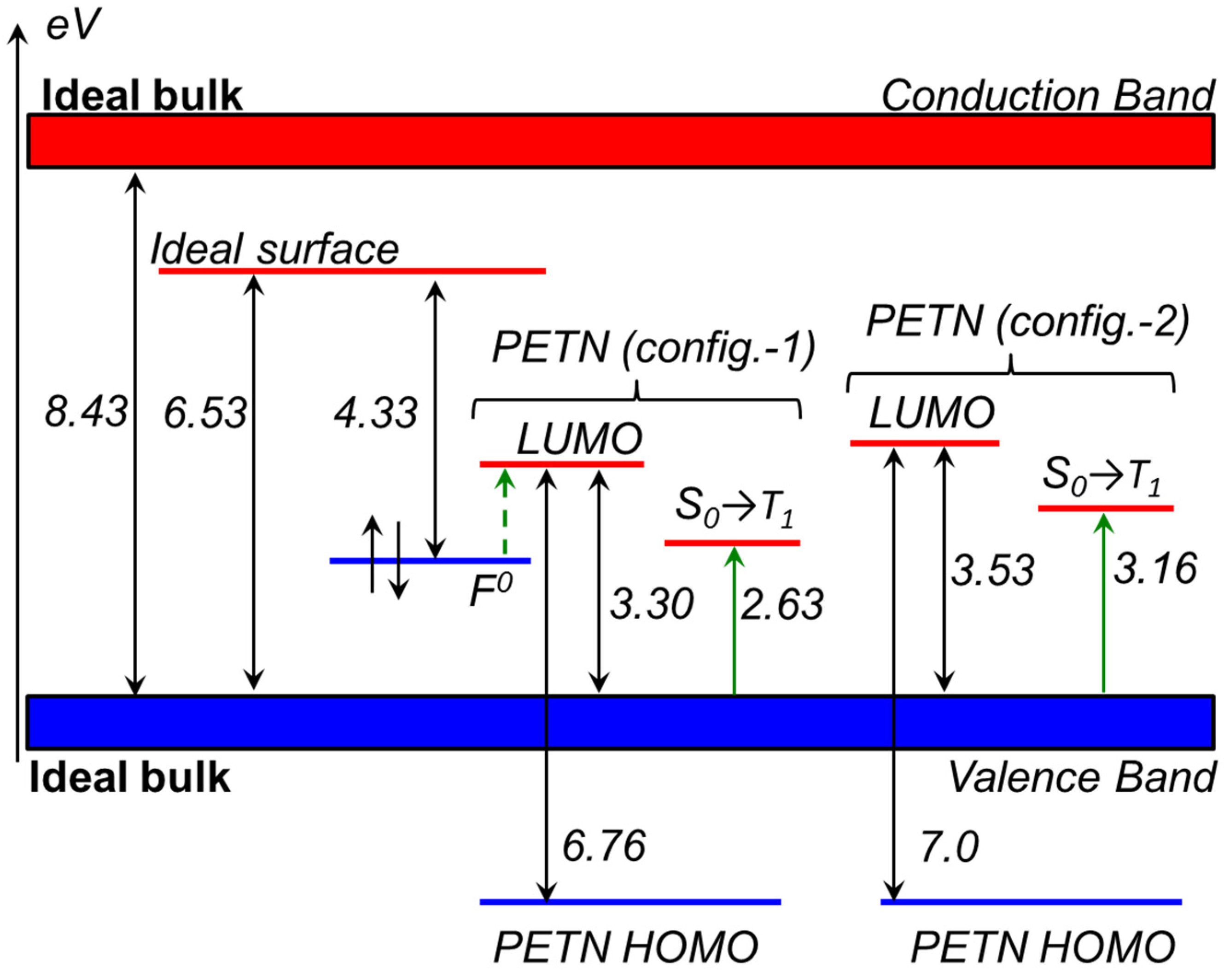
2.1.3. Optical Absorption of the α-Al2O3 (0001)-PETN Interface
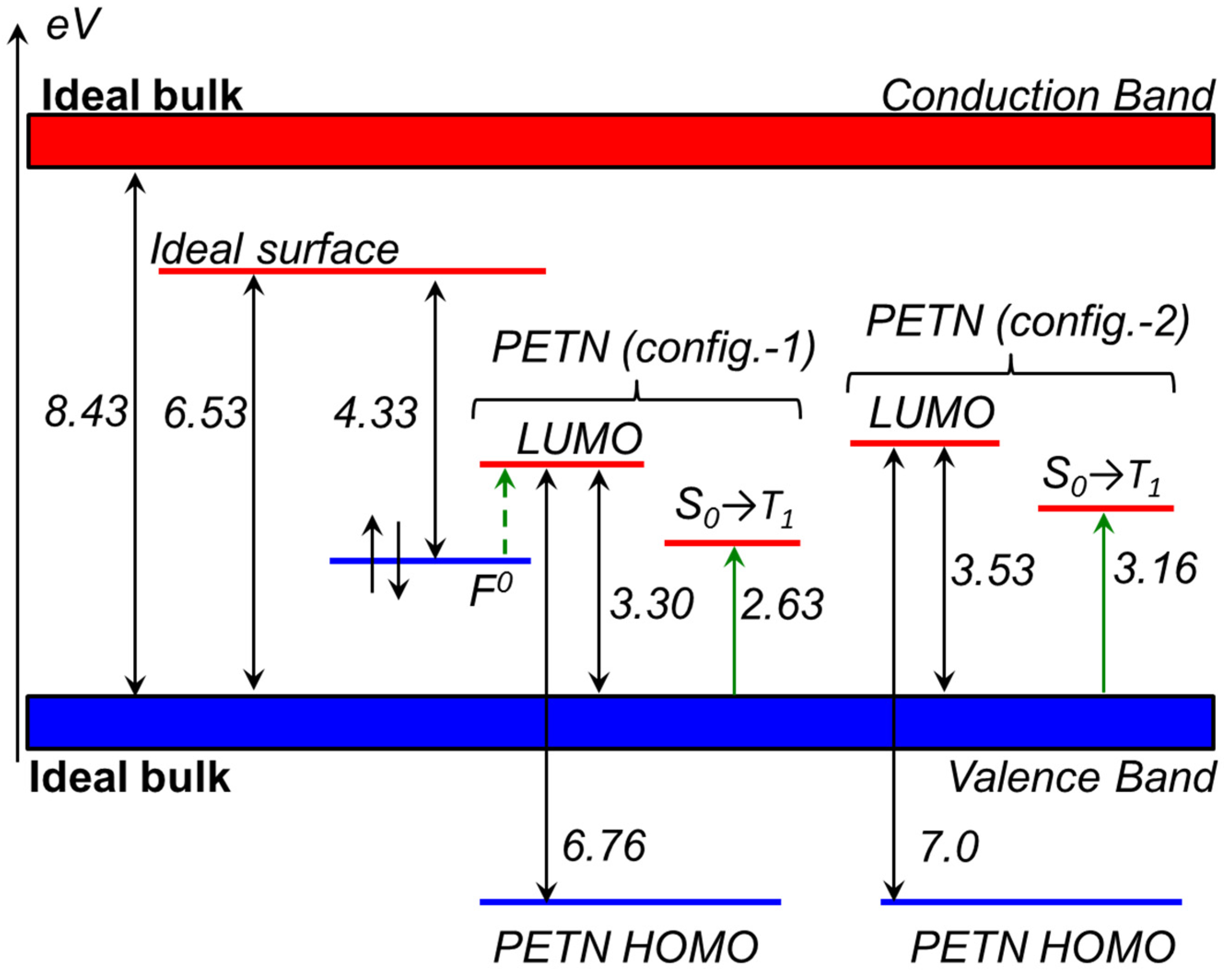
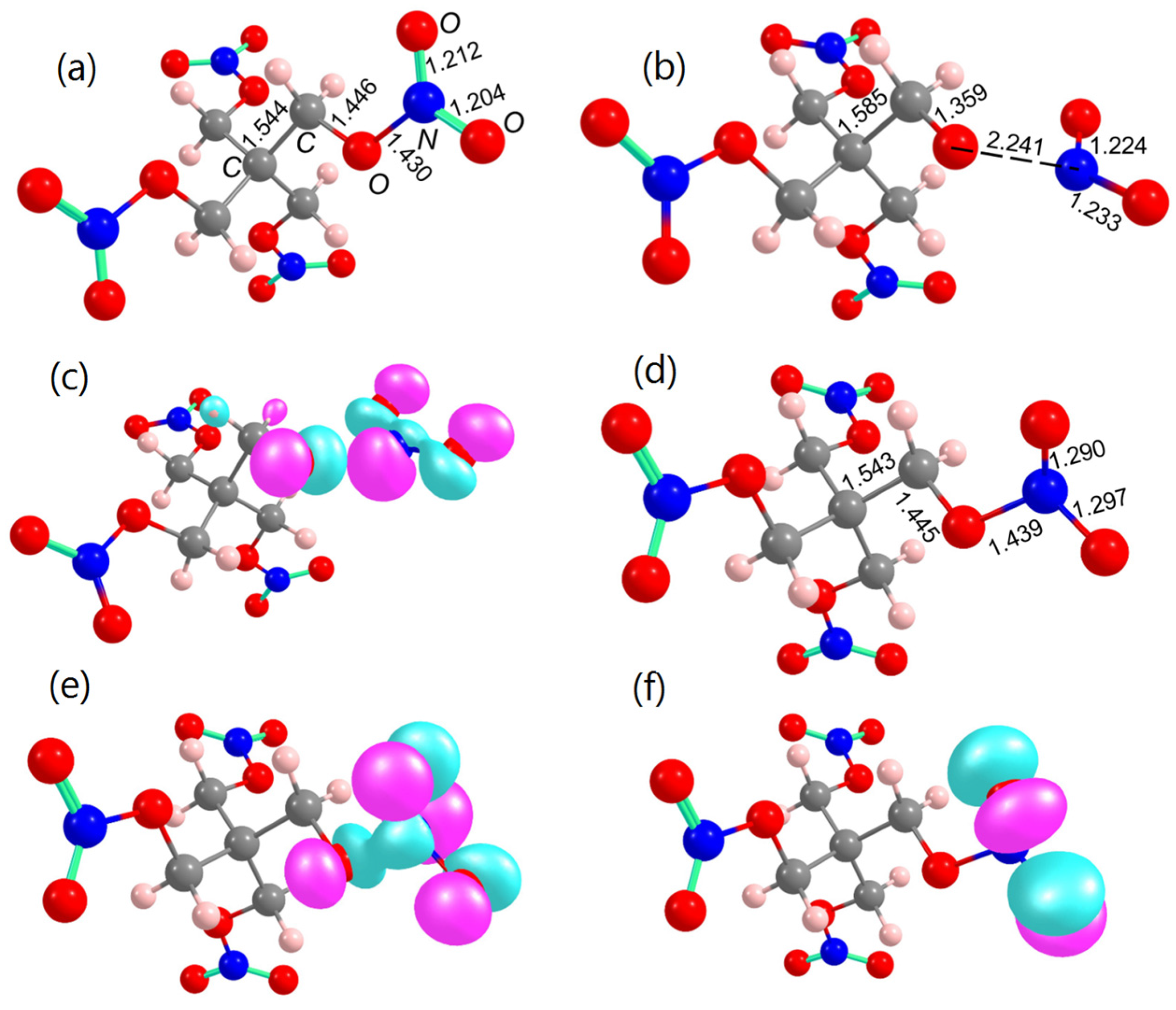
2.2. Decomposition of Charged and Excited PETN Molecules
3. Methods
3.1. Details of Calculations
3.2. Details of Experiment
4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and notes
- Akimov, A.V.; Neukirch, A.J.; Prezhdo, O.V. Theoretical insights into photoinduced charge transfer and catalysis at oxide interfaces. Chem. Rev. 2013, 113, 4496–4565. [Google Scholar] [PubMed]
- Duncan, W.R.; Prezhdo, O.V. Theoretical studies of photoinduced electron transfer in dye-sensitized TiO2. Annu. Rev. Phys. Chem. 2007, 58, 143–843. [Google Scholar] [PubMed]
- Gregg, B.A. Interfacial processes in the dye-sensitized solar cell. Coord. Chem. Rev. 2004, 248, 1215–1224. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Irie, H.; Ohwaki, T. Nitrogen-doped titanium dioxide as visible-light-sensitive photocatalyst: Designs, developments, and prospects. Chem. Rev. 2014, 114, 9824–9852. [Google Scholar] [PubMed]
- Naddo, T.; Che, Y.; Zhang, W.; Balakrishnan, K.; Yang, X.; Yen, M.; Zhao, J.; Moore, J.S.; Zang, L. Detection of explosives with a fluorescent nanofibril film. J. Am. Chem. Soc. 2007, 129, 6978–6979. [Google Scholar] [CrossRef] [PubMed]
- Tu, R.; Liu, B.; Wang, Z.; Gao, D.; Wang, F.; Fang, Q.; Zhang, Z. Amine-capped ZnS-Mn2+ nanocrystals for fluorescence detection of trace TNT explosive. Anal. Chem. 2008, 80, 3458–3465. [Google Scholar] [CrossRef] [PubMed]
- Kartha, K.K.; Babu, S.S.; Srinivasan, S.; Ajayaghosh, A. Attogram sensing of trinitrotoluene with a self-assembled molecular gelator. J. Am. Chem. Soc. 2012, 134, 4834–4841. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, S.; Zheng, C.; Zhang, X.; Emge, T.J.; Li, J. New microporous metal-organic framework demonstrating unique selectivity for detection of high explosives and aromatic compounds. J. Am. Chem. Soc. 2011, 133, 4153–4155. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Swager, T.M. Porous shape persistent fluorescent polymer films: An approach to TNT sensory materials. J. Am. Chem. Soc. 1998, 120, 5321–5322. [Google Scholar] [CrossRef]
- Sohn, H.; Sailor, M.J.; Magde, D.; Trogler, W.C. Detection of nitroaromatic explosives based on photoluminescent polymers containing metalloles. J. Am. Chem. Soc. 2003, 125, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.E.; Knapp, M.J. Discrimination of nitroaromatics and explosives mimics by a fluorescent Zn(salicylaldimine) sensor array. J. Am. Chem. Soc. 2008, 130, 5422–5423. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ding, J.; Song, N.; Lu, J.; Tao, Y. Development of a new s-tetrazine-based copolymer for efficient solar cells. J. Am. Chem. Soc. 2010, 132, 13160–13161. [Google Scholar] [CrossRef] [PubMed]
- Pinnaduwage, L.A.; Gehl, A.; Hedden, D.L.; Muralidharan, G.; Thundat, T.; Lareau, R.T.; Sulchek, T.; Manning, L.; Rogers, B.; Jones, M.; et al. Explosives: A microsensor for trinitrotoluene vapour. Nature 2003, 425. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.S. Recent advances in trace explosives detection instrumentation. Sens. Imaging 2007, 8, 9–38. [Google Scholar]
- Rose, A.; Zhu, Z.; Madigan, C.F.; Swager, T.M.; Bulovic, V. Sensitivity gains in chemosensing by lasing action in organic polymers. Nature 2005, 434, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Andrew, T.L.; Swager, T.M. A fluorescence turn-on mechanism to detect high explosives RDX and PETN. J. Am. Chem. Soc. 2007, 129, 7254–7255. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.N.; Arechederra, R.L.; Minteer, S.D. Nitroaromatic actuation of mitochondrial bioelectrocatalysis for self-powered explosive sensors. J. Am. Chem. Soc. 2008, 130, 15272–15273. [Google Scholar] [CrossRef] [PubMed]
- Aluker, E.D.; Krechetov, A.G.; Mitrofanov, A.Y.; Sakharchuk, Y.P. Model of the photostimulated fragmentation of PETN molecules in selective photoinitiation. Russ. J. Phys. Chem. B 2011, 5, 821–823. [Google Scholar] [CrossRef]
- Aluker, E.D.; Krechetov, A.G.; Mitrofanov, A.Y.; Nurmukhametov, D.R.; Kuklja, M.M. Laser initiation of energetic materials: Selective photoinitiation regime in pentaerythritol tetranitrate. J. Phys. Chem. C 2011, 115, 6893–6901. [Google Scholar] [CrossRef]
- Aluker, E.D.; Krechetov, A.G.; Mitrofanov, A.Y.; Zverev, A.S.; Kuklja, M.M. Understanding limits of the thermal mechanism of laser initiation of energetic materials. J. Phys. Chem. C 2012, 116, 24482–24486. [Google Scholar]
- Aluker, E.D.; Krechetov, A.G.; Mitrofanov, A.Y.; Zverev, A.S.; Kuklja, M.M. Topography of photochemical initiation in molecular materials. Molecules 2013, 18, 14148–14160. [Google Scholar] [CrossRef] [PubMed]
- Tsyshevsky, R.V.; Rashkeev, S.N.; Kuklja, M.M. Defect states at organic–inorganic interfaces: Insight fromfirst principles calculations for pentaerythritol tetranitrate on MgO surface. Surf. Sci. 2015, 637–638, 19–28. [Google Scholar]
- Tsyshevsky, R.V.; Sharia, O.; Kuklja, M.M. Energies of electronic transitions of PETN molecules and crystals. J. Phys. Chem. C 2014, 118, 9324–9335. [Google Scholar]
- McLeod, J.A.; Kurmaev, E.Z.; Sushko, P.V.; Boyko, T.D.; Levitsky, I.A.; Moewes, A. Selective response of mesoporous silicon to adsorbants with nitro groups. Chem. Eur. J. 2012, 18, 2912–2922. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.C.; Boatz, J.A.; Thompson, D.L. First-principles calculations of the adsorption of nitromethane and 1,1-diamino-2,2-dinitroethylene (FOX-7) molecules on the Al(111) surface. J. Phys. Chem. B 2003, 107, 8953–8964. [Google Scholar] [CrossRef]
- Sorescu, D.C.; Boatz, J.A.; Thompson, D.L. First-principles calculations of the adsorption of nitromethane and 1,1-diamino-2,2-dinitroethylene (FOX-7) molecules on the α-Al2O3 (0001) Surface. J. Phys. Chem. B 2005, 109, 1451–1463. [Google Scholar] [PubMed]
- Zepeda-Ruiz, L.A.; Maiti, A.; Gee, R.; Gilmer, G.H.; Weeks, B.L. Size and habit evolution of PETN crystals: A lattice monte carlo study. J. Cryst. Growth 2006, 291, 461–467. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Kunz, A.B. Electronic structure of molecular crystals containing edge dislocations. J. Appl. Phys. 2001, 89, 4962–4970. [Google Scholar] [CrossRef]
- Tsyshevsky, R.V.; Sharia, O.; Kuklja, M.M. Thermal decomposition mechanisms of nitroesters: Ab Initio modeling of pentaerythritol tetranitrate. J. Phys. Chem. C 2013, 117, 18144–18153. [Google Scholar] [CrossRef]
- French, R.H. Electronic band structure of Al2O3, with comparison to Alon and AIN. J. Am. Ceram. Soc. 1990, 73, 477–489. [Google Scholar] [CrossRef]
- Gomes, J.R.B.; Lodziana, Z.; Illas, F. Adsorption of small palladium clusters on the relaxed α-Al2O3(0001) surface. J. Phys. Chem. B 2003, 107, 6411–6424. [Google Scholar] [CrossRef]
- Łodziana, Z.; Nørskov, J.K. Adsorption of Cu and Pd on α-Al2O3 (0001) surfaces with different stoichiometries. J. Chem. Phys. 2001, 115. [Google Scholar] [CrossRef] [Green Version]
- Kotomin, E.A.; Popov, A.I. Radiation-induced point defects in simple oxides. Nucl. Instrum. Methods Phys. 1998, 141, 1–15. [Google Scholar]
- Carrasco, J.; Gomes, J.R.B.; Illas, F. Theoretical study of bulk and surface oxygen and aluminum vacancies in α-Al2O3. Phys. Rev. B 2004, 69. [Google Scholar] [CrossRef]
- In this research, we did not study F+-centers, however, we expect that its absorption band, which typically has the similar or a slightly higher energy than F0-center, will fall in the observed range of 4.5–5.1 eV.
- Ivakin, Y.D.; Danchevskaya, M.N.; Ovchinnikova, O.G.; Murav’eva, G.P.; Kreisberg, V.A. The kinetics and mechanism of doped corundum structure formation in an water fluid. Rus. J. Phys. Chem. B 2009, 3, 1019–1034. [Google Scholar] [CrossRef]
- Mullen, P.A.; Orloff, M.K. Ultraviolet absorption spectrum of pentaerythritol tetranitrate. J. Phys. Chem. 1973, 77, 910–911. [Google Scholar]
- Yu, Z.; Bernstein, E.R. Decomposition of pentaerythritol tetranitrate [C(CH2ONO2)4] following electronic excitation. J. Chem. Phys. 2011, 135. [Google Scholar] [CrossRef]
- Cooper, J.K.; Grant, C.D.; Zhang, J.Z. Experimental and TD-DFT Study of optical absorption of six explosive molecules: RDX, HMX, PETN, TNT, TATP, and HMTD. J. Phys. Chem. A 2013, 117, 6043–6051. [Google Scholar] [CrossRef] [PubMed]
- The fact that our optical measurements of the Al2O3-PETN composites did not register the absorption band corresponding to the transition from F0-center (HOMO) to PETN (LUMO) is explained by a simple reason. The absorption was measured in the range from 190 to 1100 nm (1.12–6.52 eV), and the excitation of 1.2 falls right at the edge of the measurement interval.
- Carrasco, J.; Lopez, N.; Sousa, C.; Illas, F. First-principles study of the optical transitions of F centers in the bulk and on the (0001) surface of α-Al2O3. Phys. Rev. B 2005, 72. [Google Scholar] [CrossRef]
- Tsyshevsky, R.V.; Sharia, O.; Kuklja, M.M. Optical absorption energies of molecular defects in pentaerythritol tetranitrate crystals: Quantum chemical modeling. J. Phys. Chem. C 2014, 118, 26530–26542. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. A self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schroder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 2004, 92. [Google Scholar] [CrossRef]
- Thonhauser, T.; Cooper, V.R.; Li, S.; Puzder, A.; Hyldgaard, P.; Langreth, D.C. Van der Waals density functional: Self-consistent potential and the nature of the van der Waals bond. Phys. Rev. B 2007, 76. [Google Scholar] [CrossRef]
- Roman-Perez, G.; Soler, J.M. Efficient implementation of a van der Waals density functional: Application to double-wall carbon nanotubes. Phys. Rev. Lett. 2009, 103. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Futhmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar]
- Kresse, G.; Furthmuller, F. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kirfel, A.; Eichhorn, K. Accurate structure analysis with synchrotron radiation. The electron density in Al2O3 and Cu2O. Acta Cryst. 1990, 46, 271–284. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Gaussian 09; Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- Ahmad, S.R.; Cartwright, M. Laser Ignition of Energetic Materials; John Wiley & Sons: New York, NY, USA, 2014. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors upon request.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial State | Formation Energy | Activation Barrier | Reaction Energy |
|---|---|---|---|
| [PETN] | 0 | 35.0 | 35.0 |
| (PETN)- | −2.4 (EA of isolated molecule) | 18.0 | 18.0 |
| (PETN)* | 3.88–4.22 (Vertical excitation) | 4.5 | −28.7 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsyshevsky, R.V.; Zverev, A.; Mitrofanov, A.; Rashkeev, S.N.; Kuklja, M.M. Photochemistry of the α-Al2O3-PETN Interface. Molecules 2016, 21, 289. https://doi.org/10.3390/molecules21030289
Tsyshevsky RV, Zverev A, Mitrofanov A, Rashkeev SN, Kuklja MM. Photochemistry of the α-Al2O3-PETN Interface. Molecules. 2016; 21(3):289. https://doi.org/10.3390/molecules21030289
Chicago/Turabian StyleTsyshevsky, Roman V., Anton Zverev, Anatoly Mitrofanov, Sergey N. Rashkeev, and Maija M. Kuklja. 2016. "Photochemistry of the α-Al2O3-PETN Interface" Molecules 21, no. 3: 289. https://doi.org/10.3390/molecules21030289