
Synthesis of Naphthalene-Based Push-Pull Molecules with a Heteroaromatic Electron Acceptor

Abstract
:1. Introduction
- (i)
- The donor group. The most efficient donor groups are those exhibiting positive resonance effect (+R). Of those, N,N-dialkylamino groups proved to be one of the most efficient and frequently utilized in D-π-A molecules.
- (ii)
- The π-system. As the π-link between the A and D groups in D-π-A molecules usually serves an aromatic/heteroaromatic ring alone or combined with a conjugated alkene or alkyne structural element. It has been shown, that a conjugation with an alkene structural element leads to higher A–D interactions than the acetylene one [1,3].
- (iii)
- The acceptor. Groups with negative resonance (–R) and/or inductive effects (–I), such as cyano, nitro and carbonyl are the most efficient acceptors. As the A group, five or six-membered electron-deficient heterocycles, such as thiazole, benzo[d]thiazole, imidazole, pyrazine, pyridine etc. bearing one or more electron-withdrawing substituents, can also be applied [1,4].
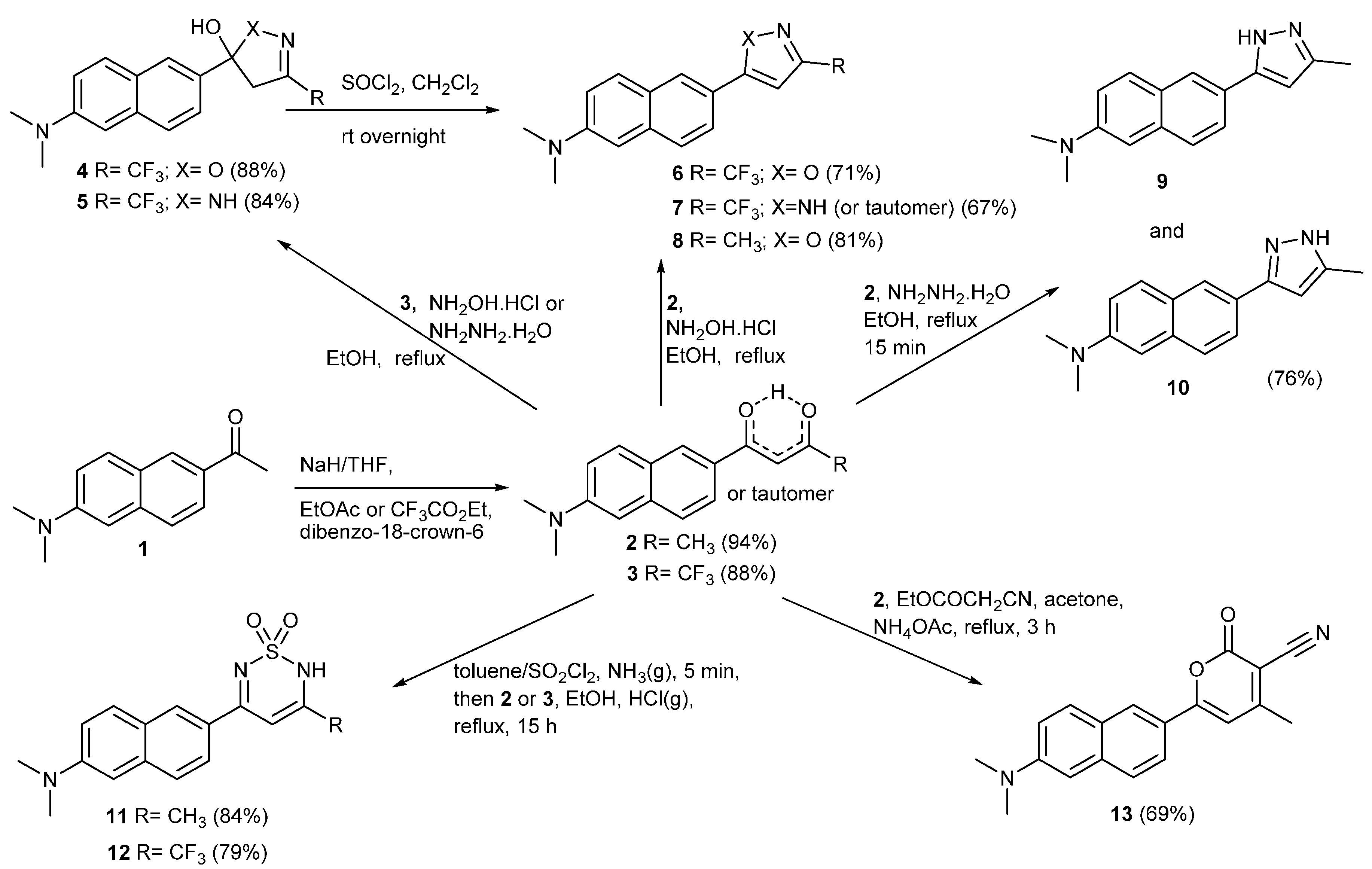
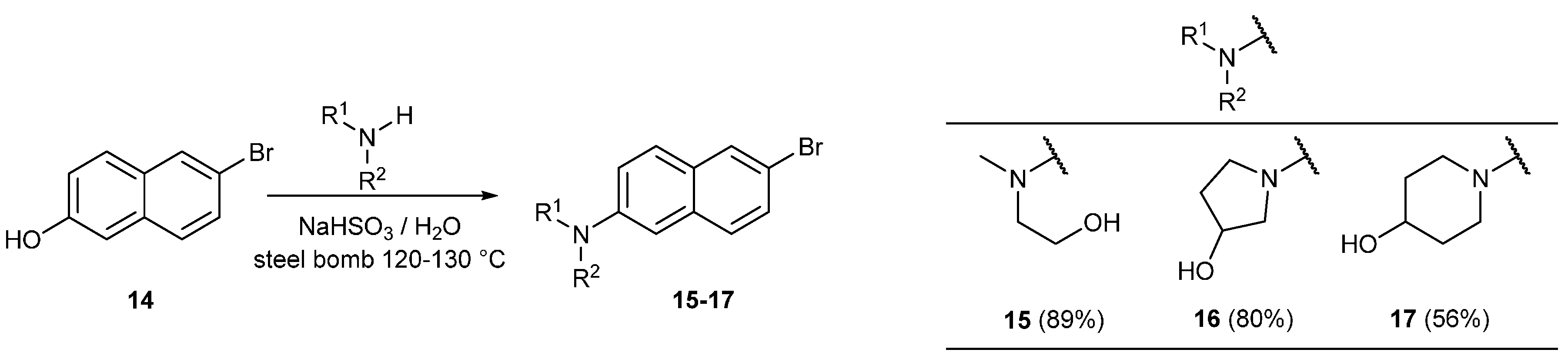
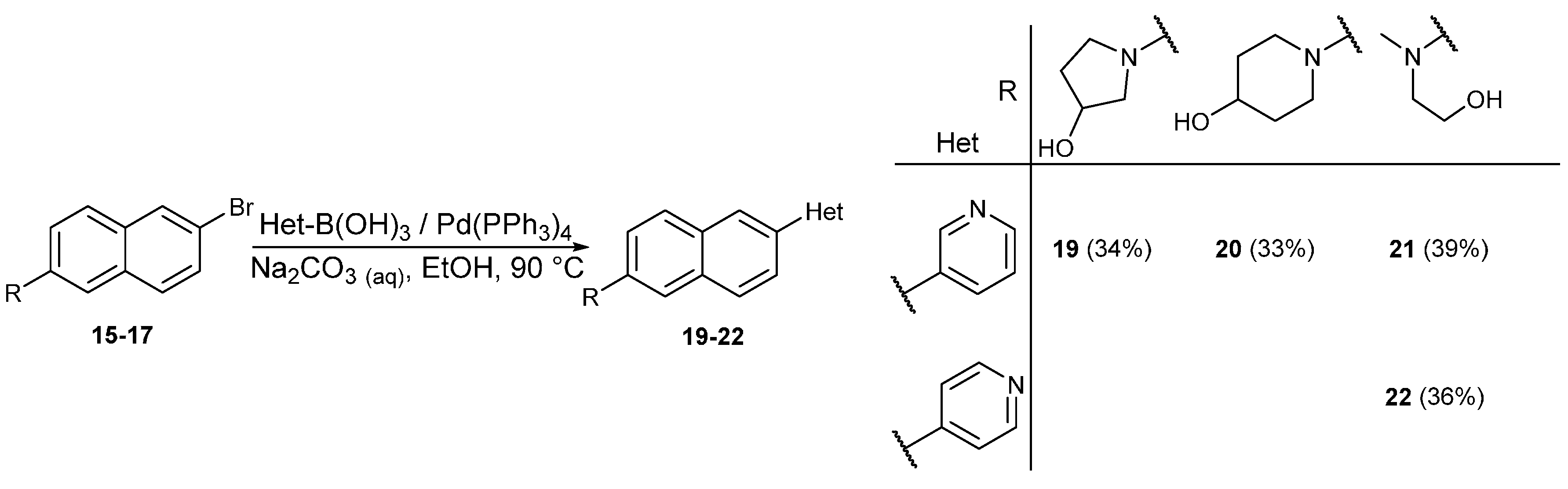
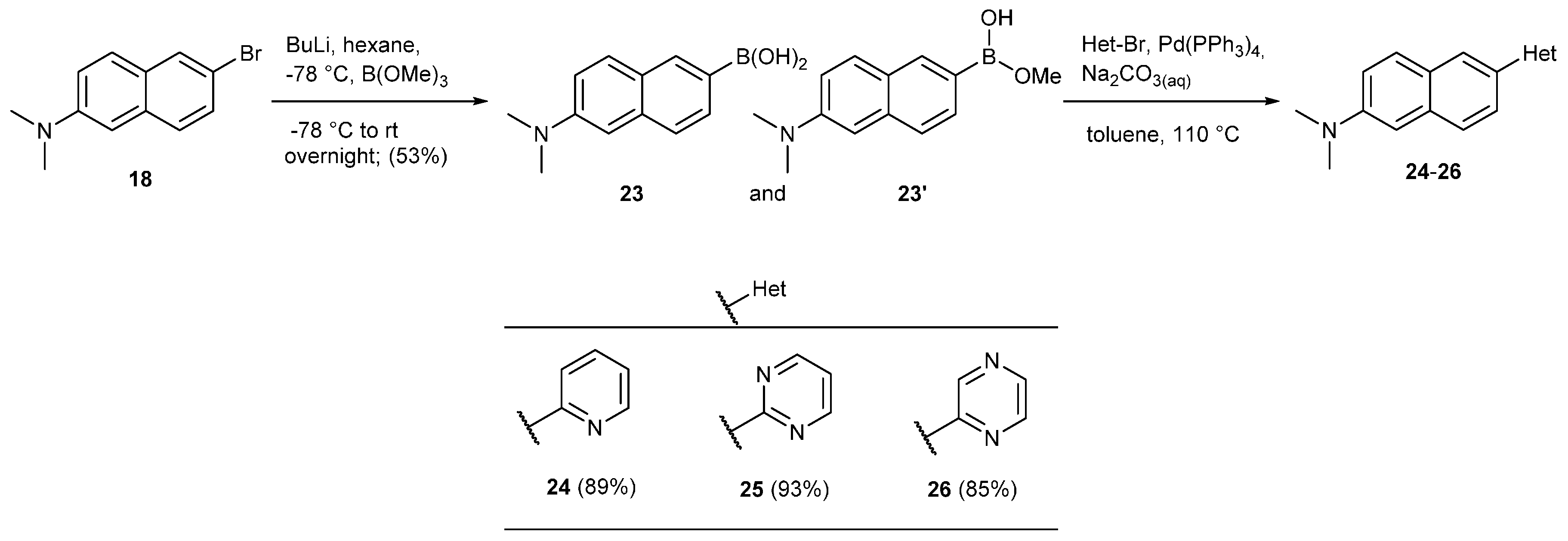
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 1-(6-(Dimethylamino)naphthalen-2-yl)butan-1,3-dione (2)
3.3. Synthesis of 1-(6-(Dimethylamino)naphthalen-2-yl)-4,4,4-trifluorobutan-1,3-dione (3)
3.4. Synthesis of 5-(6-(Dimethylamino)naphthalen-2-yl)-3-(trifluoromethyl)-4,5-dihydroisoxazol-5-ol (4)
3.5. Synthesis of N,N-Dimethyl-6-(3-(trifluoromethyl)isoxazol-5-yl)naphthalen-2-amine (6)
3.6. Synthesis of 5-(6-(Dimethylamino)naphthalen-2-yl)-3-(trifluoromethyl)-4,5-dihydro-1Hpyrazol-5-ol (5) and N,N-dimethyl-6-(3-(trifluoromethyl)-1H-pyrazol-5-yl)naphthalen-2-amine (7)
3.7. Synthesis of N,N-Dimethyl-6-(3-methylisoxazol-5-yl)naphthalen-2-amine (8)
3.8. Synthesis of N,N-Dimethyl-6-(3-methyl-1H-pyrazol-5-yl)naphthalen-2-amine (9) and N,N-dimethyl-6-(5-methyl-1H-pyrazol-3-yl)naphthalen-2-amine (10)
3.9. General Procedure for the Synthesis of Cyclic Sulfonamides
3.9.1. N,N-Dimethyl-6-(3-methyl-1,1-dioxido-2H-1,2,6-thiadiazin-5-yl)naphthalen-2-amine (11)
3.9.2. Synthesis of 6-[1,1-Dioxido-3-(trifluoromethyl)-2H-1,2,6-thiadiazin-5-yl]-N,N-dimethyl-naphthalen-2-amine (12)
3.10. Synthesis of 6-[6-(Dimethylamino)naphthalen-2-yl]-4-methyl-2-oxo-2H-pyran-3-carbonitrile (13)
3.11. General Procedure for the Bucherer Reaction
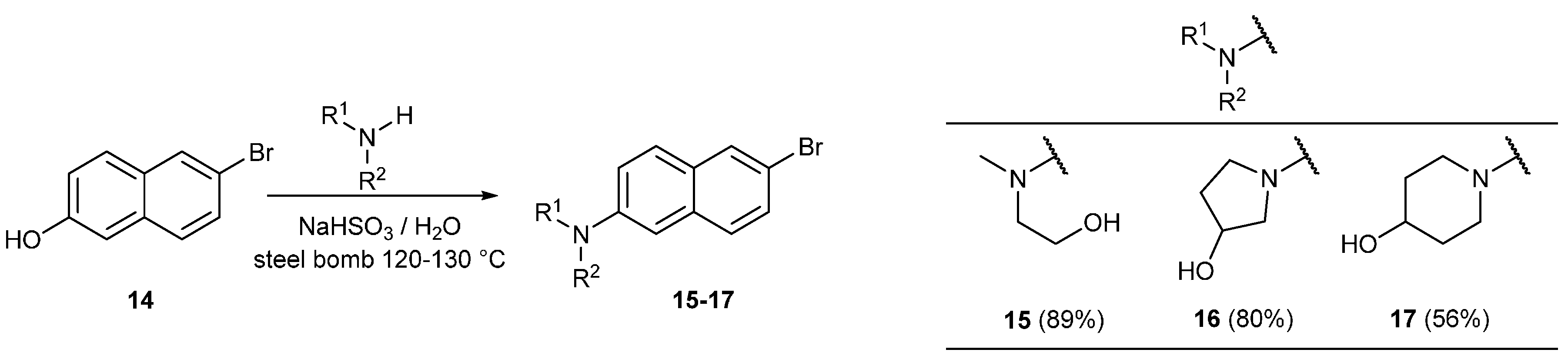
3.11.1. Synthesis of 2-((6-Bromonaphthalen-2-yl)(methyl)amino)ethan-1-ol (15)
3.11.2. Synthesis of 1-(6-Bromonaphthalen-2-yl)pyrrolidin-3-ol (16)
3.11.3. Synthesis of 1-(6-Bromonaphthalen-2-yl)piperidin-4-ol (17)
3.12. General Procedure for the Suzuki Coupling
3.12.1. Synthesis of 1-(6-(Pyridin-3-yl)naphthalen-2-yl)pyrrolidin-3-ol (19)
3.12.2. Synthesis of 1-(6-(Pyridin-3-yl)naphthalen-2-yl)piperidin-4-ol (20)
3.12.3. Synthesis of 2-(Methyl(6-(pyridin-3-yl)naphthalen-2-yl)amino)ethan-1-ol (21)
3.12.4. Synthesis of 2-(Methyl(6-(pyridin-4-yl)naphthalen-2-yl)amino)ethan-1-ol (22)
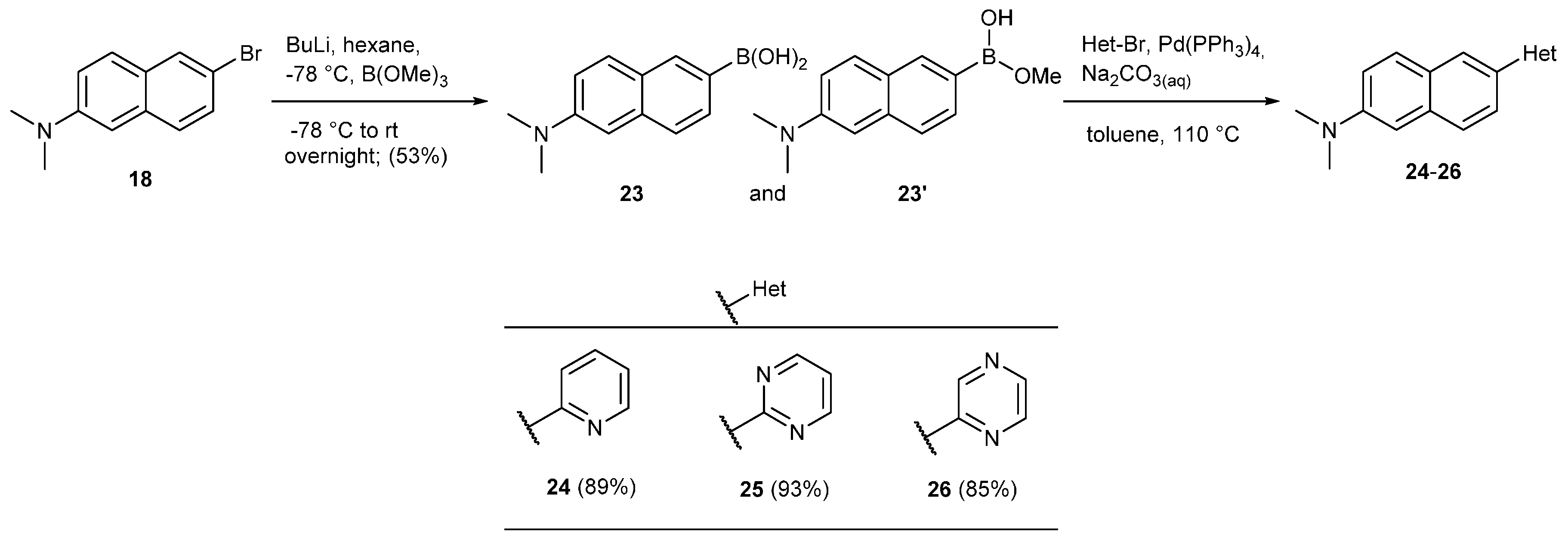
3.12.5. Synthesis of N,N-Dimethyl-6-(pyridin-2-yl)naphthalen-2-amine (24)
3.12.6. Synthesis of N,N-Dimethyl-6-(pyrimidin-2-yl)naphthalen-2-amine (25)
3.12.7. Synthesis of N,N-Dimethyl-6-(pyrazin-2-yl)naphthalen-2-amine (26)
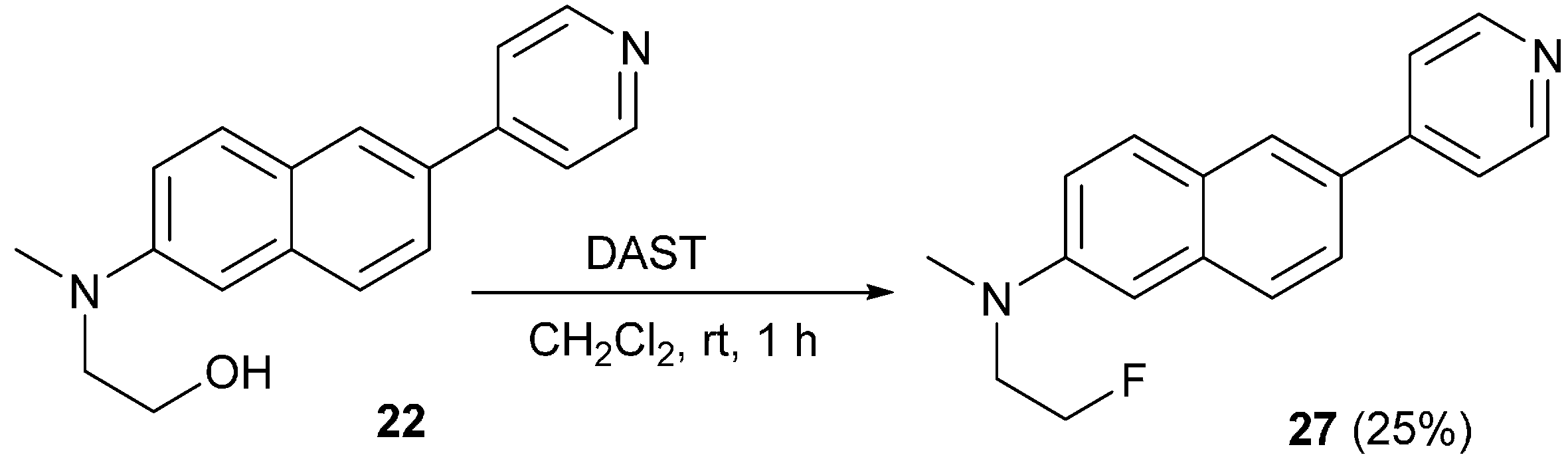
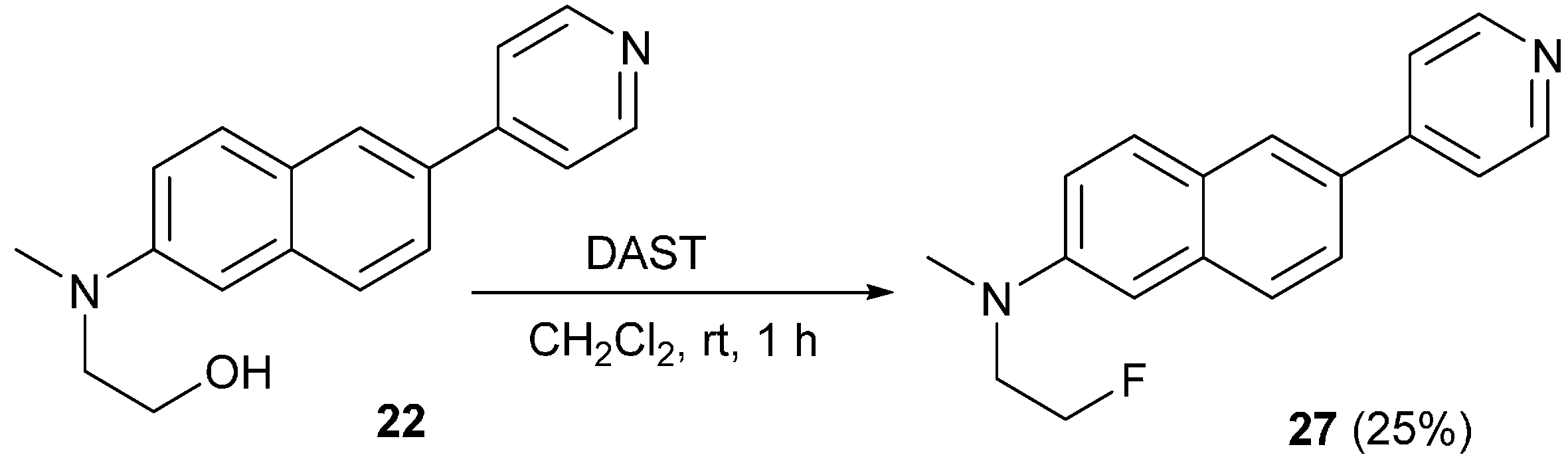
3.13. Synthesis of N-(2-Fluoroethyl)-N-methyl-6-(pyridin-4-yl)naphthalen-2-amine (27)
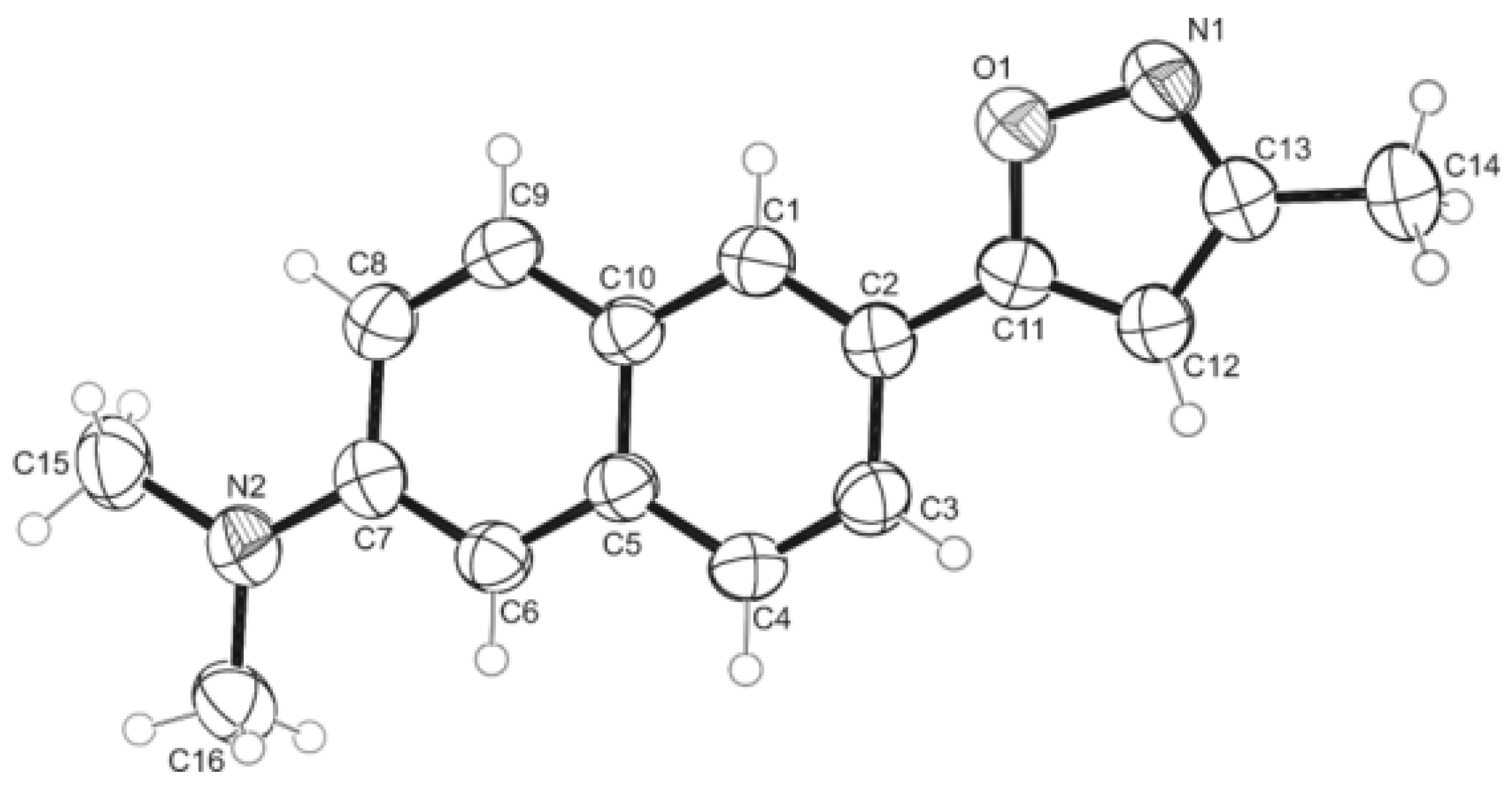
3.14. Single Crystal X-ray Structure Analysis for Compound 8
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bureš, F. Fundamental aspects of property tuning in push–pull molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
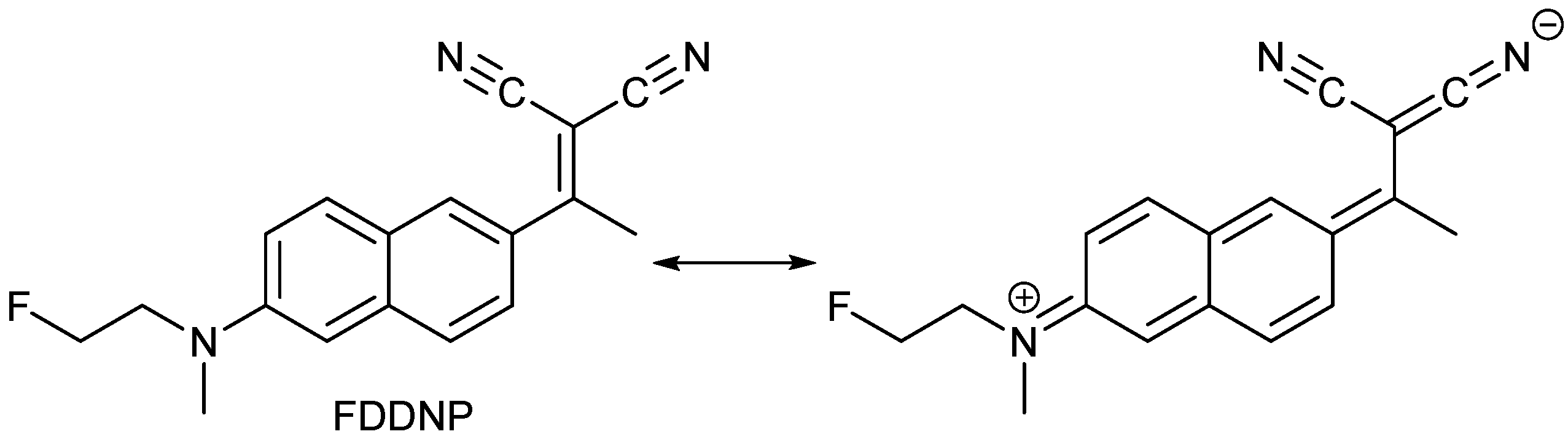
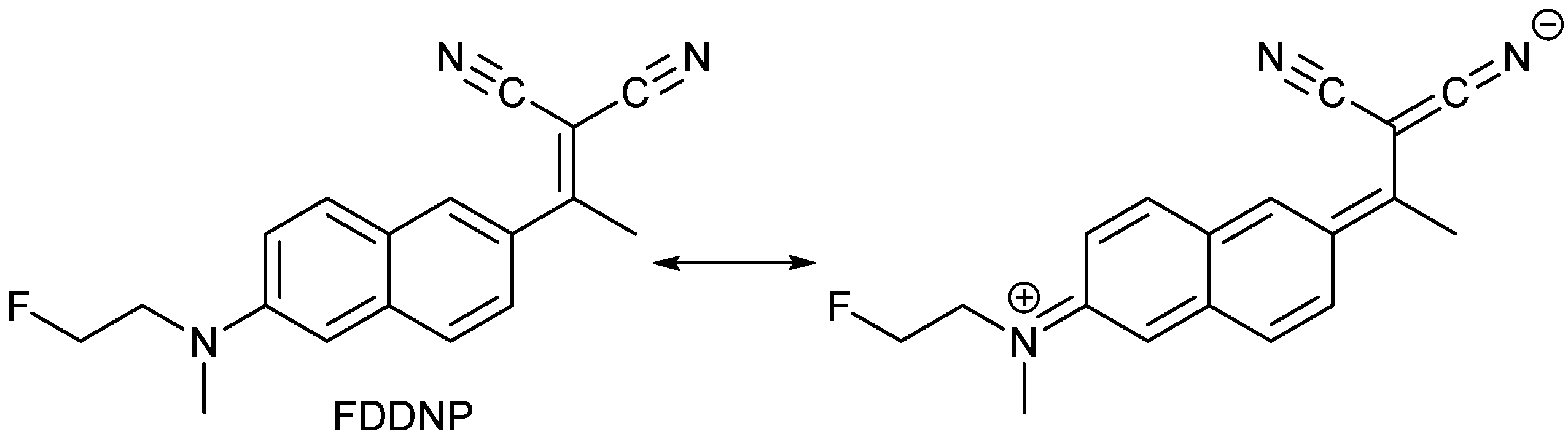
- Petrič, A.; Johnson, S.A.; Pham, H.V.; Li, Y.; Čeh, S.; Golobič, A.; Agdeppa, E.D.; Timbol, G.; Liu, J.; Keum, G.; et al. Dicyanovinylnaphthalenes for neuroimaging of amyloids and relationships of electronic structures and geometries to binding affinities. Proc. Natl. Acad. Sci. USA 2012, 109, 16492–16497. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.R.; Twieg, R.J.; Lee, V.Y.; Swanson, S.A.; Betterton, K.M.; Miller, R.D. Nonlinear Optical Chromophores with Large Hyperpolarizabilities and Enhanced Thermal Stabilities. J. Am. Chem. Soc. 1993, 115, 12599–12600. [Google Scholar] [CrossRef]
- Škofic, P.; Dambrot, C.; Koželj, M.; Golobič, A.; Barrio, J.R.; Petrič, A. Syntheses of 4-(2-naphthyl)pyridine derivatives from DDNP. Acta Chim. Slov. 2005, 52, 391–397. [Google Scholar]
- Jacobson, A.; Petrič, A.; Hogenkamp, D.; Sinur, A.; Barrio, J.R. 1,1-Dicyano-2-[6-(dimethylamino)-naphthalen-2-yl]propene (DDNP): A solvent polarity and viscosity sensitive fluorophore for fluorescence microscopy. J. Am. Chem. Soc. 1996, 118, 5572–5579. [Google Scholar] [CrossRef]
- Barrio, J.R.; Satyamurthy, N.; Huang, S.-C.; Petrič, A.; Small, G.W.; Kepe, V. Dissecting molecular mechanisms in the living brain of dementia patients. Acc. Chem. Res. 2009, 42, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Barrio, J.R.; Petrič, A.; Satyamurthy, N.; Small, G.W.; Cole, G.M.; Huang, S.-C. Compositions for Labeling β-Amyloid Plaques and Neurofibrillary Tangles. U.S. Patent US 7.341.709 B2, 11 March 2008. [Google Scholar]
- Agdeppa, E.D.; Kepe, V.; Liu, J.; Flores-Torres, S.; Satyamurthy, N.; Petrič, A.; Cole, G.M.; Small, G.W.; Huang, S.-C.; Barrio, J.R. Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer’s disease. J. Neurosci. 2001, 21, U19–U23. [Google Scholar]
- Shoghi-Jadid, K.; Small, G.W.; Agdeppa, E.D.; Kepe, V.; Ercoli, L.M.; Siddarth, P.; Read, S.; Satyamurthy, N.; Petrič, A.; Huang, S.-C.; et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am. J. Geriatr. Psychiatry 2002, 10, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Cui, M. Past and Recent Progress of Molecular Imaging Probes for β-Amyloid Plaques in the Brain. Curr. Med. Chem. 2014, 21, 82–112. [Google Scholar] [CrossRef] [PubMed]
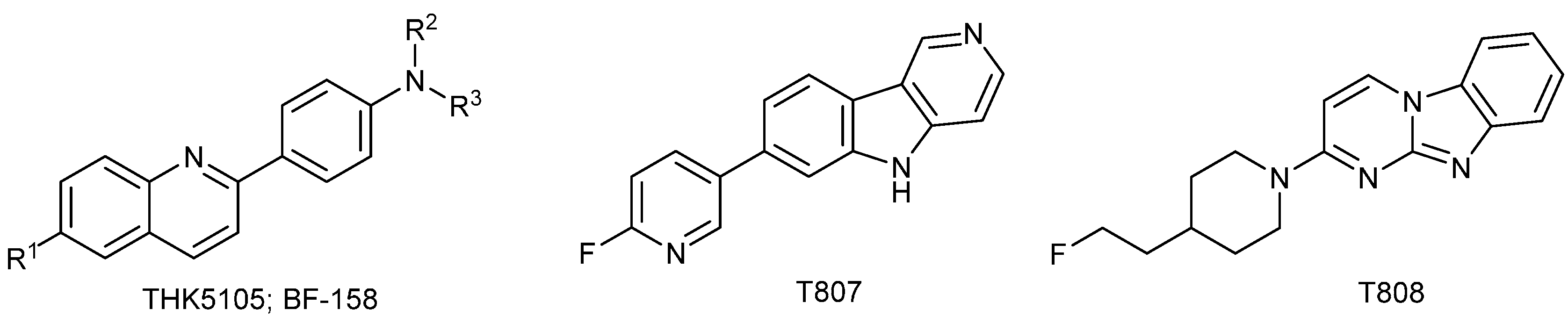
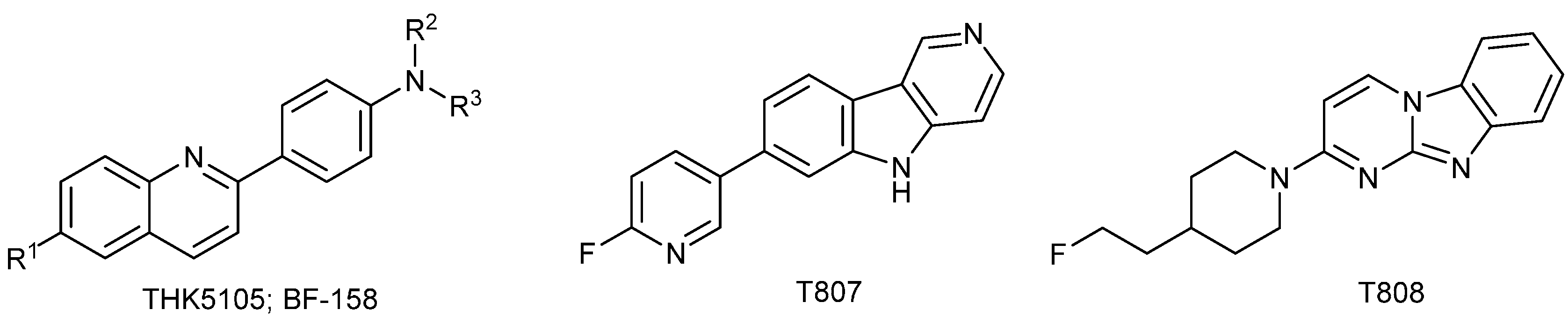
- Okamura, N.; Suemoto, T.; Furumoto, S.; Suzuki, M.; Shimadzu, H.; Akatsu, H.; Yamamoto, T.; Fujiwara, H.; Nemoto, M.; Maruyama, M.; et al. Quinoline and Benzimidazole Derivatives: Candidate Probes for in Vivo Imaging of Tau Pathology in Alzheimer’s Disease. J. Neurosci. 2005, 25, 10857–10862. [Google Scholar] [CrossRef] [PubMed]
- Fodero-Tavoletti, M.T.; Okamura, N.; Furumoto, S.; Mulligan, R.S.; Connor, A.R.; McLean, C.A.; Cao, D.; Rigopoulos, A.; Cartwright, G.A.; O’Keefe, G.; et al. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 2011, 134, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.-F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Arteaga, J.; Cashion, D.K.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; et al. A Highly Selective and Specific PET Tracer for Imaging of Tau Pathologies. J. Alzheimers Dis. 2012, 31, 601–612. [Google Scholar] [PubMed]
- Okamura, N.; Furumoto, S.; Harada, R.; Tago, T.; Yoshikawa, T.; Fodero-Tavoletti, M.; Mulligan, R.S.; Villemagne, V.L.; Akatsu, H.; Yamamoto, T.; et al. Novel 18F-Labeled Arylquinoline Derivatives for Noninvasive Imaging of Tau Pathology in Alzheimer’s Disease. J. Nucl. Med. 2013, 54, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Okamura, N.; Furumoto, S.; Fodero-Tavoletti, M.T.; Mulligan, R.S.; Harada, R.; Yates, P.; Pejoska, S.; Kudo, Y.; Masters, C.L.; Yanai, K.; et al. Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain 2014, 137, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Okamura, N.; Furumoto, S.; Furukawa, K.; Ishiki, A.; Tomita, N.; Hiraoka, K.; Watanuki, S.; Shidahara, M.; Miyake, M.; et al. [18F]THK-5117 for assessing neurofibrillary pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Lou, K.; Wang, W. Near-infrared fluorescent probes for imaging of amyloid plaques in Alzheimer’s disease. Acta Pharm. Sin. B 2015, 5, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Petrič, A.; Barrio, J.R. The syntheses of 1-(2-napthyl)piperazine derivatives. Novel spiperone-containing probes. Acta Chim. Slov. 1998, 45, 475–486. [Google Scholar]
- Petrič, A.; Špes, T.; Barrio, J.R. Novel fluorescent reactive dyes as intermediated for the preparation of UV and vis wavelength fluorescent probes. Monatshefte Chem. 1998, 129, 777–786. [Google Scholar]
- Petrič, A.; Jacobson, A.F.; Barrio, J.R. Functionalization of a viscosity-sensitive fluorophore for probing of biological systems. Bioorganic Med. Chem. Lett. 1998, 8, 1455–1460. [Google Scholar] [CrossRef]
- Wiley, R.H.; Hexner, P.E. 3,5-Dimethylpyrazole. Org. Synth. 1951, 31, 43–44. [Google Scholar]
- Elguero, J.; Goya, P.; Martinez, A. Ring Transformation of 1,2,6-Thiadiazine 1,1-Dioxides into Pyrazoles. A Convenient Synthesis of N-Alkylsulfamides. Heterocycles 1989, 29, 245–248. [Google Scholar]
- Nicolaou, K.C.; Bulger, P.G.; Šarlah, D. Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Seki, M. Recent Advances in Pd/C-Catalyzed Coupling Reactions. Synthesis 2006, 18, 2975–2992. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y.; Wang, B. A highly fluorescent water-soluble boronic acid reporter for saccharide sensing that shows ratiometric UV changes and significant fluorescent changes. Tetrahedron 2005, 61, 9111–9117. [Google Scholar] [CrossRef]
- Collect Software. Nonius, B.V., Delft, The Netherlands. 1998. [Google Scholar]
- Otwinowski, Z.; Minor, W. Methods in Enzymology, Volume 276: Macromolecular Crystallography, Part A; Carter, C.W., Jr., Sweet, R.M., Eds.; Academic Press: New York, NY, USA, 1997; pp. 307–326. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for Windows—A Version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Sample Availability: Not available.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λab (nm) | ε × 10−4 (L·mol−1·cm−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C6H14 | CH2Cl2 | DMF | MeOH | H2O | C6H14 | CH2Cl2 | DMF | MeOH | H2O | |
| DDNP a | 412 | 438 | - | 426 | - | 2.5 | 2.0 | - | 2.0 | - |
| FDDNP b | - | 431 | - | - | - | - | - | - | - | - |
| 2 | 377 | 394 | - | 392 | 371 | 2.9 | 2.8 | - | 3.1 | 1.3 |
| 3 | 384 | 401 | - | 384 | 388 | 2.0 | 2.0 | - | 1.9 | 1.2 |
| 4 | 334 | 345 | - | 339 | 336 | 2.2 | 2.3 | - | 2.2 | 1.1 |
| 5 | 333 | 339 | - | 334 | 328 | 2.8 | 2.7 | - | 2.7 | 1.5 |
| 6 | 329 | 345 | - | 339 | 340 | 5.9 | 2.4 | - | 1.7 | 0.4 |
| 7 | 320 | 327 | - | 324 | 324 | 1.9 | 2.0 | - | 1.9 | 0.7 |
| 8 | 336 | 342 | - | 339 | 330 | 2.5 | 2.4 | - | 2.1 | 0.6 |
| 9/10 | 315 | 320 | - | 311 | 308 | 2.3 | 2.3 | - | 2.4 | 1.5 |
| 11 | 399 | 425 | - | 375 | 364 | 2.0 | 2.2 | - | 2.2 | 1.6 |
| 12 | 412 | 454 | - | 389 | 383 | 2.1 | 2.5 | - | 2.5 | 1.9 |
| 13 | 441 | 473 | - | 462 | - | 2.8 | 4.7 | - | 4.3 | - |
| 19 | - | - | 335 | - | - | - | - | 4.5 | - | - |
| 20 | - | - | 319 | - | - | - | - | 6.7 | - | - |
| 24 | 334 | 339 | - | 337 | 325 | 2.4 | 2.3 | - | 2.1 | 1.2 |
| 25 | 350 | 354 | - | 352 | 339 | 2.6 | 2.2 | - | 2.1 | 1.3 |
| 26 | 354 | 363 | - | 363 | 343 | 2.5 | 2.6 | - | 2.0 | 1.6 |
| 27 | 328 c | 339 | 342 | 346 | - | 2.0 c | 1.8 | 1.9 | 1.8 | - |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šarlah, D.; Juranovič, A.; Kožar, B.; Rejc, L.; Golobič, A.; Petrič, A. Synthesis of Naphthalene-Based Push-Pull Molecules with a Heteroaromatic Electron Acceptor. Molecules 2016, 21, 267. https://doi.org/10.3390/molecules21030267
Šarlah D, Juranovič A, Kožar B, Rejc L, Golobič A, Petrič A. Synthesis of Naphthalene-Based Push-Pull Molecules with a Heteroaromatic Electron Acceptor. Molecules. 2016; 21(3):267. https://doi.org/10.3390/molecules21030267
Chicago/Turabian StyleŠarlah, David, Amadej Juranovič, Boris Kožar, Luka Rejc, Amalija Golobič, and Andrej Petrič. 2016. "Synthesis of Naphthalene-Based Push-Pull Molecules with a Heteroaromatic Electron Acceptor" Molecules 21, no. 3: 267. https://doi.org/10.3390/molecules21030267