The Diels-Alder Cycloaddition Reaction of Substituted Hemifullerenes with 1,3-Butadiene: Effect of Electron-Donating and Electron-Withdrawing Substituents

Abstract
: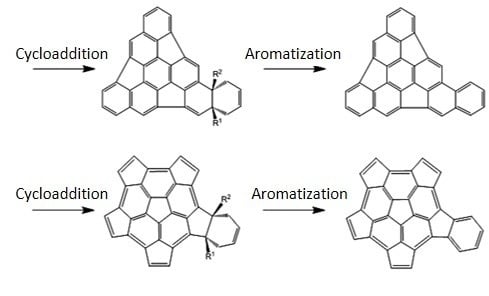
1. Introduction
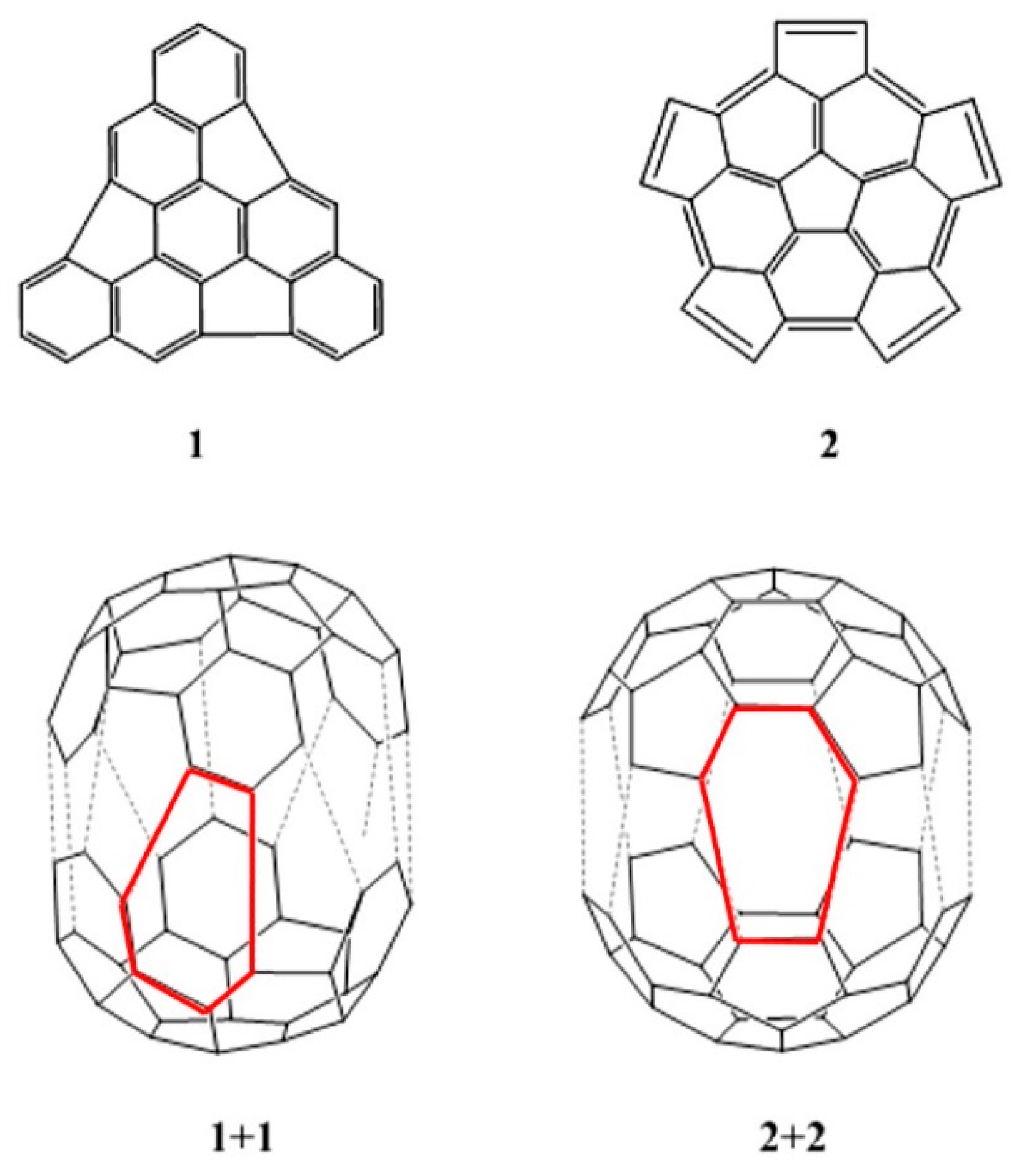
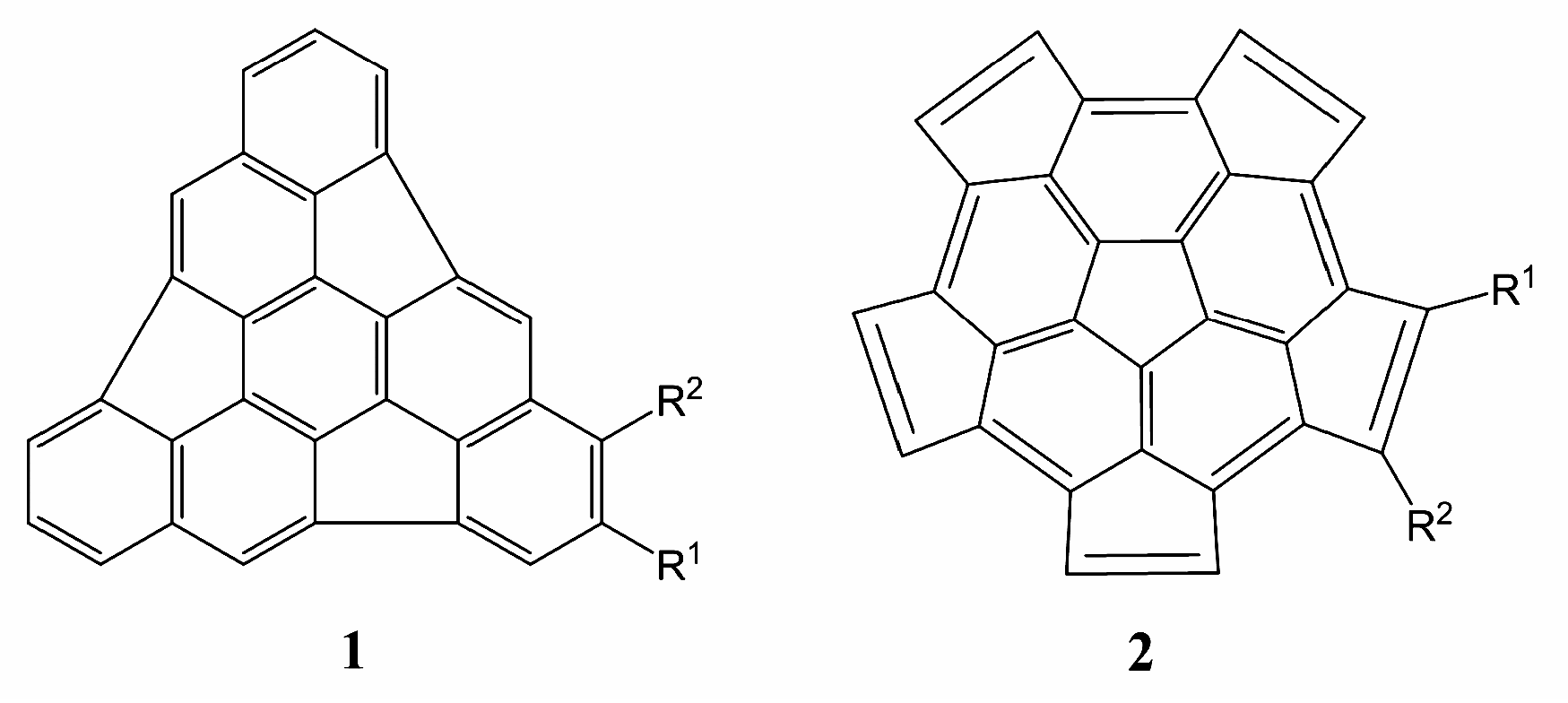
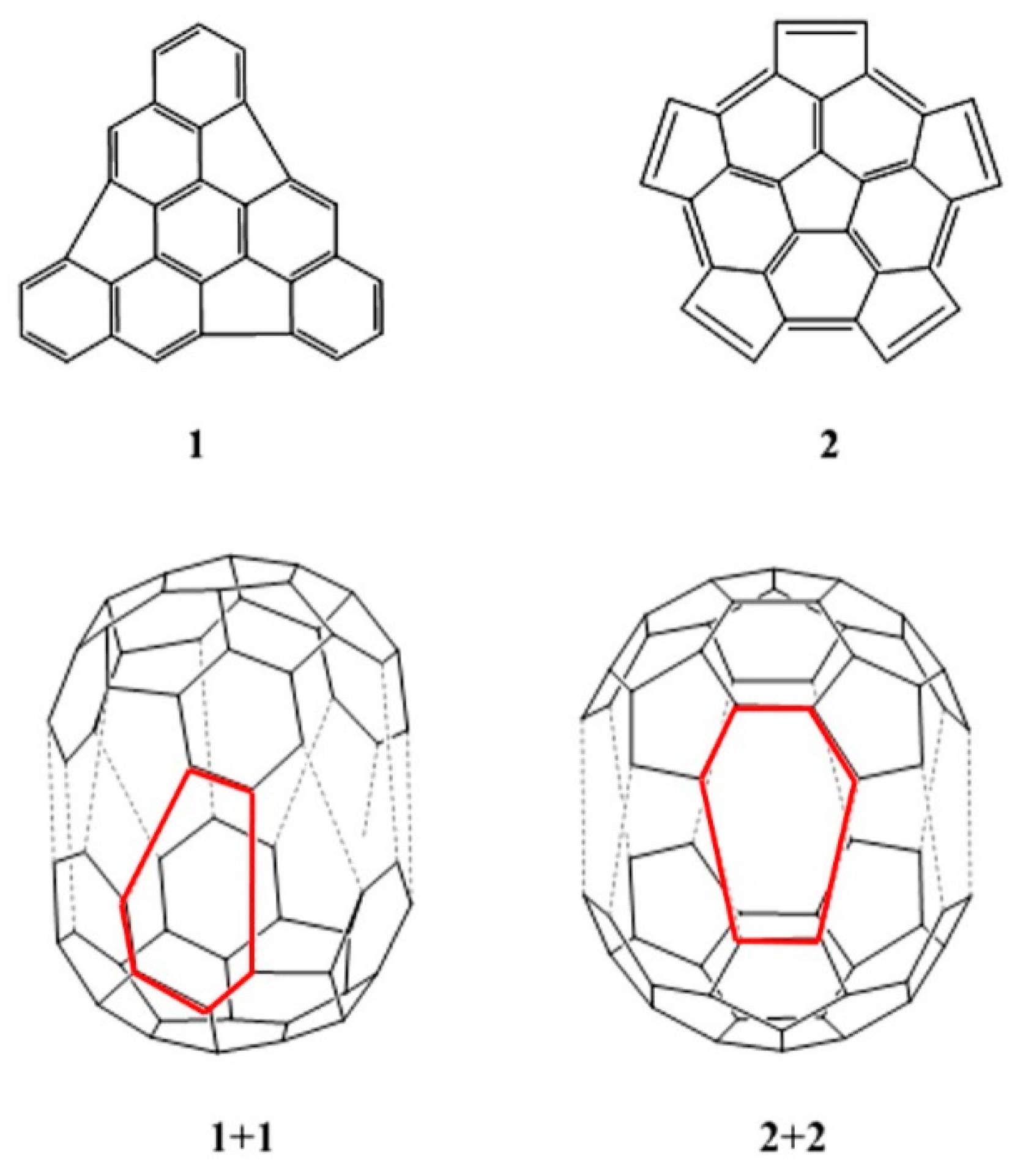
2. Results and Discussion
2.1. Frontier Molecular Orbitals


{kind=link}
{kind=link}
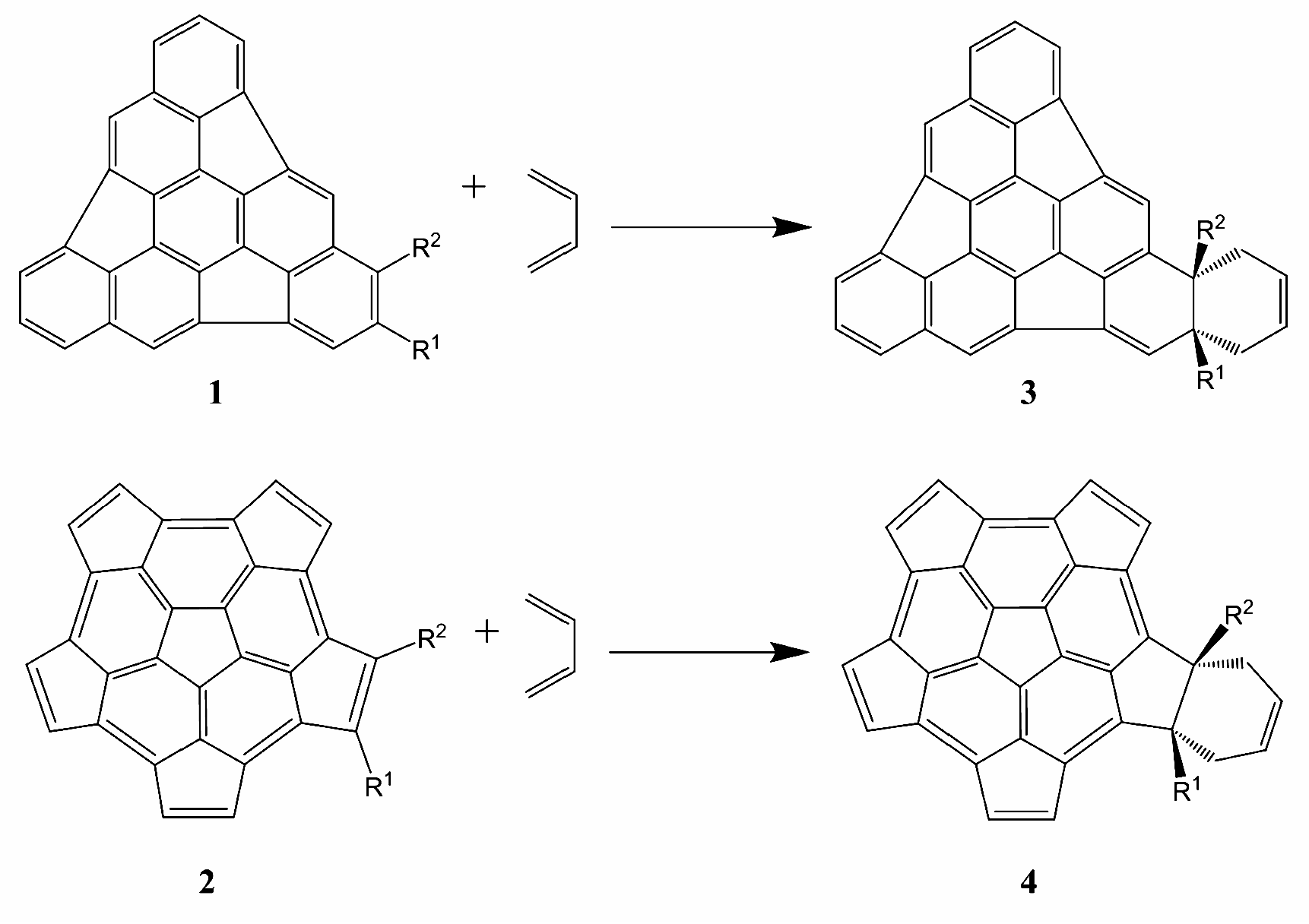
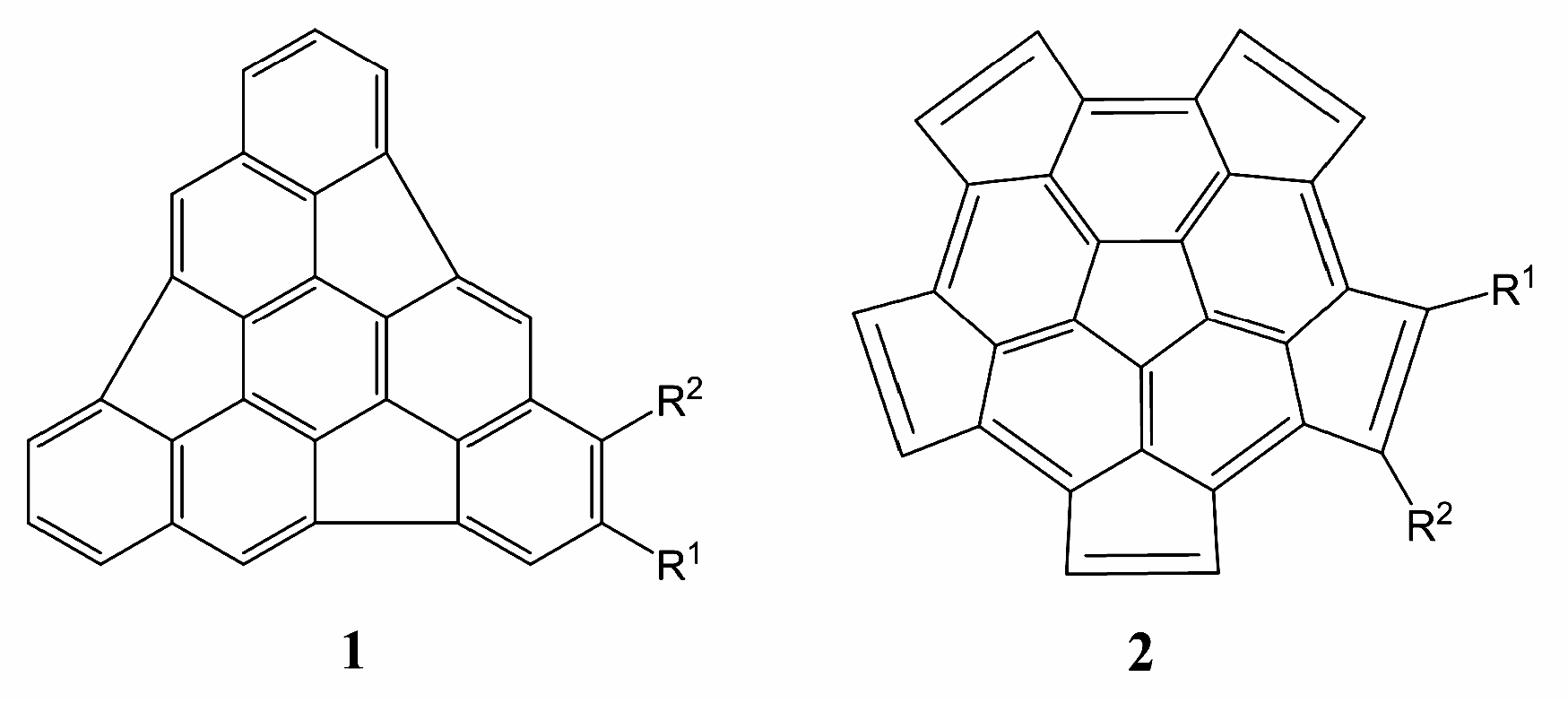{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituent R1 | E(HOMO) | E(LUMO) | ΔE(NEDDA) | ΔE(IEDDA) | δΔE | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | |
| NH2 | −6.51 | −6.78 | 0.84 | 0.72 | 9.62 | 9.49 | 9.96 | 10.23 | 0.35 | 0.73 |
| OMe | −7.03 | −7.06 | 0.66 | 0.62 | 9.43 | 9.39 | 10.48 | 10.51 | 1.05 | 1.12 |
| OH | −6.97 | −7.05 | 0.65 | 0.53 | 9.43 | 9.31 | 10.42 | 10.50 | 0.99 | 1.20 |
| Me | −7.08 | −7.10 | 0.72 | 0.68 | 9.49 | 9.45 | 10.53 | 10.55 | 1.04 | 1.10 |
| i-Pr | −7.06 | −7.09 | 0.70 | 0.68 | 9.48 | 9.46 | 10.51 | 10.53 | 1.03 | 1.08 |
| H | −7.18 | −7.18 | 0.67 | 0.67 | 9.45 | 9.45 | 10.63 | 10.63 | 1.18 | 1.18 |
| F | −7.40 | −7.26 | 0.37 | 0.48 | 9.15 | 9.25 | 10.85 | 10.71 | 1.70 | 1.46 |
| COOH | −7.40 | −7.31 | −0.01 | 0.26 | 8.77 | 9.03 | 10.85 | 10.75 | 2.08 | 1.72 |
| CF3 | −7.62 | −7.42 | −0.05 | 0.33 | 8.72 | 9.11 | 11.07 | 10.87 | 2.35 | 1.76 |
| CHO | −7.48 | −7.35 | −0.37 | 0.19 | 8.40 | 8.96 | 10.93 | 10.80 | 2.53 | 1.83 |
| CN | −7.79 | −7.52 | −0.52 | 0.09 | 8.24 | 8.86 | 11.24 | 10.96 | 3.00 | 2.10 |
| NO2 | −7.91 | −7.60 | −0.73 | −0.18 | 8.04 | 8.59 | 11.36 | 11.04 | 3.31 | 2.45 |
2.2. Thermodynamic Parameters
| Substituent R1 | ΔrG° (kcal·mol−1) | ΔrH° (kcal·mol−1) | TΔrS° (kcal·mol−1) | |||
|---|---|---|---|---|---|---|
| R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | |
| NH2 | 2.43 | −3.93 | −13.87 | −19.10 | −16.30 | −15.17 |
| OMe | 1.44 | −4.64 | −15.29 | −19.79 | −16.72 | −15.15 |
| OH | −9.10 | −9.33 | −24.94 | −24.51 | −15.84 | −15.18 |
| Me | 2.43 | −6.54 | −13.38 | −21.75 | −15.80 | −15.21 |
| i-Pr | 15.88 | 2.04 | −1.07 | −14.18 | −16.96 | −16.22 |
| H | −13.05 | −13.05 | −27.88 | −27.88 | −14.82 | −14.82 |
| F | −18.56 | −15.38 | −33.99 | −30.30 | −15.43 | −14.93 |
| COOH | 3.66 | −5.01 | −12.41 | −20.06 | −16.07 | −15.05 |
| CF3 | −1.23 | −8.47 | −17.54 | −24.26 | −16.30 | −15.78 |
| CHO | 4.30 | −4.82 | −10.47 | −19.43 | −14.77 | −14.61 |
| CN | 3.45 | −5.43 | −11.90 | −20.46 | −15.36 | −15.02 |
| NO2 | −7.70 | −10.50 | −23.81 | −25.46 | −16.10 | −14.96 |

| Substituent R1 | ΔrG° (kcal·mol−1) | ΔrH° (kcal·mol−1) | TΔrS° (kcal·mol−1) | |||
|---|---|---|---|---|---|---|
| R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | |
| NH2 | −48.33 | −28.37 | −24.23 | −7.66 | 24.11 | 20.71 |
| OMe | −54.12 | −31.89 | −28.05 | −10.85 | 26.07 | 21.04 |
| OH | −38.72 | −25.92 | −14.94 | −5.58 | 23.78 | 20.35 |
| Me | −53.70 | −30.18 | −29.28 | −9.46 | 24.42 | 20.72 |
| i-Pr | −74.94 | 39.90 | −46.76 | −17.25 | 28.18 | 22.65 |
| H | −10.48 | −10.48 | 6.51 | 6.51 | 16.99 | 16.99 |
| F | −28.48 | −17.54 | −7.38 | 1.42 | 21.10 | 18.96 |
| COOH | −30.95 | −16.12 | −4.73 | 5.32 | 26.22 | 21.44 |
| CF3 | −39.97 | −20.27 | −13.68 | 1.06 | 26.29 | 21.33 |
| CHO | −28.53 | −16.68 | −3.95 | 4.04 | 24.57 | 20.72 |
| CN | −28.48 | −17.17 | −6.31 | 2.40 | 22.17 | 19.57 |
| NO2 | −40.66 | −19.16 | −15.27 | 1.79 | 25.40 | 20.96 |
2.3. Kinetic Parameters
| Substituent R1 | r1 | r2 | Δr | |||
|---|---|---|---|---|---|---|
| R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | |
| NH2 | 3.48 | 3.28 | 1.88 | 1.91 | 1.60 | 1.37 |
| OMe | 3.27 | 2.98 | 1.88 | 1.93 | 1.39 | 1.05 |
| OH | 2.94 | 3.34 | 1.91 | 1.91 | 1.03 | 1.43 |
| Me | 2.26 | 2.38 | 2.26 | 2.17 | 0 | 0.21 |
| i-Pr | 2.93 | 2.44 | 1.79 | 2.11 | 1.14 | 0.33 |
| H | 2.27 | 2.27 | 2.27 | 2.27 | 0 | 0 |
| F | 2.30 | 2.33 | 2.27 | 2.22 | 0.03 | 0.11 |
| COOH | 3.06 | 2.68 | 1.97 | 1.99 | 1.09 | 0.69 |
| CF3 | 2.27 | 2.34 | 2.27 | 2.21 | 0.01 | 0.13 |
| CHO | 2.69 | 2.69 | 1.97 | 1.98 | 0.72 | 0.71 |
| CN | 2.24 | 2.64 | 2.24 | 2.02 | 0 | 0.62 |
| NO2 | 2.19 | 3.04 | 1.78 | 1.96 | 0.41 | 1.08 |
| Substituent R1 | Ea (kcal·mol−1) | Δ‡G° (kcal·mol−1) | Δ‡H° (kcal·mol−1) | TΔ‡S° (kcal·mol−1) | ||||
|---|---|---|---|---|---|---|---|---|
| R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | R2 = R1 | R2 = H | |
| NH2 | 28.06 | 24.03 | 41.38 | 36.61 | 27.28 | 23.54 | −14.11 | −13.07 |
| OMe | 29.21 | 26.82 | 42.63 | 40.32 | 28.40 | 25.87 | −14.22 | −14.45 |
| OH | 27.99 | 24.03 | 40.70 | 36.46 | 27.37 | 23.47 | −13.34 | −12.99 |
| Me | 30.73 | 26.60 | 44.42 | 39.92 | 29.86 | 25.91 | −14.55 | −14.01 |
| i-Pr | 46.86 | 31.34 | 61.52 | 45.17 | 45.67 | 30.52 | −15.85 | −14.66 |
| H | 22.61 | 22.61 | 35.52 | 35.52 | 22.02 | 22.02 | −13.51 | −13.51 |
| F | 25.60 | 24.67 | 38.39 | 37.65 | 25.12 | 24.14 | −13.27 | −13.51 |
| COOH | 23.60 | 22.88 | 37.52 | 36.34 | 22.98 | 22.25 | −14.54 | −14.09 |
| CF3 | 25.23 | 23.22 | 39.30 | 37.32 | 24.42 | 22.50 | −14.88 | −14.82 |
| CHO | 27.54 | 22.98 | 40.88 | 36.14 | 26.88 | 22.37 | −14.01 | −13.78 |
| CN | 27.18 | 23.55 | 40.58 | 36.58 | 26.56 | 23.03 | −14.03 | −13.56 |
| NO2 | 19.80 | 19.03 | 35.78 | 32.18 | 18.22 | 18.51 | −17.56 | −13.67 |
3. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fringuelli, F.; Taticchi, A.A. The Diels-Alder Reaction: Selected Practical Methods; John Wiley& Sons Ltd.: West Sussex, UK, 2002; pp. 1–25. [Google Scholar]
- Bear, B.R.; Sparks, S.M.; Shea, K.J. The type 2 intramolecular Diels-Alder reaction: Synthesis and chemistry of bridgehead alkenes. Angew. Chem. Int. Ed. 2001, 40, 820–849. [Google Scholar] [CrossRef]
- Jørgensen, K.A. Catalytic asymmetric Hetero-Diels-Alder reactions of carbonyl compounds and imines. Angew. Chem. Int. Ed. 2000, 39, 3558–3588. [Google Scholar] [CrossRef]
- Clar, E.; Zander, M. Syntheses of coronene and 1:2–7:8-dibenzocoronene. J. Chem. Soc. 1957, 4616–4619. [Google Scholar] [CrossRef]
- Rao, K.V.; George, S.J. Synthesis and controllable self-assembly of a novel coronene bisimide amphiphile. Org. Lett. 2010, 12, 2656–2659. [Google Scholar]
- Alibert-Fouet, S.; Seguy, I.; Bobo, J.-F.; Destruel, P.; Bock, H. Liquid-crystalline and electron-deficient coronene oligocarboxylic esters and imides by twofold benzogenic Diels–Alder reactions on perylenes. Chem. Eur. J. 2007, 13, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- Konishi, A.; Hirao, Y.; Matsumoto, K.; Kurata, H.; Kubo, T. Facile synthesis and lateral π-expansion of bisanthenes. Chem. Lett. 2013, 42, 592–594. [Google Scholar] [CrossRef]
- Li, J.; Jiao, C.; Huang, K.-W.; Wu, J. Lateral extension of π-conjugation along the bay regions of bisanthene via Diels-Alder cycloaddition reaction. Chem. Eur. J. 2011, 17, 14672–14680. [Google Scholar] [CrossRef] [PubMed]
- Fort, E.H.; Donovan, P.M.; Scott, L.T. Diels-Alder reactivity of polycyclic aromatic hydrocarbon bay regions: Implications for metal-free growth of single-chirality carbon nanotubes. J. Am. Chem. Soc. 2009, 131, 16006–16007. [Google Scholar] [CrossRef] [PubMed]
- Fort, E.H.; Scott, L.T. One-step conversion of aromatic hydrocarbon bay regions into new unsubstituted benzene rings. a reagent for the low-temperature, metal-free growth of single-chirality carbon nanotubes. Angew. Chem. Int. Ed. 2010, 49, 6626–6628. [Google Scholar] [CrossRef] [PubMed]
- Rabideau, P.; Sygula, A. Chapter 1: Polynuclear Aromatic Hydrocarbons with Curved Surfaces in Advances in Theoretically Interesting Molecules; Thummel, R.P., Ed.; JAI PRESS INC.: Greenwich, CT, USA, 1995; Volume 3, pp. 1–36. [Google Scholar]
- Scott, L.T. Methods for the chemical synthesis of fullerenes. Angew. Chem. Int. Ed. 2004, 43, 4994–5007. [Google Scholar] [CrossRef] [PubMed]
- Woodward, R.B.; Fukunaga, T.; Kelly, R.C. Triquinacene. J. Am. Chem. Soc. 1964, 86, 3162–3164. [Google Scholar] [CrossRef]
- Sastry, G.N.; Jemmis, E.D.; Mehta, G.; Shah, S.R. Synthetic strategies towards C60. Molecular mechanics and MNDO study on sumanene and related structures. J. Chem. Soc. Perkin Trans. 1993, 2, 1867–1871. [Google Scholar] [CrossRef]
- Abdourazak, A.H.; Marcinow, Z.; Sygula, A.; Sygula, R.; Rabideau, P.W. Buckybowls 2. Toward the total synthesis of Bukminsterfullerene (C60): Benz[5,6]-as-indaceno-[3,2,1,8,7-mnopqr]indeno[4,3,2,1-cdef]chrysene. J. Am. Chem. Soc. 1995, 117, 6410–6411. [Google Scholar] [CrossRef]
- Geneste, F.; Moradpour, A.; Dive, G.; Peeters, D.; Malthete, J.; Sadoc, J.-F. Retrosynthetic analysis of fullerene C60: Structure, stereochemistry, and calculated stability of C30 fragments. J. Org.Chem. 2002, 67, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Mojica, M.; Méndez, F.; Alonso, J.A. Growth of fullerene fragments using the diels-alder cycloaddition reaction: First step towards a C60 synthesis by dimerization. Molecules 2013, 18, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Plater, J.; Rzepa, H.S.; Stoppa, F.; Stossel, S. Selective π-facial binding of metal cations to triindenotriphenylene as a possible catalytic route to C60 precursors: A MNDO, PM3 and ab initio SCF-MO study. J. Chem. Soc. Perkin Trans. 1994, 2, 399–400. [Google Scholar] [CrossRef]
- Karousis, N.; Tagmatarchis, N.; Tasis, D. Current progress on the chemical modification of carbon nanotubes. Chem. Rev. 2010, 110, 5366–5397. [Google Scholar] [CrossRef] [PubMed]
- Tesferikas, V.M.; Scott, L.T. Geodesic polyarenes by flash vacuum pyrolysis. Chem. Rev. 2006, 106, 4868–4884. [Google Scholar]
- Sygula, A. Chemistry on a Half-Shell: Synthesis and derivatization of buckybowls. Eur. J. Org. Chem. 2011, 1611–1625. [Google Scholar] [CrossRef]
- Flemig, I. Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons Ltd.: London, UK, 2009; pp. 224–242. [Google Scholar]
- Scott, L.T. Polycyclic Aromatic hydrocarbon bowls, baskets, balls, and tubes: Challenging targets for chemical synthesis. Polycycl. Aromat. Compd. 2010, 30, 247–259. [Google Scholar] [CrossRef]
- Gaussian 09, Revision B.01, software for electronic structure calculation. Gaussian, Inc.: Wallingford, CT, USA, 2010.
- Suarez-Moreno, G.V.; González-Zamora, E.; Méndez, F. Oxazole as an Electron-Deficient Diene in the Diels-Alder Reaction. Org. Lett. 2011, 13, 6358–6361. [Google Scholar] [CrossRef] [PubMed]
- Méndez, F.; García-Garibay, M.A. A hard-soft acid-base and DFT analysis of singlet-triplet gaps and the addition of singlet carbenes to alkenes. J. Org. Chem. 1999, 64, 7061–7066. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Lee, K.H.; Lee, C.; Kang, J.; Park, S.S.; Lee, J.; Lee, S.K.; Bohme, D.K. Preferential site of attack on fullerene cations: Frontier orbitals and rate coefficients. J. Phys. Chem. A 2006, 110, 11730–11733. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.I.; Moncada, J.L.; Larenas, J.M. The dual descriptor to measure local reactivity on buckminster fullerenes: An analysis within the framework of conceptual DFT. J. Mol. Model. 2010, 16, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Petrukhina, M.A.; Andreini, K.W.; Mack, J.; Scott, L.T. X-ray quality geometries of geodesic polyarenes from theoretical calculations: What levels of theory are reliable? J. Org. Chem. 2005, 70, 5713–5716. [Google Scholar] [CrossRef] [PubMed]
- Osuna, S.; Morera, J.; Cases, M.; Morokuma, K.; Solà, M. Diels−Alder reaction between cyclopentadiene and C60: An analysis of the performance of the ONIOM method for the study of chemical reactivity in fullerenes and nanotubes. J. Phys. Chem. A 2009, 113, 9721–9726. [Google Scholar] [CrossRef] [PubMed]
- Ikuma, N.; Susami, Y.; Oshima, T. Kinetics and regioselectivity in the Diels–Alder reaction of fulleroids vs. methanofullerene and C60. Org. Biomol. Chem. 2010, 8, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Website of the Dacapo Code. Available online: https://wiki.fysik.dtu.dk/dacapo (accessed on 5 December 2015).
- Krätschmer, W.; Lamb, L.D.; Fostiropoulos, K.; Huffman, D.R. Solid C60: A new form of carbon. Nature 1990, 347, 354–358. [Google Scholar] [CrossRef]
- Mojica, M.; Alonso, J.A.; Méndez, F. Synthesis of fullerenes. J. Phys. Org. Chem. 2013, 26, 526–539. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mojica, M.; Méndez, F.; Alonso, J.A. The Diels-Alder Cycloaddition Reaction of Substituted Hemifullerenes with 1,3-Butadiene: Effect of Electron-Donating and Electron-Withdrawing Substituents. Molecules 2016, 21, 200. https://doi.org/10.3390/molecules21020200
Mojica M, Méndez F, Alonso JA. The Diels-Alder Cycloaddition Reaction of Substituted Hemifullerenes with 1,3-Butadiene: Effect of Electron-Donating and Electron-Withdrawing Substituents. Molecules. 2016; 21(2):200. https://doi.org/10.3390/molecules21020200
Chicago/Turabian StyleMojica, Martha, Francisco Méndez, and Julio A. Alonso. 2016. "The Diels-Alder Cycloaddition Reaction of Substituted Hemifullerenes with 1,3-Butadiene: Effect of Electron-Donating and Electron-Withdrawing Substituents" Molecules 21, no. 2: 200. https://doi.org/10.3390/molecules21020200