
Theoretical Study of Molecular Structure and Physicochemical Properties of Novel Factor Xa Inhibitors and Dual Factor Xa and Factor IIa Inhibitors


Abstract
:1. Introduction
2. Results and Discussion
2.1. DFT Calculations of Molecular Structures
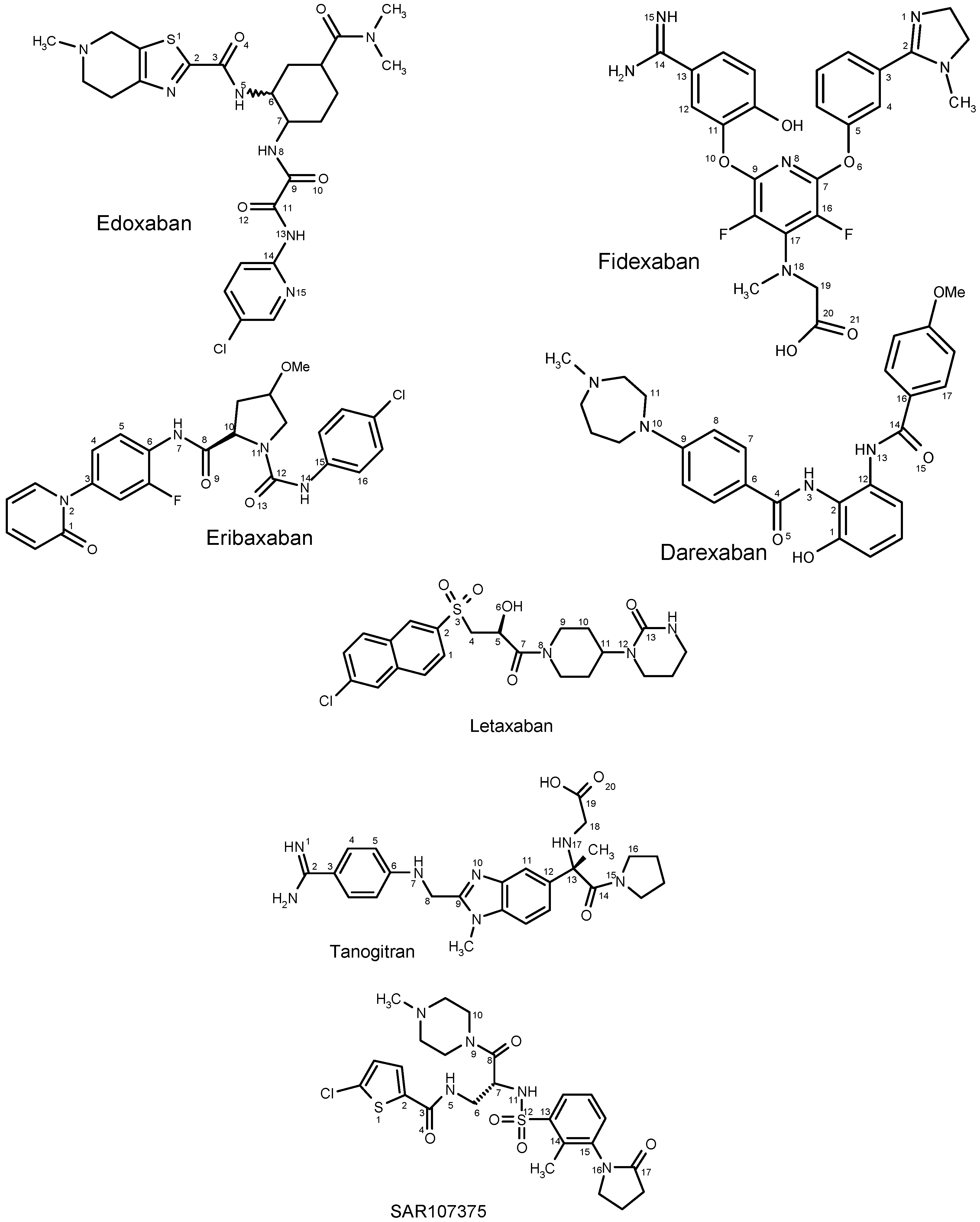

{kind=link}
{kind=link}
{kind=link}
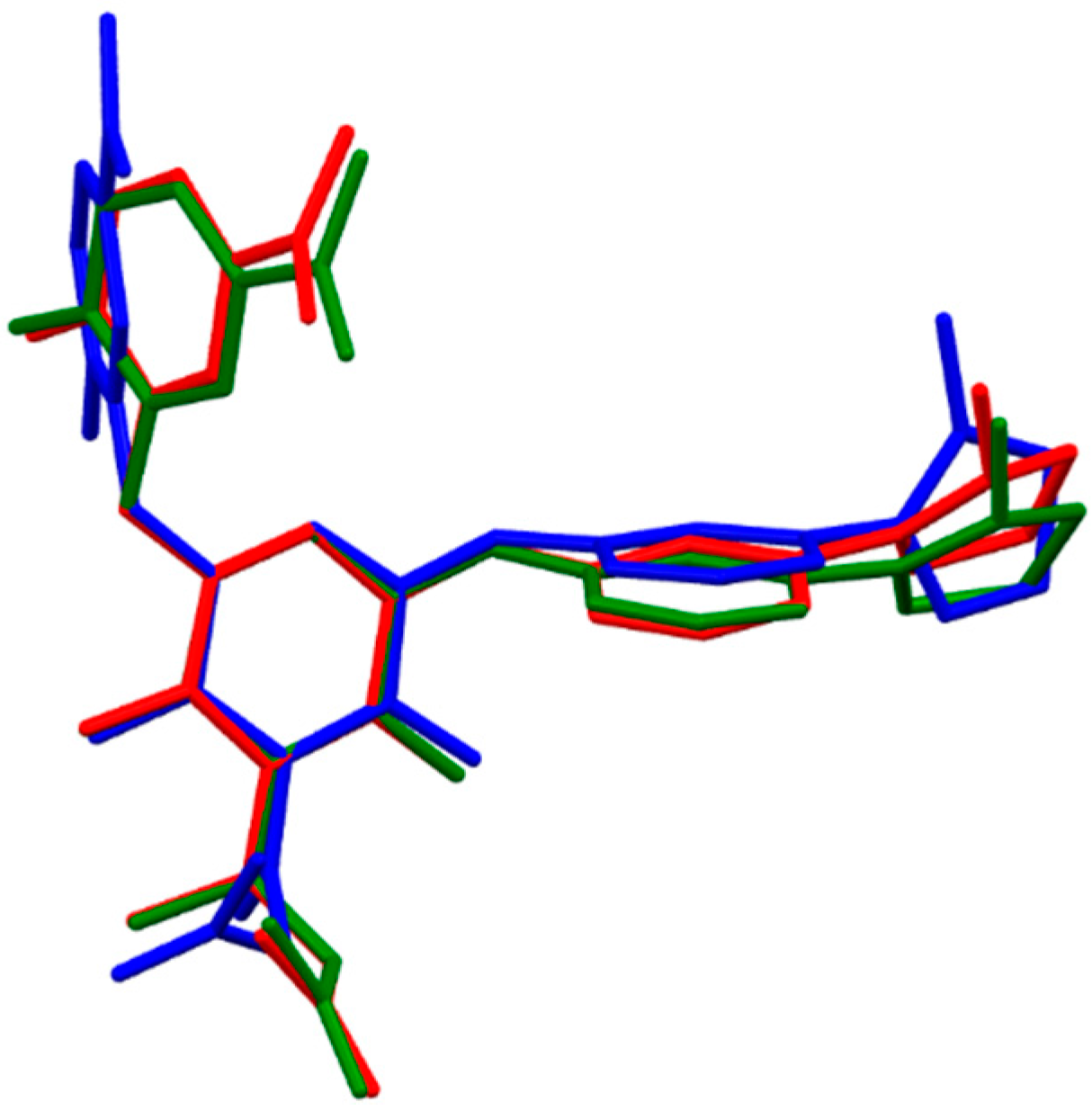{kind=link}
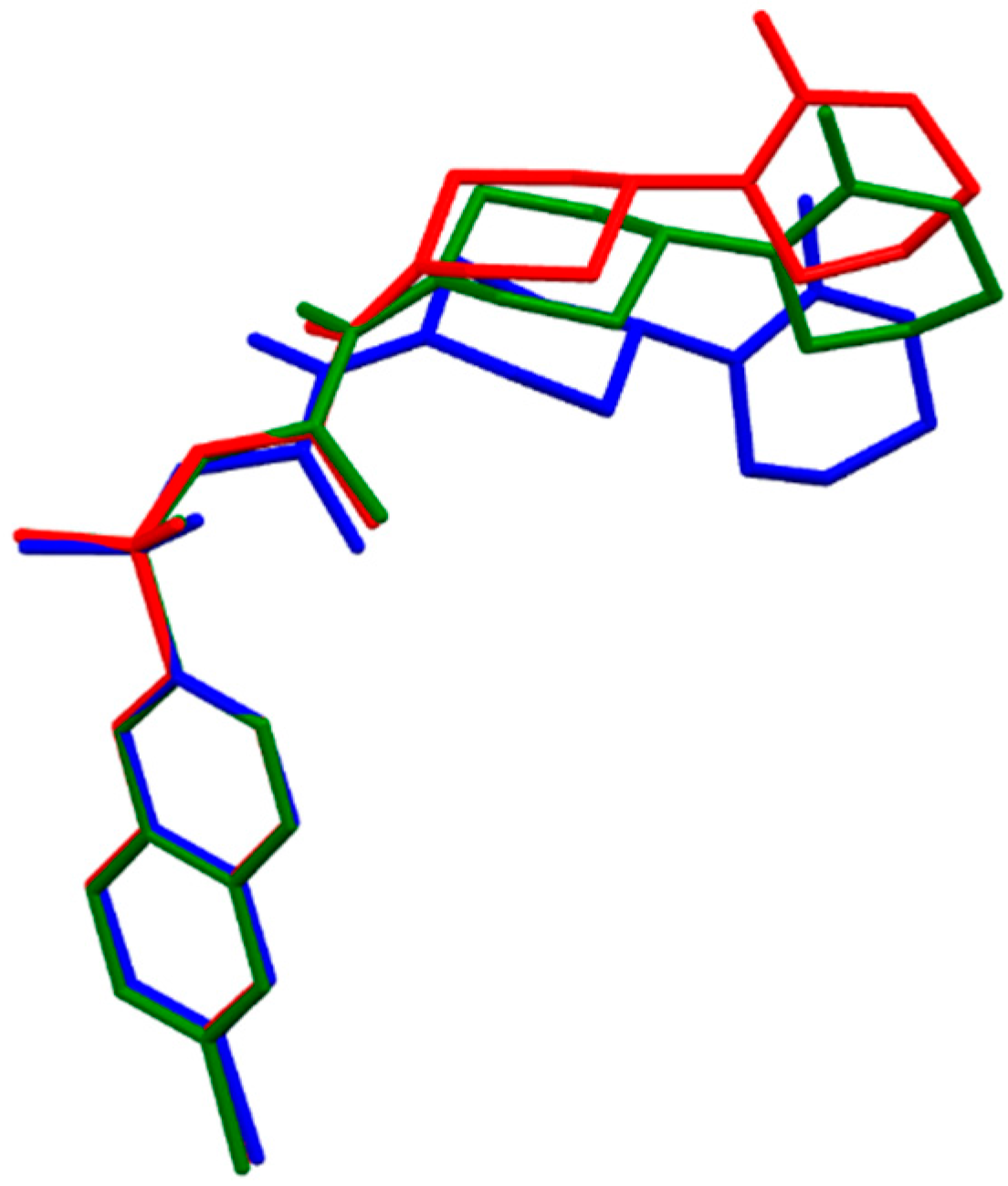{kind=link}
| Dihedral Angle a | X-ray b | B3LYP/6-31++g(p,d) | B97D/6-31++g(p,d) | B3LYP–CPCM | B97D–CPCM |
|---|---|---|---|---|---|
| (S,R,S)-Edoxaban | |||||
| α[S(1)-C(2)-C(3)-O(4)] | −2.59 | −2.77 | −0.44 | −0.38 | |
| β[S(1)-C(2)-C(3)-N(5)] | 176.73 | 176.50 | 179.30 | 179.13 | |
| γ[C(2)-C(3)-N(5)-C(6)] | 173.81 | 173.90 | 175.62 | 175.40 | |
| δ[C(3)-N(5)-C(6)-C(7)] | 134.17 | 132.08 | 122.43 | 132.05 | |
| ε[C(6)-C(7)-N(8)-C(9)] | −147.47 | −154.20 | −120.05 | −144.15 | |
| ζ[C(7)-N(8)-C(9)-O(10)] | 1.30 | 2.45 | −0.50 | 3.10 | |
| η[C(7)-N(8)-C(9)-C(11)] | −178.53 | −177.58 | 179.45 | −177.16 | |
| θ[N(8)-C(9)-C(11)]-O(12)] | 1.90 | 2.83 | −0.10 | 1.20 | |
| μ[N(8)-C(9)-C(11)]-N(13)] | −178.53 | −177.57 | 179.92 | −179.06 | |
| ν[C(9)-C(11)]-N(13)-C(14)] | −179.38 | −179.90 | 179.69 | −179.71 | |
| ξ[C(11)]-N(13)-C(14)-N(15)] | 179.16 | 179.45 | −179.78 | −179.34 | |
| (R,R)-Eribaxaban | 2PHB | ||||
| α[C(1)-N(2)-C(3)-C(4)] | −82.79 | −58.39 | −56.15 | −96.18 | −66.62 |
| β[C(5)-C(6)-N(7)-C(8)] | 65.14 | 4.19 | −12.63 | 5.85 | −17.44 |
| γ[C(6)-N(7)-C(8)-O(9)] | −0.09 | −4.81 | 1.63 | −2.73 | 0.77 |
| δ[C(6)-N(7)-C(8)-C(10)] | 179.50 | 176.37 | −175.78 | 178.01 | −176.90 |
| ε[N(7)-C(8)-C(10)-N(11)] | 1.67 | 7.42 | 65.65 | 4.84 | 63.15 |
| ζ[C(10)-N(11)-C(12)-O(13)] | −0.39 | 155.21 | 164.51 | 161.65 | 169.76 |
| η[C(10)-N(11)-C(12)-N(14)] | 179.24 | −26.05 | −16.00 | −19.78 | −10.78 |
| θ[N(11)-C(12)-N(14)]-C(15)] | 168.30 | 179.48 | −178.64 | −179.29 | −177.55 |
| μ[C(12)-N(14)]-C(15)-C(16)] | −155.47 | 176.92 | 174.63 | 175.98 | 170.63 |
| H-bond MeO···H-N, Å | 2.9338 | 1.8796 | 3.0505 | 1.8373 | |
| Fidexaban | 1FJS | ||||
| α[N(1)-C(2)-C(3)-C(4)] | −87.79 | −41.41 | −39.50 | −46.24 | −43.11 |
| β[C(4)-C(5)-O(6)-C(7)] | 147.64 | 154.48 | 161.22 | 162.20 | 161.65 |
| γ[C(5)-O(6)-C(7)-N(8)] | 115.45 | 115.13 | 121.46 | 115.40 | 121.26 |
| δ[N(8)-C(9)-O(10)-C(11)] | 32.16 | −19.64 | −106.05 | −12.40 | −20.05 |
| ε[C(9)-O(10)-C(11)-C(12)] | −120.44 | −57.75 | 9.65 | −69.17 | −69.45 |
| ζ[C(12)-C(13)-C(14)-N(15)] | −4.70 | −21.84 | −28.47 | −25.67 | −25.18 |
| η[C(16)-C(17)-N(18)-C(19)] | −83.09 | −132.13 | 57.97 | −133.91 | 54.12 |
| θ[C(17)-N(18)-C(19)-C(20)] | 54.20 | 85.14 | 59.75 | 85.54 | 80.03 |
| μ[N(18)-C(19)-C(20)-O(21)] | 92.47 | 157.26 | −170.74 | 164.70 | 162.60 |
| H-bond OH···O, Å | 2.1810 | 2.1949 | 2.2247 | 2.2777 | |
| H-bond C(=O)OH···N, Å | No interaction | 1.5433 | No interaction | No interaction | |
| Darexaban | |||||
| α[C(1)-C(2)-N(3)-C(4)] | 42.45 | −27.23 | 44.41 | −33.72 | |
| β[C(2)-N(3)-C(4)-O(5)] | −3.16 | 15.93 | −5.24 | 15.21 | |
| γ[C(2)-N(3)-C(4)-C(6)] | 176.75 | −165.63 | 174.85 | −165.63 | |
| δ[ N(3)-C(4)-C(6)-C(7)] | 0.40 | 38.24 | −1.49 | 36.32 | |
| ε[C(8)-C(9)-N(10)-C(11)] | −15.09 | −19.65 | −11.84 | −15.62 | |
| ζ[C(2)-C(12)-N(13)-C(14)] | −55.13 | −69.03 | −55.92 | −67.97 | |
| η[C(12)-N(13)-C(14)-O(15)] | 6.59 | −6.97 | 5.36 | −8.64 | |
| θ[C(12)-N(13)-C(14)-C(16)] | −173.43 | 167.34 | −175.12 | 166.62 | |
| μ[N(13)-C(14)-C(16)-C(17)] | 156.76 | 167.28 | 157.74 | 164.95 | |
| H-bond OH···O, Å | 1.6197 | 1.5871 | 1.6089 | 1.5627 | |
| H-bond NH···O, Å | 1.8333 | 2.9243 | 1.8388 | 3.0555 | |
| (S)-Letaxaban | 3KL6 | ||||
| α[C(1)-C(2)-S(3)-C(4)] | 106.69 | 95.99 | 99.47 | 96.34 | 93.17 |
| β[C(2)-S(3)-C(4)-C(5)] | −76.01 | −84.63 | −76.11 | −77.49 | −55.63 |
| γ[S(3)-C(4)-C(5)-O(6)] | 71.15 | 73.11 | 74.21 | 72.76 | 85.44 |
| δ[S(3)-C(4)-C(5)-C(7)] | −170.80 | −167.10 | −166.10 | −168.99 | −156.90 |
| ε[C(4)-C(5)-C(7)-N(8)] | 177.43 | 165.01 | 163.51 | 144.15 | 168.58 |
| ζ [C(5)-C(7)-N(8)]-C(9)] | 178.57 | 176.96 | 171.50 | 179.43 | 171.71 |
| η[C(10)-C(11)-N(12)-C(13)] | 103.92 | 124.08 | 130.11 | 113.62 | 110.42 |
| (R)-Tanogitran | |||||
| α[N(1)-C(2)-C(3)-C(4)] | −20.46 | −18.73 | −24.41 | −23.77 | |
| β[C(5)-C(6)-N(7)-C(8)] | 165.54 | −157.23 | 166.83 | −158.63 | |
| γ[C(6)-N(7)-C(8)-C(9)] | 79.48 | 49.24 | 80.05 | 49.22 | |
| δ[N(7)-C(8)-C(9)-N(10)] | −125.14 | −115.94 | −123.21 | −118.50 | |
| ε[C(11)-C(12)-C(13)-C(14)] | 31.69 | 38.45 | 34.23 | 38.52 | |
| ζ[C(11)-C(12)-C(13)-N(17)] | −87.36 | −80.82 | −83.75 | −80.04 | |
| η[C(12)-C(13)-N(17)-C(18)] | −177.45 | −177.20 | −174.71 | −174.96 | |
| ν[C(13)-N(17)-C(18)-C(19)] | −164.74 | −163.43 | −164.14 | −164.20 | |
| θ[N(17)-C(18)-C(19)-O(20)] | 16.24 | 17.08 | 15.28 | 16.42 | |
| μ[C(12)-C(13)-C(14)-N(15)] | 70.94 | 64.97 | 72.13 | 66.74 | |
| ξ[C(13)-C(14)-N(15)-C(16)] | −177.86 | −171.37 | −178.37 | −174.10 | |
| (S)-SAR107375 | |||||
| α[S(1)-C(2)-C(3)-O(4)] | −4.12 | −5.85 | −2.08 | −0.81 | |
| β[S(1)-C(2)-C(3)-N(5)] | 176.91 | 175.47 | 178.21 | −179.81 | |
| γ[C(2)-C(3)-N(5)-C(6)] | −178.37 | −169.31 | 179.46 | −172.09 | |
| δ[C(3)-N(5)-C(6)-C(7)] | 101.01 | 90.58 | 115.31 | 92.59 | |
| ε[N(5)-C(6)-C(7)-C(8)] | −60.05 | −57.45 | −62.20 | −59.89 | |
| ζ[C(6)-C(7)-C(8)-N(9)] | −71.35 | −78.07 | −73.08 | −82.25 | |
| η[C(7)-C(8)-N(9)-C(10)] | 169.79 | 172.87 | 173.19 | 177.96 | |
| ν[N(5)-C(6)-C(7)-N(11)] | 58.89 | 63.57 | 56.60 | 61.56 | |
| θ[C(6)-C(7)-N(11)-S(12)] | 106.96 | 129.39 | 105.71 | 135.19 | |
| μ[C(7)-N(11)-S(12)-C(13)] | 65.20 | 52.66 | 62.55 | 53.58 | |
| ξ[N(11)-S(12)-C(13)-C(14)] | 62.25 | 61.92 | 63.94 | 66.16 | |
| ρ[C(14)-C(15)-N(16)-C(17)] | 117.02 | 141.33 | 98.19 | 113.44 | |
| σ[N(9)-C(8)-C(7)-N(11)] | 168.87 | 160.95 | 166.80 | 156.52 | |
| d[O(4)…S(1)], Å | 2.9491 | 2.9876 | 2.9378 | 2.9795 |
| Drug | ΔECPCM, kJ/mol | Gas-phase Dipole Moment, Debye (D) | ||
|---|---|---|---|---|
| B3LYP–CPCM | B97D–CPCM | B3LYP/6-31++g(p,d) | B97D/6-31++g(p,d) | |
| Edoxaban | −77.48 | −73.67 | 9.54 | 9.16 |
| Eribaxaban | −75.99 | −68.57 | 1.86 | 6.61 |
| Fidexaban | −90.60 | −37.07 | 3.33 | 5.93 |
| Darexaban | −65.66 | −72.67 | 9.89 | 11.87 |
| Letaxaban | −95.09 | −93.43 | 8.45 | 7.39 |
| Tanogitran | −101.56 | −100.15 | 5.75 | 5.31 |
| SAR107375 | −89.02 | −79.57 | 6.74 | 4.62 |
2.1.1. Edoxaban

2.1.2. Eribaxaban

2.1.3. Fidexaban
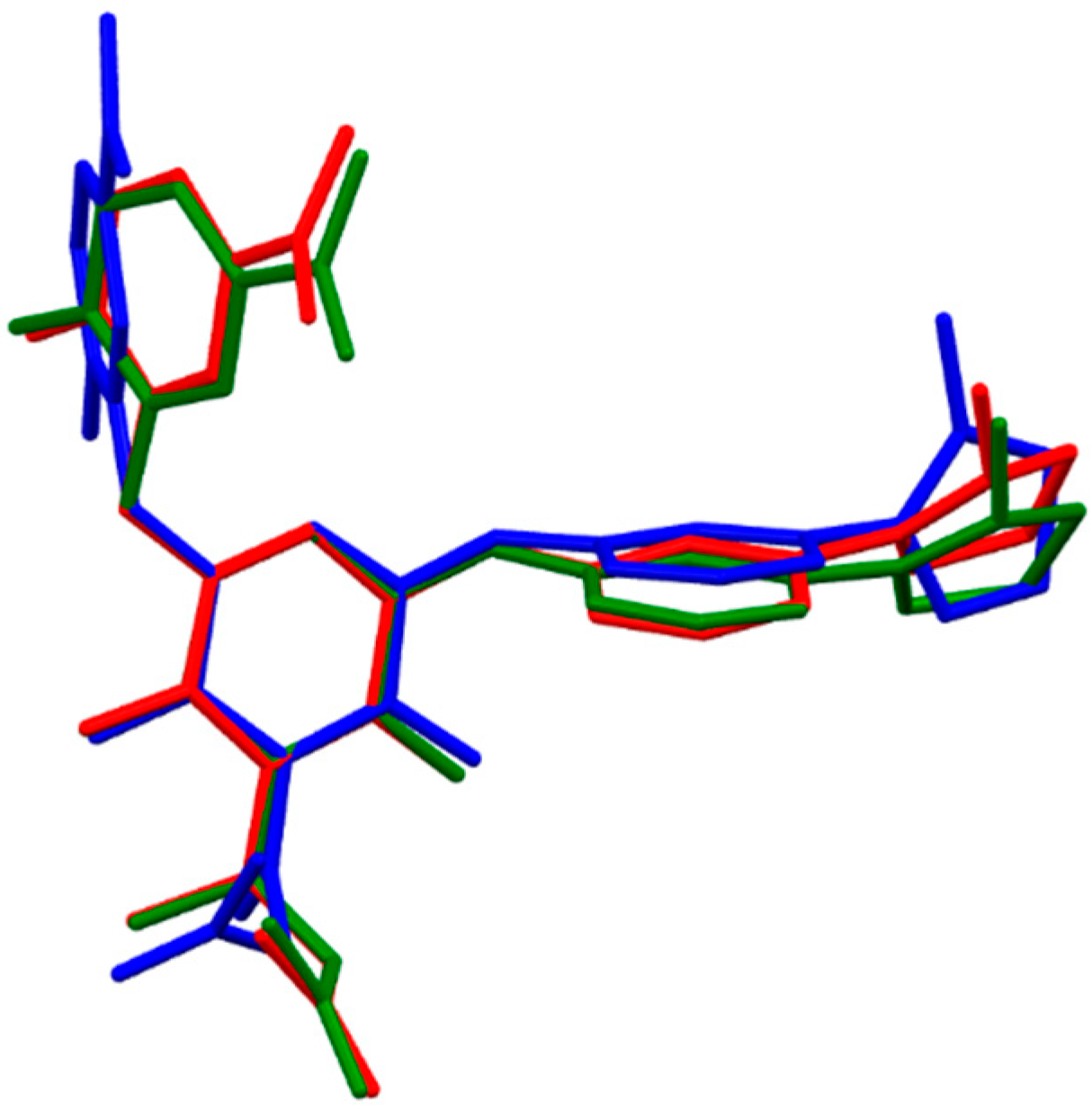
2.1.4. Darexaban
2.1.5. Letaxaban
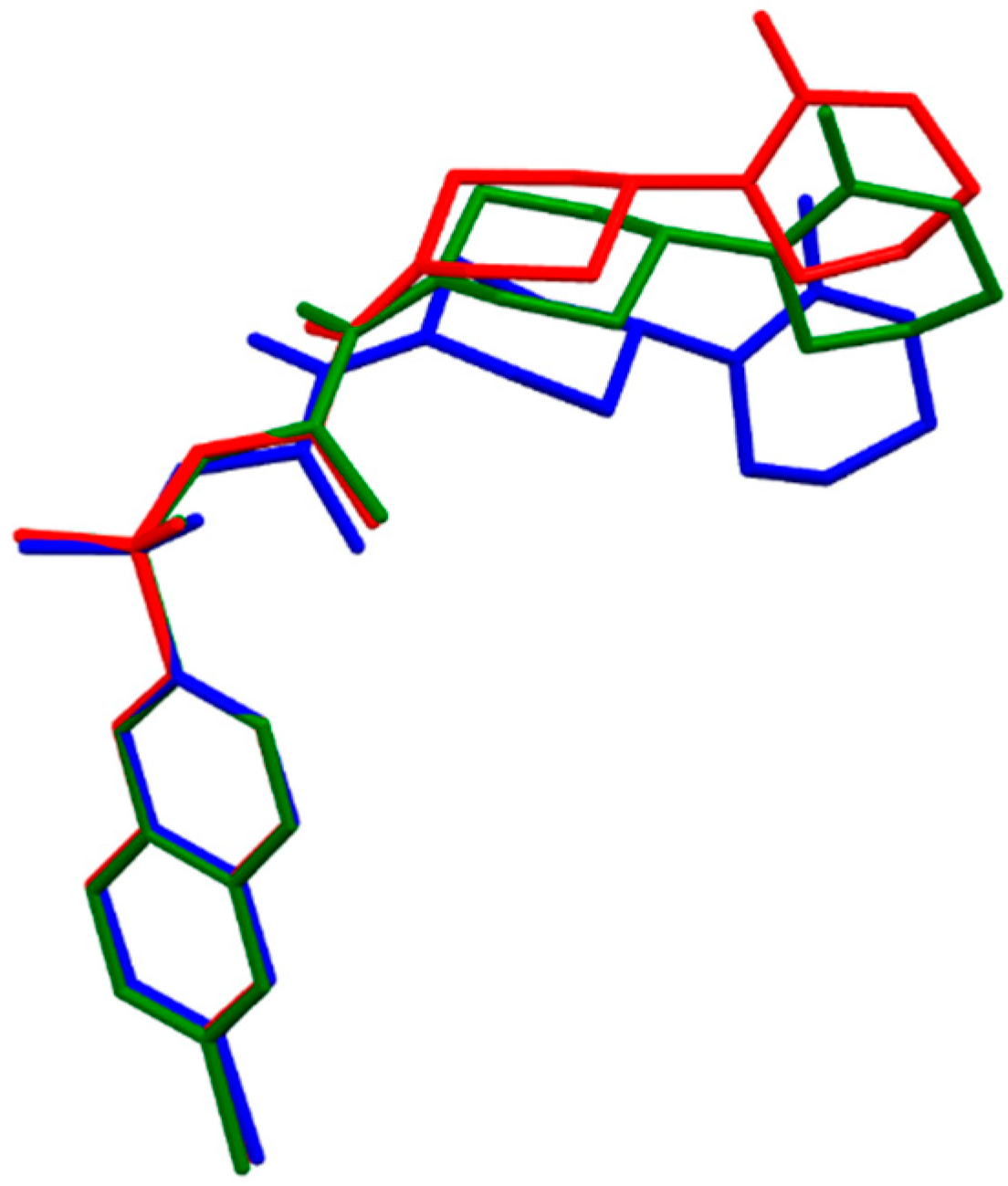
2.1.6. Tanogitran (Dual)
2.1.7. SAR107375 (Dual)
2.2. Dissociation Constants
| Drug | pKa | pKa | % Ionized Form | ||
|---|---|---|---|---|---|
| Exp. | Acid Function a | Basic Function b | Acid Function | Basic Function | |
| Edoxaban | 6.7 (FDA label) c | 11.08 | 7.23 | 0 | 40 |
| Eribaxaban | 9.04 | 2 | |||
| Fidexaban | 3.54 | 12.28 | 100 | 100 | |
| Darexaban | 8.76 | 8.11 | 4 | 83 | |
| Letaxaban | 12.93 | 0 | |||
| Tanogitran | 1.98 | 11.53 | 100 | 100 | |
| SAR107375 | 8.35 | 6.52 | 10 | 11 | |
2.3. Lipophilicity and Solubility
| Drug | ALOGPS | Log D, (pH = 7.4) | ALOGPS, Solubility |
|---|---|---|---|
| Edoxaban | 1.61 | −4.68 (11.4 mg/L) | |
| Eribaxaban | 3.35 | −4.83 (7.2 mg/L) | |
| Fidexaban | 3.07 | −0.79 | −4.34 (24.2 mg/L) |
| Darexaban | 4.03 | 3.24 | −4.21 (29.1 mg/L) |
| Letaxaban | 1.55 | −3.34 (0.2 g/L) | |
| Tanogitran | 1.58 | −3.84 | −3.54 (0.14 g/L) |
| SAR107375 | 1.15 | −4.42 (21.4 mg/L) |
| Drug | %ABS | Volume | PSA | NROTB | n ON Acceptors | n OHNH Donors | Formula Weight |
|---|---|---|---|---|---|---|---|
| Edoxaban | 61.9 | 469.39 | 136.62 | 6 | 11 a | 3 | 548.07 a |
| Eribaxaban | 77.0 | 407.20 | 92.67 | 5 | 8 | 2 | 484.91 |
| Fidexaban | 54.6 | 440.66 | 157.60 | 9 | 11 a | 5 | 526.55 a |
| Darexaban | 76.5 | 438.49 | 94.13 | 6 | 8 | 3 | 474.61 |
| Letaxaban | 72.1 | 403.34 | 107.02 | 5 | 8 | 2 | 479.99 |
| Tanogitran | 57.5 | 437.41 | 149.36 | 9 | 10 | 6 a | 477.57 |
| SAR107375 | 67.9 | 473.92 | 119.12 | 8 | 10 | 2 | 568.18 a |
2.4. Absorption, Polar Surface Area, and “Rule of Five” Properties
2.5. Selection Criteria for Drug-Like Properties of fXa Inhibitors
| Drug | %ABS | Volume Å3 | PSA Å2 | Clog S | Clog P | Formula Weight, Da | Ki b, nmol/L | Bioavailability c % |
|---|---|---|---|---|---|---|---|---|
| Rivaroxaban a | 77.8 | 351.74 | 88.18 | −4.64 | 1.74 | 435.89 | 0.4 | 80 |
| Apixaban a | 69.8 | 406.55 | 110.77 | −3.83 | 2.23 | 459.51 | 0.08 | 50 |
| Otamixaban a | 62.7 | 407.73 | 130.73 | −5.35 | 2.12 | 446.51 | 0.4 | |
| Betrixaban a | 70.1 | 392.76 | 107.41 | −4.44 | 2.86 | 451.91 | 0.117 | 47 |
| Razaxaban a | 66.5 | 423.70 | 120.04 | −4.07 | 3.90 | 528.47 (viol.) | 0.19 | |
| DX-9065a a | 65.3 | 410.68 | 123.50 | −4.49 | 2.61 | 444.53 | 41 | 3 |
| Edoxaban | 61.9 | 469.39 | 136.62 | −4.68 | 1.61 | 548.07 (viol.) | 0.56 | 62 |
| Eribaxaban | 77.0 | 407.20 | 92.67 | −4.83 | 3.35 | 484.91 | 0.32 | |
| Fidexaban | 54.6 | 440.66 | 157.60 | −4.34 | 3.07 | 526.55 (viol) | 0.11 | |
| Darexaban | 76.5 | 438.49 | 94.13 | −4.21 | 4.03 | 474.61 | 31 | |
| Letaxaban | 72.1 | 403.34 | 107.02 | −3.34 | 1.55 | 479.99 | 1.8 | 50 |
| Molecular Weight | 430–550 |
| Octanol/water partition coefficient (clog P) | 1–4 |
| Aqueous solubility (clog S) | (−3.3)–(−5.3) |
| Polar surface area (PSA, Å2) | 90–160 |
| Volume (Vol, Å3) | 350–470 |
| Percent of oral absorption (%ABS) | 55–76 |
3. Computational Details
4. Conclusions
- i)
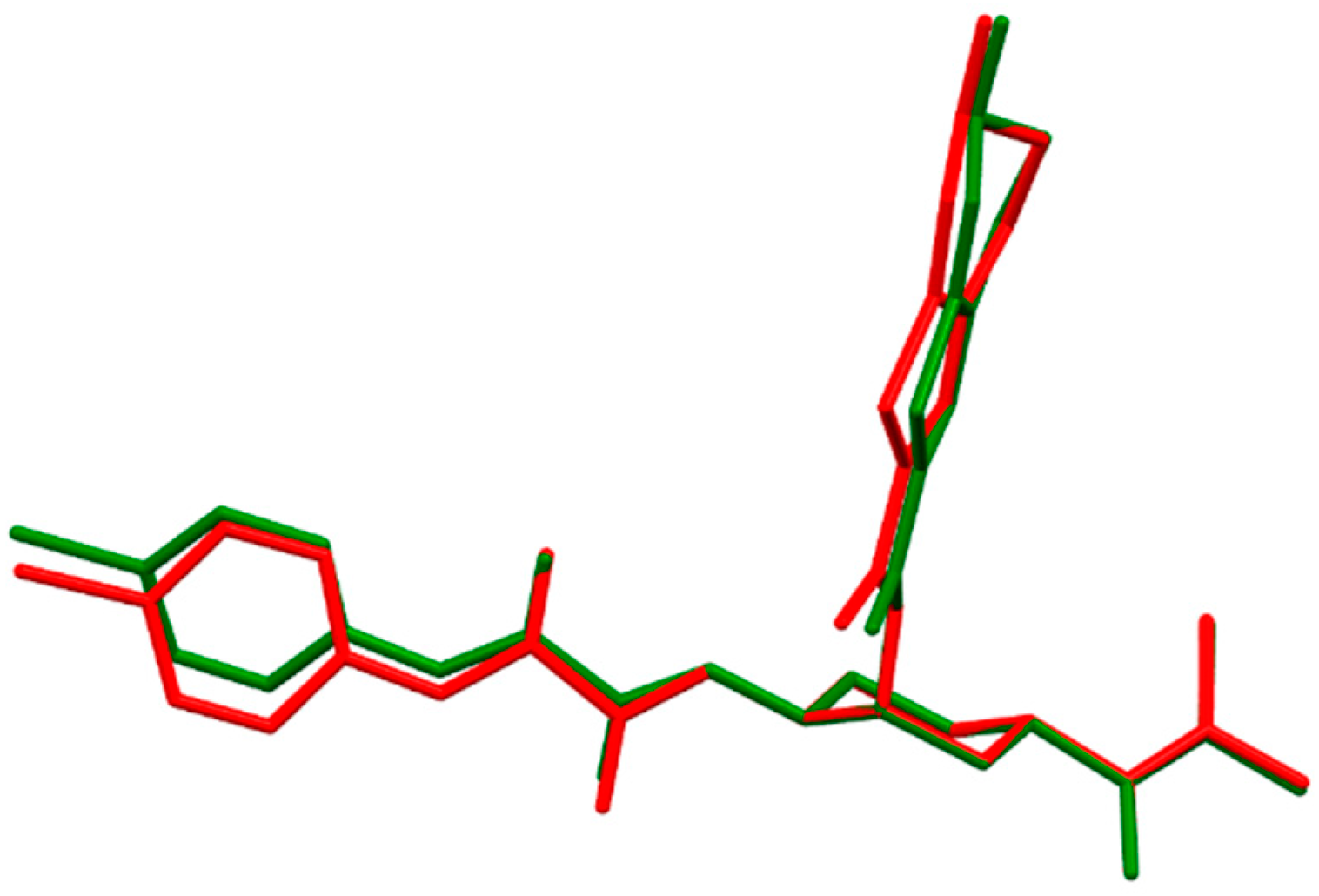
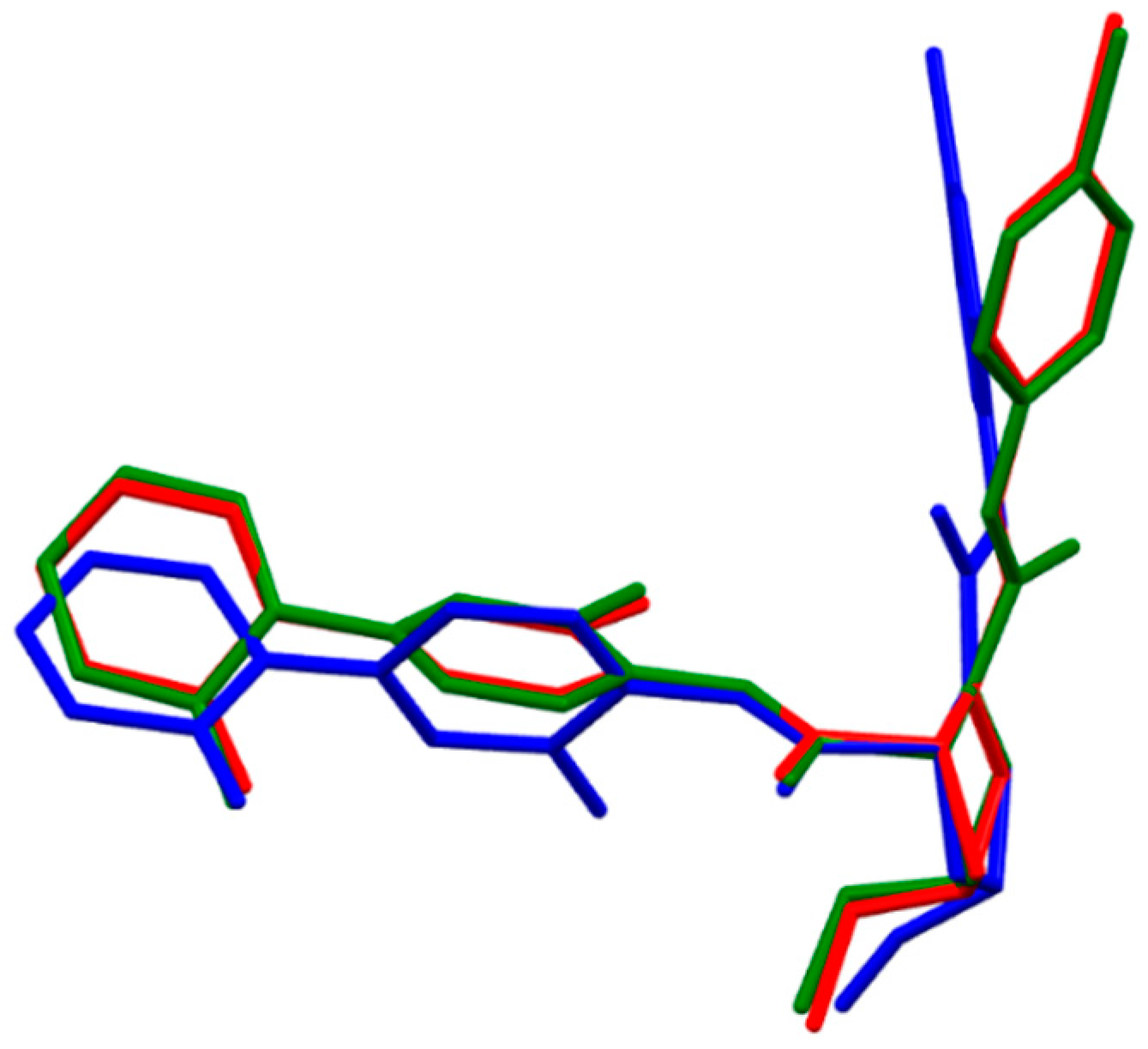
- The fully optimized most stable conformers of these drugs possess a characteristic l-shaped structure. Examination of the spatial models of the B3LYP and B97D optimized structures indicated that the equilibrium geometries computed using the B3LYP and B97D functionals are in some cases different.
- ii)
- Water had a remarkable effect on the geometry of the studied anticoagulants. The anticoagulant drugs exhibit considerable stability in this solvent, as expected.
- iii)
- Eribaxaban, and letaxaban are present in neutral undissociated form at pH 7.4. Fidexaban and tanogitran exist as zwitterionic structures.
- iv)
- A trend in the compound lipophilicity was also observed. It is lowest for the dual inhibitors tanogitran and SAR107375 and increase for the clinically approved fXa inhibitor edoxaban.
- v)
- The studied anticoagulants were only slightly soluble in water, but their computed solubility between 7 and 200 mg/L is sufficient for fast absorption.
- vi)
- The dual inhibitor SAR107375 represents an improvement in structural, physicochemical and pharmacokinetic characteristics over tanogitran. At blood pH, SAR107375 predominantly exists in neutral form. In contrast to tanogitran, it is better absorbed and more lipophilic and active after oral application.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vermeer, C.; Schurgers, L.J. A comprehensive review of vitamin K and vitamin K antagonists. Hematol. Oncol. Clin. N. Am. 2000, 14, 339–353. [Google Scholar]
- Becattini, C.; Vedovati, M.C.; Agnelli, G. Old and new oral anticoagulants for venous thromboembolism and atrial fibrillation: A review of the literature. Thromb. Res. 2012, 129, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, F. New oral anticoagulants: Comparative pharmacology with vitamin K antagonists. Clin. Pharmacokinet. 2013, 52, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.J.P.; Smallheer, J.M.; Cheney, D.L.; Knabb, R.M.; Wexler, R.R. Factor Xa inhibitors: Next-generation antithrombotic agents. J. Med. Chem. 2010, 53, 6243–6274. [Google Scholar] [PubMed]
- Levy, J.H.; Spyropoulos, A.C.; Samama, C.M.; Douketis, J. Direct oral anticoagulants: New drugs and new concepts. JACC Cardiovasc. Interv. 2014, 7, 1333–1351. [Google Scholar] [PubMed]
- Schwienhorst, A. Direct thrombin inhibitors—A survey of recent developments. Cell. Mol. Life Sci. 2006, 63, 2773–2791. [Google Scholar] [PubMed]
- Graefe-Mody, E.U.; Schühly, U.; Rathgen, K.; Stähle, H.; Leitner, M.J.; Jilma, B. Pharmacokinetics and pharmacodynamics of BIBT 986, a novel small molecule dual inhibitor of thrombin and factor Xa. J. Thromb. Haemost. 2006, 4, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- He, L.W.; Dai, W.Ch.; Li, N.G. Development of orally active thrombin inhibitors for the treatment of thrombotic disorder diseases. Molecules 2015, 20, 11046–11062. [Google Scholar] [CrossRef] [PubMed]
- Giardino, E.C.; Haertlein, B.J.; de Garavilla, L.; Costanzo, M.J.; Damiano, B.P.; Andrade-Gordon, P.; Maryanoff, B.E. Cooperative antithrombotic effect from the simultaneous inhibition of thrombin and factor Xa. Blood Coagul. Fibrinol. 2010, 21, 128–134. [Google Scholar]
- Nar, H. The role of structural information in the discovery of direct thrombin and factor Xa inhibitors. Trends Pharmacol. Sci. 2012, 33, 279–288. [Google Scholar] [PubMed]
- Remko, M. Molecular structure, lipophilicity, solubility, absorption, and polar surface area of novel anticoagulant agents. J. Mol. Struct. 2009, 916, 76–85. [Google Scholar]
- Remko, M.; Broer, R.; Remková, A. A comparative study of the molecular structure, lipophilicity, solubility, acidity, absorption and polar surface area of coumarinic anticoagulants and direct thrombin inhibitors. RSC Adv. 2014, 4, 8072–8084. [Google Scholar]
- Sebastiani, D.; Röthlisberger, U.; Cavalli, A.; Folkers, G.; Recanatini, M.; Scapozza, L.; Rovira, C.; Raber, J.; Llano, J.; Eriksson, L.A.; et al. Quantum Medicinal Chemistry; Carloni, P., Alber, F., Eds.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Ooms, F. Molecular modeling and computer aided drug design. Examples of their applications in medicinal chemistry. Curr. Med. Chem. 2000, 7, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [PubMed]
- Minor, Ch.; Tellor, K.B.; Armbruster, A.L. Edoxaban, a novel oral factor Xa inhibitor. Ann. Pharmacother. 2015, 49, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.; Fourme, R.; André, D.; Renaud, M. Crystal structure of cyclohexane I and II. Acta Crystallogr. B 1973, 29, 131–138. [Google Scholar] [CrossRef]
- Kohrt, J.T.; Bigge, C.F.; Bryant, J.W.; Casimiro-Garcia, A.; Chi, L.; Cody, WL.; Dahring, T.; Dudley, D.A.; Filipski, K.J.; Haarer, S.; et al. The Discovery of (2R,4R)-N-(4-chlorophenyl)-N-(2-fluoro-4-(2-oxopyridin-1(2H)-yl)phenyl)-4-methoxypyrrolidine-1,2-dicarboxamide (PD 0348292), an orally efficacious factor Xa inhibitor. Chem. Biol. Drug Des. 2007, 70, 100–112. [Google Scholar] [PubMed]
- Ahrens, I.; Peter, K.; Lip, G.Y.; Bode, C. Development and clinical applications of novel oral anticoagulants. Part II. Drugs under clinical investigation. Discov. Med. 2012, 13, 445–450. [Google Scholar] [PubMed]
- Phillips, G.B.; Buckman, B.O.; Davey, D.D.; Eagen, K.A.; Guilford, W.J.; Hinchman, J.; Ho, E.; Koovakkat, S.; Liang, A.; Light, D.R.; et al. Discovery of N-[2-[5-[Amino(imino)methyl]-2-hydroxyphenoxy]-3,5-difluoro-6-[3-(4,5-dihydro-1-methyl-1H-imidazol-2-yl)phenoxy]pyridin-4-yl]-N-methylglycine (ZK-807834): A potent, selective, and orally active inhibitor of the blood coagulation enzyme factor Xa. J. Med. Chem. 1998, 41, 3557–3562. [Google Scholar] [PubMed]
- Adler, M.; Davey, D.D.; Phillips, G.B.; Kim, S.H.; Jancarik, J.; Rumennik, G.; Light, D.R.; Whitlow, M. Preparation, characterization, and the crystal structure of the inhibitor ZK-807834 (CI-1031) complexed with factor Xa. Biochemistry 2000, 39, 12534–12542. [Google Scholar] [PubMed]
- Hirayama, F.; Koshio, H.; Ishihara, T.; Hachiya, S.; Sugasawa, K.; Koga, Y.; Seki, N.; Shiraki, R.; Shigenaga, T.; Iwatsuki, Y.; et al. Discovery of N-[2-Hydroxy-6-(4-methoxybenzamido)phenyl]-4-(4-methyl-1,4-diazepan-1-yl)benzamide (Darexaban, YM150) as a potent and orally available factor Xa inhibitor. J. Med. Chem. 2011, 54, 8051–8065. [Google Scholar] [PubMed]
- Fujimoto, T.; Imaeda, Y.; Konishi, N.; Hiroe, K.; Kawamura, M.; Textor, G.P.; Aertgeerts, K.; Kubo, K. Discovery of a tetrahydropyrimidin-2(1H)-one derivative (TAK-442) as a potent, selective, and orally active factor Xa inhibitor. J. Med. Chem. 2010, 53, 3517–3531. [Google Scholar] [PubMed]
- Remko, M.; Herich, P.; Gregáň, F.; Kožíšek, J. Structure, acidity and basicity of a benzene disulfonamide inhibitor of carbonic anhydrase. J. Mol. Struct. 2014, 1059, 124–131. [Google Scholar] [CrossRef]
- Remko, M.; Kožíšek, J.; Semanová, J.; Gregáň, F. Synthesis, crystal and molecular structure of two biologically active aromatic sulfonamides and their hydrochloride salts. J. Mol. Struct. 2010, 973, 18–26. [Google Scholar] [CrossRef]
- Leitner, J.M.; Jilma, B.; Mayr, F.B.; Cardona, F.; Spiel, A.O.; Firbas, C.; Rathgen, K.; Stähl, H.; Schühly, U.; Graefe-Mody, E.U. Pharmacokinetics and pharmacodynamics of the dual FII/FX inhibitor BIBT 986 in endotoxin-induced coagulation. Clin. Pharmacol. Ther. 2007, 81, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Meneyrol, J.; Follmann, M.; Lassalle, G.; Wehner, V.; Barre, G.; Rousseaux, T.; Altenburger, J.-M.; Petit, F.; Bocskei, Z.; Schreuder, H.; et al. 5-Chlorothiophene-2-carboxylic acid [(S)-2-[2-methyl-3-(2-oxopyrrolidin-1-yl)benzenesulfonylamino]-3-(4-methylpiperazin-1-yl)-3-oxopropyl]amide (SAR107375), a selective and potent orally active dual thrombin and factor Xa inhibitor. J. Med. Chem. 2013, 56, 9441–9456. [Google Scholar] [PubMed]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Remko, M. Molecular structure, pKa, lipophilicity, solubility and absorption of biologically active aromatic and heterocyclic sulfonamides. J. Mol. Struct. 2010, 944, 34–42. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, R.; Li, P.; Zhang, H. On the properties of S···O and S···π noncovalent interactions: The analysis of geometry, interaction energy and electron density. New J. Chem. 2015, 39, 1611–1618. [Google Scholar]
- Hilal, S.; Karickhoff, S.W.; Carreira, L.A. A Rigorous test for SPARC’s chemical reactivity models: Estimation of more than 4300 ionization pKas. Quant. Struc. Act. Real. 1995, 14, 348–355. [Google Scholar]
- SPARC. Available online: http://archemcalc.com/sparc/ (accessed on 15 September 2015).
- Malkia, A.; Murtomaki, L.; Urtti, A.; Kontturi, K. Drug permeation in biomembranes: In vitro and in silico prediction and influence of physicochemical properties. Eur. J. Pharm. Sci. 2004, 23, 13–47. [Google Scholar] [PubMed]
- Drug Bank. Available online: http://www.drugbank.ca/drugs/DB09075 (accessed on 10 January 2016).
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [PubMed]
- Xing, L.; Glen, R.C. Novel Methods for the Prediction of logP, pKa, and logD. J. Chem. Inf. Comput. Sci. 2002, 42, 796–805. [Google Scholar] [PubMed]
- Veber, D.F.; Johnson, R.S.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kapple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar]
- Zhao, Y.H.; Abraham, M.H.; Lee, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B. Cooper, Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [PubMed]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Lecumberri, R.; Terleira-Fernández, A.I.; Vargas-Castrillón, E.; Rocha, E. Potential role of new anticoagulants for prevention and treatment of venous thromboembolism in cancer patients. Vasc. Health Risk Manag. 2013, 9, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorrisen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2015, 31, 405–412. [Google Scholar] [PubMed]
- Toschi, V.; Lettino, M. Inhibitors of propagation of coagulation: Factors V and X. Br. J. Clin. Pharmacol. 2011, 72, 563–580. [Google Scholar] [PubMed]
- Masotti, L.; Campanini, M. Pharmacology of new oral anticoagulants: Mechanism of action, pharmacokinetics, pharmacodynamics. Italian J. Med. 2013, 7, 1–7. [Google Scholar]
- Weitz, J.I.; Eikelboom, J.W.; Samama, M.M. New antithrombotic drugs, antithrombotic therapy and prevention of thrombosis: American college of chest physicians, evidence-based clinical practice guidelines. Chest 2012, 141 (Suppl. S2), e120S–e151S. [Google Scholar] [PubMed]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [PubMed]
- Kontogiorgis, C.A.; Hadjipavlou-Litina, D. Current trends in quantitative structure activity relationships on fxa inhibitors: Evaluation and comparative analysis. Med. Res. Rev. 2004, 24, 687–747. [Google Scholar] [PubMed]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-sham density functional theory: Predicting and understanding chemistry. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; Wiley-VCH: New York, NY, USA, 2000; Volume 15, pp. 1–86. [Google Scholar]
- Gaussian 09, version 9.0; Gaussian, Inc.: Wallingford, CT, USA, 2011.
- Becke, D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Cossi, M.; Rega, N.; Scalman, I.G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remko, M.; Remková, A.; Broer, R. Theoretical Study of Molecular Structure and Physicochemical Properties of Novel Factor Xa Inhibitors and Dual Factor Xa and Factor IIa Inhibitors. Molecules 2016, 21, 185. https://doi.org/10.3390/molecules21020185
Remko M, Remková A, Broer R. Theoretical Study of Molecular Structure and Physicochemical Properties of Novel Factor Xa Inhibitors and Dual Factor Xa and Factor IIa Inhibitors. Molecules. 2016; 21(2):185. https://doi.org/10.3390/molecules21020185
Chicago/Turabian StyleRemko, Milan, Anna Remková, and Ria Broer. 2016. "Theoretical Study of Molecular Structure and Physicochemical Properties of Novel Factor Xa Inhibitors and Dual Factor Xa and Factor IIa Inhibitors" Molecules 21, no. 2: 185. https://doi.org/10.3390/molecules21020185