
An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines

Department of Chemistry, Section of Organic Chemistry and Biochemistry, University of Ioannina, 451 10 Ioannina, Greece
Molecules 2016, 21(2), 154; https://doi.org/10.3390/molecules21020154
Submission received: 17 September 2015
/
Revised: 23 December 2015
/
Accepted: 5 January 2016
/
Published: 28 January 2016
(This article belongs to the Special Issue Design and Synthesis of Novel Conjugated and Non Conjugated Small Molecules)
Abstract
:Pyrrolo[1,4]benzodiazepines are tricyclic compounds that are considered “privileged structures” since they possess a wide range of biological activities. The first encounter with these molecules was the isolation of anthramycin from cultures of Streptomyces, followed by determination of the X-ray crystal structure of the molecule and a study of its interaction with DNA. This opened up an intensive synthetic and biological study of the pyrrolo[2,1-c][1,4]benzodiazepines that has culminated in the development of the dimer SJG-136, at present in Phase II clinical trials. The synthetic efforts have brought to light some new synthetic methodology, while the contemporary work is focused on building trimeric pyrrolo[2,1-c][1,4]benzodiazepines linked together by various heterocyclic and aliphatic chains. It is the broad spectrum of biological activities of pyrrolo[1,2-a][1,4]benzodiazepines that has maintained the interest of researchers to date whereas several derivatives of the even less studied pyrrolo[1,2-d][1,4]benzodiazepines were found to be potent non-nucleoside HIV-1 reverse transcriptase inhibitors. The present review is an update on the synthesis of pyrrolo[2,1-c][1,4]benzodiazepines since the last major review of 2011, while the overview of the synthesis of the other two tricyclic isomers is comprehensive.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
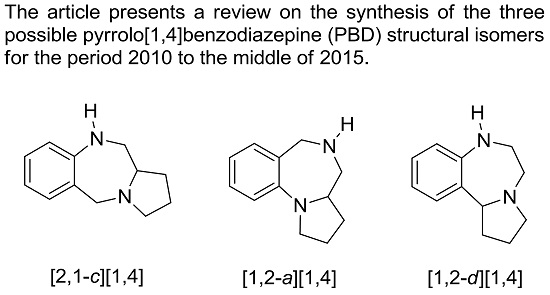
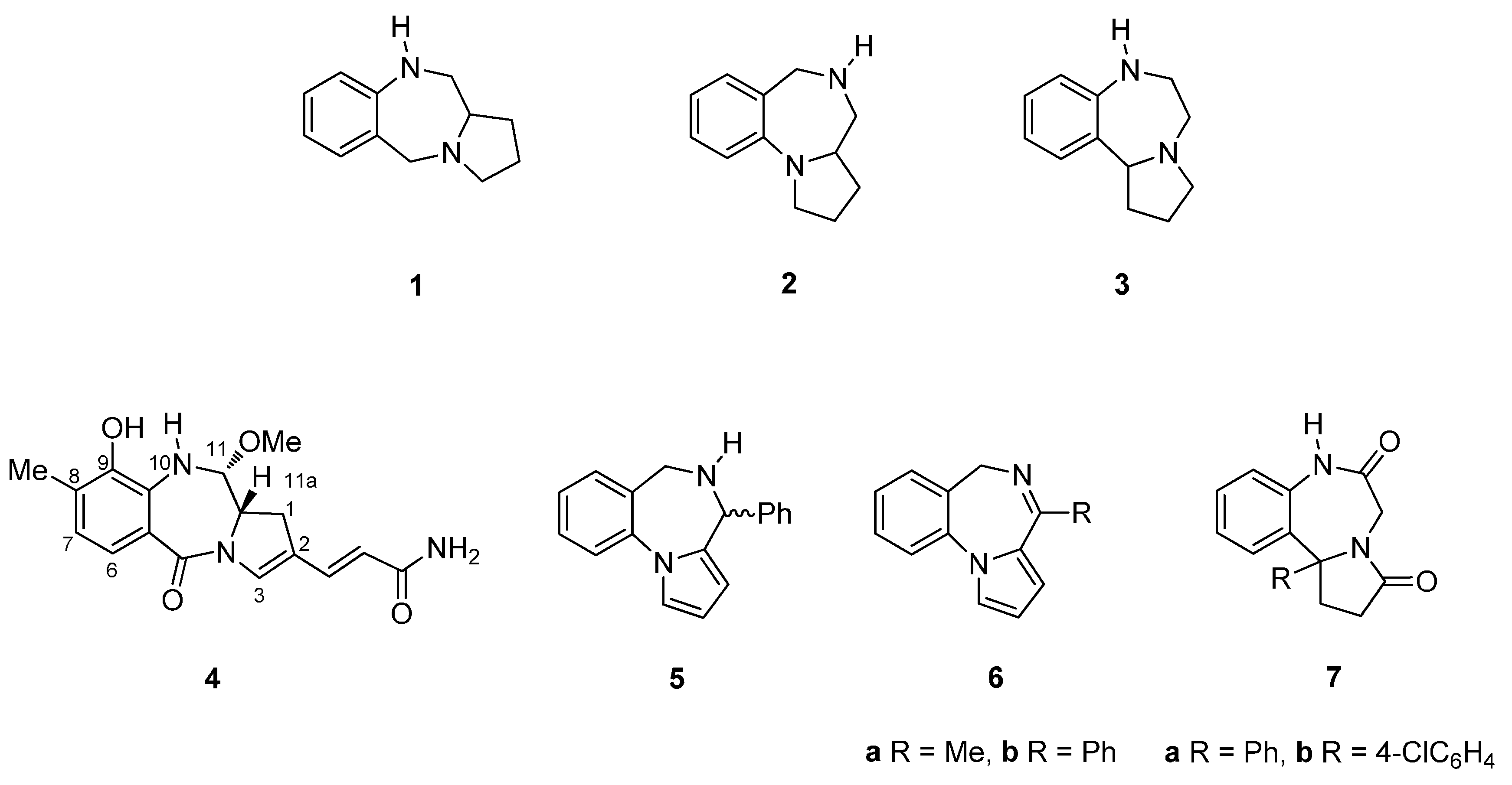
The pyrrolo[1,4]benzodiazepine (PBD) tricyclic ring system is represented by three possible structural isomers [2,1-c][1,4] 1, [1,2-a][1,4] 2 and [1,2-d][1,4] 3 (Figure 1). The first derivative of any of these ring systems to be synthesized was a pyrrolo[2,1-c][1,4]benzodiazepine, anthramycin (4), reported by Leimgruber et al., in 1968 [1]. Three years later Cheeseman and Rafiq [2] published the synthesis of the first two pyrrolo[1,2-a][1,4]benzodiazepines 5 and 6a,b. Then in 1977, Yamawaki et al. [3] described the preparation of the first two pyrrolo[1,2-d][1,4]benzodiazepines 7a,b. For the purposes of the present review, synthetic chemistry publications from the primary literature were retrieved from the SciFinder and Scopus databases. For the period 2010 to the middle of 2015, thirty-two pyrrolo[2,1-c][1,4]benzodiazepine-related publications were retrieved, whereas for the period 1960 to the present, twenty-five pyrrolo[1,2-a][1,4]benzodiazepine and seven pyrrolo[1,2-d][1,4]benzodiazepine publications were downloaded. This is the first review on the synthesis of PBDs that includes all three structural isomers.
Figure 1.
Structure of the three pyrrolo[1,4]benzodiazepine isomers 1–3 and the first derivatives 4–7.
Figure 1.
Structure of the three pyrrolo[1,4]benzodiazepine isomers 1–3 and the first derivatives 4–7.
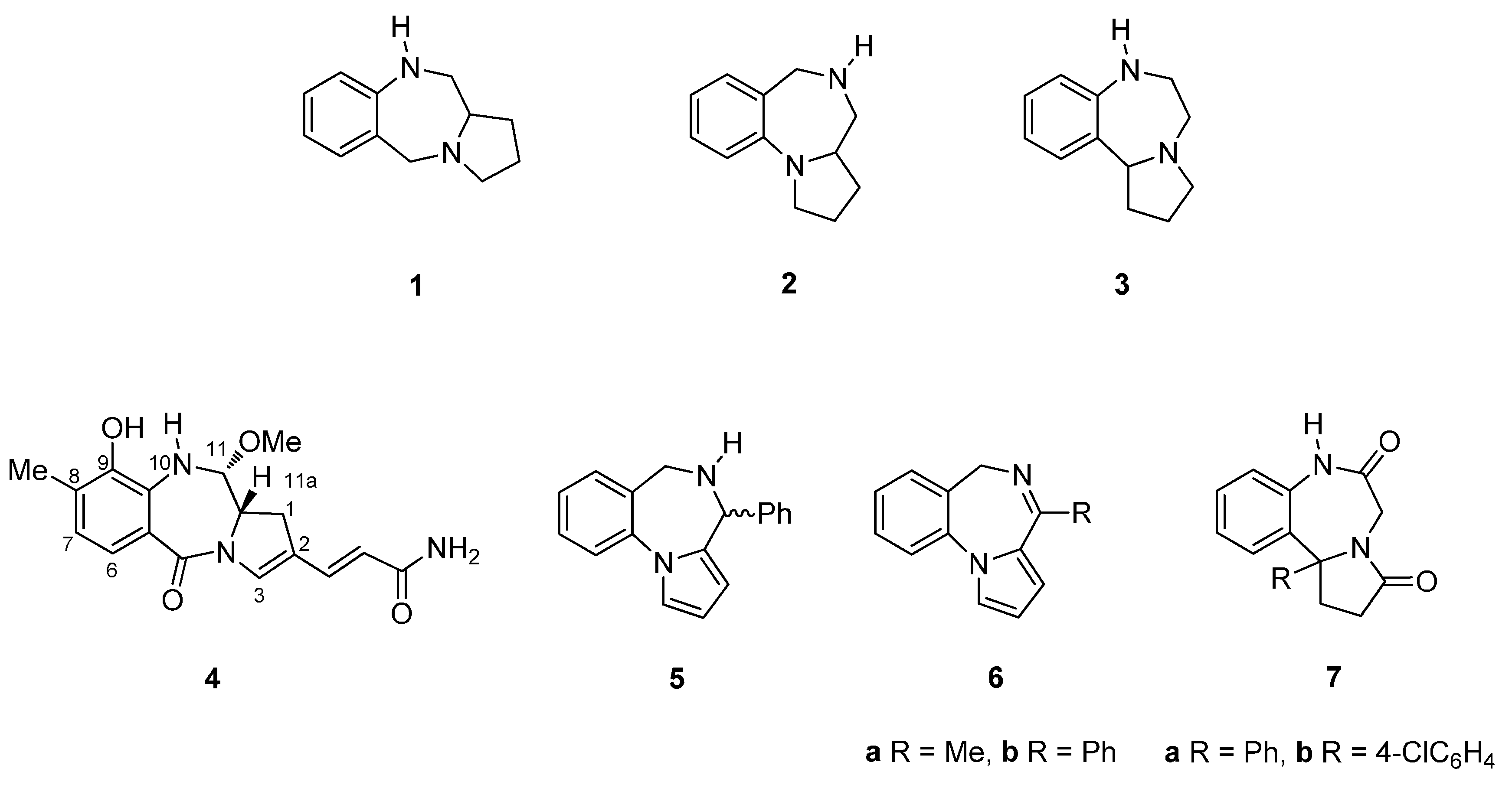
1.1. Pyrrolo[2,1-c][1,4]benzodiazepines
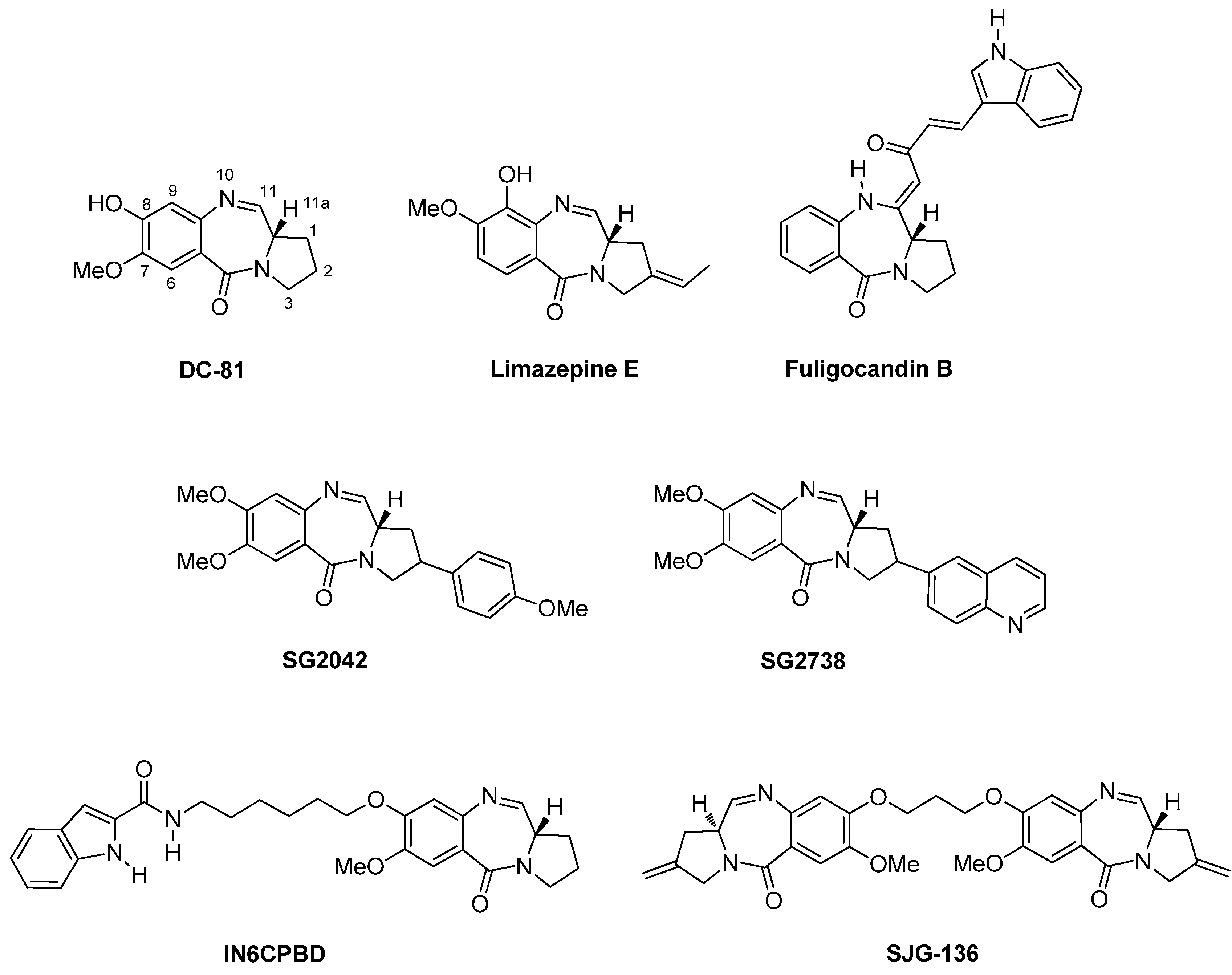
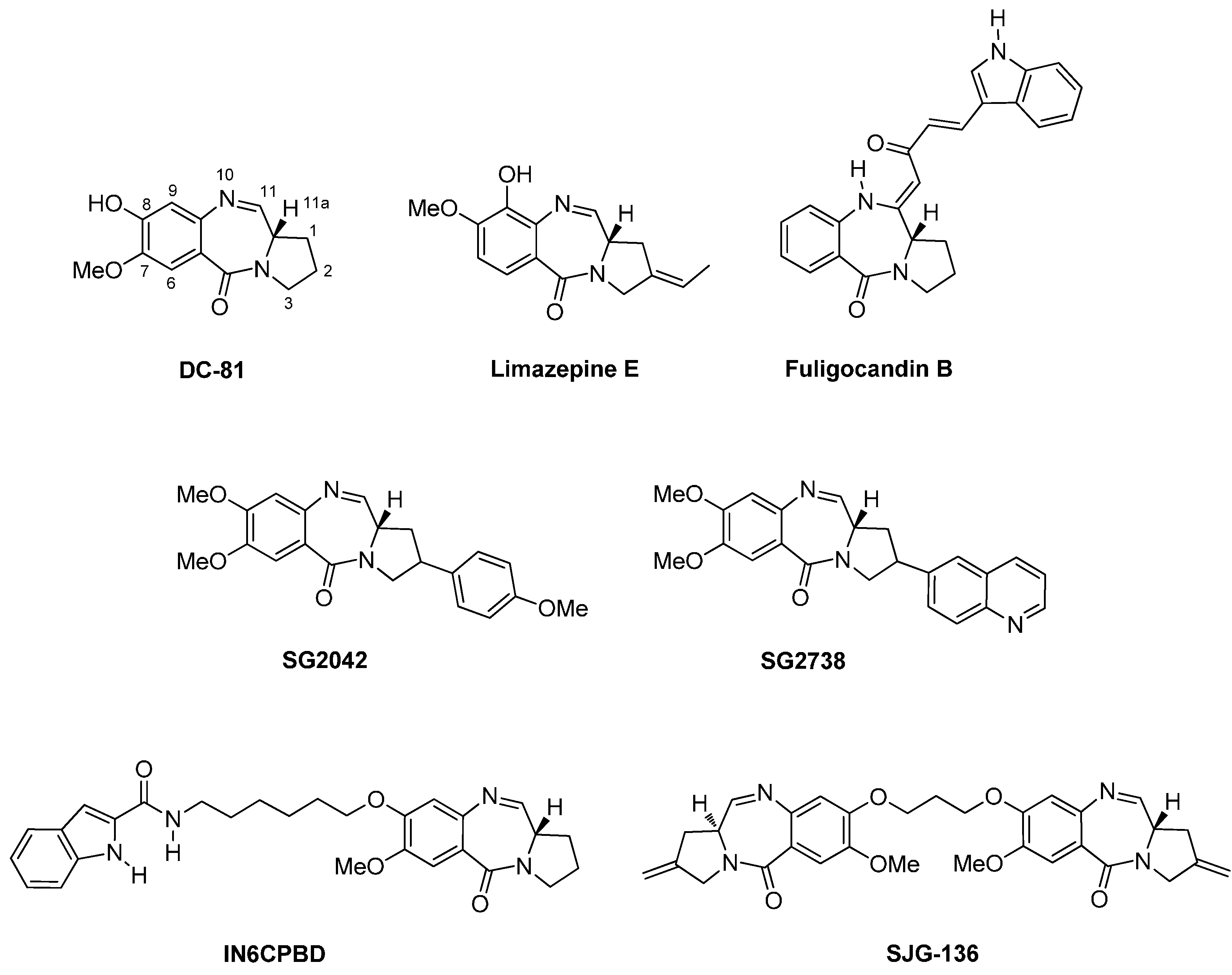
Since the isolation of anthramycin (4, Figure 1) from cultures of Streptomyces by Leimgruber et al., in 1965 [4], the chemistry and biology of pyrrolo[2,1-c][1,4]benzodiazepines took the lead as the most intensively studied isomer of the three. Other important PBDs isolated from Streptomyces species are sibiromycin, tomaymycin, the neothramycins and DC-81 [5]. Their mode of action relies on the right-handed helical conformation due to the (S)-configuration at C-11a, found from an X-ray crystal structure analysis of anthramycin (4) [6], enabling the molecules to adapt in the minor groove of DNA, selectivity at the 5′-purine-G-purine sequences, by alkylating the C2 amino group of guanine [7]. Anthramycin was the most promising of these natural products having activity against gastrointestinal and breast cancers, lymphomas and sarcomas. Although possessing low haematological toxicity, clinical use is limited because of dose-limiting cardiotoxicity. More recently limazepine E was isolated from a culture broth of Micrococcus [8] and fuligocandin B from the myxomycete Fuligo candida [9] (Figure 2).
The unwanted side-effects of the naturally occurring PBDs led to prolific research on the synthesis of analogues, known as PBD monomers. Two notable examples that have been tested in vivo against various human tumour xenografts are SG2042 and SG2738 possessing the C10-C11 imine structure (Figure 2), but at the end of the line no critical DNA damage was observed. Moreover, structure-activity relationship (SAR) studies have shown that linking primarily position C8 of DC-81 with other moieties such as other DNA intercalators or pyrrole and imidazole polyamide analogues of dystamycin and neotropsin, would produce PBD conjugates possessing two pharmacophoric heads with improved in vitro DNA binding affinity, sequence selectivity and/or cytotoxic efficacy. One promising outcome of this research was the work of Wang and co-workers [10] who designed and synthesized the indole-PBD conjugate IN6CPBD (Figure 2) and found that this compound exhibited higher cytotoxicity than DC-81 against human melanoma A375 cells and displayed enhanced DNA sequence selectivity. A significant breakthrough in the search of PBD antitumour agents was the discovery of PBD dimers, designed to be capable of creating cross-links in the DNA by forming covalent bonds to guanine bases at each end of the molecule. This work culminated in the development of C2-exo-methylene PBD dimer SJG-136 (Figure 2), at present in Phase II clinical trials.
The extensive literature on the isolation of PBD natural products and the synthesis of PBD monomers has been reviewed first by Thurston and Bose in 1994 [11]. An update including PBD conjugates and PBD dimers was reported recently by Antonow and Thurston [12]. In the present review the synthetic chemistry literature from 2010 to mid-2015 is covered.
Figure 2.
DC-81, Limazepine E and Fuligocandin B are natural products, SG2042 and SG2738 are PBD monomers, IN6CPBD is a PBD conjugate and SJG-136 is a PBD dimer.
Figure 2.
DC-81, Limazepine E and Fuligocandin B are natural products, SG2042 and SG2738 are PBD monomers, IN6CPBD is a PBD conjugate and SJG-136 is a PBD dimer.
Recently, a review by Hartley [13] described the development of PBD as antitumour agents while a review by Gerratana [14] reported the biosynthesis, synthesis, and biological activities of PBDs. The synthetic chemistry section of the latter article does not provide coverage of the work published since the last review [12]. The evaluation of the antitumour activity of PBDs is dominated by in vitro cytotoxicity against tumour cell lines, and, DNA thermal denaturation experiments and DNA footprinting assays, used to measure the DNA-binding affinity and find the sequence selectivity of these compounds.
The publications [15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35], cover the period 2010 to the middle of 2015, and refer solely to biological studies of PBDs. Three publications [36,37,38] are devoted to spectroscopic studies on PBDs whereas one publication deals with computational studies of PBDs [39] and another with the isolation of natural PBDs [40].
1.2. Pyrrolo[1,2-a][1,4]benzodiazepines
The comparatively small number of publications on the [1,2-a] and [1,2-d] isomers is a reflection of the lesser interest in the biology of these compounds. Nevertheless, it is known that pyrrolo[1,2-a][1,4]benzodiazepines exhibit a wide spectrum of biological effects, such as antinociceptive CNS [41,42], anti-inflammatory [41], analgesic [43] and fungicidal [44] activity, and are potent sedative [41,45,46], anticonvulsant [45,46], myorelaxant [45,47] and psychotropic [43,47] agents.
1.3. Pyrrolo[1,2-d][1,4]benzodiazepines
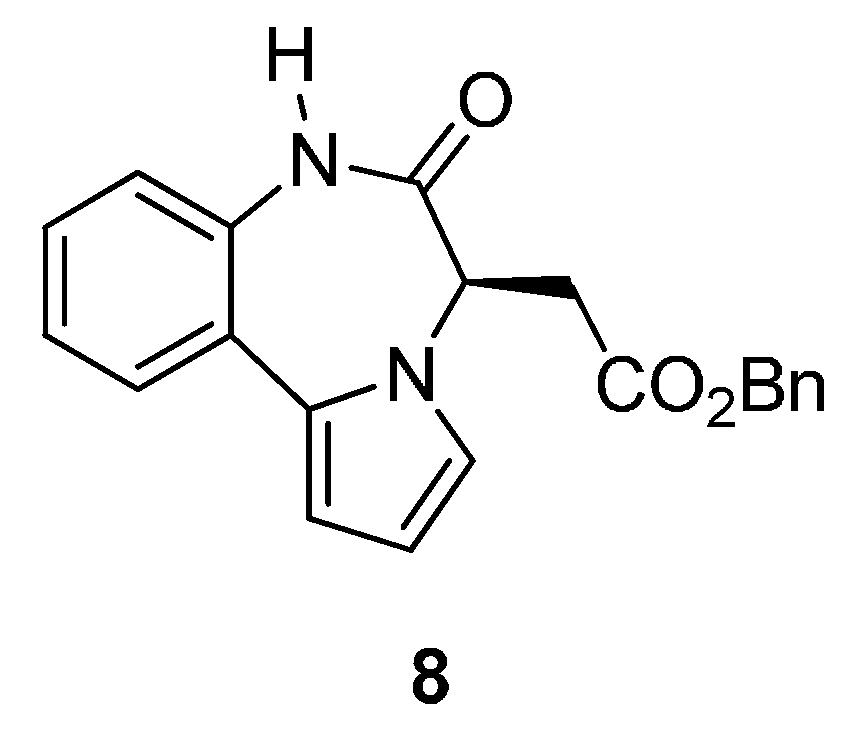
Among the small number of publications on the pyrrolo[1,2-d][1,4]benzodiazepine ring system, one reference has been made to PBD 8 (Figure 3), as an important non-nucleoside HIV-1 reverse transcriptase inhibitor with IC90 = 0.29 μg/mL [48].
Figure 3.
A biologically active pyrrolo[1,2-d][1,4]benzodiazepine.

1.4. General Information
The review presents the synthetic routes to PBDs from simple starting materials according to the cyclisation procedure taking place to produce the PBD and also by the type of PBD synthesized.
2. Synthesis of Pyrrolo[1,4]benzodiazepines
2.1. Pyrrolo[2,1-c][1,4]benzodiazepines
The most common features of the pyrrolo[2,1-c][1,4]benzodiazepines presented in this review are: (i) an aromatic or non-aromatic pyrrole ring; (ii) one carbonyl group on the diazepine ring that is at position 5 of the PBD, then N10 could be a secondary amine, substituted tertiary amine or a N10-C11 imine; (iii) two carbonyl groups on the diazepine ring that are at positions 5 and 11 on the PBD, then these compounds are considered “dilactams” and (iv) monomers with various medium sized substituents, monomers that possess an extended C8-O-linked chain whose structure can possess various combinations of alkyl, ether, amide, carboxylic ester, carbodithioic ester, α,β-unsaturated carbonyl, carbocyclic and heterocyclic substitution (these as named “PBD conjugates”) and dimers that are linked via the O-atom at their C8 positions where linkers can be either a linear alkyl chain or a combination of alkyl and carbocyclic entities. C7-linked PBD conjugates and PBD trimers have not been reported in the past five years.
The most important contributions towards the chemistry and biological activity of pyrrolo[2,1-c][1,4]benzodiazepines have been from Antonow, Bose, Hurley, Kamal and Thurston, among which the last two authors have published the largest number of scientific papers in this field of research.
2.1.1. Pyrrolo[2,1-c][1,4]benzodiazepine-5-one Monomers or Dimers with a Non-Aromatic Pyrrole Ring
During the last five years, three novel [2,1-c] PBD syntheses have been reported in this category of compounds, (Section “Reductive cyclisation of N-(2-azidobenzoyl)pyrrolidine-2-carboxaldehydes”), (Section “Cyclisation of N-(2-azidobenzoyl)pyrrolidine-2-carboxaldehydes”), although, a large number of new compounds containing [2,1-c] PBD scaffolds in their structure have been synthesized in the search of new effective anticancer agents. These include PDB conjugates and PDB monomers that possess an extended C8–O-linked pharmacophore, the most used strategy.
In this sense, the established cyclisation of N-(2-aminobenzoyl)pyrrolidine-2-carboxaldehyde diethyl thioacetals, described first by the Kamal group [49], is by far the most frequently used method for synthesizing these compounds. Several groups such as alkyl, carbonyl and thioacetal and ring structures including estradiol, chalcone, anisole, naphthyl, piperazine, 1,2,3-triazole, carbazole, benzo[c,d]indole, naphtha[1,8-c,d]isothiazole, bisindole, imidazo[1,2-a]pyridine and 1,3-benzothiazole, have been chosen in various combinations to form the second pharmacophore, that is connected to the C8–O of the PBD nucleus by the synthetic methodology presented in the following section.
Cyclocondensation of N-(2-Aminobenzoyl)pyrrolidine-2-Carboxaldehyde Diethyl Thioacetals
All of the PBDs produced from the reactions of this subsection have an N10–C11 imine functionality in their structures. The imine group is known to interact electrophilically with the C2–NH2 group of a guanine within the minor groove of DNA and therefore there is a strong interest to synthesize analogues of these PBDs in order to study this interaction.
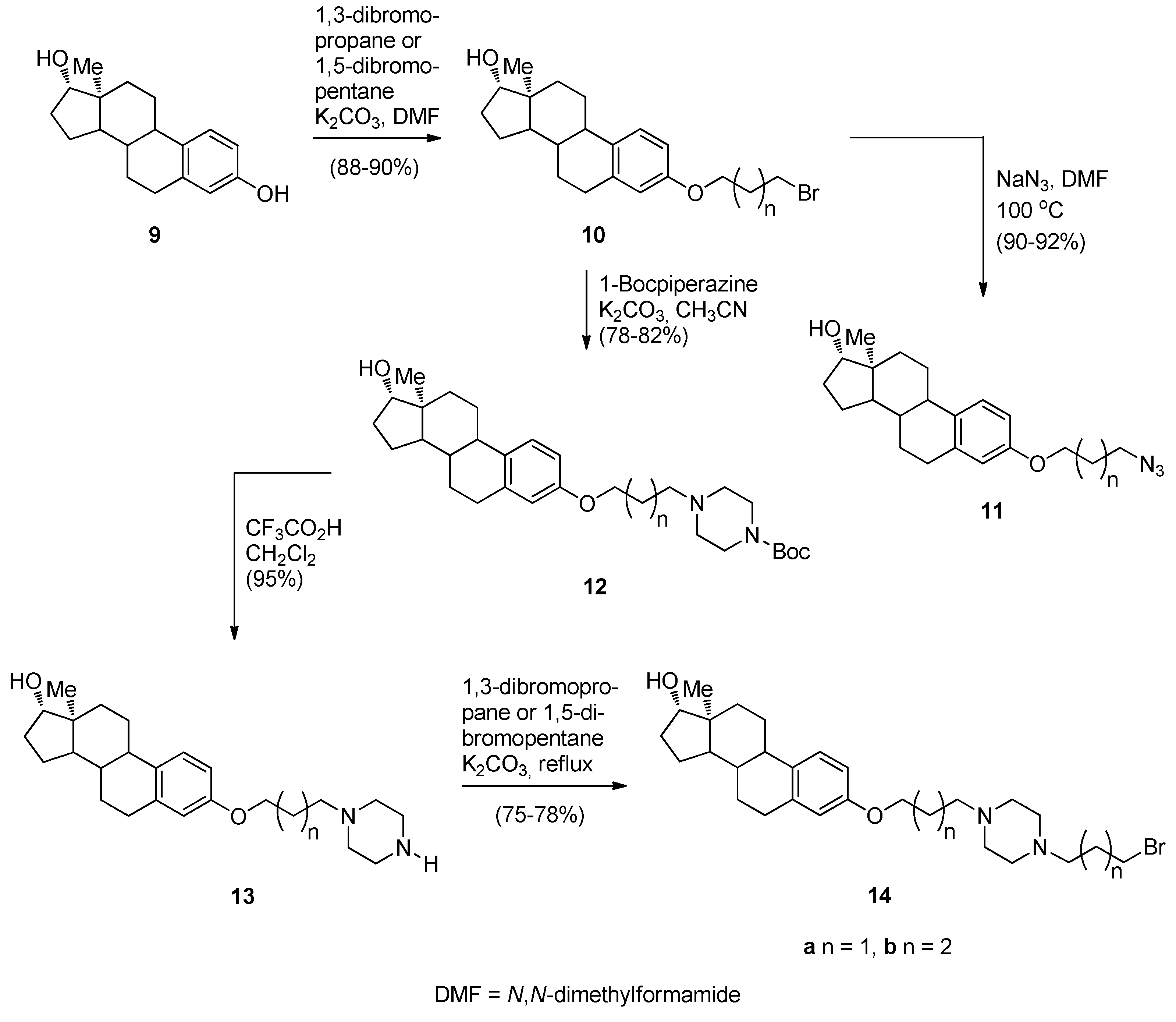
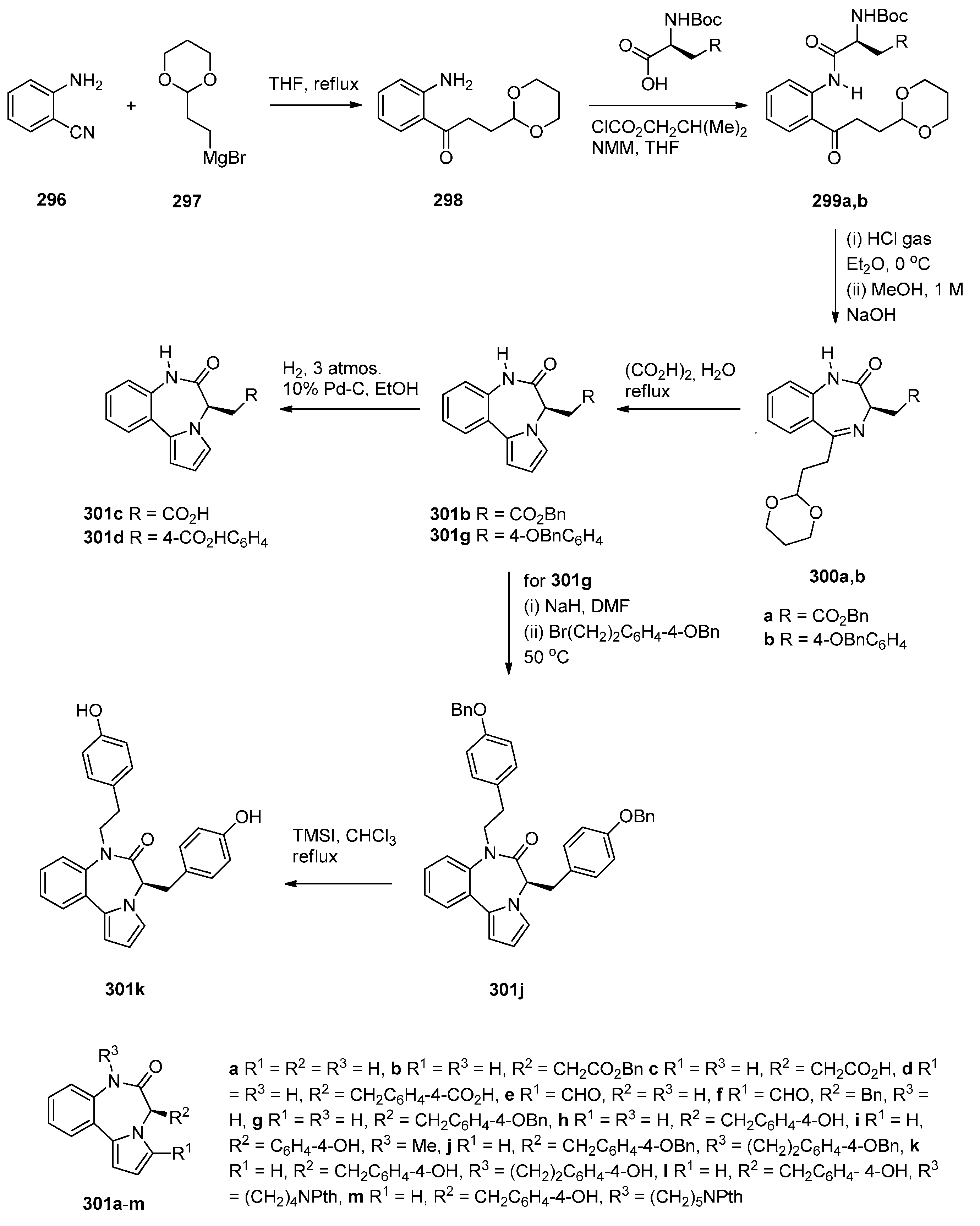
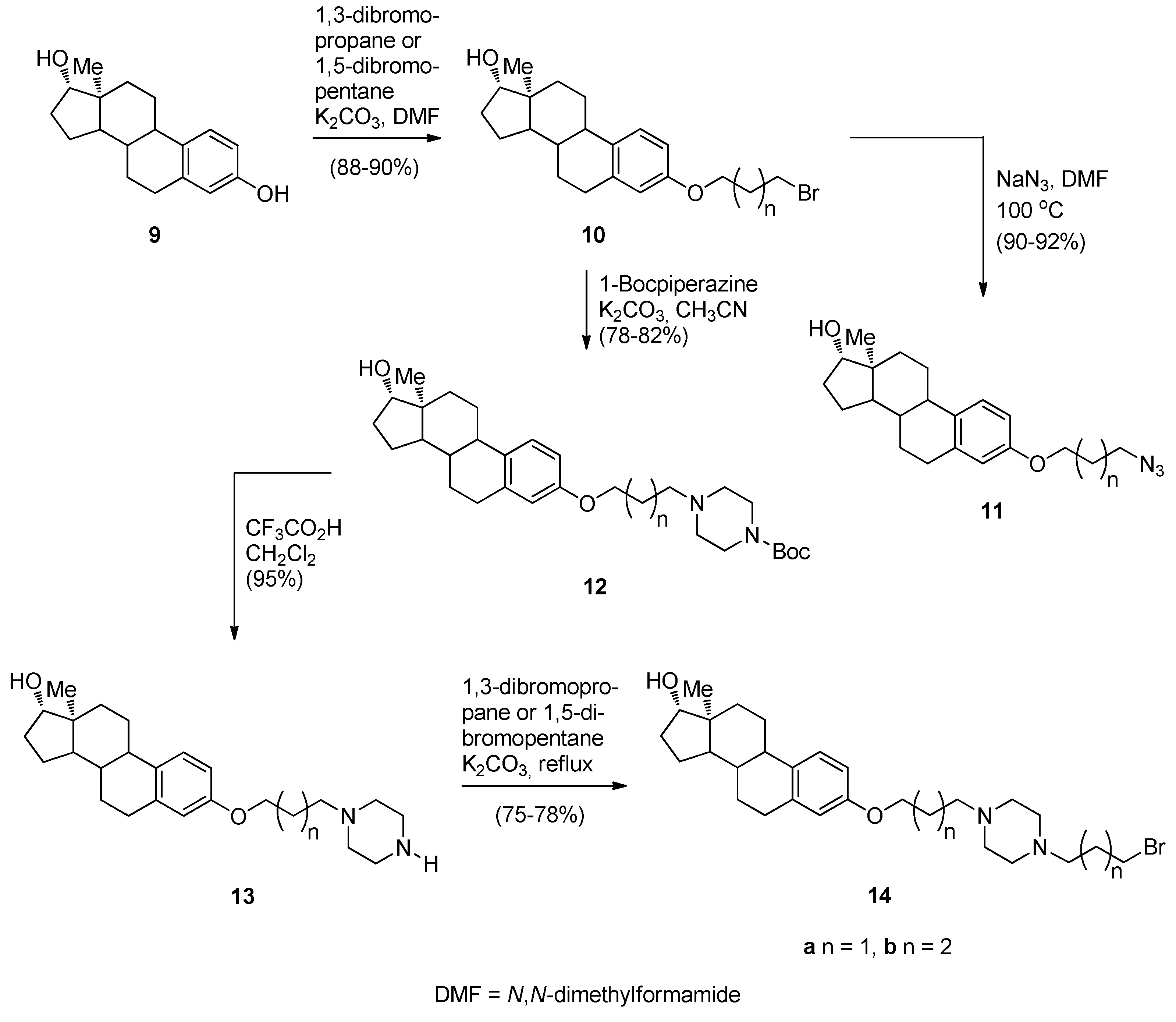
Continuing their efforts in search of new effective anticancer agents, after having established themselves in the field of [2,1-c] PBDs, Kamal et al. [50] have designed, synthesized and studied the biological activities of several novel estradiol PBD conjugates 17a–f, 19a–f and 22a–f (Scheme 1, Scheme 2 and Scheme 3). The convergent approach described in these schemes is the most common method of preparing PBD conjugates of this type. The synthetic strategy necessitated attaching to the phenolic side of estradiol (E2), stable bromoalkane spacers or a piperazine or 1,2,3-triazole moieties which are then linked to DC-81 (Figure 2). In the first step towards this goal estradiol derivatives 10a,b, 11a,b and 14a,b were synthesized as outlined in Scheme 1. Estradiol 9 was alkylated with appropriate dibromoalkanes under basic conditions to afford the alkyl bromides 10a,b which were transformed into azides 11a,b or converted into the protected piperazine Boc derivatives 12a,b and then deprotected to provide secondary amines 13a,b. These amines were monoalkylated as above to the corresponding alkyl bromides 14a,b.
Scheme 1.
Synthesis of estradiol derivatives.
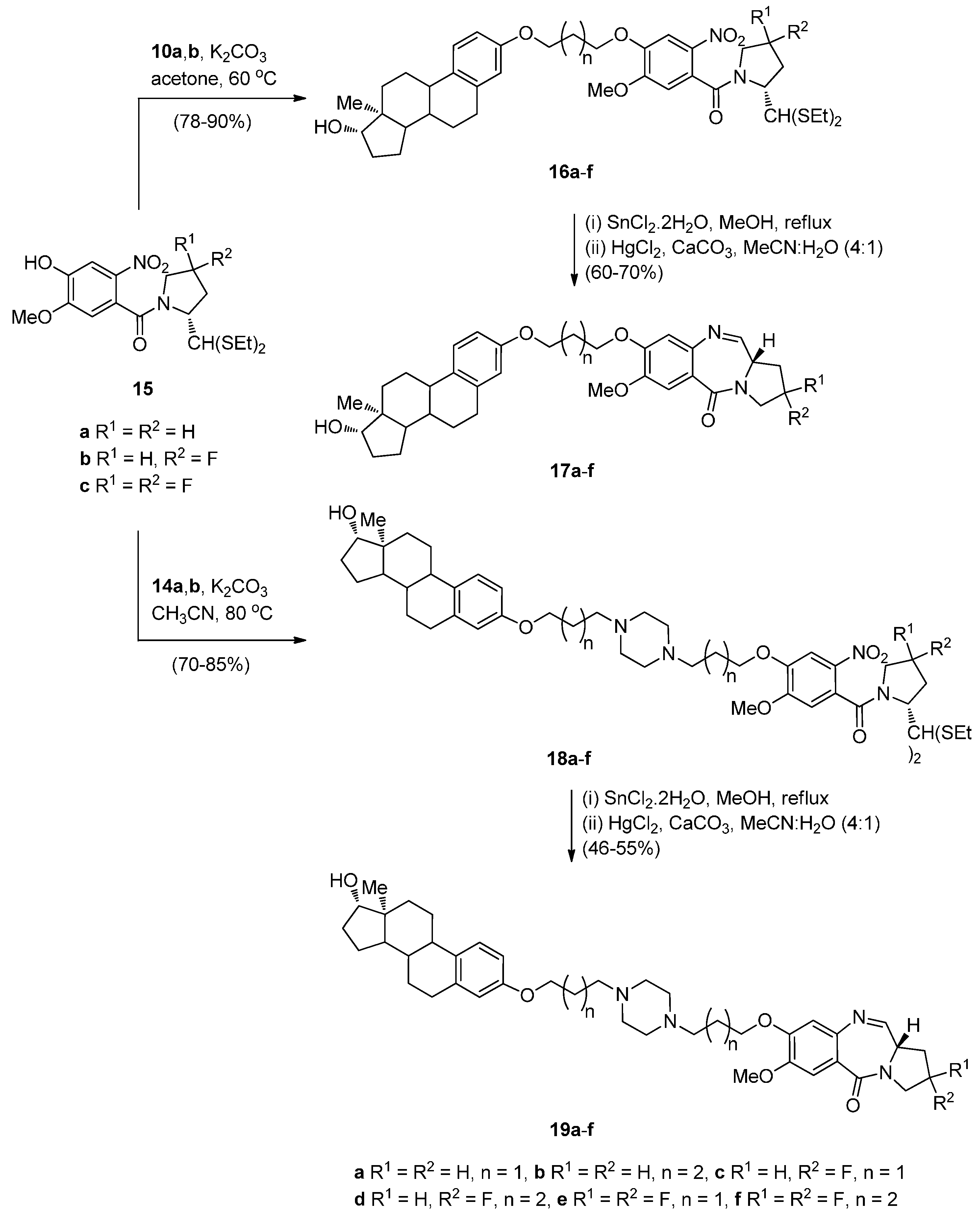
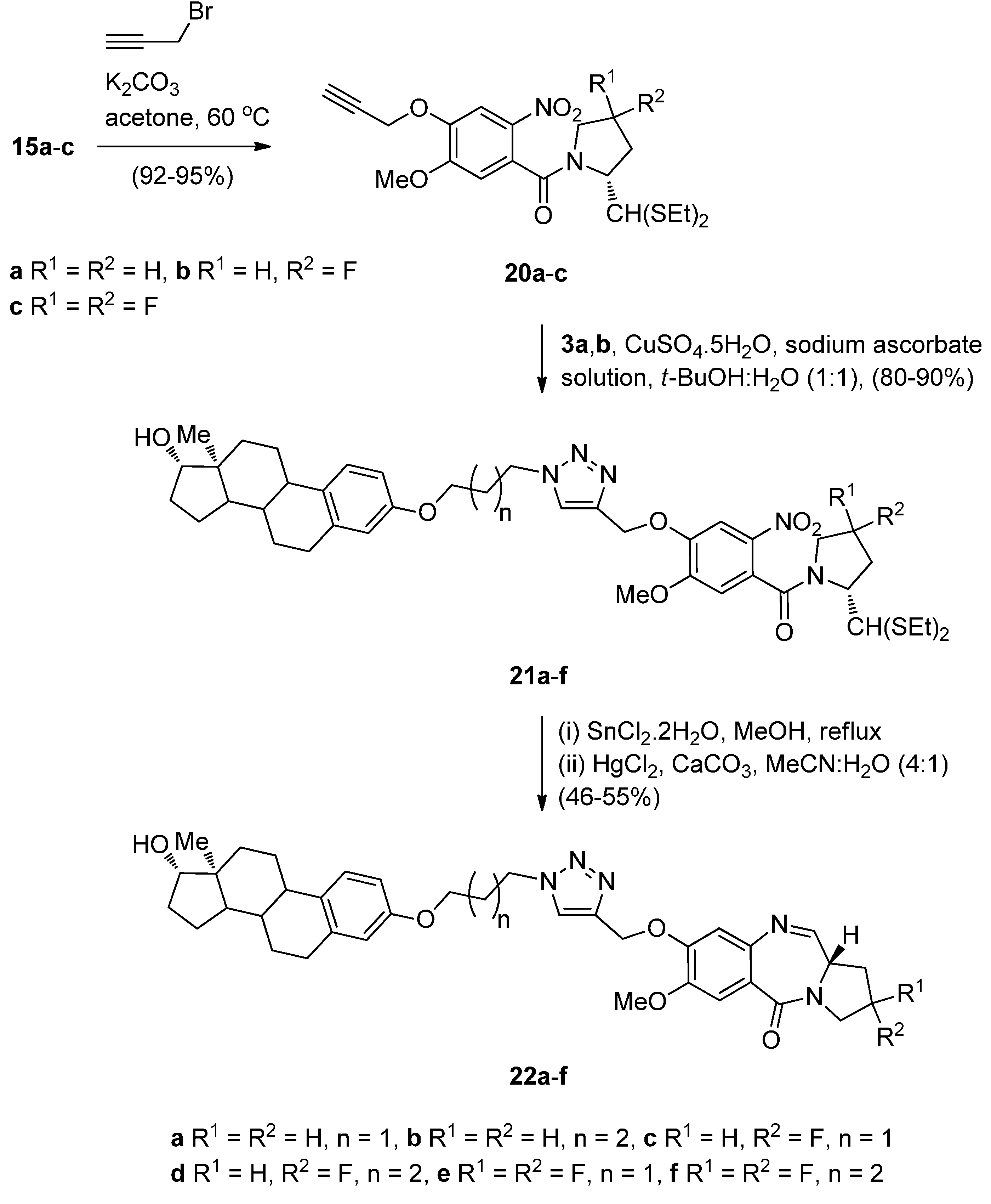
The synthesis of estradiol PBD conjugates 17a–f and 19a–f required in the first step the alkylation of simple nitro-thioacetals 15a–c, previously reported [49], with the corresponding alkyl bromides 10,b and 14a,b to produce substituted nitro-thioacetals 16a–f and 18a–f, respectively (Scheme 2). The method of preparing PBDs by reduction and deprotection of N-(2-nitrobenzoyl)pyrrolidine-2-carboxaldehyde diethyl thioacetals was also widely used up to 2010 [10]. The advantages of the diethyl thioacetal protective group are the ease with which it is introduced to an aldehyde precursor, its stability that enables, if required, a variety of chemical transformations to take place to nitro thioacetal scaffolds of type 15 and that no racemization occurs at the C11-a position of the final PBD structures. In the present synthesis, as with most syntheses of this type since 2011, the phenolic hydroxyl group of simple nitro-thioacetal scaffolds such as 15 is alkylated (i.e., 15a–c to 16a–f and 18a–f above) before reduction, deprotection and cyclisation to the final PBD. Reductive methods usually employ tin(II) chloride dihydrate (SnCl2·2H2O) in methanol and heating to reflux, as in the reduction of 16a–f and 18a–f, although, on several occasions hydrogenation with 10% palladium-on-carbon catalyst in methanol has been effective [10]. In general, regardless of the reduction method, after work-up, the resulting amino-thioacetals are obtained crude in 80%–90% yields and are directly used in the next step, due to potential stability problems. Thus, the crude amino-thioacetals from the tin(II) chloride dihydrate reduction of 16a–f and 18a–f were treated with mercury(II) chloride (HgCl2) and calcium carbonate and the resulting aminoaldehyde spontaneously cyclized to afford the estradiol PBD conjugates 17a–f and 19a–f, in 46%–70% overall yields. Although these established reaction conditions work well for this type of cyclisation method, the use of highly toxic mercury(II) chloride for deprotecting the thioacetal group does not however conform with green chemistry protocols. Nevertheless, this reagent remains in use even if efficient deprotection of thioacetals by less toxic iron(III) chloride hexahydrate or bismuth triflate has been reported [10]. Moreover, it seems that the use of the thioacetal protective group has not yet been overcome due to its efficiency, even if handling ethyl mercaptan during the protection/deprotection steps is inconvenient due to its pungent smell.
Scheme 2.
Synthesis of C8-O-substituted PBD conjugates 19a–f.

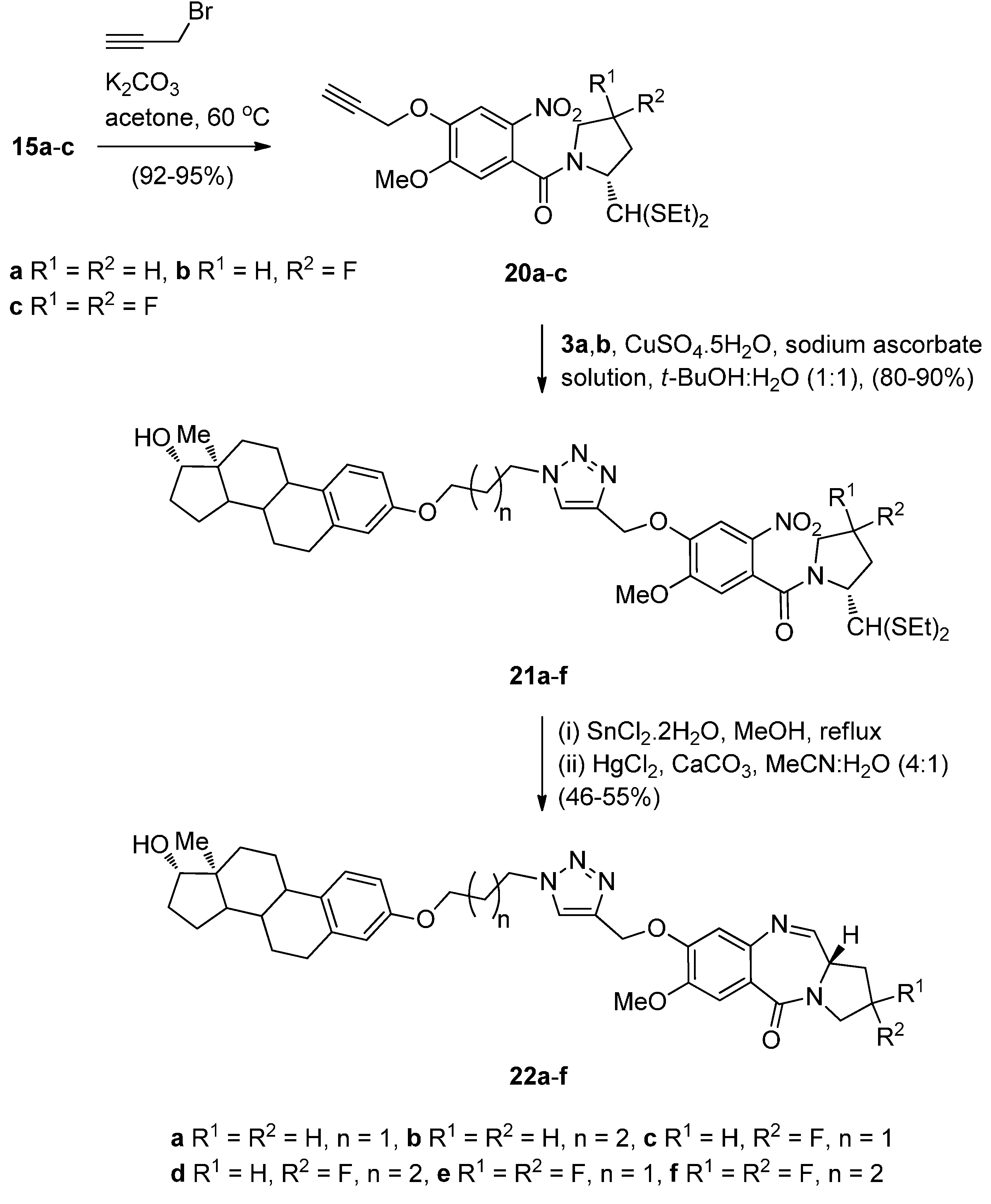
Compounds 15a–c were alkylated with propargyl bromide in base to afford the terminal alkynes intermediates 20a–c in high yields (Scheme 3). These were further reacted with azides 11a,b to undergo the “click” reaction in the presence of in situ generated Cu(I) ions, to produce the estradiol 1,2,3-triazolenitrothioacetals 21a–f in over 80% yields. Although this “click” chemistry has been used for the first time in 2011 to synthesize C8–triazole linked PBD conjugates of type 22, the same author has previously reported [10] triazole linked PBD dimers and trimers where the cycloaddition step proceeded in comparable yields. Under the established reaction conditions, described above, compounds 21a–f were converted into estradiol PBD conjugates 22a–f, in moderate yields. PBD conjugates 22a,b are derivatives where the estradiol pharmacophore is linked to the C8-position of DC-81.
Scheme 3.
Synthesis of C8-O-substituted PBD conjugates 22a–f.
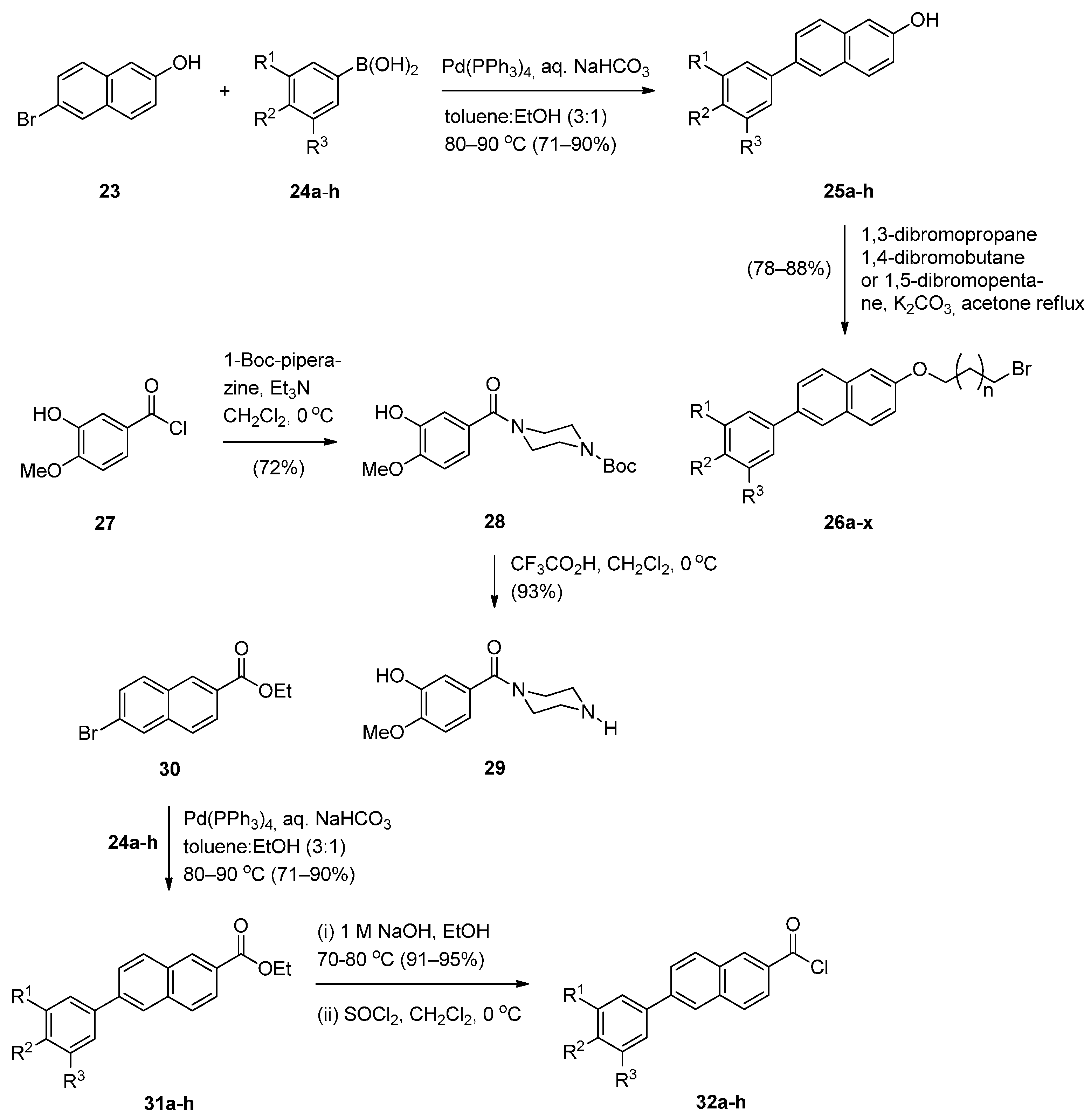
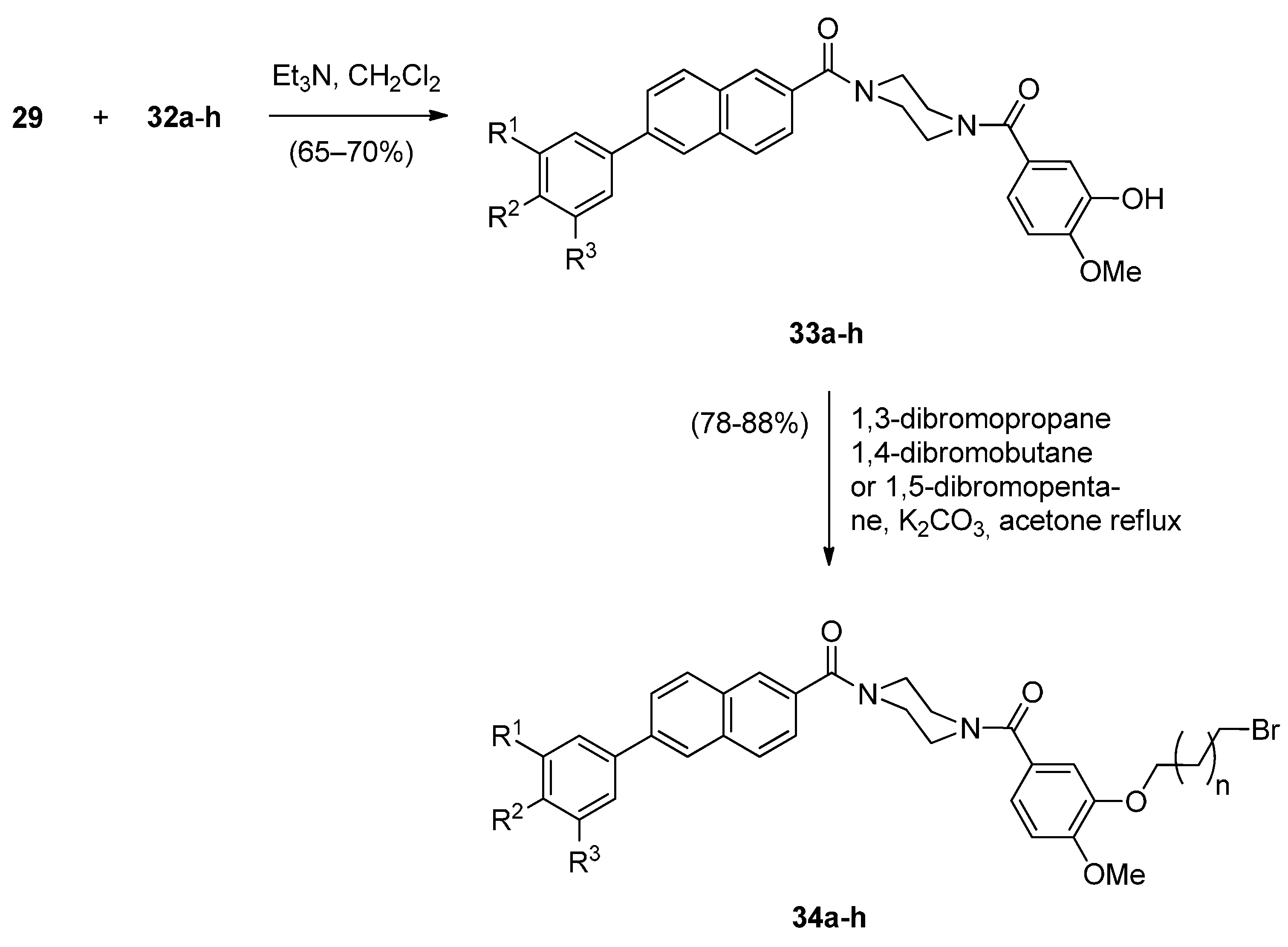
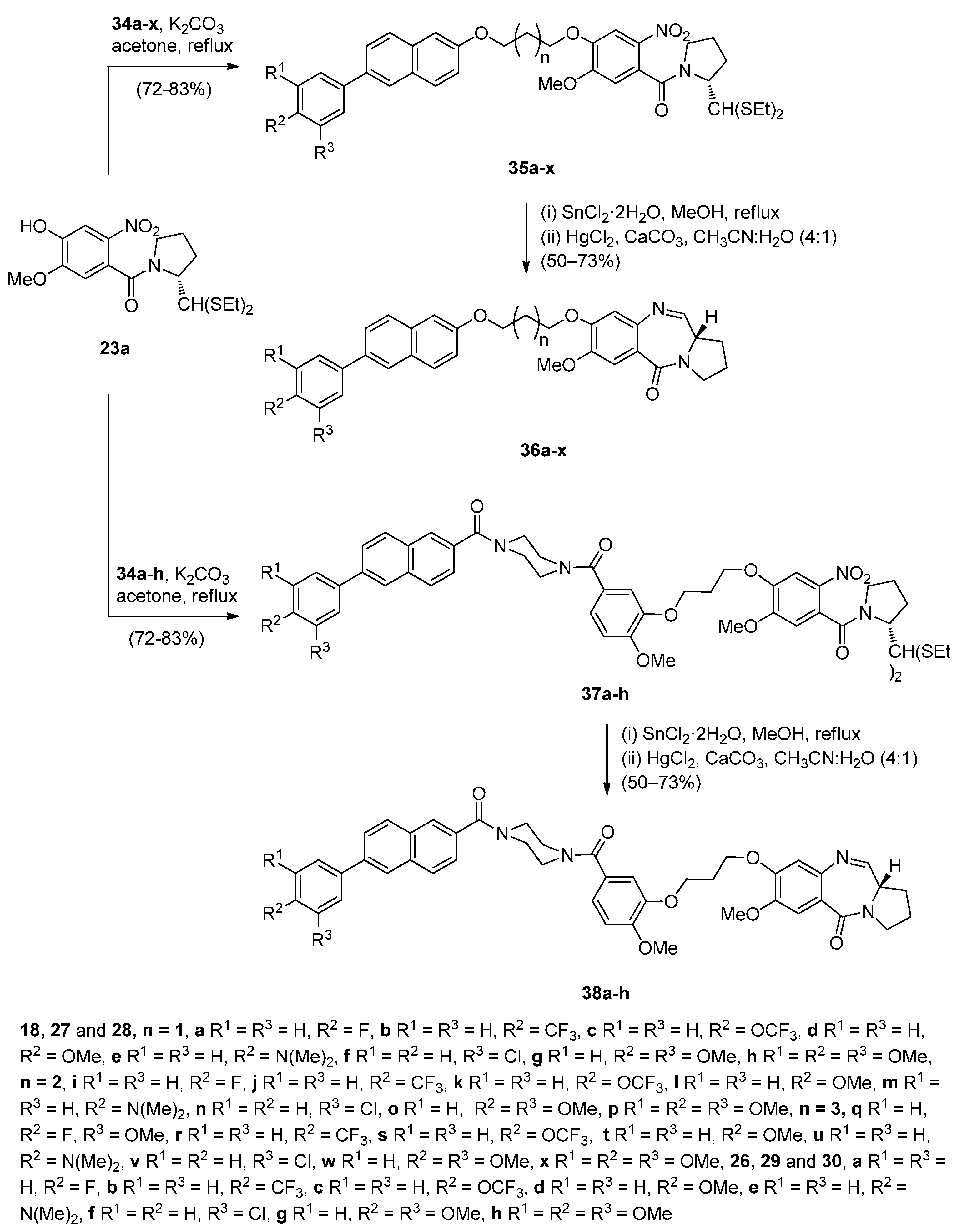
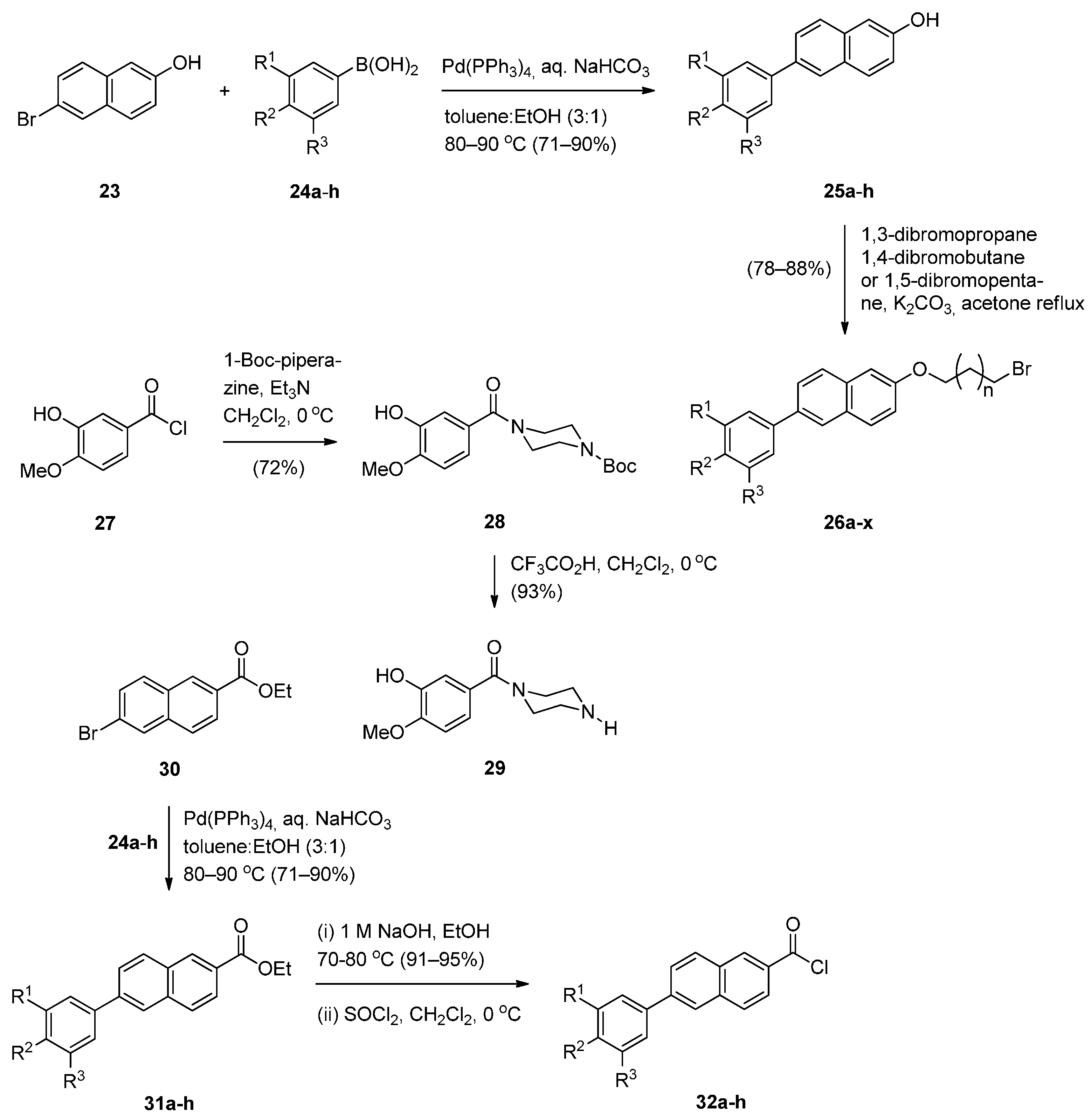
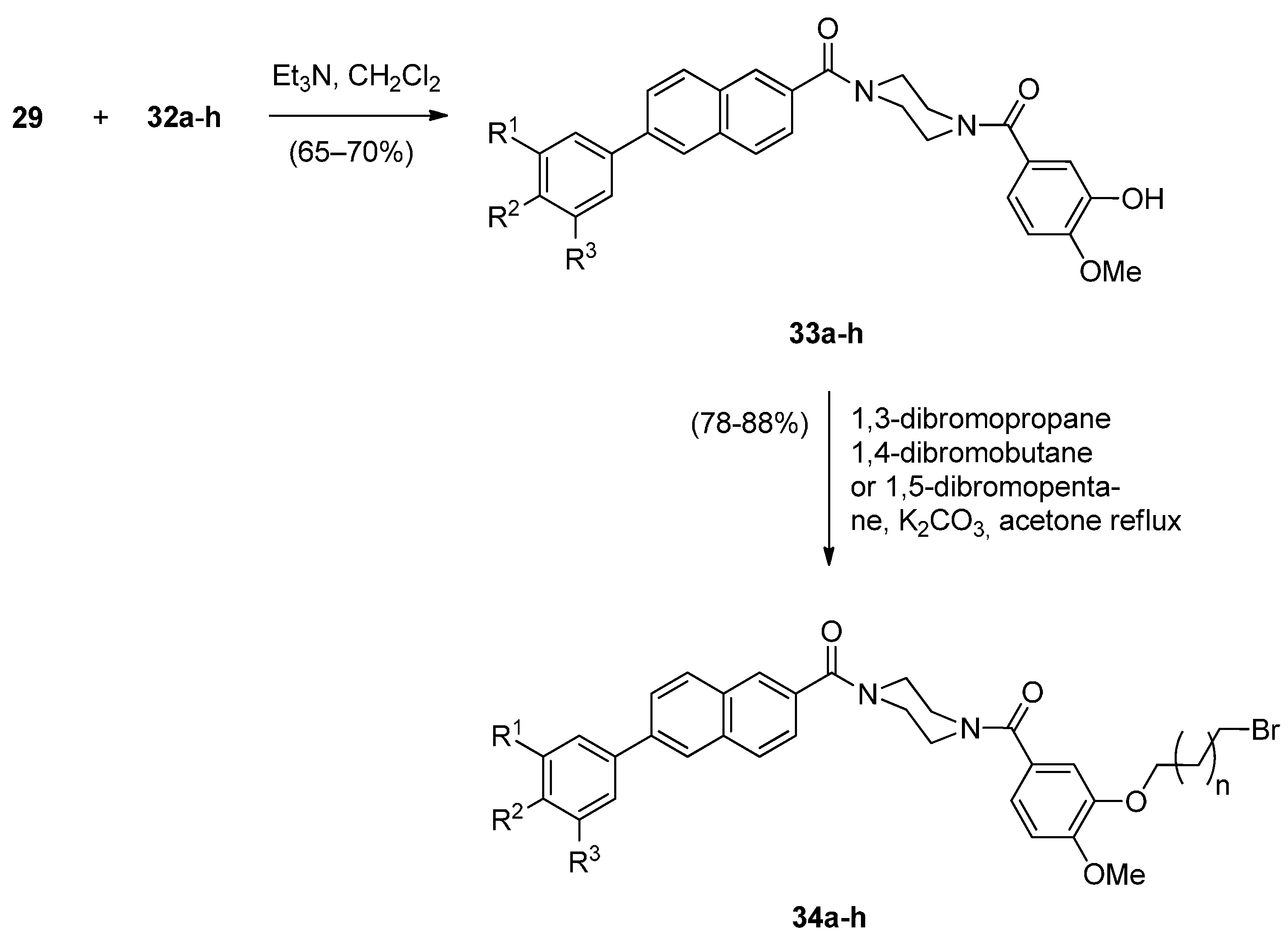
The significant anticancer activity of both piperazine and naphthalene derivatives as well as piperazine-containing PBDs, motivated Kamal et al. [51] to explore various aryl-substituted naphthalene derivatives linked to the C8-position of DC-81 through stable alkane linkers and also by incorporating a piperazine moiety, as pharmacophores in the design of novel monomeric PBD conjugates 36a–x and 38a–h. The precursors for the synthesis of these PBDs are the bromoalkyl derivatives 26a–x and 34a–h that are prepared as shown in Scheme 4 and Scheme 5. The difference between the synthetic strategies towards PBDs 36a–x and 38a–h (Scheme 6) and PBDs 22a–f (Scheme 3) is the method by which the non-PBD pharmacophores are linked to the hydroxyl group of the nitro-thioacetal precursors 15a–c. Bromoalkyl derivatives 26a–x were prepared in two steps from 6-bromo-2-naphthol 23 phenylboronic acids 24a–h by a Suzuki palladium-catalyzed cross coupling reaction followed by an alkylation reaction with appropriate dibromoalkanes. 3-Bromopropyl 2-naphthyl ethers 34a–h were prepared by acyl substitution of acid chlorides 32a–h by 2-methoxy-5-(piperazin-1-ylcarbonyl)phenol 29 to give diamides 33a–h which were alkylated by 1,3-dibromopropane (Scheme 5).
Scheme 4.
Synthesis of 2-methoxy-5-(piperazin-1-ylcarbonyl)phenol 29 and acid chlorides 32a–h.
Scheme 5.
Synthesis of bromoalkyl derivatives 34a–h.
Scheme 6.
Synthesis of C8-O-substituted PBD conjugates 38a–h.
In the final two steps towards target C8-substituted PBDs 36a–x and 38a–h (Scheme 6) the hydroxyl group of dithioacetal 15a was alkylated by the bromoalkyl derivatives 26a–x and 34a–h to provide the respective nitro-thioacetals 35a–x and 37a–h, which upon reduction of the nitro group to amino with tin(II) chloride dihydrate followed by mercury(II) chloride and calcium carbonate deprotection and subsequent spontaneous cyclisation of the resulting amino aldehyde, afforded the required PBD conjugates 36a–x and 38a–h, in yields ranging from 78% to 88%. The cyclisation of nitro-thioacetals 21a–f (Scheme 3) by a similar manner gave much lower yields (46%–55%) of PBD conjugates 22a–f.
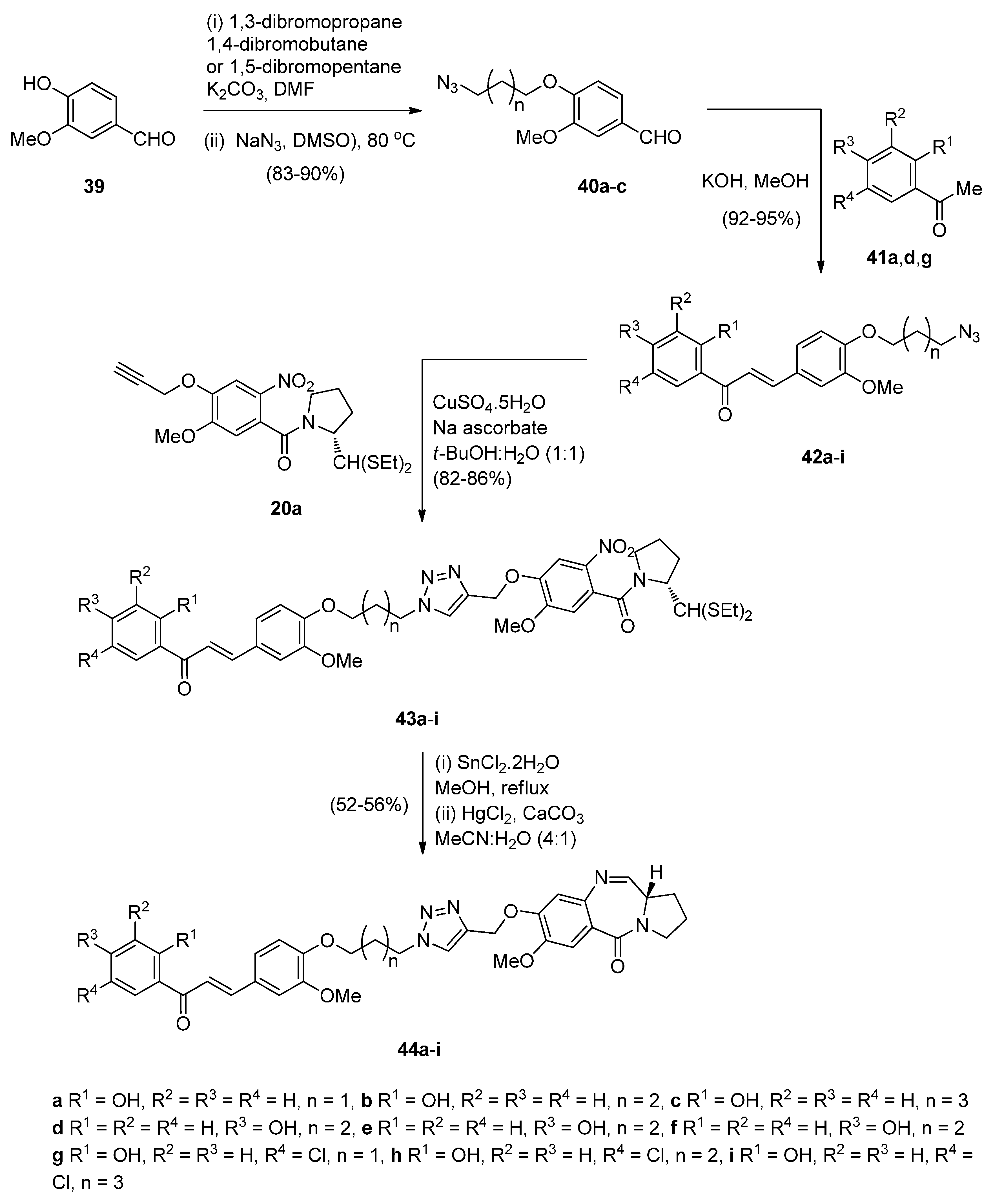
Although chalcones and 1,2,3-triazoles are known to exhibit anticancer activity, chalcone has been further involved as a scaffold in structural modifications for the development of anticancer agents. With this in mind, Kamal et al. [52] set out to study the synthesis and biological evaluation of monomeric triazolochalcone-PBD conjugates 44a–i and 49a,b (Scheme 7 and Scheme 8). The synthetic strategy relies upon two key intermediates, that is azidochalcones 42a–e and alkyne 20a, which via 1,3-dipolar cycloadditions will provide the triazole derivatives 43a–i, that will then lead to the monomeric PBD conjugates 44a–i by the appropriate ring closure. This synthetic procedure is quite similar to that described by the same author in Scheme 3 where the non-PBD pharmacophore contains an azide group which undergoes cycloaddition with an alkyne substituted nitro thioacetal to form the connective triazole ring prior to cyclisation to the PBD conjugate. Azidochalcones 42a–e (Scheme 7) were prepared by etherification of vanillin 39 with suitable dibromoalkanes followed by azidation of the non-isolable bromoalkylethers to produce the corresponding azido precursors 40a–c which were then condensed with acetophenones 41a,d,g. At this stage the “click” chemistry protocol was introduced between alkyne 20a and azides 42a–e to afford nitro intermediates 43a–i. Reduction, deprotection and cyclocondensation by the established reaction conditions (Scheme 7) afforded the PBD conjugates 44a–i.
Scheme 7.
Synthesis of C8-O-substituted PBD conjugates 44a–i.

Scheme 8.
Synthesis of C2-substituted PBD conjugates 49a–b.

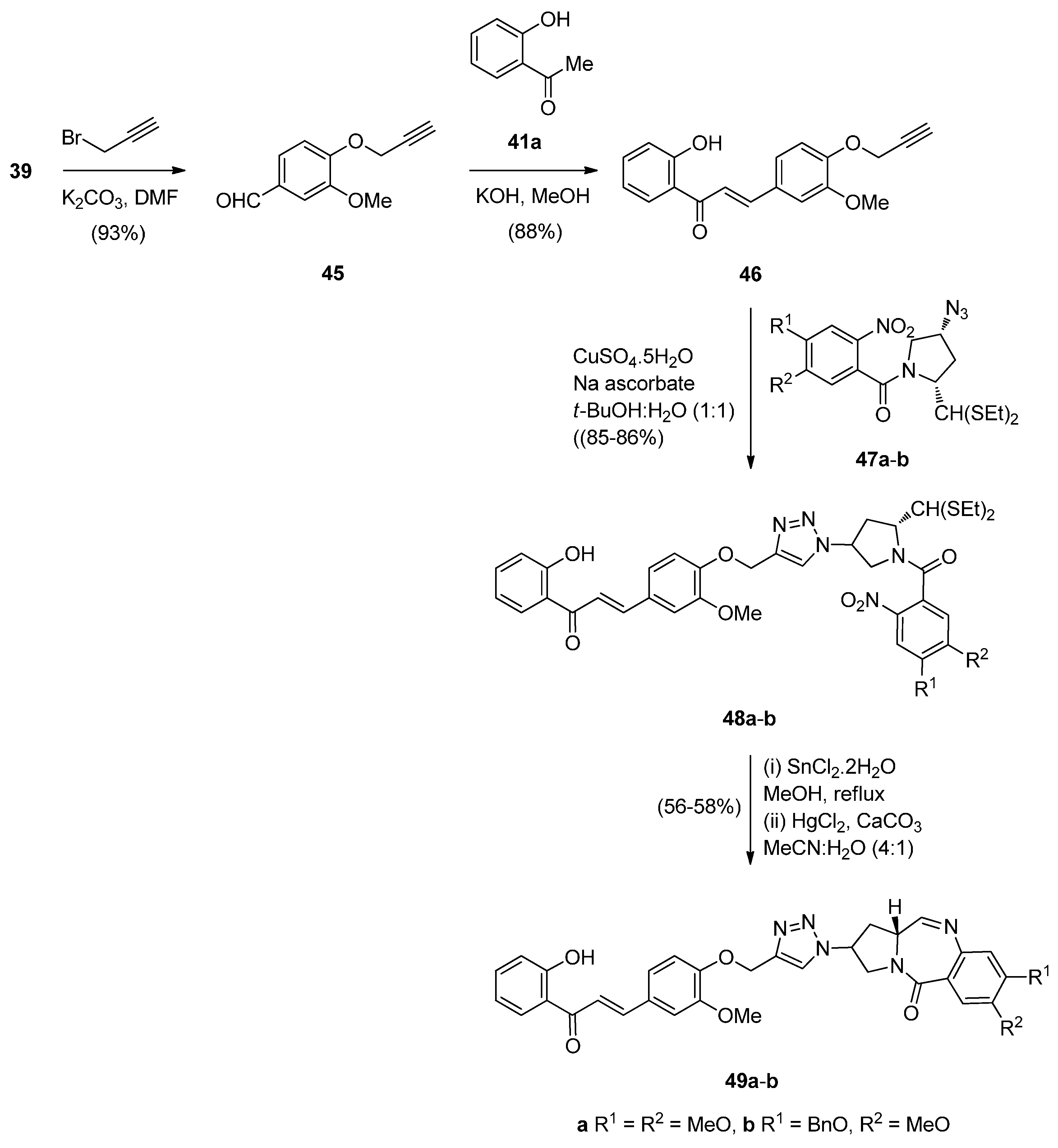
The authors diversified this synthetic methodology for the synthesis of PBD conjugates 49a,b (Scheme 8) by tethering the alkyne moiety on the non-PBD pharmacophore and introducing the azide group on the pyrrolidine ring of the nitro-thioacetal precursor. Thus, cycloaddition of alkyne 46 with azides 47a,b provided 1,2,3-triazoles 49a,b which cyclized under the established reaction conditions of the authors, to provide PBD conjugates 49a,b in 52%–56% yields. The moderate yields of these cyclisations are comparable to the yields (45%–55%) of PBD conjugates 22a–f (Scheme 3).
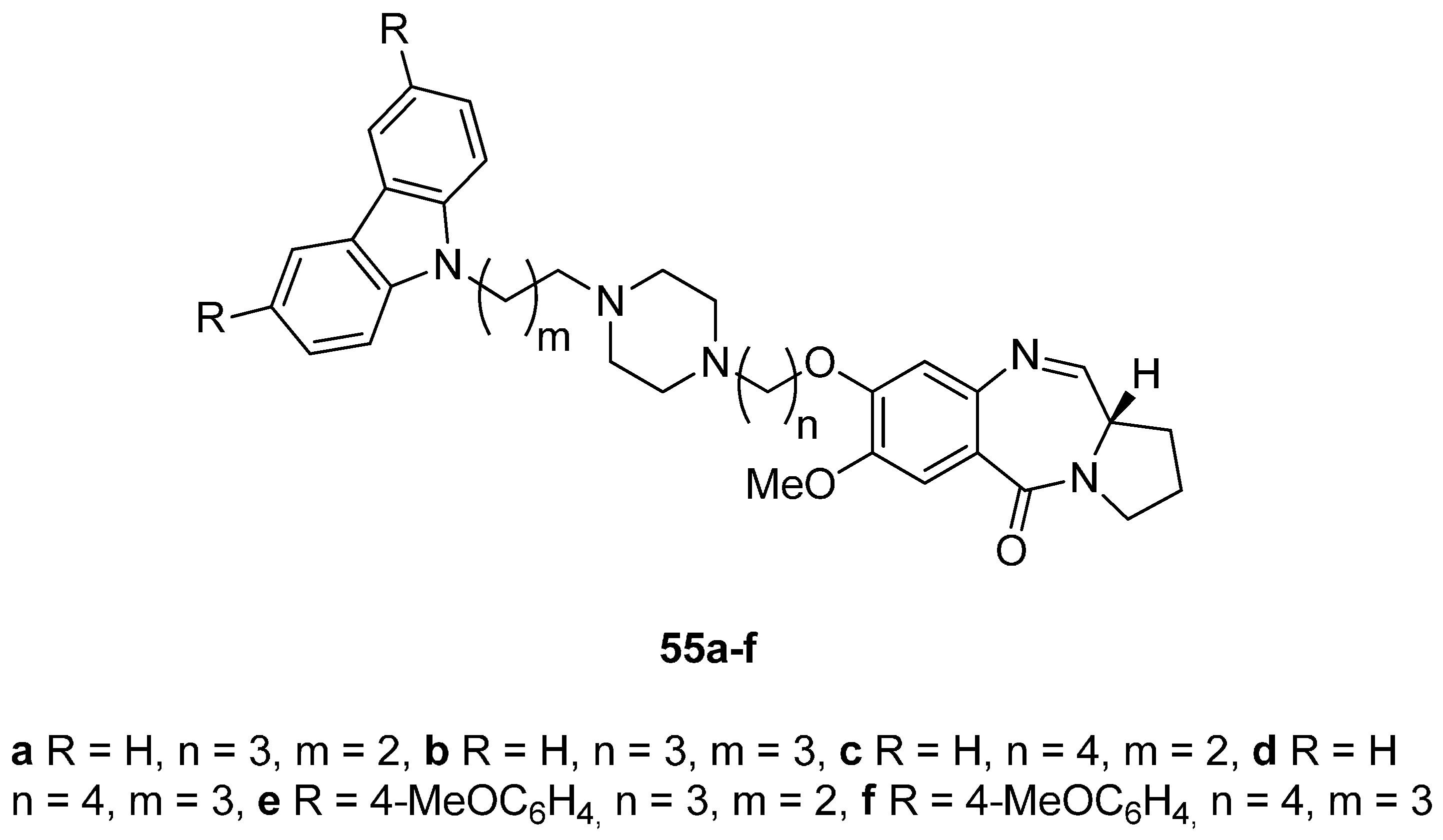
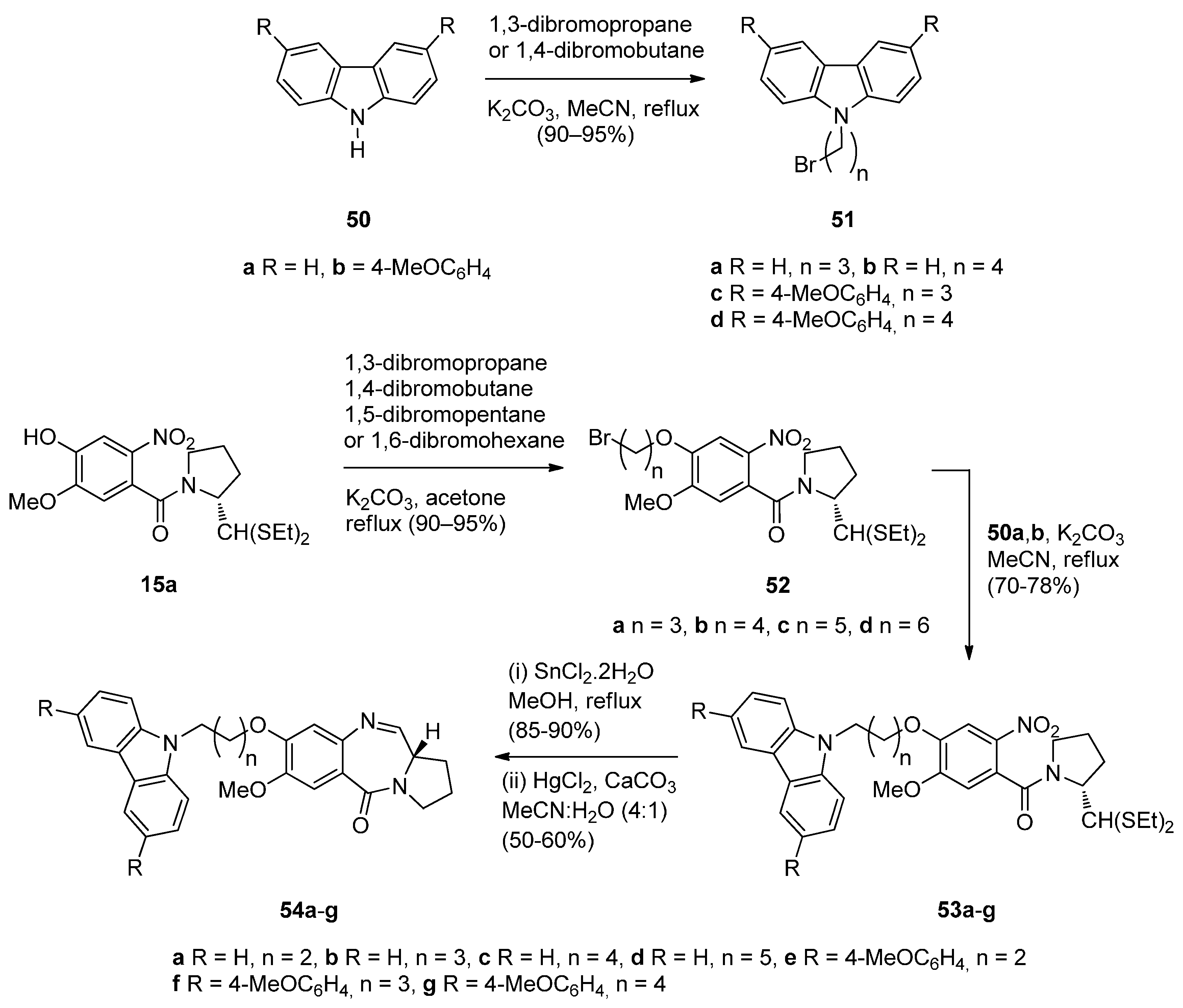
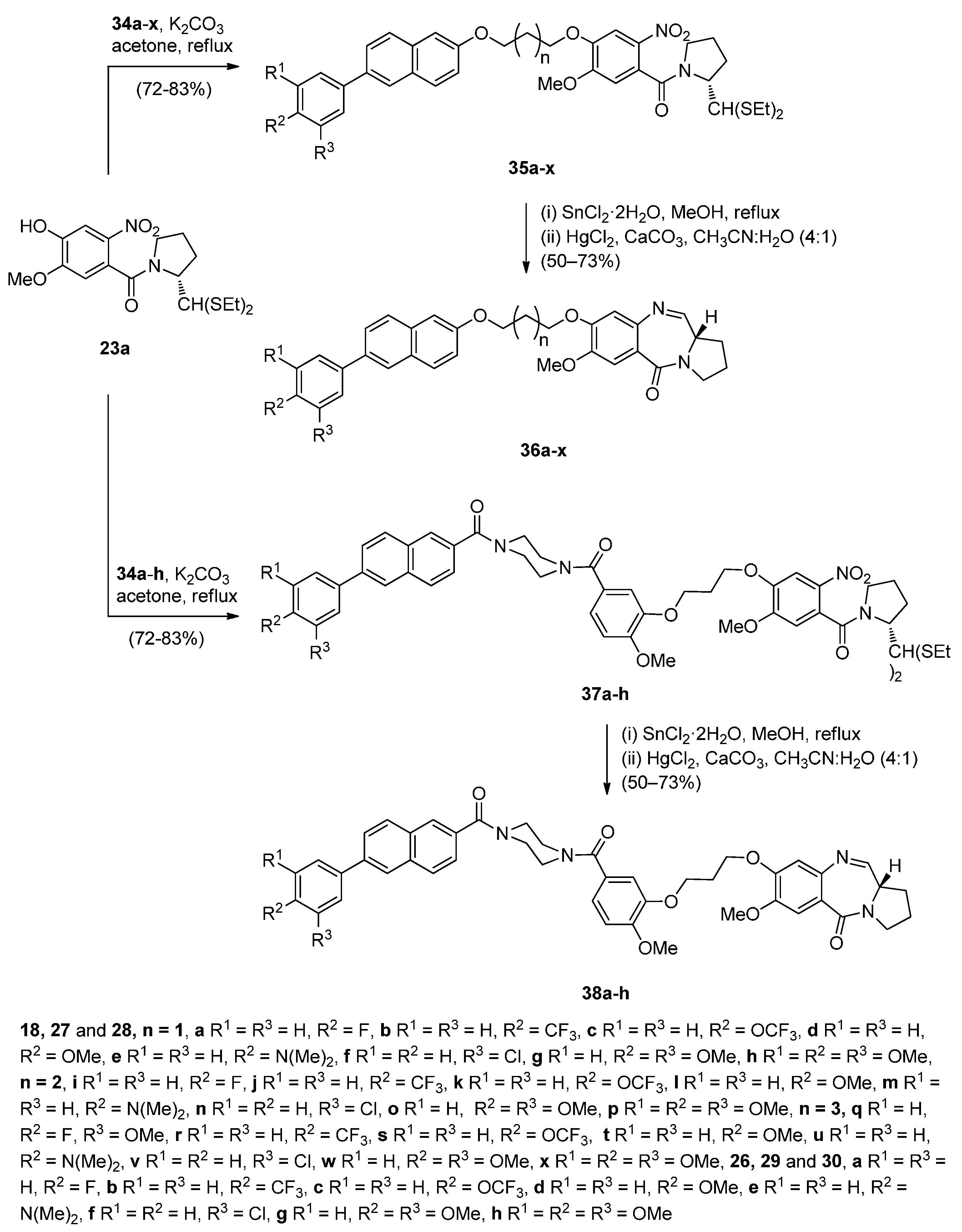
Carbazoles belong to the unusual class of DNA binding agents while the carbazole scaffold is found in many synthetic anticancer agents. These facts encouraged Kamal et al. [53] to synthesise a series of monomeric C8–carbazole PBD conjugates 54a–g and 55a–f (Scheme 9 and Figure 4) and to determine their DNA binding ability by thermal denaturation studies that were supported by molecular docking studies. The carbazole bromoalkyl spacers 51a–d were prepared by N-alkylation of carbazoles 50a,b with suitable dibromoalkanes. The thioacetal 15a, previously reported [48], was alkylated by the appropriate dibromoalkane, according to Thurston et al. [54] to provide bromoalkyl nitro-thioacetals 52a–d which further reacted with carbazoles 50a,b and nitro-thioacetals 53a–g. Cyclisation of these compounds by the authors’ established reaction conditions (Scheme 9) produced the C8–carbazole PBD conjugates 54a–g in 56%–58% yields that are comparable to similar type of cyclisations leading to PBD conjugates 22a–f (45%–55%) (Scheme 3) and PBD conjugates 44a–i (52%–56%) (Scheme 7).
By reacting 52a,b first with 1-Boc-piperazine followed by deprotection of the Boc adduct to the secondary amine, then alkylation of this amine by bromoalkylcarbazoles 51a–d, reduction of the nitro group of these products to amino and lastly deprotection and cyclisation, by a manner analogous to the steps of Scheme 9, furnished C8–carbazole-piperizinal PBD conjugates 55a–f (Figure 4) in good yields.
Scheme 9.
Synthesis of C8-O-substituted PBD conjugates 54a–g.
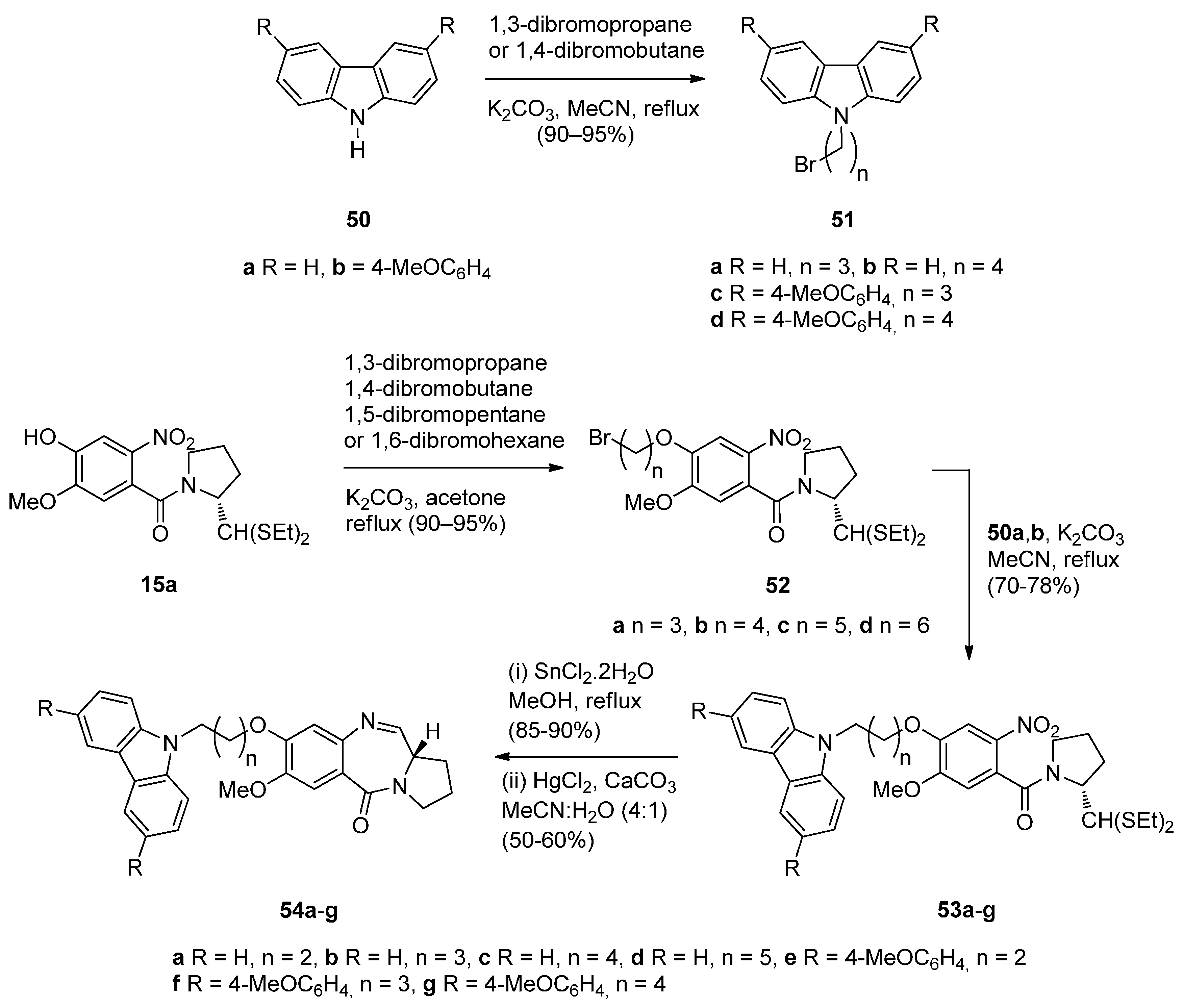
Figure 4.
C8-O-substituted PBD conjugates 55a–f.
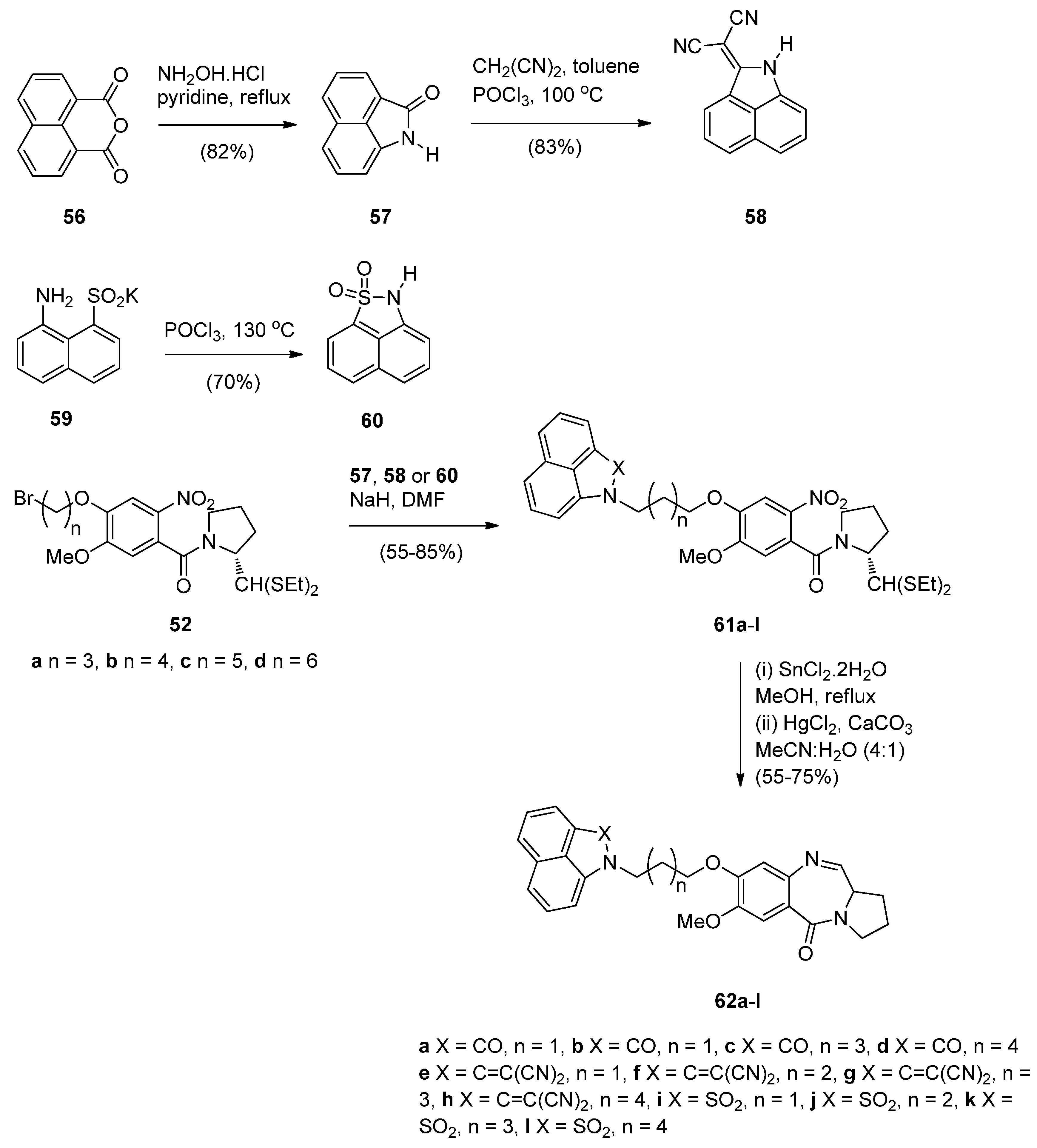
In view of a recent report that describes certain benzo[c,d]indol-2(1H)one derivatives as novel DNA intercalators and as efficient antitumour agents, Kamal et al. [55] designed and synthesised a series of monomeric C8–benzo[c,d]indol-2(1H)one PBD conjugates 62a–l as potential anticancer agents. The DNA binding ability of these conjugates was evaluated as well as their anticancer activity. The synthetic strategy for these PBDs (Scheme 10) involves first the preparation of benzo[c,d]indol-2(1H)-one (57), benzo[c,d]indol-2(1H)-ylidenemalononitrile (58) and 2H-naphtho-[1,8-c,d]isothiazole 1,1-dioxide (60) from 1H,3H-benzo[d,e]isochromene-1,3-dione (56) and [(8-amino-1-naphthyl)sulfonyl]potassium (59). In the next steps, indolone derivatives 57, 58 and 60 were alkylated by bromoalkane-thioacetals 52a–d, prepared earlier [54], to afford the respective nitro- thioacetals 61a–l. By using the authors’ established reaction conditions compounds 61a–l were converted into the corresponding C8–substituted PBD conjugates 62a–l in 55%–75% yields that are on average slightly better than yields of similar cyclisations previously described such as PBDs 22a–f (45%–55%) (Scheme 3), PBDs 44a–i (52%–56%) (Scheme 7) and PBDs 54a–g (50%–60%) (Scheme 9).
Scheme 10.
Synthesis of C8-O-substituted PBD conjugates 62a–l.

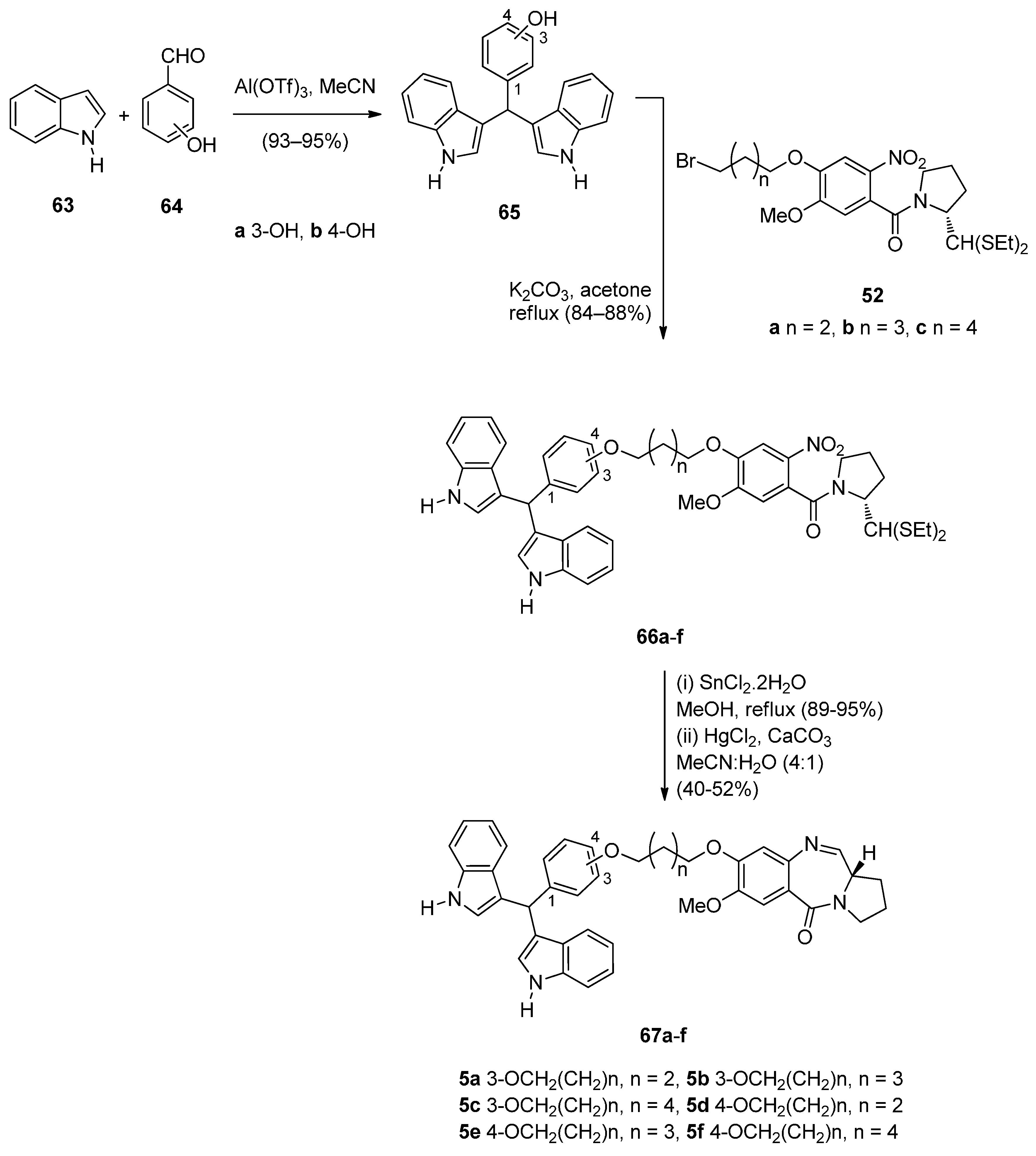
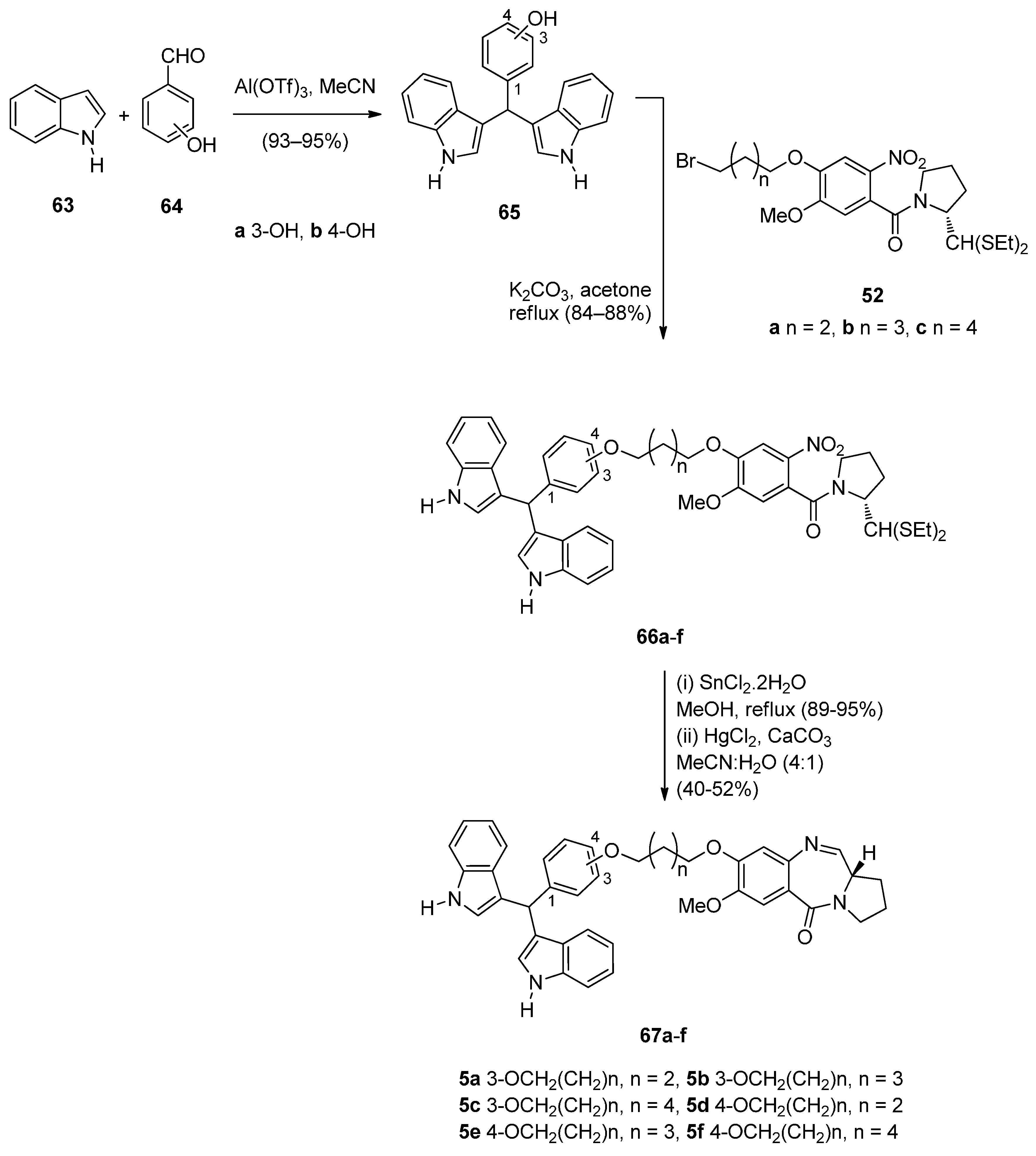
Several bisindolyl methanes have recently been found to exhibit various anticancer effects and it was for this reason that Kamal et al. [56] decided to select and synthesize the six monomeric bisindole-PBDs 67a–f and to study their anticancer properties. In the first step of the synthetic route towards these PBDs (Scheme 11), 3(or 4)-(di-1H-indol-3-ylmethyl)phenols 65a,b were prepared from the reaction of 3-hydroxybenzaldehyde (64a) or 4-hydroxybenzaldehyde (64b) and 1H-indole (63) using aluminium triflate [Al(OTf)3] as a catalyst. Alkylation of phenolic bisindoles 65a,b by the bromoalkyl derivatives 52a–c [54], afforded nitro-thioacetals 66a–f. Next, by applying the authors’ established reaction conditions [49] compounds 66a–f were transformed into the C8-linked bisindole PBD conjugates 67a–f.
Scheme 11.
Synthesis of C8-O-substituted PBD conjugates 67a–f.
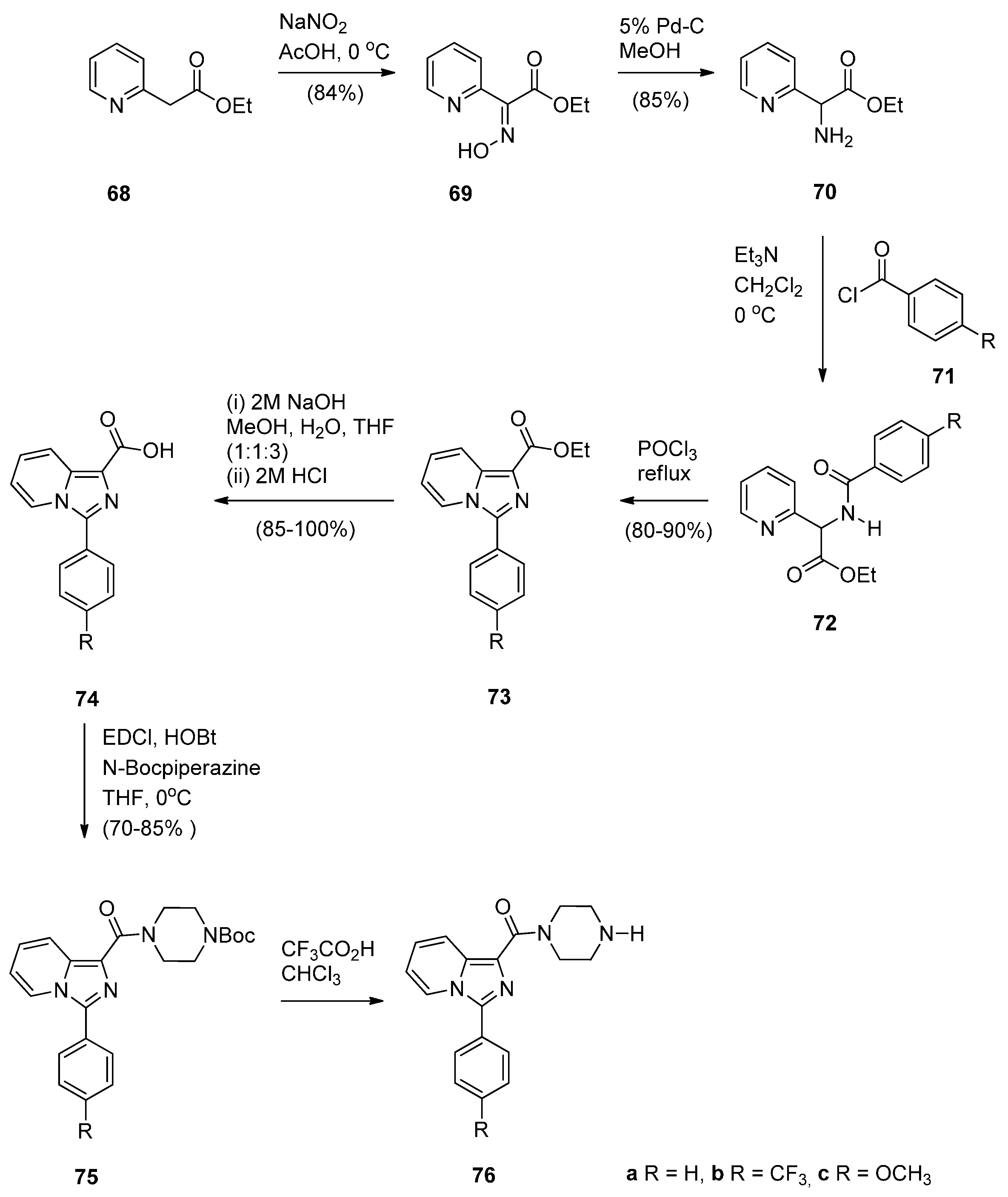
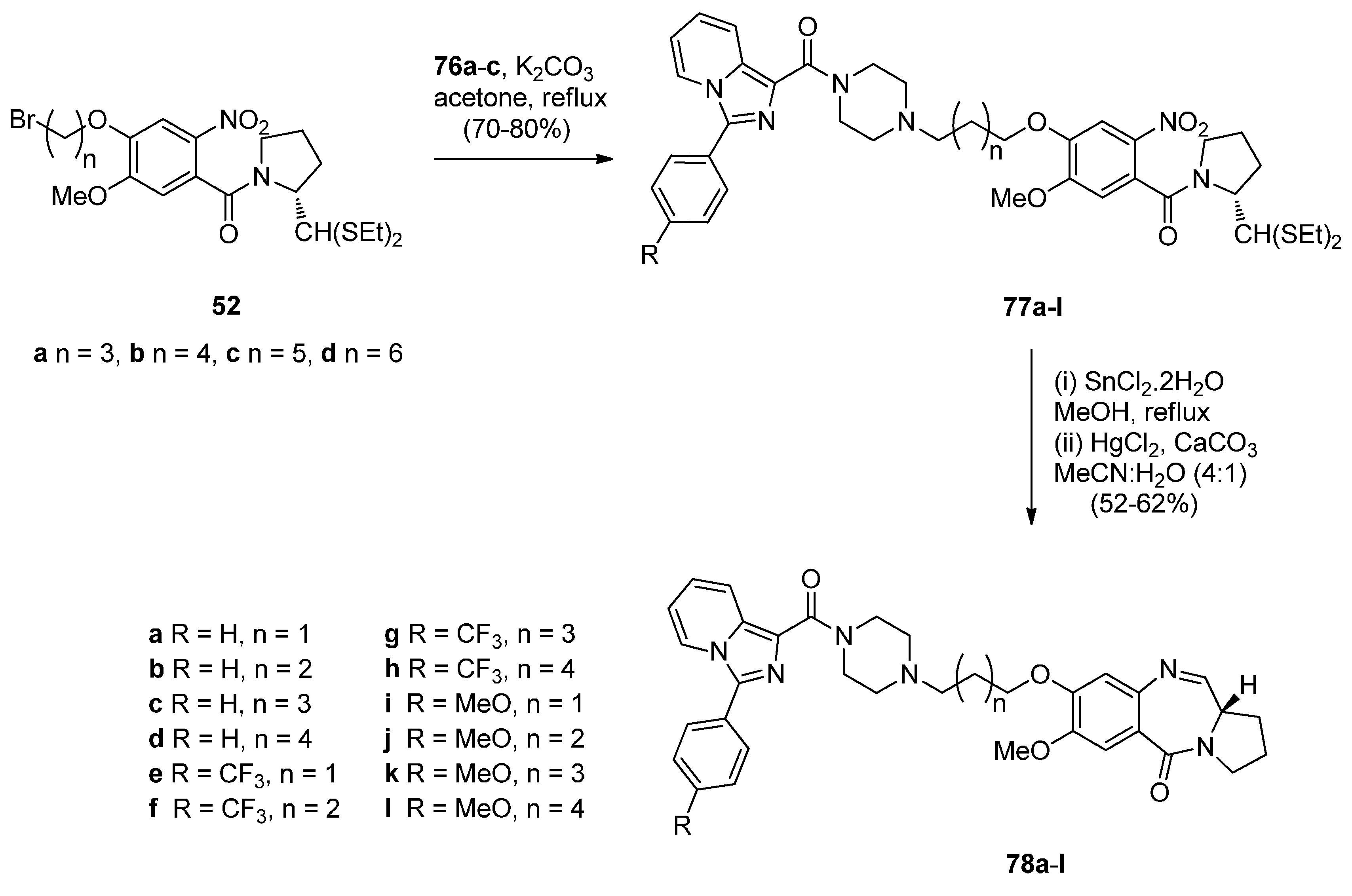
As part of their work on the synthesis and study of PBD conjugates as DNA interactive drugs, Kamal et al. [57] became interested in imidazo[1,5-a]pyridine, a known pharmacological scaffold, and prepared a new class of imidazo[1,5-a]pyridine-piperazine linked PBD conjugates 79a–l, in order to evaluate their antitumour potential. The synthetic strategy involves preparing the 3-ary-1-(piperazin-1-ylcarbonyl)imidazo[1,5-a]pyridines 76a–c (Scheme 12) and the 1-(4-bromoalkoxy-5-methoxy-2-nitrobenzoyl]pyrrolidines 77a–d (Scheme 13) and then linking the two to produce eventually the target compounds 79a–l. The synthesis of precursors 76a–c requires in the first step diazotisation of ethyl 2-(2-pyridyl) acetate 68 to give ethyl 2-hydroxyimino-2-(2-pyridyl) acetate (69). Catalytic hydrogenation of the latter afforded amino intermediate 70 which upon treatment with substituted acid halides 71a–c gave the corresponding amides 72a–c. Cyclisation of amides 72a–c with phosphorous oxychloride provided imidazo[1,5-a]pyridines 66a–c and then de-esterification of these, afforded the corresponding acids 74a–c. Coupling of acids 74a–c with 1-Boc-piperazine gave amides 75a–c. Deprotection of 75a–c produced the desired precursors 76a–c as shown in Scheme 12. The route to PBD conjugates 78a–l (Scheme 13) was carried out by applying established chemistry described by these authors [49] (vide supra).
Scheme 12.
Synthesis of imidazo[1,5-a]pyridine precursors 76a–c.
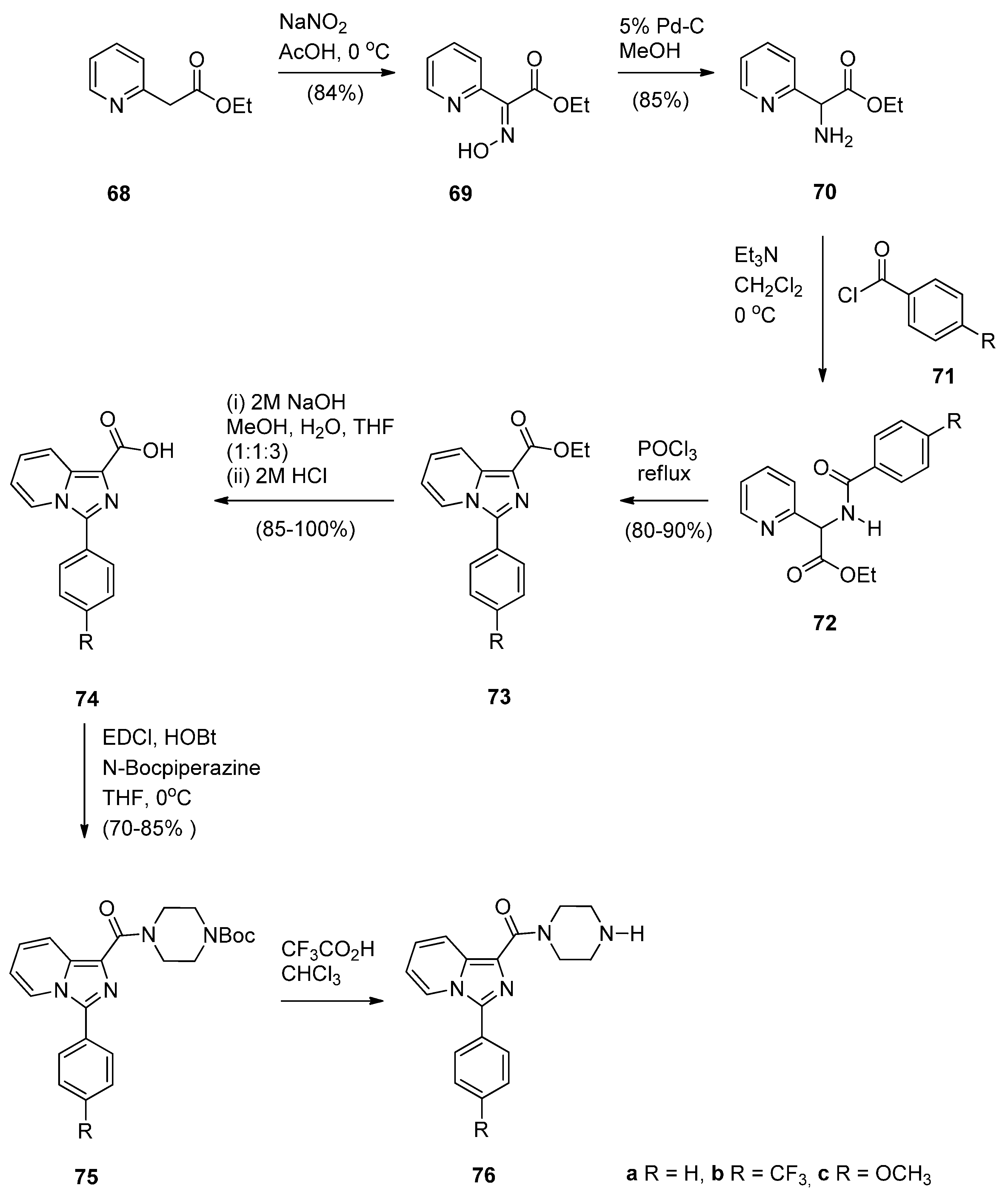
Scheme 13.
Synthesis of C8-O-substituted PBD conjugates 78a–l.
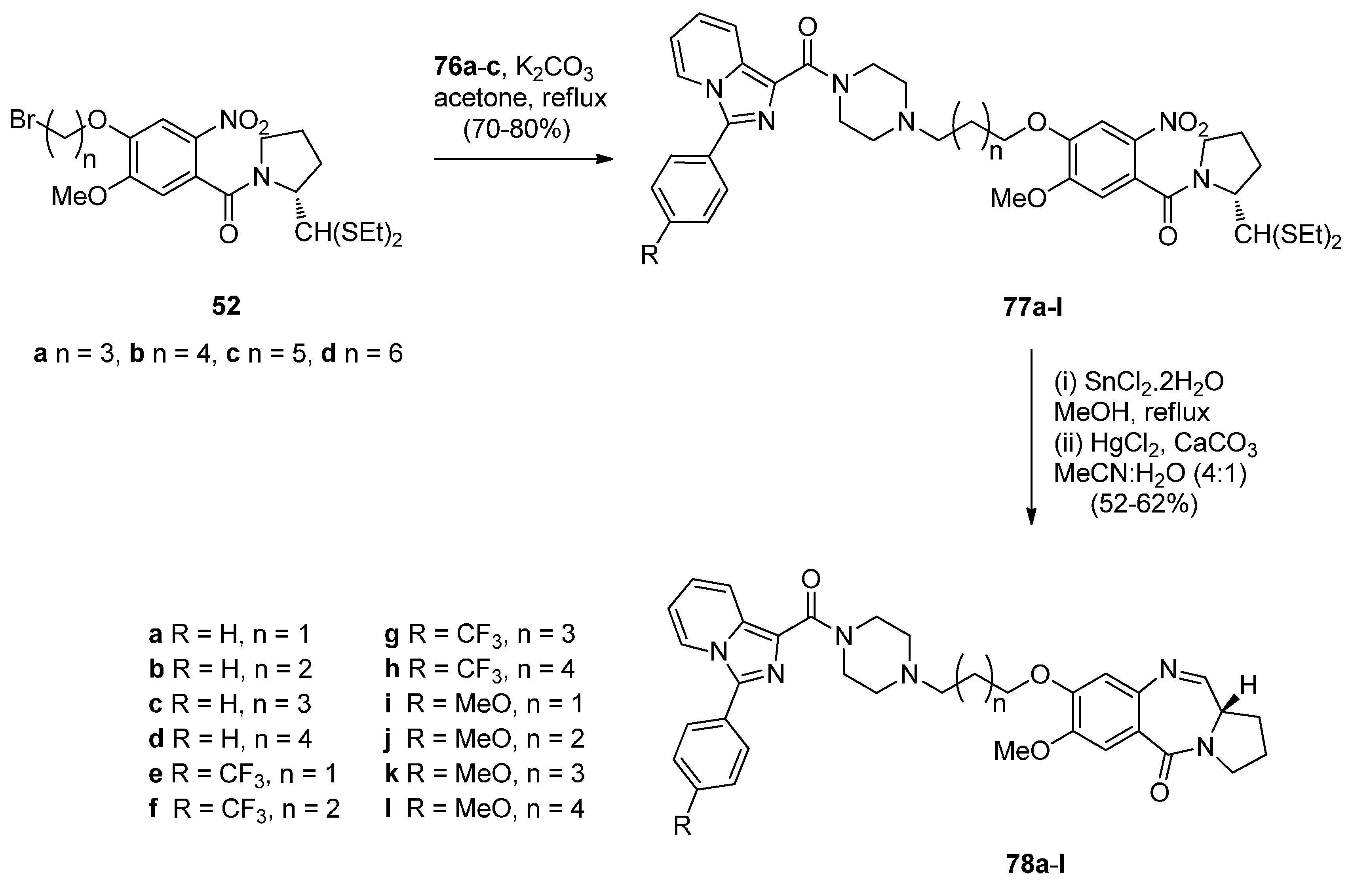
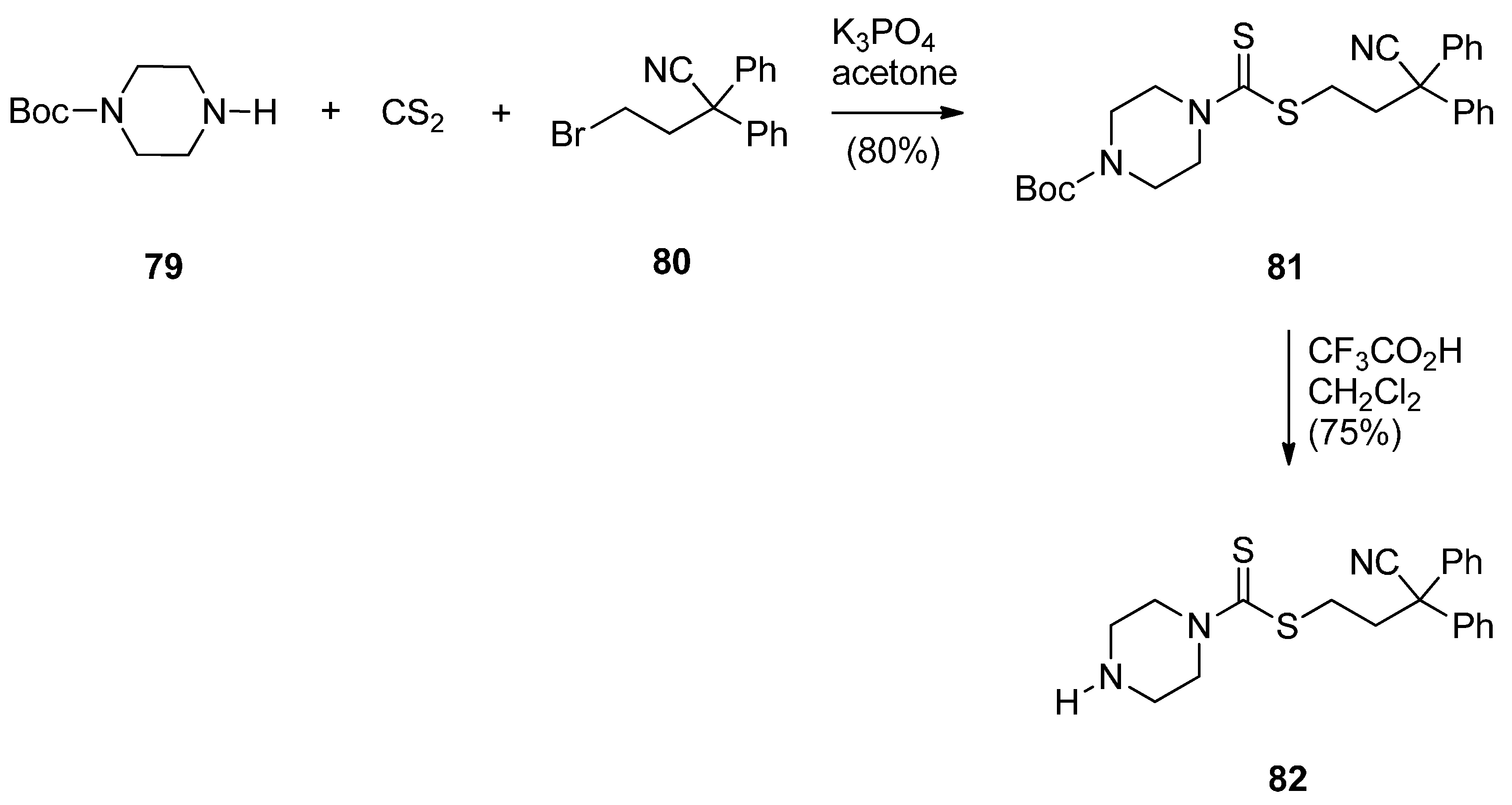
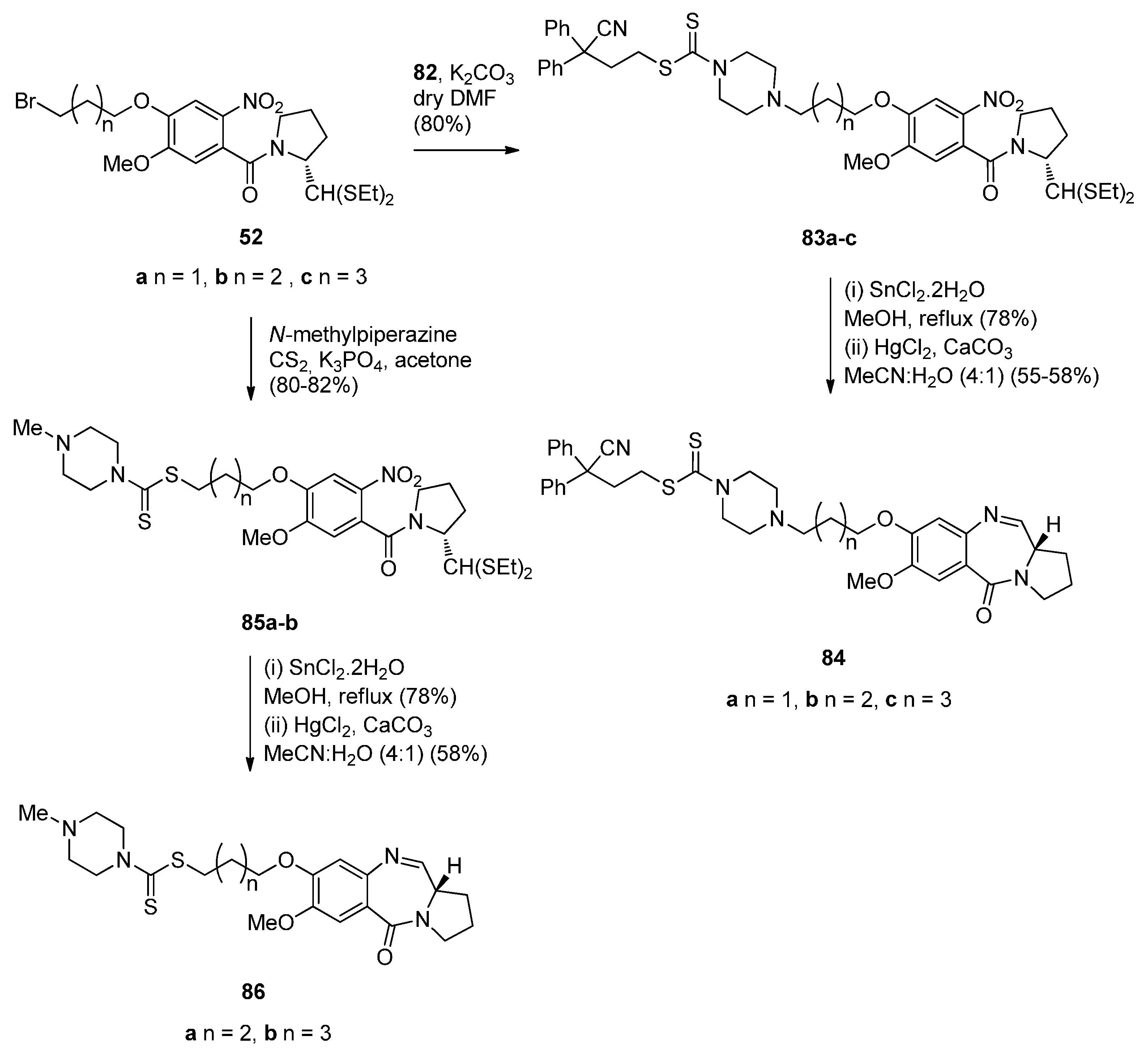
A novel series of C8-linked dithiocarbamate/piperazine bridged PBD conjugates 84a–c and 86a,b were prepared (Scheme 14 and Scheme 15) and evaluated for their cytotoxic potential and DNA-binding ability by Kamal et al. [58]. The synthesis of target compounds 84a–c required in the first step an SN2 reaction between carbodithioate 82 and bromoalkyl nitro-thioacetals 52a–c, prepared previously [54], which gave nitro-thioacetals 83a–c, in very good yields. Precursor 82 was prepared by addition of carbon disulfide to 1-Boc-piperazine 79, alkylation by propylbromide 80 to produce conjugate 81 and then deprotection with trifluoroacetic acid (Scheme 14).
Scheme 14.
Synthesis of carbodithioate derivative 82.

Scheme 15.
Synthesis of C8-O-substituted PBD conjugates 84a–c and 86a,b.
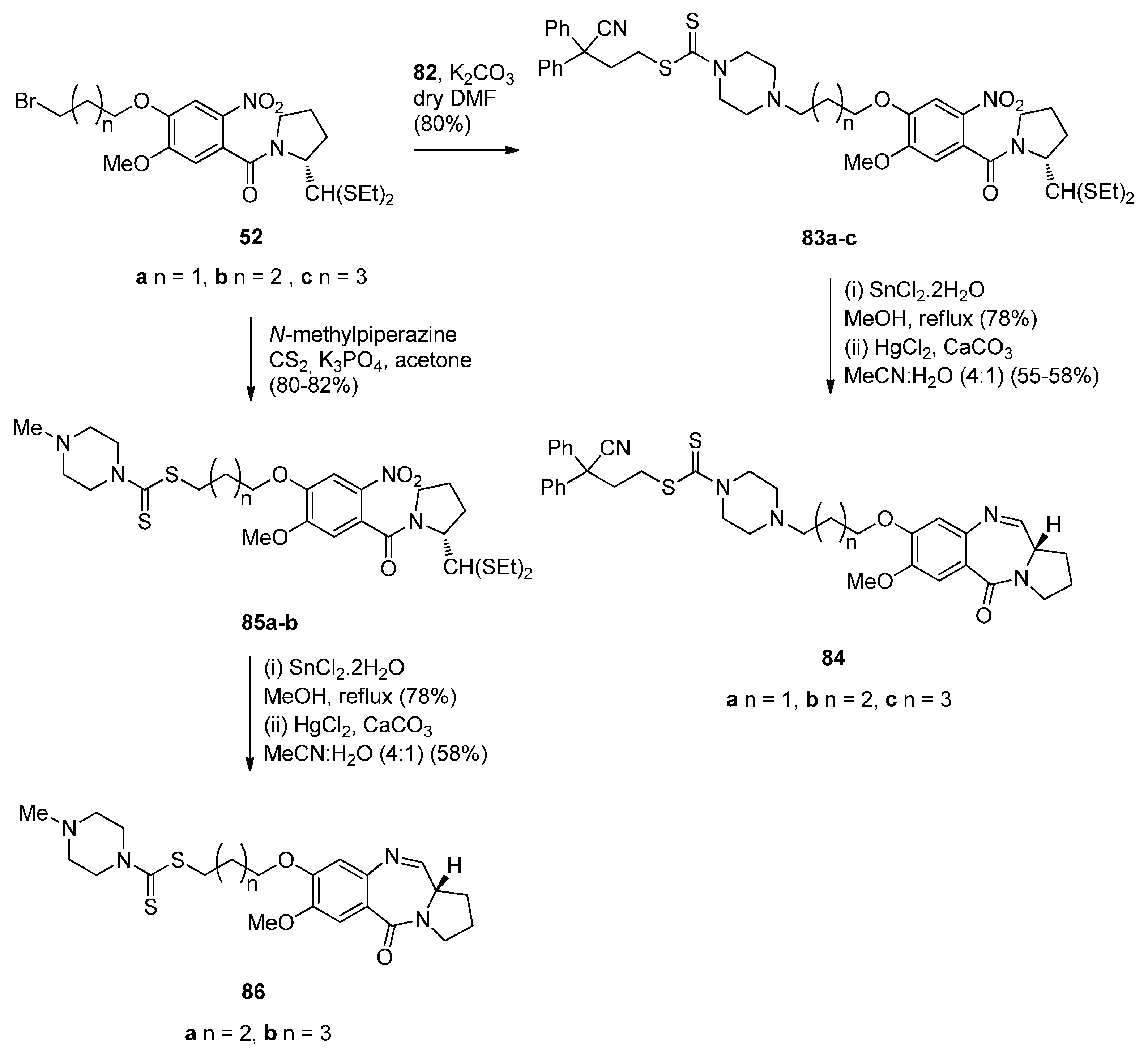
O-substituted nitro-thioacetals 83a–c prepared by alkylating 82 by 52a–c and O-substituted nitro-thioacetals 85a,b prepared by alkylating N-methylpiperazine with 52b,c were efficiently reduced by stannous (II) chloride dihydrate (SnCl2·2H2O) to afford the corresponding amino-thioacetals which were deprotected using mercuric(II) chloride (HgCl2) and calcium carbonate (CaCO3) and cyclocondensed, according to the authors’ established reaction procedures [48] (Scheme 15), to afford PBD conjugates 84a–c and 86a,b in 55%–58% and 58% yields, respectively. These yields are comparable to yields of previous similar cyclisations that give PBDs 22a–f (45%–55%) (Scheme 3), PBDs 44a–i (52%–56%) (Scheme 7) PBDs 54a–g (50%–60%) (Scheme 9), 67a–f (40%–52%) (Scheme 11) and 79a–l (52%–62%) (Scheme 13), with the exception of PBDs 62a–l (55%–75%) where yields are on average slightly higher.
This synthetic approach to the PBD conjugates that is, by using a bromoalkyl nitro-thioacetal (i.e., 52) and then coupling with a non-PBD pharmacophore, has been previously described by these authors [53] (Scheme 9), [55] (Scheme 10) and [57] (Scheme 13).
The last two synthetic steps described so far from the work of Kamal et al. [47,49,50,51,53] and [54,55,56], all involve the reduction of the nitro group of an N-(2-nitrobenzoyl)pyrrolidine-2-carboxaldehyde diethyl thioacetal derivative to the corresponding amino derivative and workup of the reaction mixture without characterizing the product or checking its purity (TLC). The authors however report that the nitro compounds are efficiently reduced to amino compounds and due to potential stability problems they proceed to the next step. The yields of these crude N-(2-amino-benzoyl)pyrrolidine-2-carboxaldehyde diethyl thioacetal derivatives are given in three papers [51] (85%–90%), [54] (89%–95%) and [56] (78%).
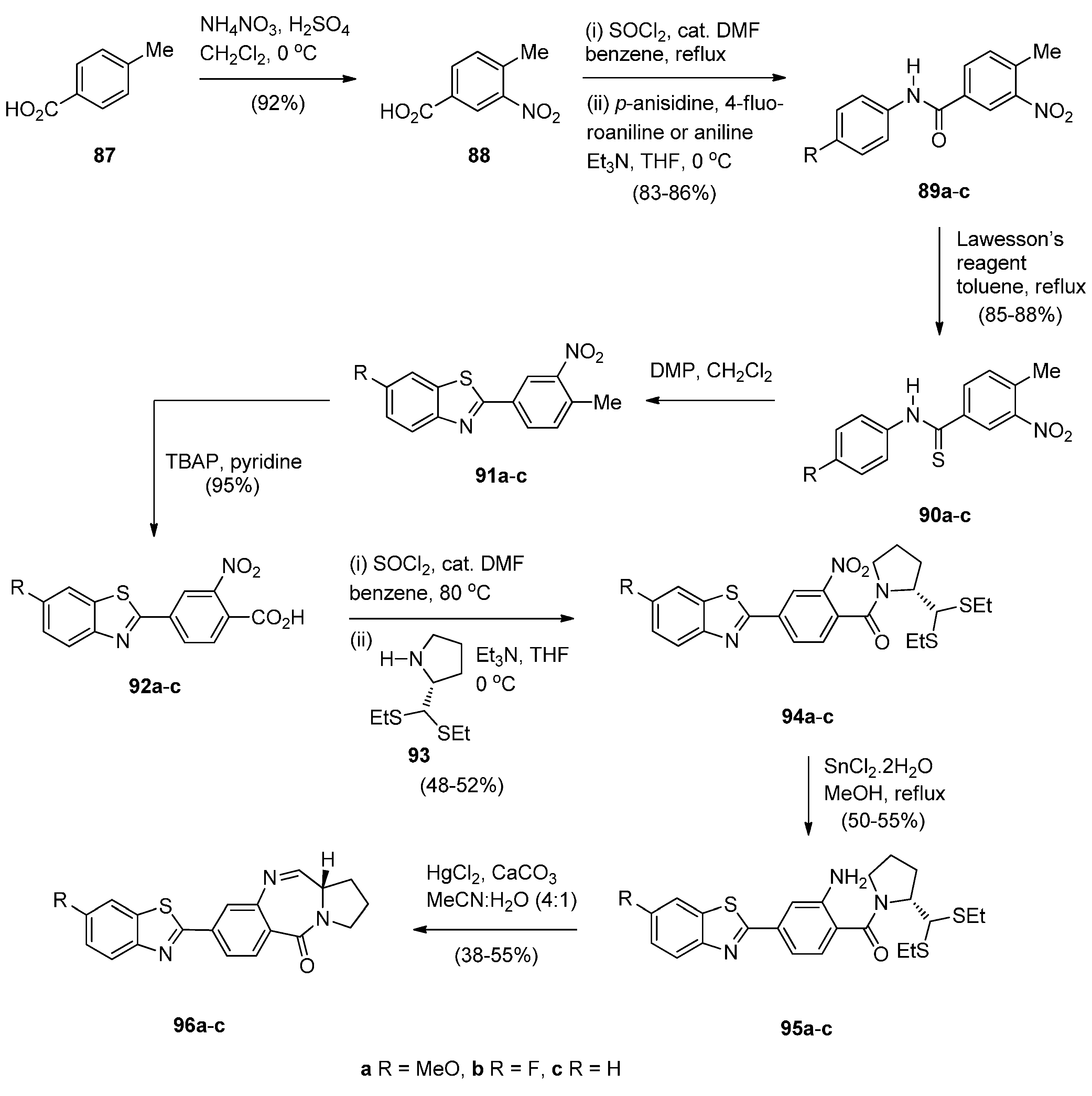
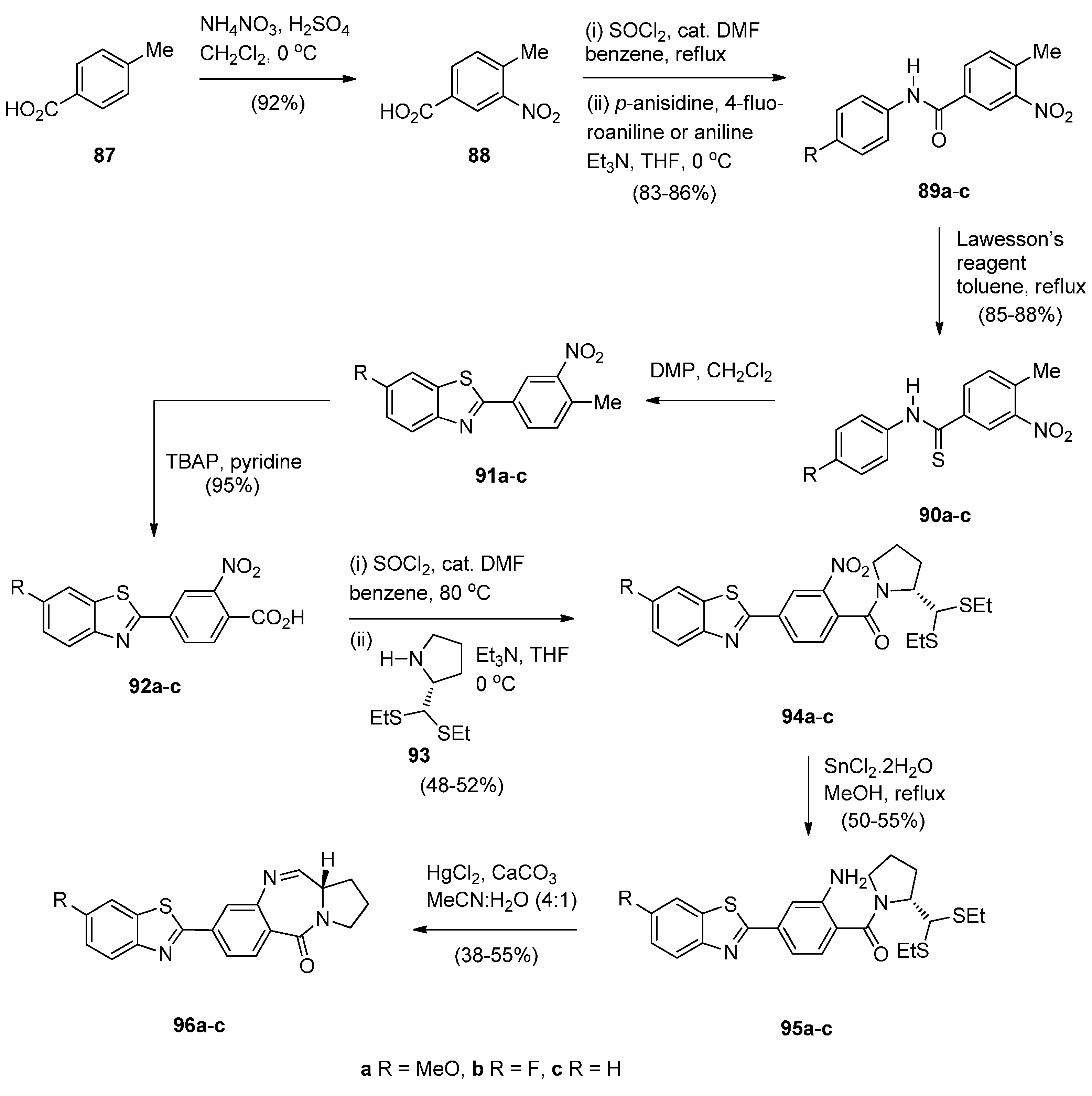
Bose et al. [59] became interested in synthesising benzothiazole (BTA) PBD conjugates 95a–c after the recognition of 4-(1,3-benzothiazol-2-yl)-2-methylaniline and two of its derivatives as novel anticancer drug candidates. The synthetic plan required first the synthesis of the key intermediate 4-(6-substituted-1,3-benzothiazol-2-yl)-2-nitrobenzoic acids 92a–c from 4-methylbenzoic acid (87) (Scheme 16). Thus, 87 was nitrated to nitro derivative 88 which was then converted into the corresponding acid chloride that was used in crude form in the acyl substitution with p-anisidine, 4-fluoroaniline or aniline to afford amides 89a–c. Amides 89a–c were then treated with Lawesson’s reagent to afford thioamides 90a–c which were cyclised using Dess-Martin periodinane (DMP), to yield benzothiazoles 91a–c. The methyl group of compounds 91a–c was oxidised with tetrabutylammonium permanganate (TBAP) to afford the corresponding benzothiazolyl-2-benzoic acids 92a–c. The next step required the coupling of the acid chlorides of 92a–c with (2S)-2-[bis(ethylthio)methyl]pyrrolidine (93) to afford the amides 94a–c. Although 93 has been synthesized earlier by Langley and Thurston [60] here it was prepared in better yields with excellent optical purity in five steps from (S)-proline. Following established reaction conditions [49], reduction of nitro compounds 94a–c to the corresponding amines 95a–c followed by their deprotection and spontaneous cyclodehydration, afforded the corresponding BTA PBD conjugates 96a–c, in moderate yields. This synthetic approach to the PBDs differs in the last but two steps from the syntheses in Scheme 1, Scheme 2, Scheme 3, Scheme 4, Scheme 5, Scheme 6, Scheme 7, Scheme 8, Scheme 9, Scheme 10, Scheme 11, Scheme 12, Scheme 13, Scheme 14 and Scheme 15, in that the non-PBD pharmacophore is incorporated as a substituent at position 4 of 2-nitrobenzoic acids 92a–c and is then coupled to pyrrolidine 93 to produce the N-(2-nitro-benzoyl)pyrrolidine-2-carboxaldehyde diethyl thioacetals 94a–c. Furthermore, reduction of the latter derivatives led to N-(2-aminobenzoyl)pyrrolidine-2-carboxaldehyde diethyl thioacetals 95a–c that after workup were purified by column chromatography and characterized by 1H-NMR, low resolution MS and IR spectroscopy, the first time for these type of compounds.
Scheme 16.
Synthesis of C8-substituted PBD conjugates 96a–c.
Oxidative Cyclisation of N-(2-(Alloc or Troc)aminobenzoyl)pyrrolidine-2-Methanol Derivatives
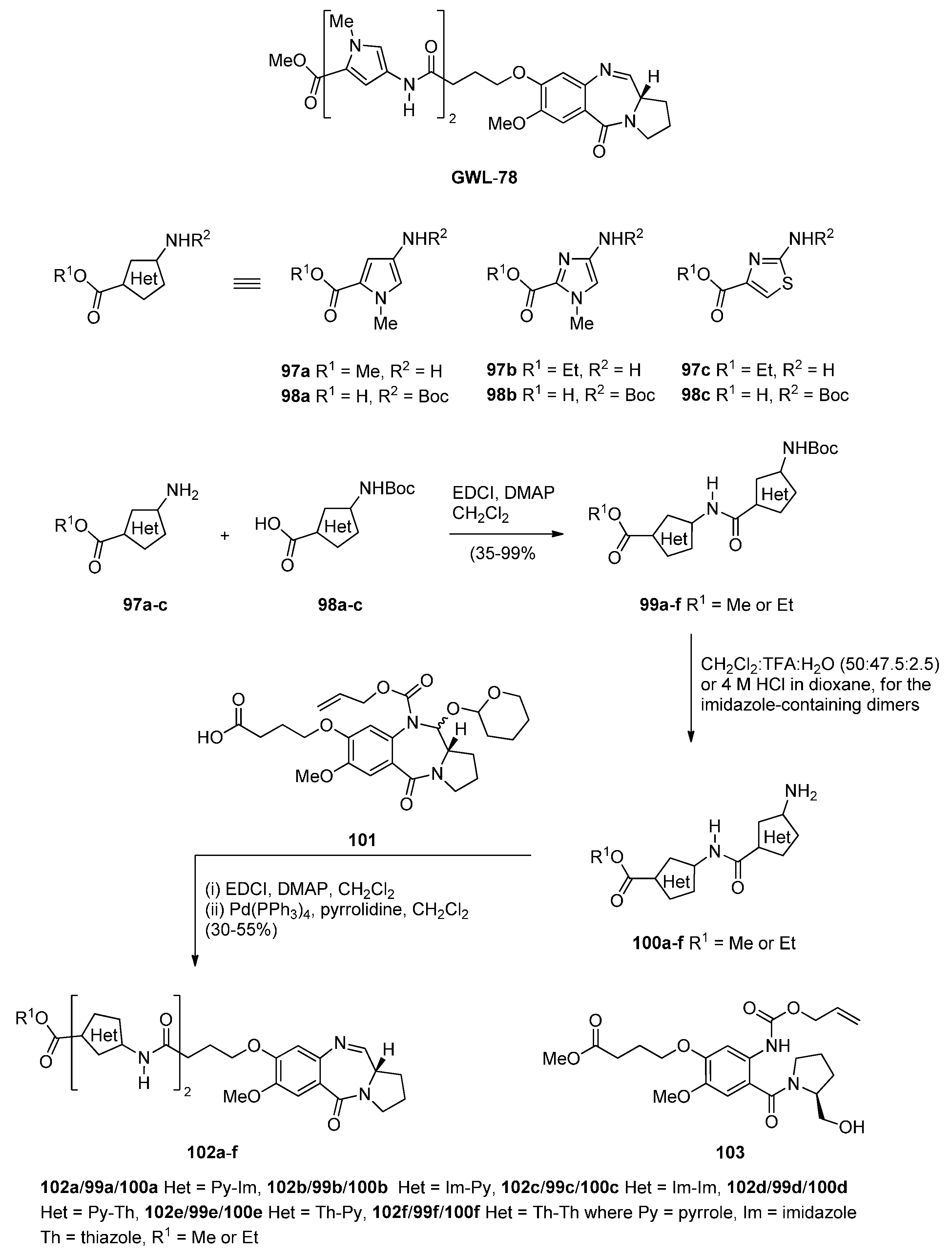
The interest in earlier work by Thurston and co-workers [61,62] who synthesied a series of C8-linked poly(N-methylpyrrole) PBD conjugates (e.g., GWL-78) and demonstrated their ability to interact with a variety of CCAAT-containing promoters leading to p53-independentcell cycle arrest, was renewed and six C8-linked poly(heterocyclic) PBD conjugates 102a–f (Scheme 17) were synthesized in order to study further this biological interaction [63]. In the synthetic route to 102a–f the starting heterocyclic amino esters 97a–c and the Boc-protected heterocyclic amino acids 98a–c were synthesized as previously reported [61]. These compounds were then coupled together using standard methodology to produce the Boc-protected imidazole 99a–c and thiazole 99d–f dimers which, without purification, were deprotected with trifluoroacetic acid to provide dimeric amino esters 100a–f. These amines were then coupled to the carboxylic acid group of previously described [62] C8-(3-carboxypropoxy)-N10-alloc-C11-O-THP protected PBD 101, under standard reaction conditions, followed by treatment with tetrakis(triphenylphosphine)palladium(0) [Pd(Ph3P)4] and pyrrolidine that promotes simultaneous removal of the alloc and THP protecting groups to afford C8-linked poly(heterocyclic) PBD imine conjugates 102a–f, in moderate to good yields. PBD 101 is prepared in three steps from N-(2-alloc protected aminobenzoyl)pyrrolidine-2-methanol derivative 103 by Swern oxidative cyclisation to an C8-(3-methoxycarbonylpropoxy)-N10-alloc-C11-OH PBD, protection to a C8-(3-methoxycarbonylpropoxy)-N10-alloc-C11-O-THP PBD and hydrolysis of the ester group to a carboxylic acid group.
Scheme 17.
Synthesis of C8-O-substituted PBD conjugates 102a–f.
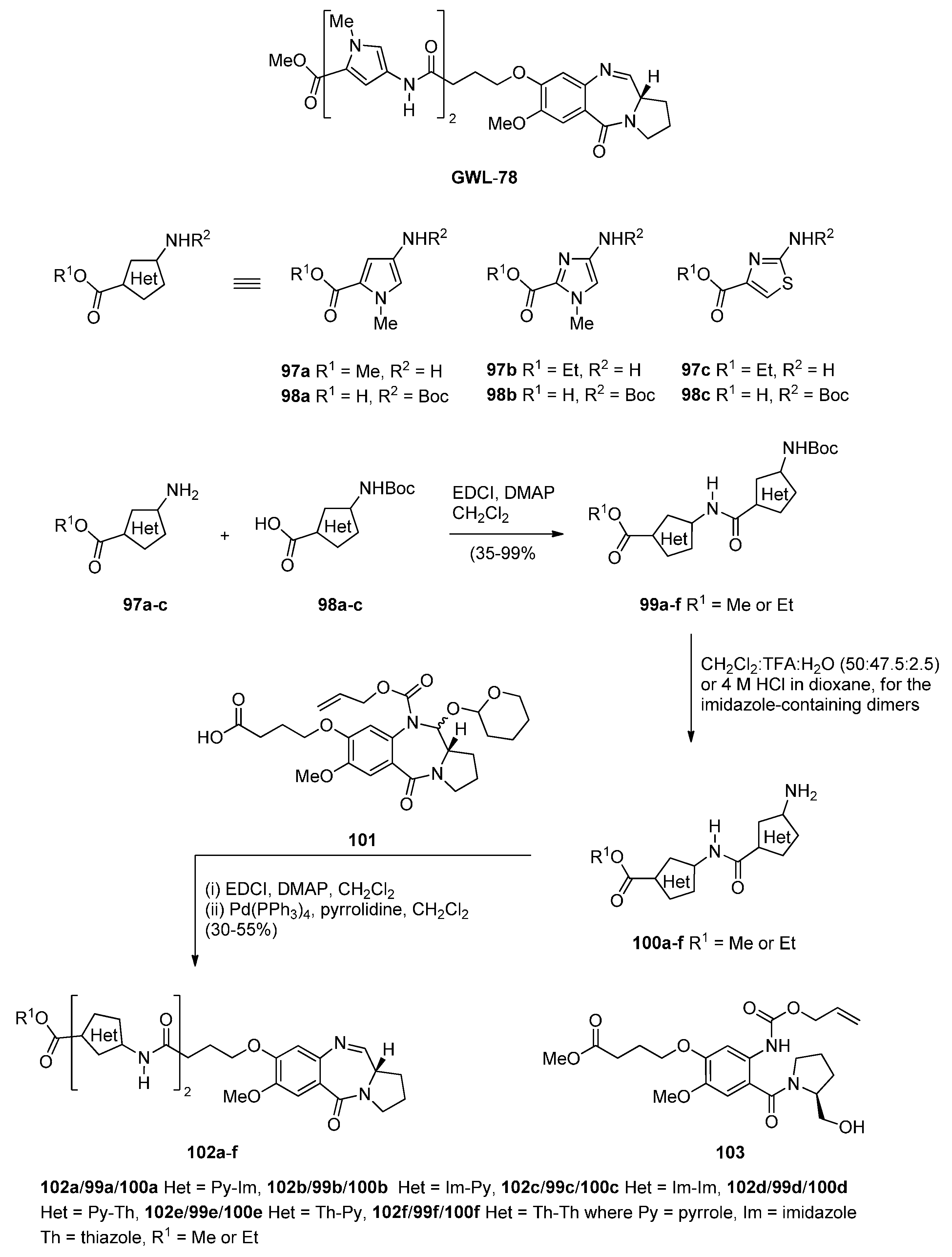
This methodology differs from those so far described in that a PBD is constructed with a C8 substituent possessing a carboxylic acid end group. The rest of the C8 moiety is then extended by coupling this carboxylic acid group with the amino group of preformed 5-membered heterocyclic aminoester dimers.
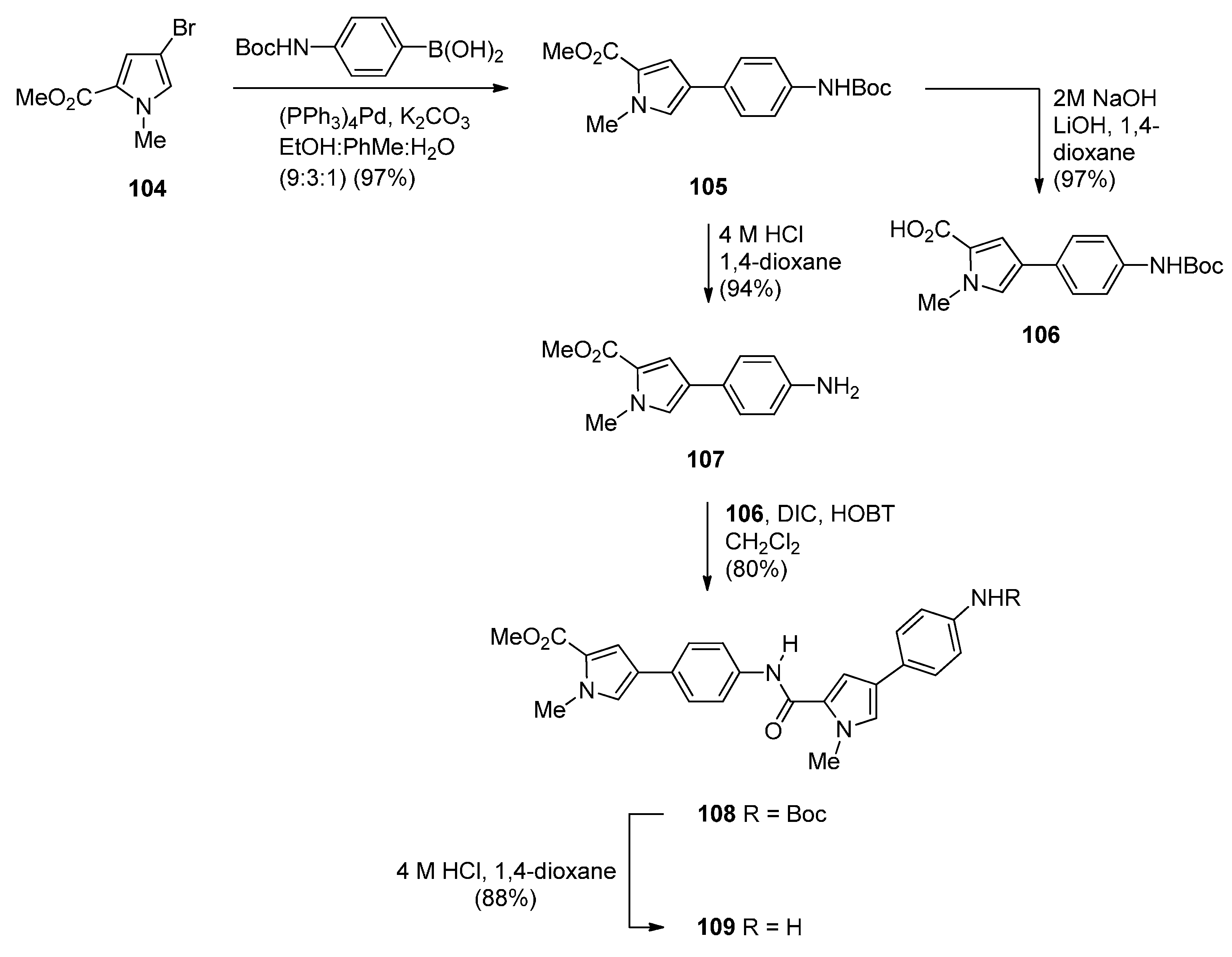
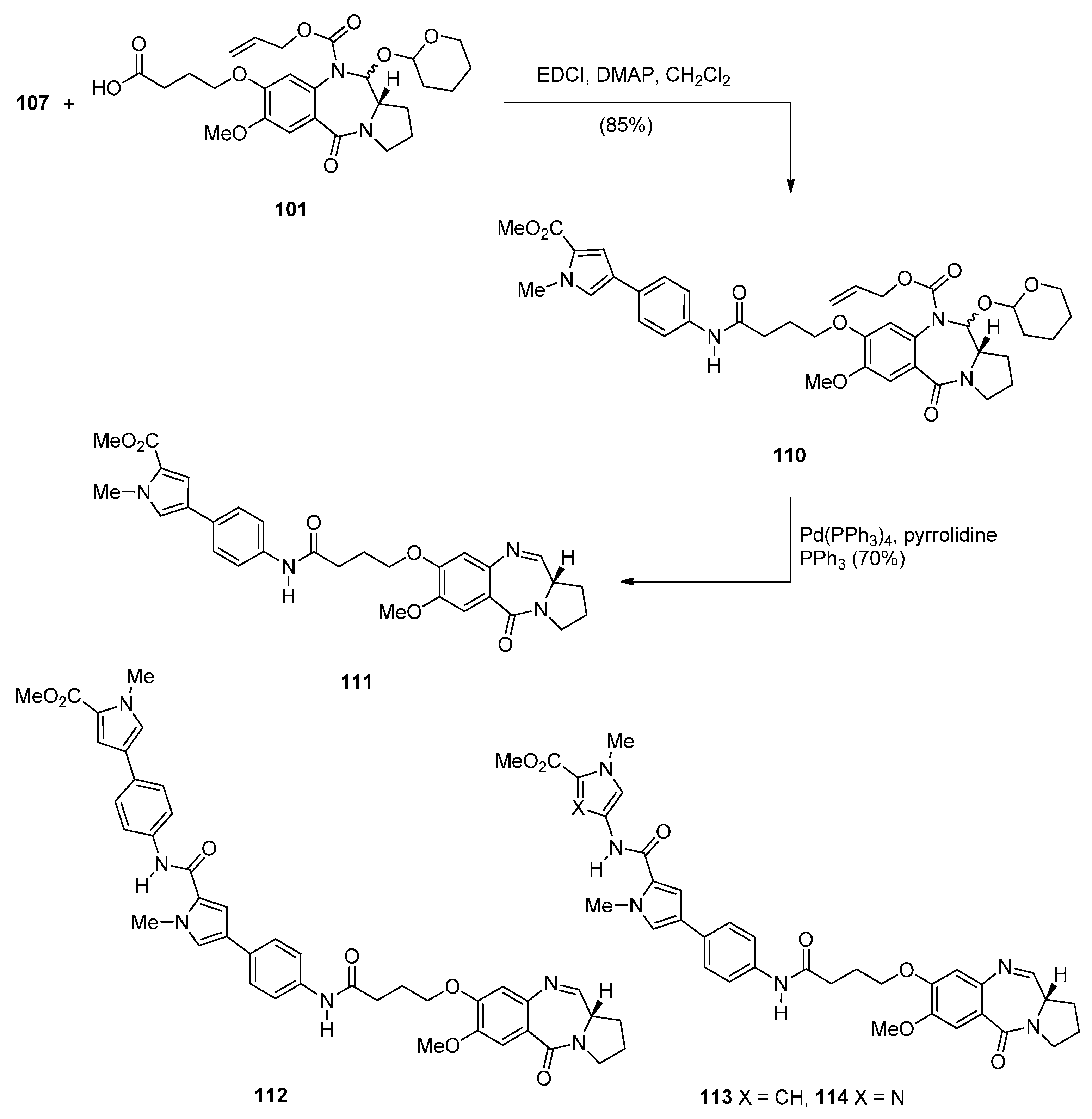
Two years later Thurston and co-workers presented [64] a continuation of their previous work [61] on the synthesis and biological study of C8-linked poly(N-methylpyrrole) PBD conjugates and used similar methodology to synthesise seven biaryl(poly)amide PBD conjugates 111–118. The simplest of these compounds, 111, was prepared in two steps from the smallest biaryl building block, namely methyl 4-(4-aminophenyl)-1-methyl-1H-pyrrole-2-carboxylate (107, Scheme 18). Coupling 107 with PBD-carboxylic acid 101 under standard reaction conditions afforded PBD adduct 110, in very good yield. The N10–alloc and C11–OTHP protecting groups of 110 were then simultaneously removed with tetrakis(triphenylphosphine)palladium(0) [(PPh3)4Pd] catalyst and pyrrolidine to afford 111 in good yield. Although PBD 101 has been synthesized earlier [61], aminoester 107 was prepared from commercially available bromoester 104 in two steps. Suzuki coupling between bromoester 104 and 4-[(tert-butoxycarbonyl)amino]phenylboronic acid took place with tetrakis(triphenylphosphine)-palladium(0) [(PPh3)4Pd] catalyst and potassium carbonate in a mixture of ethanol, toluene and water (9:3:1) under microwave irradiation for 8 minutes, to produce 4-arylpyrrole 105 in excellent yield. Deprotection of the Boc group of 105 afforded the aminoester 107. Building block 109 R=H was synthesized as shown in Scheme 18 and served to prepare biarylpolyamide PBD conjugate 112 by coupling with PBD-carboxylic acid 101 and then deprotecting the product obtained, in a manner similar to that described for compound 111 in Scheme 19. Conjugates 113–118 were prepared by analogous reactions (Scheme 19 and Scheme 20).
Scheme 18.
Synthesis of aminoester precursors 107 and 109.

Scheme 19.
Synthesis of C8-O-substituted PBD conjugates 111–114.
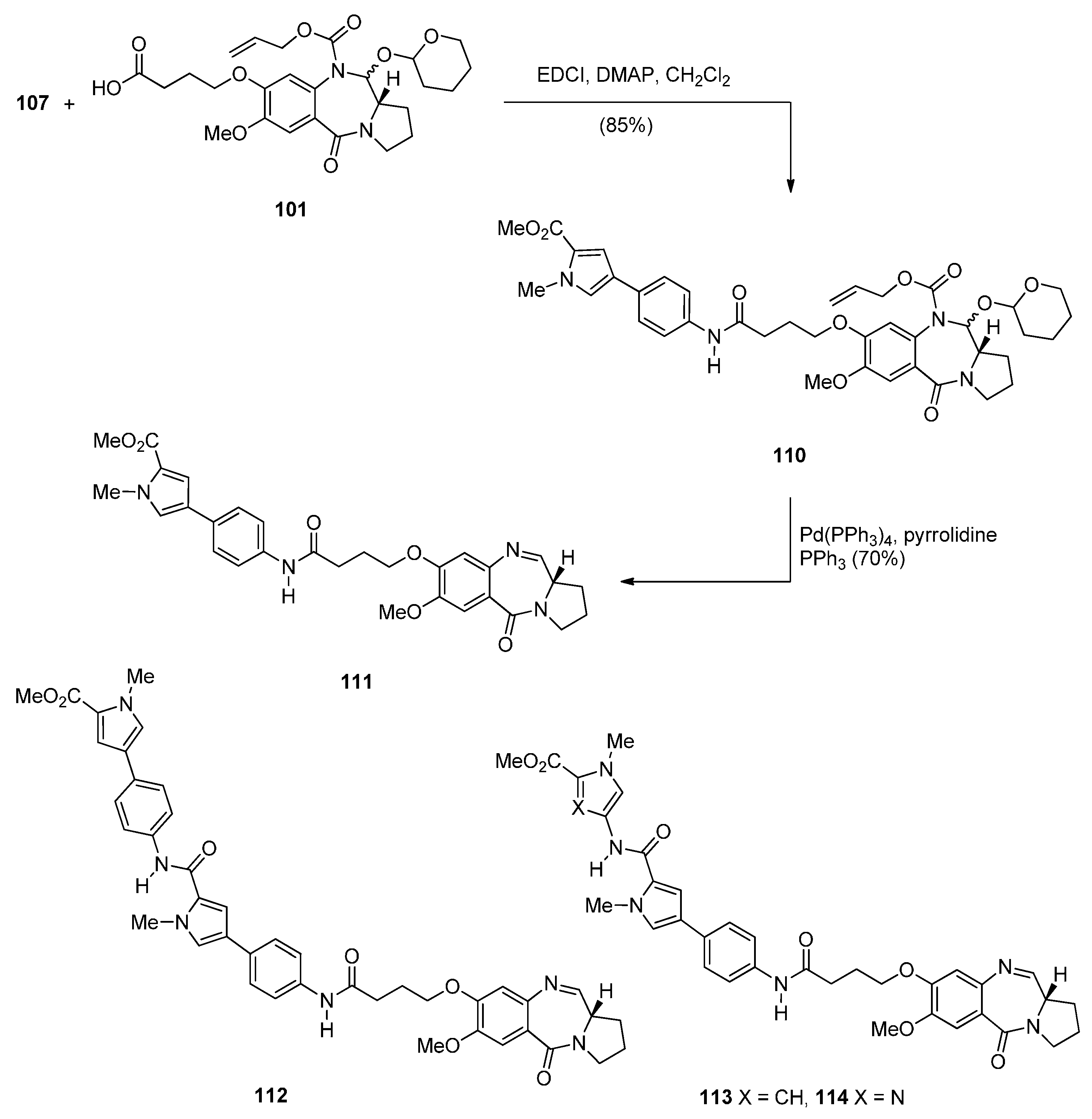
Scheme 20.
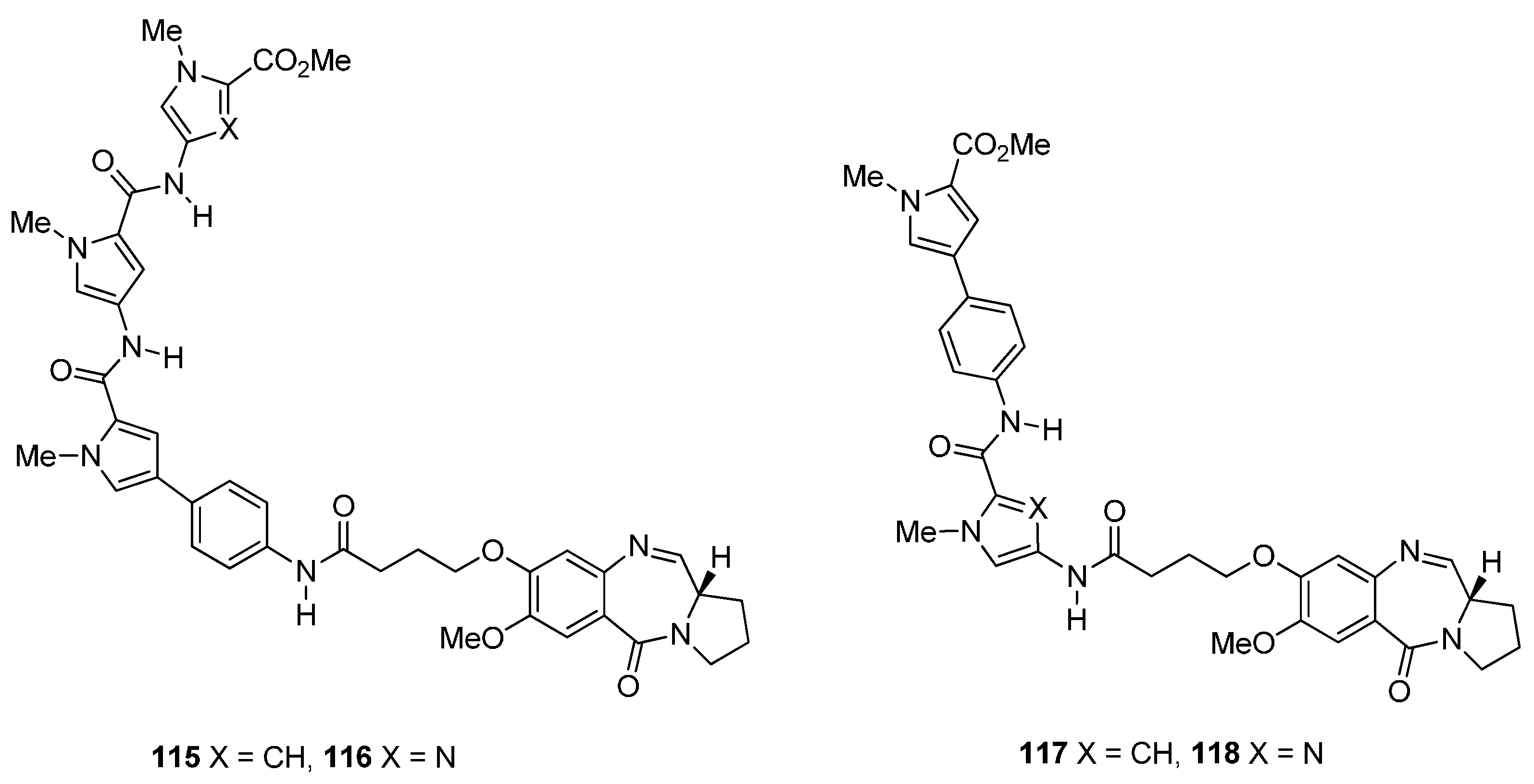
Synthesis of C8-O-substituted PBD conjugates 115–118.

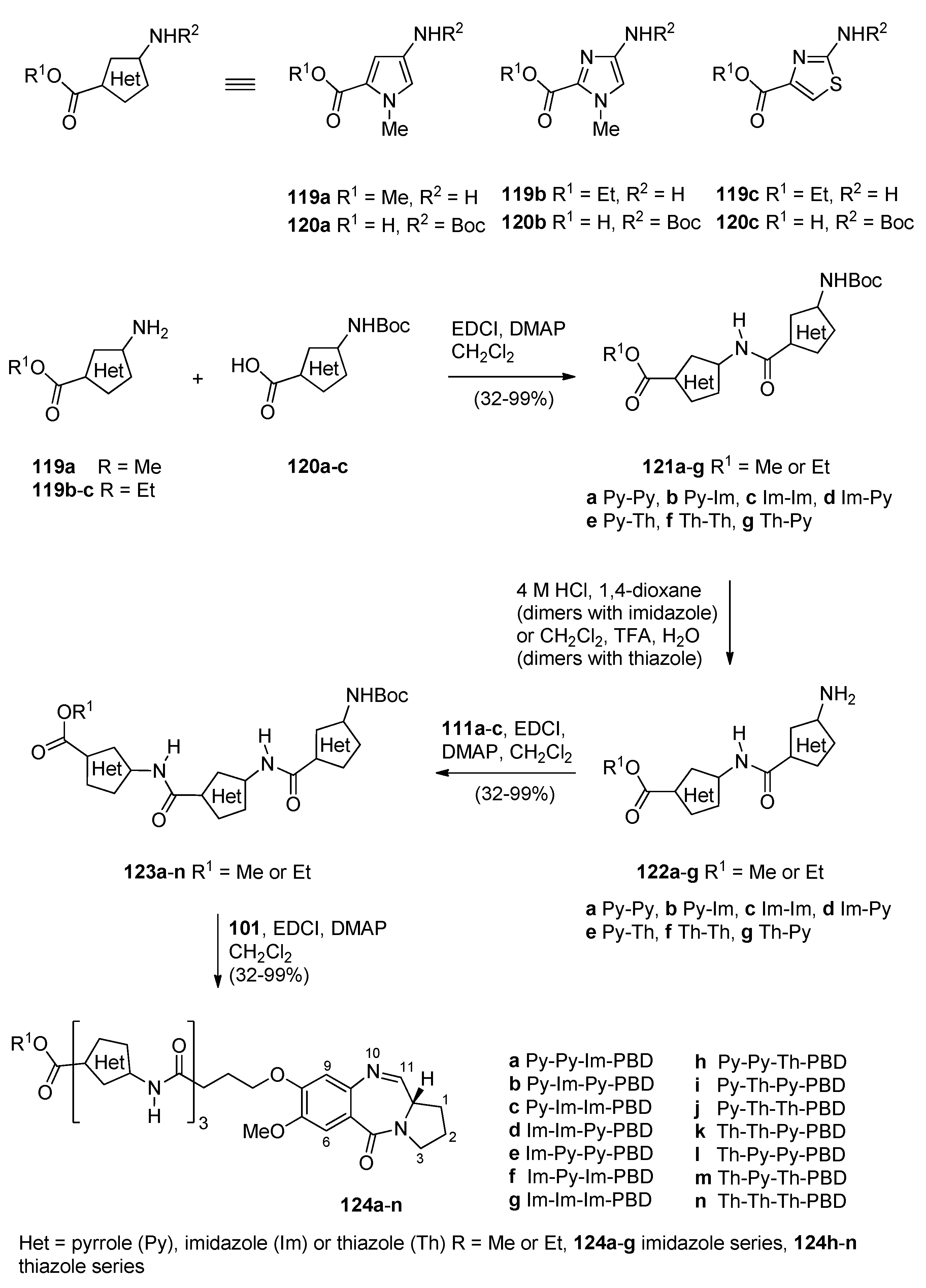
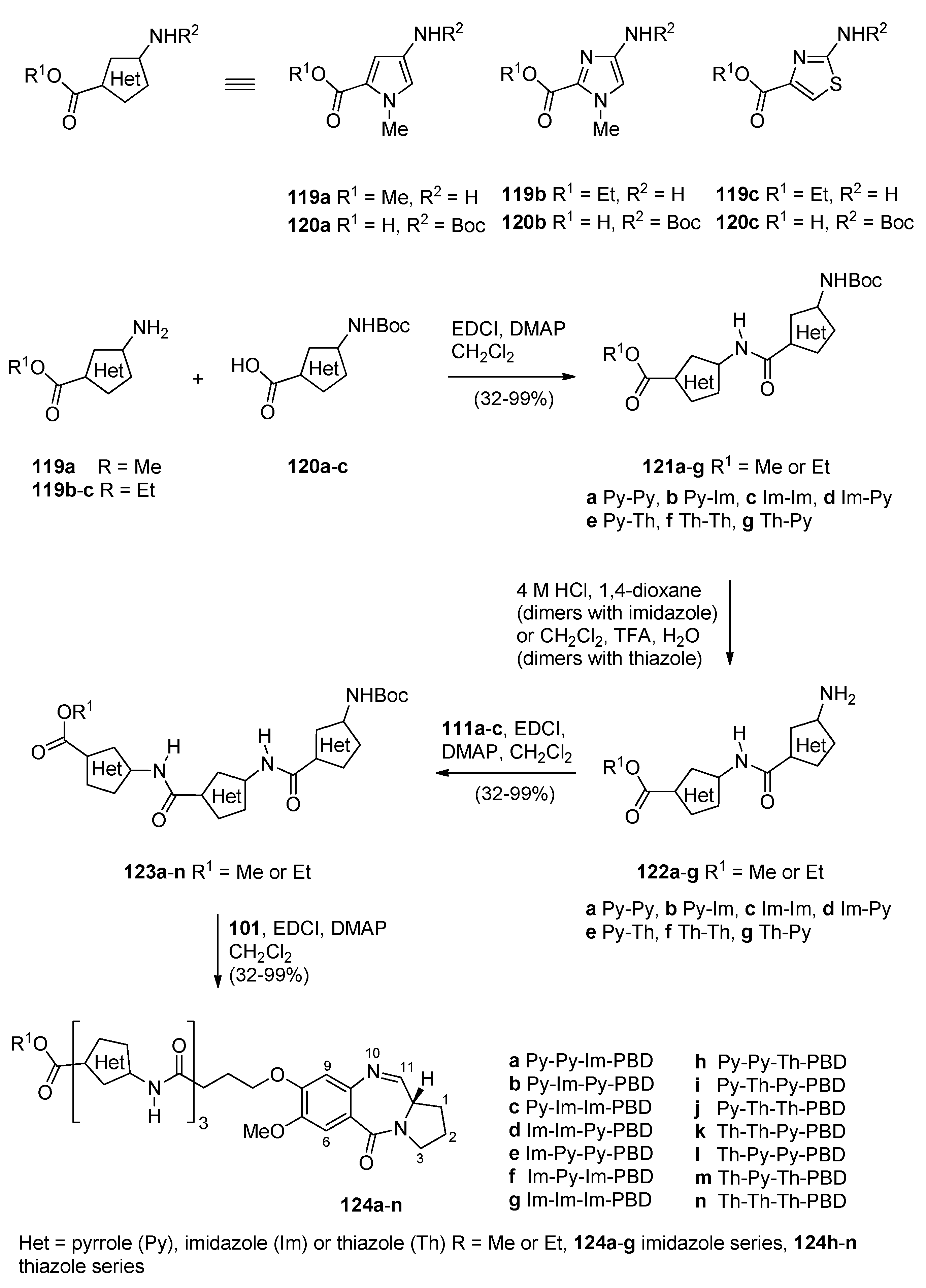
Based on recent findings that the binding of nuclear factor Y (NF-Y) to inverted CCAAT boxes (ICBs), in the Topo IIα cellular region, a target for anticancer agents, Thurston and co-workers [65] prepared a library of PBD conjugates where one PBD molecule is linked via the C8 oxygen atom to an ethane moiety which is joined to three alternative amide/5-membered heterocyclic ring units 124a–n (Scheme 21). These are potential NF-Y inhibitory molecules that were then studied as inhibitors against the replication of cancer cells. This synthetic procedure is similar to the previous two synthetic procedures as far as the preparation of PBD-carboxylic acid 93 and its coupling with preformed 5-membered heterocyclic amino ester trimers 123a–n, is concerned. Thus, for the preparation of heterocyclic trimers 123a–n the solution phase approach was used to couple amino ester heterocycles 119a–c to the Boc-protected heterocyclic acids 120a–c to provide the Boc-protected heterocyclic dimers 121a–g in sufficient yield and purity, to be used directly in the following deprotection step. It was found that 4 M hydrochloric acid in dioxane was a suitable deprotection system for the imidazole/pyrrole-containing dimers, whereas a mixture of dichloromethane–trifluoroacetic acid–water (50:47.5:2.5 v/v) was required for the thiazole/pyrrole-containing dimers. The resulting amino ester dimers 122a–g were subsequently coupled to the Boc-protected acids 120a–c to furnish the Boc-protected trimers 123a–n. The free amines of 123a–n were then coupled to the previously described [61] PBD carboxylic acid 101 to afford PBD conjugates 124a–n, in overall good yields. The synthesized PBD conjugates 124a–n underwent DNA thermal denaturation experiments to determine their DNA-binding affinity, the DNase I footprinting assay to determine both their DNA binding affinity and sequence selectivity, the electrophoretic mobility shift assay to measure the inhibition of NF-Y binding to the ICB1 and ICB2 sites and were evaluated for their in vitro toxicity at the NCI. Furthermore, following the discovery that PBD conjugate 124a is a potent of NF-Y binding to the ICB2 site, molecular modeling gave an insight of its covalent interaction with DNA.
Scheme 21.
Synthesis of C8-O-substituted PBD conjugates 124a–n.
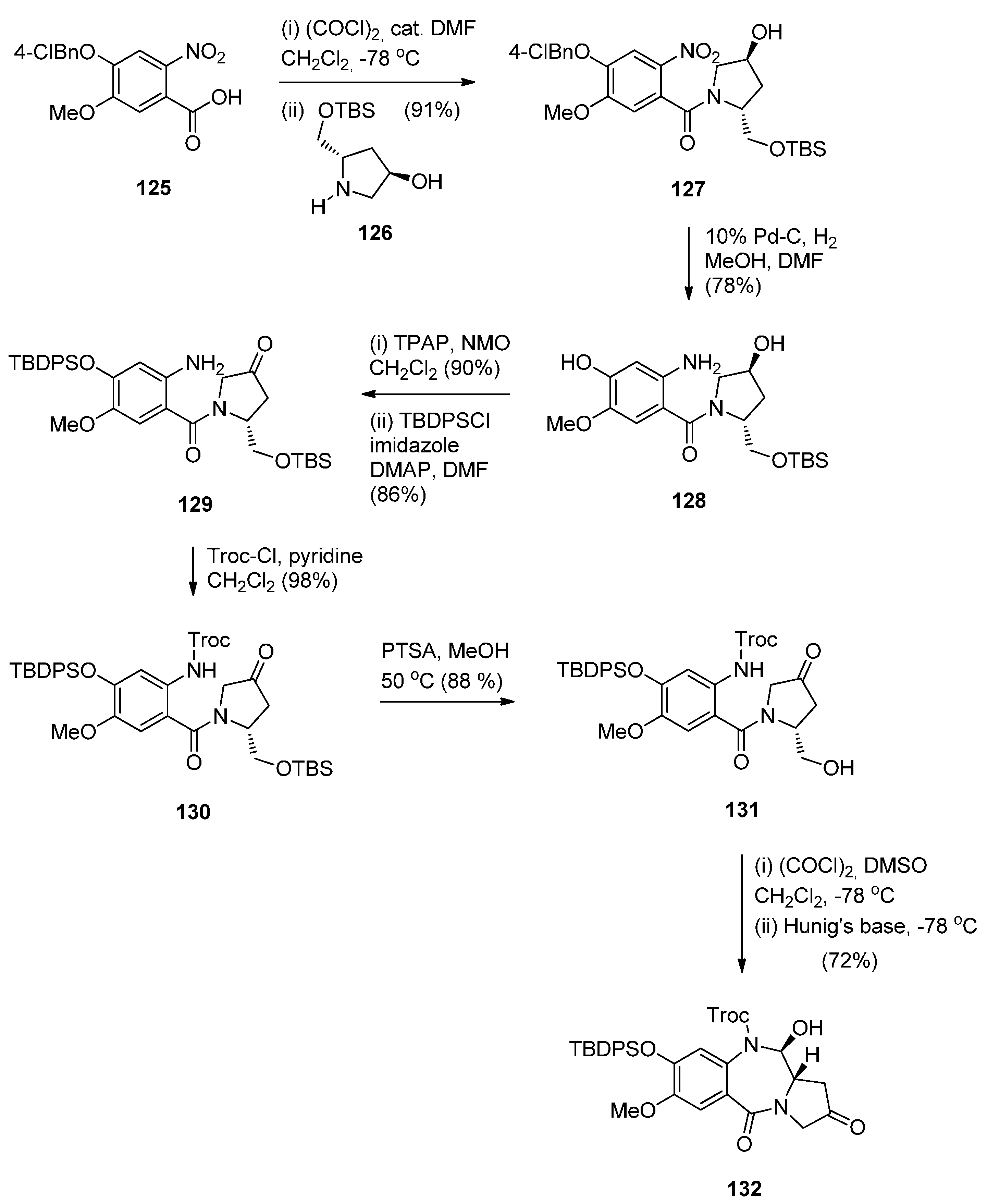
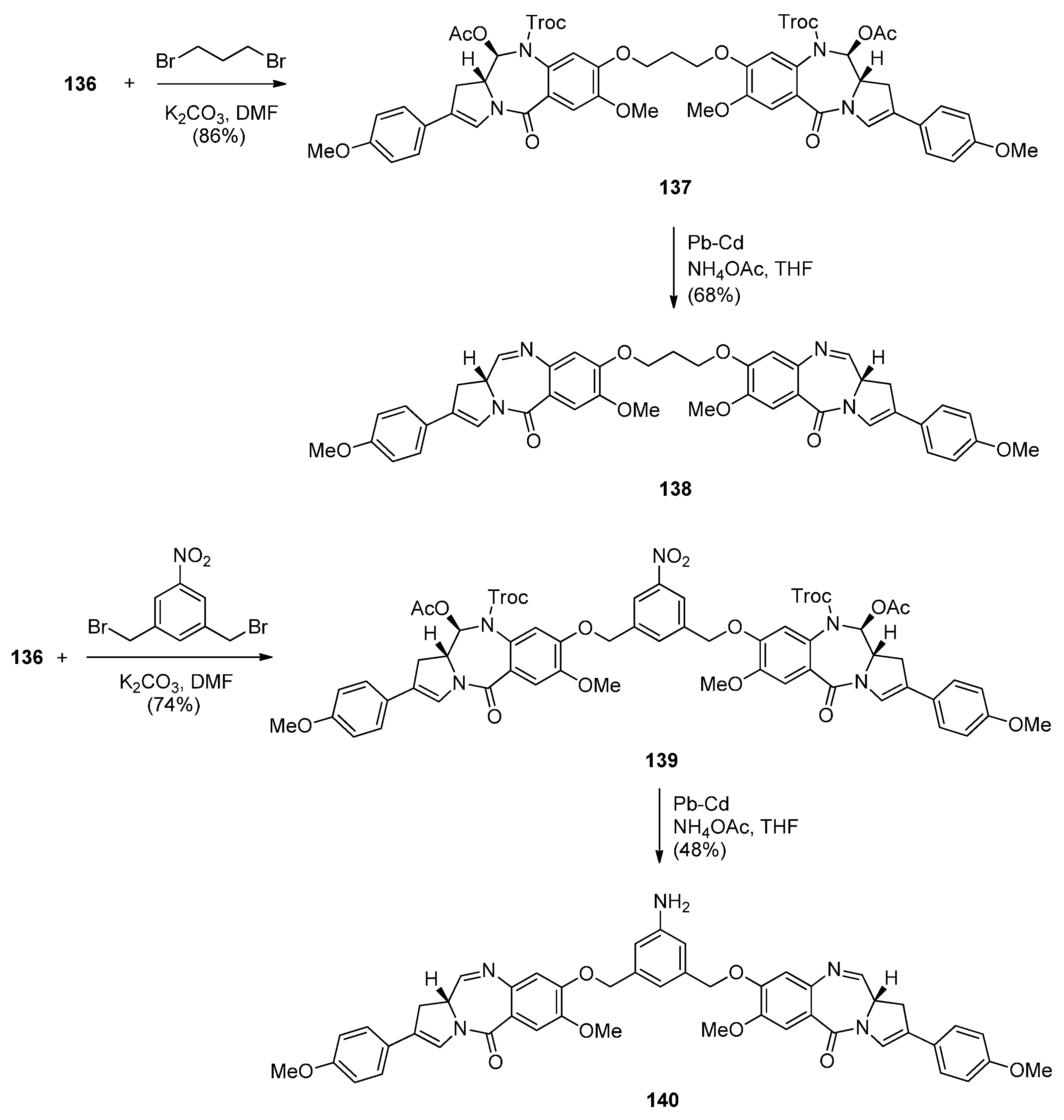
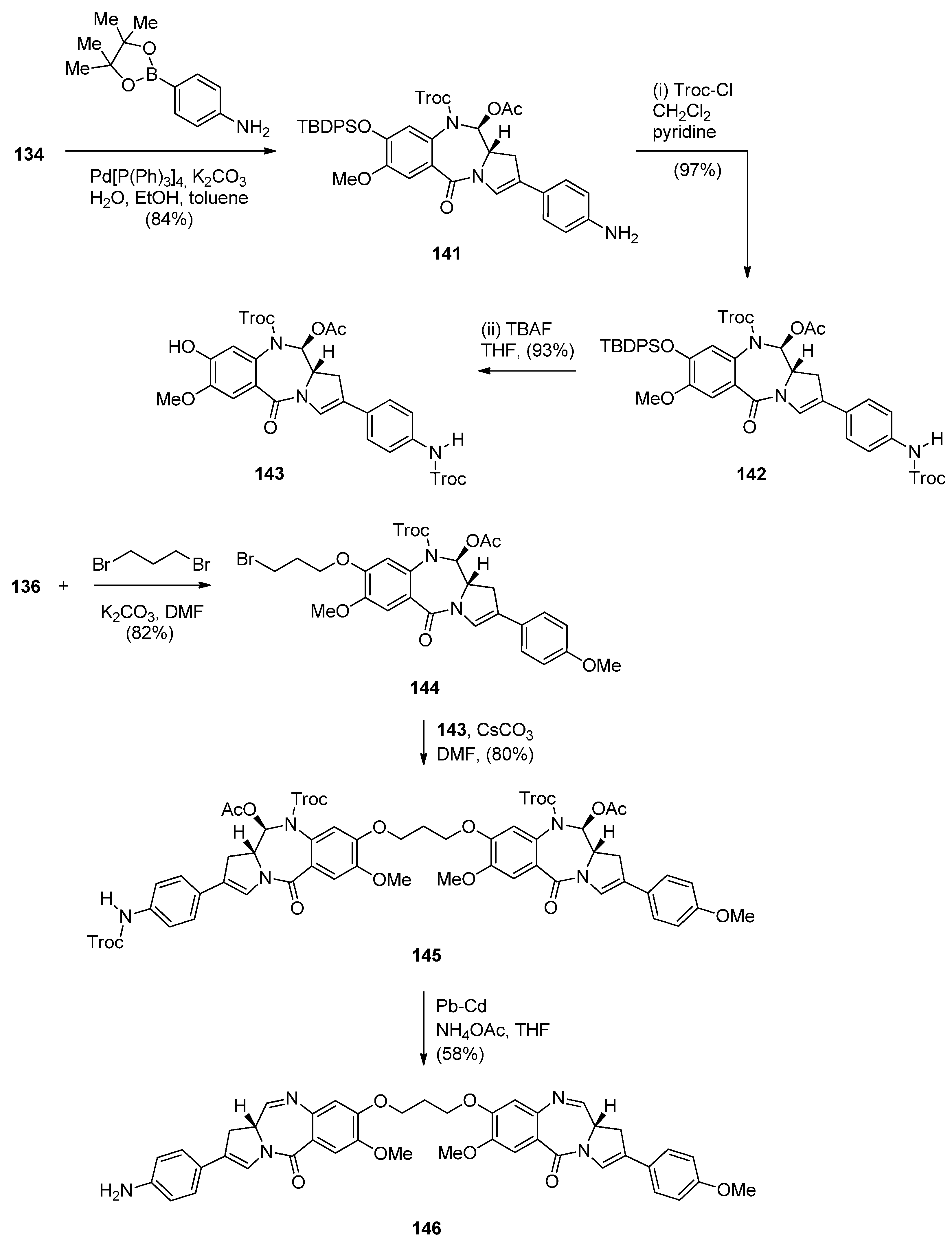
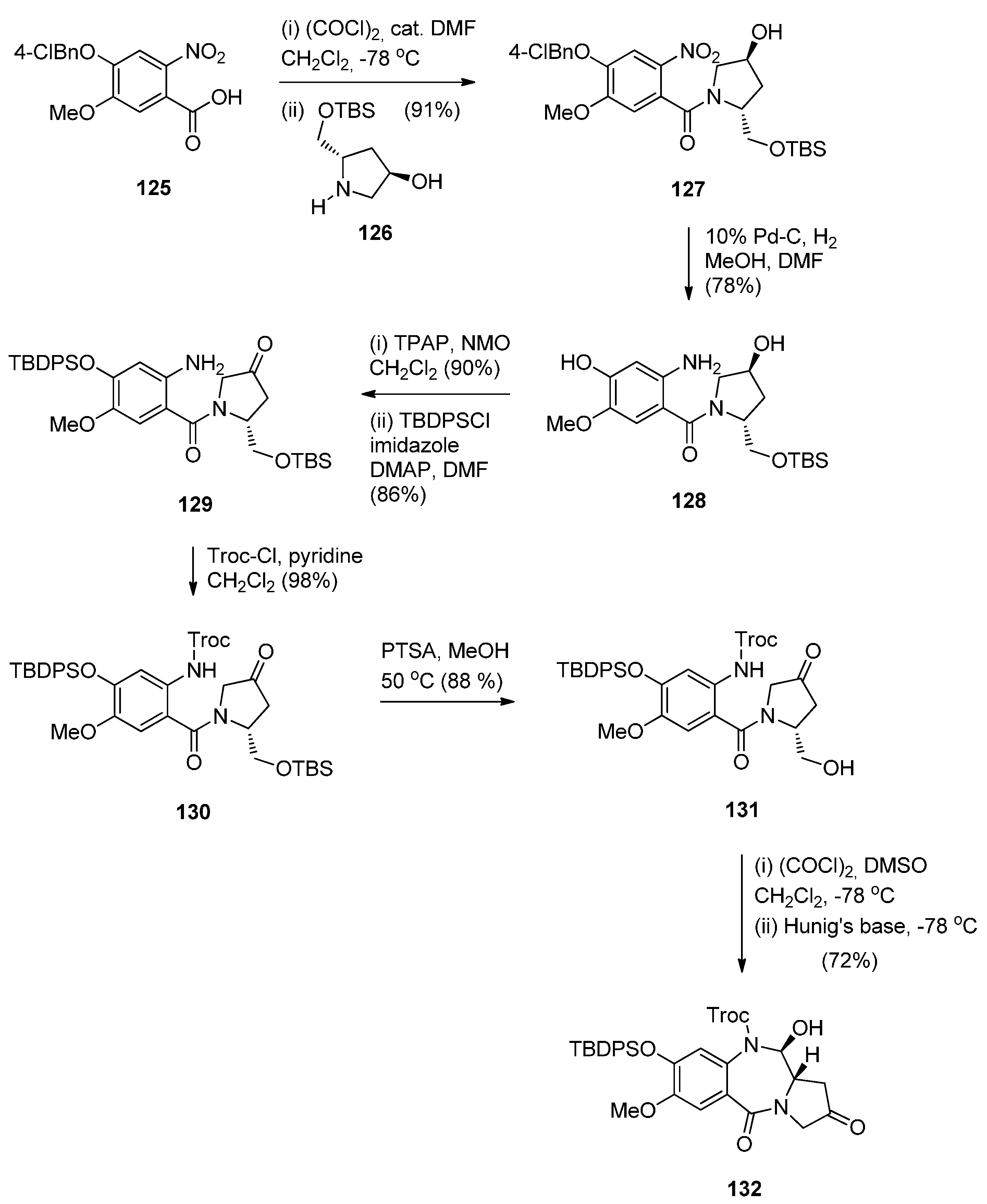
In view of the more potent cytotoxic activity of PBD dimers over PBD monomers and in order to better evaluate the properties of this class of therapeutic agents for ADCs (antibody–drug conjugates) for treating cancer, Kolakowski et al. [66] developed the versatile C2–aryl PBD monomer 136 (Scheme 22 and Scheme 23) that enables the late-stage diversification and synthesis of both symmetric and non-symmetric PBD dimers 137–140, and, 145 and 146, respectively (Scheme 24 and Scheme 25). This synthetic methodology is quite similar to the one presented by Thurston and co-workers [63] Scheme 17, [64] Scheme 19 and [65] Scheme 21, in that the PBD ring is constructed with either a 3-carboxypropyl substituent attached to the hydroxyl group at C8, i.e., 101 or without a substituent at the C8–OH group, i.e., 136. In both cases the substitution at C8 is elaborated further while imine formation at N10–C11 of the PBD ring takes place in the last synthetic step. The synthesis of the key intermediate, PBD monomer 134 (Scheme 23), began by converting the readily available carboxylic acid 125 to the corresponding acid chloride and without isolation treated with TBS-protected pyrrolidine diol 126 to yield the amide 127. Under catalytic hydrogenation the nitro group of 127 was reduced to amino and the phenol group deprotected to provide amino-phenol 128. The 3-hydroxyl group of 128 was subjected to a Ley oxidation to give the corresponding ketone whose phenolic hydroxyl group reacted with tert-butyldiphenylsilyl chloride (TBDPSC) to afford protected 129. Next 129 was protected by reaction with trichloroethyl chloroformate (Troc-Cl) to afford Troc-amine 130. Selective deprotection of the TBS group of 130 gave primary alcohol 131. Oxidative cyclisation of 131 to PBD lactol 132 was accomplished by the Swern oxidation.
Scheme 22.
Synthesis of C8-O-protected PBD precursor 132.
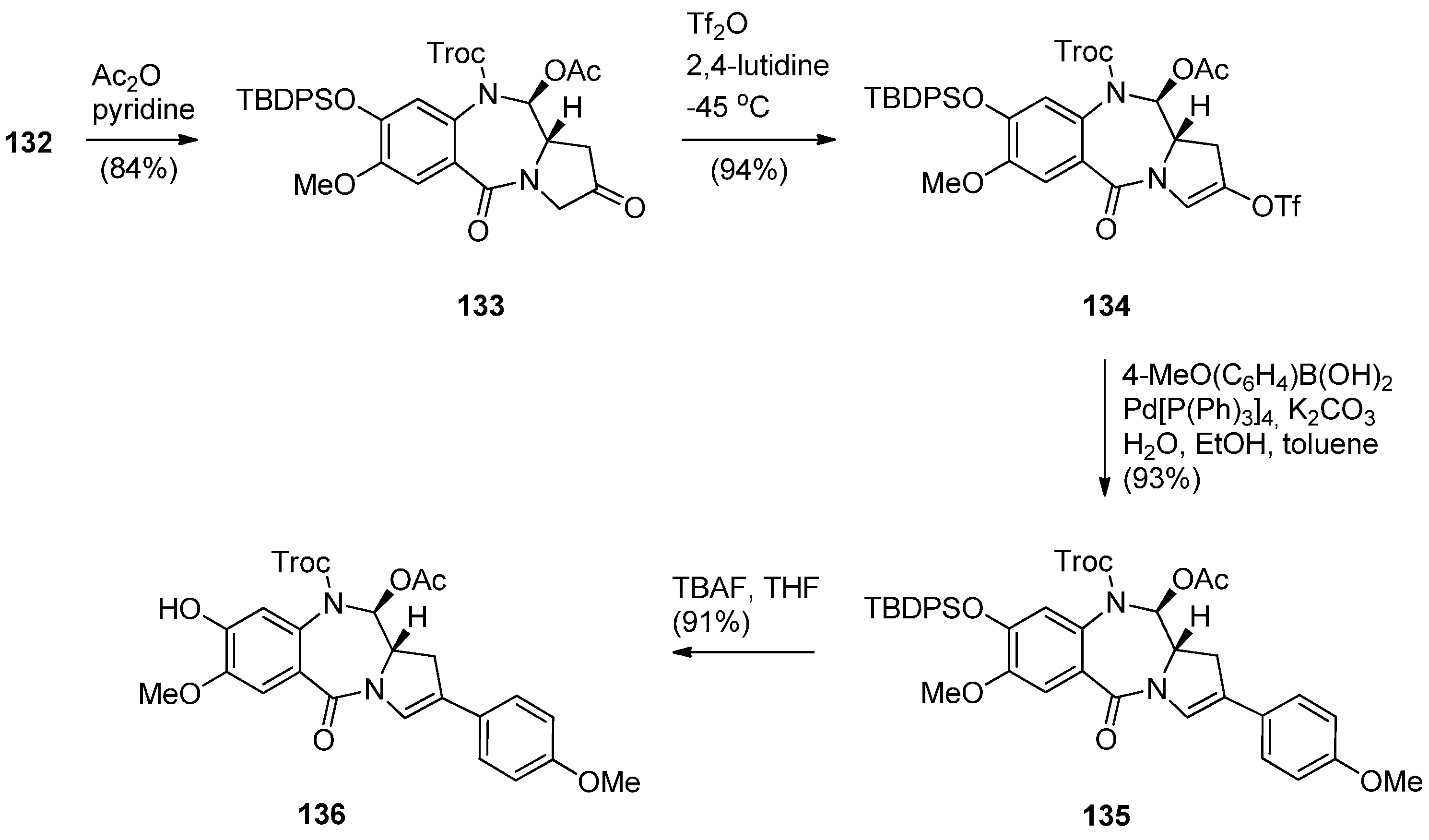
Scheme 23.
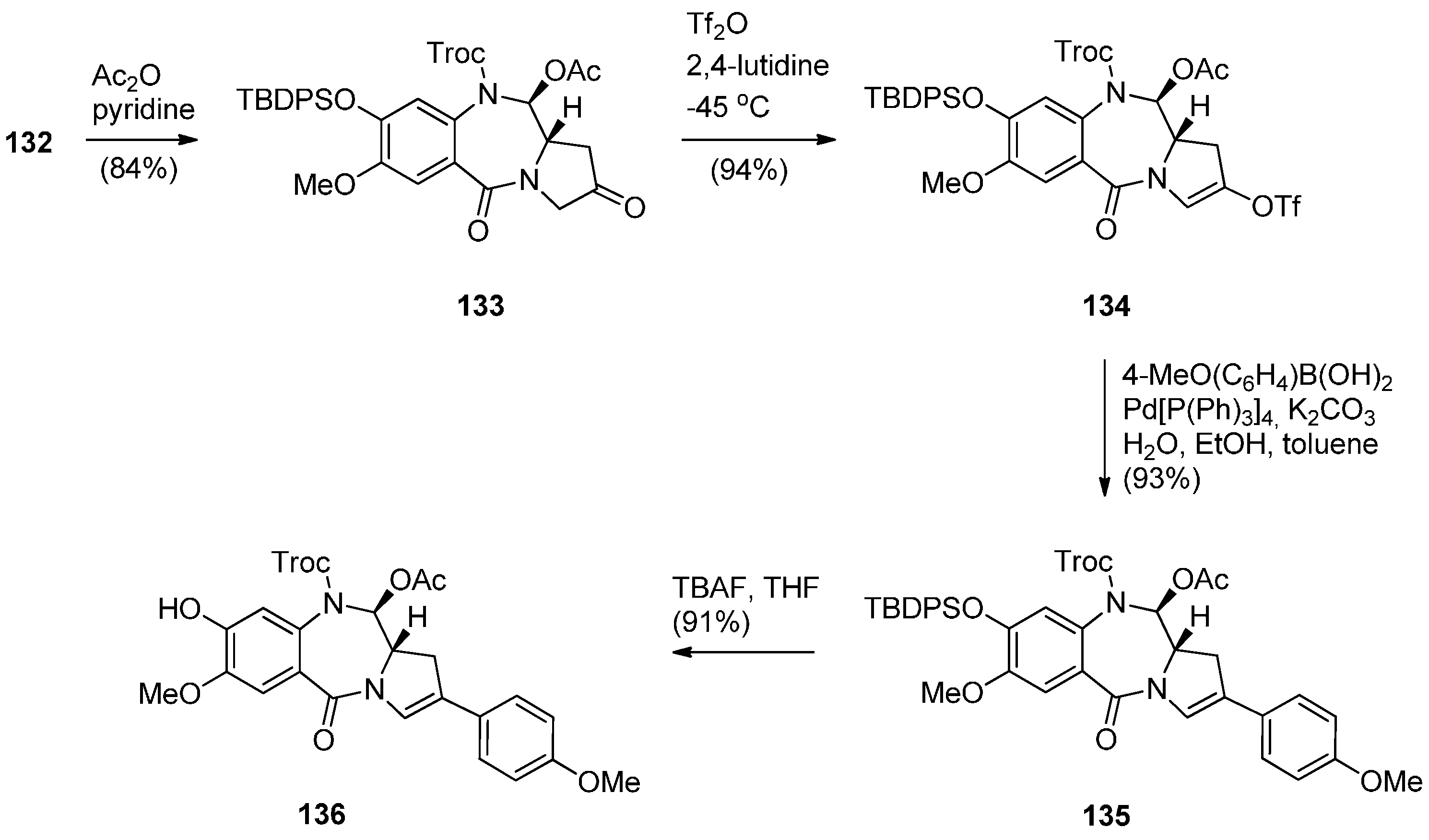
Synthesis of C8-OH N10-Troc C11-OAc PBD precursor 136.
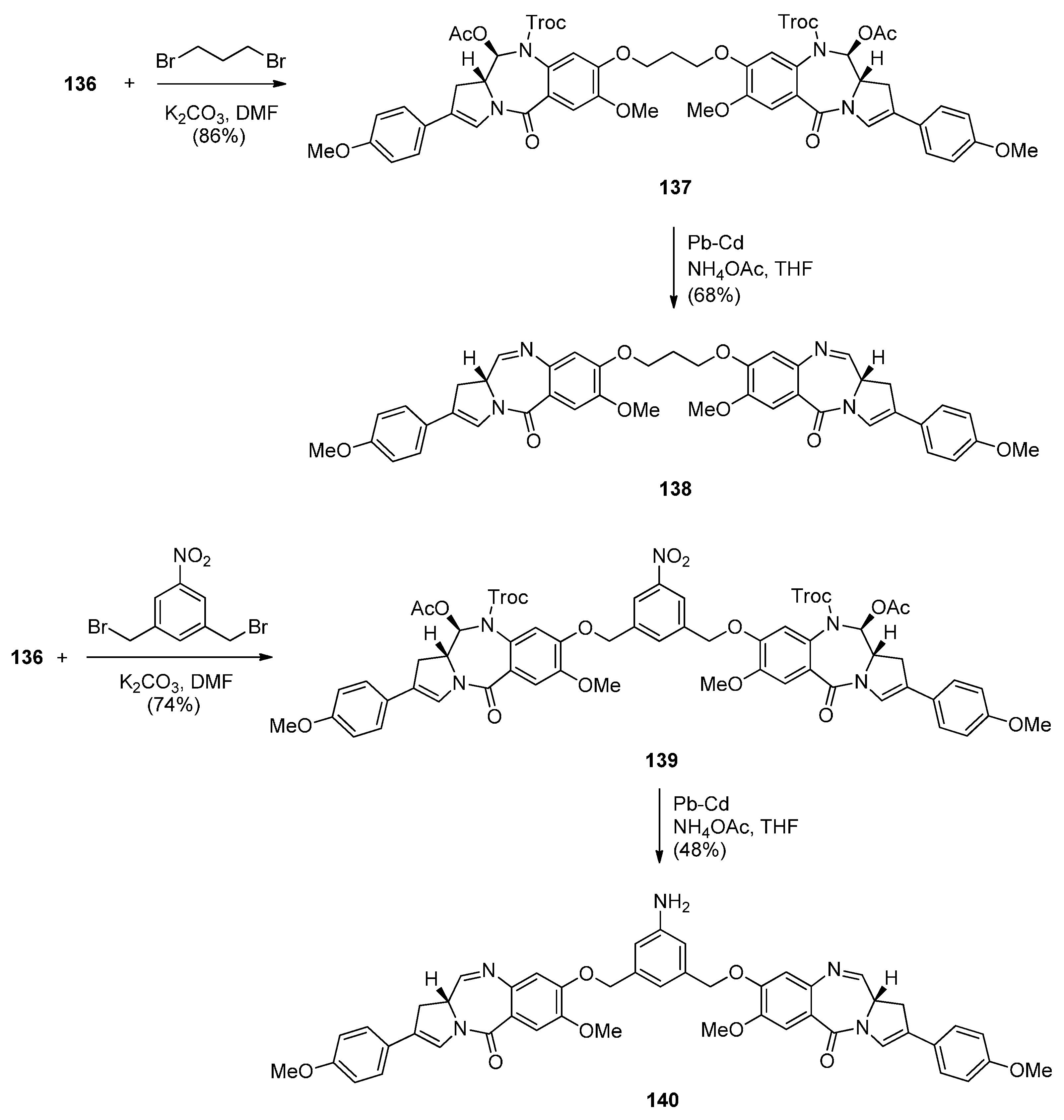
Scheme 24.
Synthesis of C8,C8′-linked PBD symmetric dimers 138 and 140.
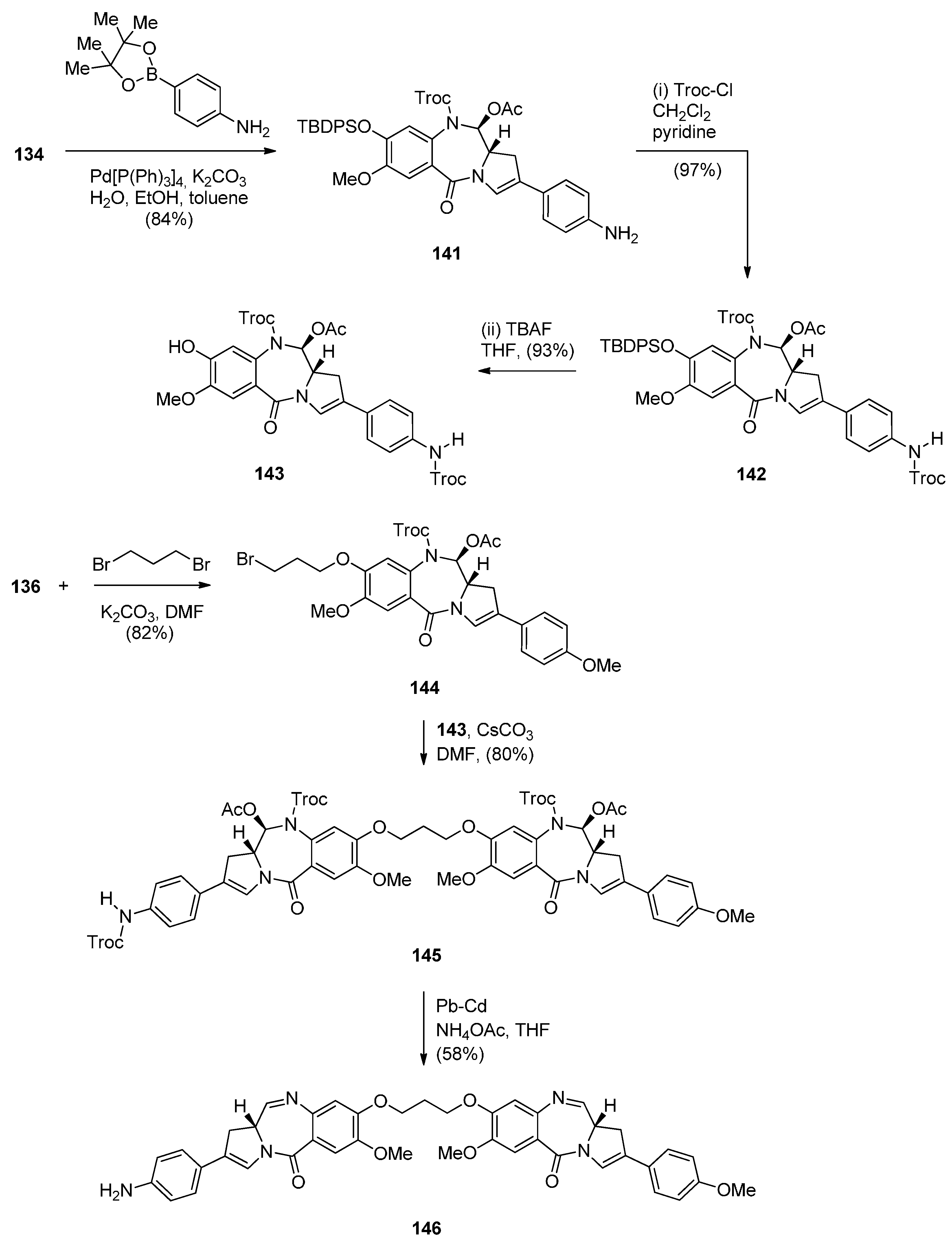
Scheme 25.
Synthesis of C8,C8′-linked PBD non-symmetric dimer 146.
In order to proceed to the synthesis of symmetrical and non-symmetrical PBD dimers key intermediate 136 (Scheme 23) was required. This was achieved by four synthetic steps from lactol 132. Acetylation of the C11–OH of 132 to 133 was followed by enol triflation to compound 134 which underwent Suzuki coupling with 4-methoxyphenylboronic acid to give 2-aryl derivative 135. Treatment of 135 with tetra–n-butylammonium fluoride (TBAF) removed the phenolic TBDPS group to yield the C8–OH PBD 136.
The following four PBD dimers 137–140 were synthesized as shown in Scheme 24. The synthetic strategy, starting from compound 125 (Scheme 22), was focused on preparing the PBD monomer 136 with a C2 4-methoxyphenyl substituent required in the PBD dimer structures and a C8–OH group used for joining two PBD monomers with an appropriate linker, to form the PBD dimer. Thus, PBD dimer 137 was obtained by alkylating PBD monomer 136 with half an equivalent of 1,3-dibromopropane in base. Deprotection of the N10–Troc and C11–acetoxy groups of PBD 137 and concomitant formation of the N10–C11 imine group took place in a one-pot procedure utilizing palladium-cadmium couple to afford the well-studied C2 p-methoxyphenyl dimer SG2202, PBD 138. Accordingly, dimerization of monomer 136 with 1,3-bis(bromomethyl)-5-nitrobenzene to PBD dimer 139 and then reduction, afforded the symmetrical PBD dimer 140.
Non-symmetric dimers 145 and 146 (Scheme 25) were prepared essentially as the above symmetrical dimers. This time apart from PBD monomer 136 the other PBD is C8–OH C2 4-NHTroc protected monomer 143. Troc protection is required to avoid alkylation of the aniline group when the two PBD monomers are connected by alkylation of their C8–OH groups. Thus, the synthetic strategy towards PBD dimer 145 (Scheme 25) requires Suzuki coupling of 134 with 4-aminophenylboronic acid pinacol ester, protection of the 4-aniline group of 141 as NHTroc derivative 142 and then deprotection of the tert-butyldiphenylsilyl (TBDPS) group of 142 to yield 143. PBD monomers 136 and 143 are linked via 1,3-dibromopropane, as shown in Scheme 25, to yield the protected non-symmetric PBD dimer 145. Deprotection of the latter compound by a lead-cadmium couple led to PBD imine dimer 146.
Reductive Cyclisation of N-(2-Azidobenzoyl)pyrrolidine-2-Carboxaldehydes
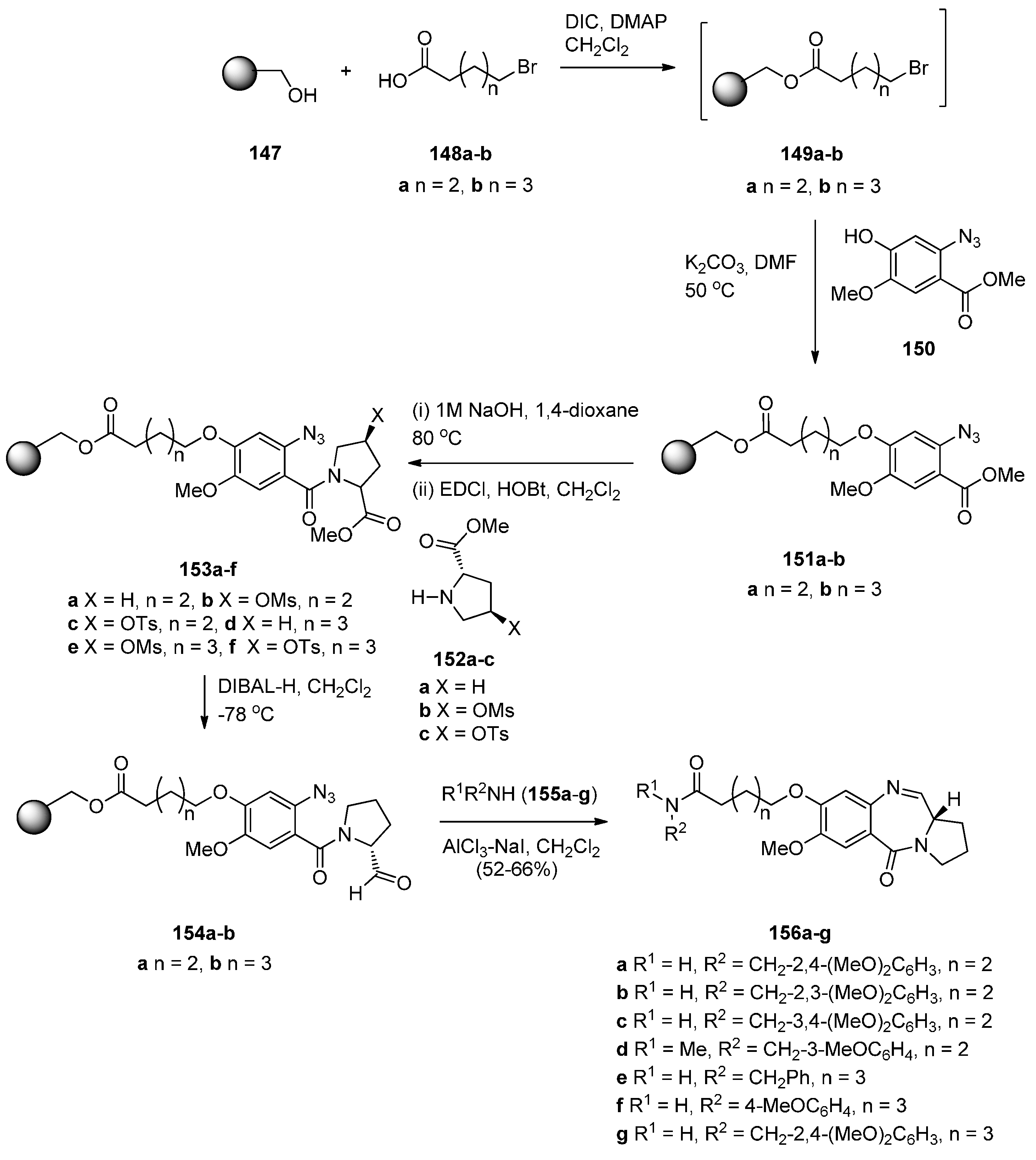
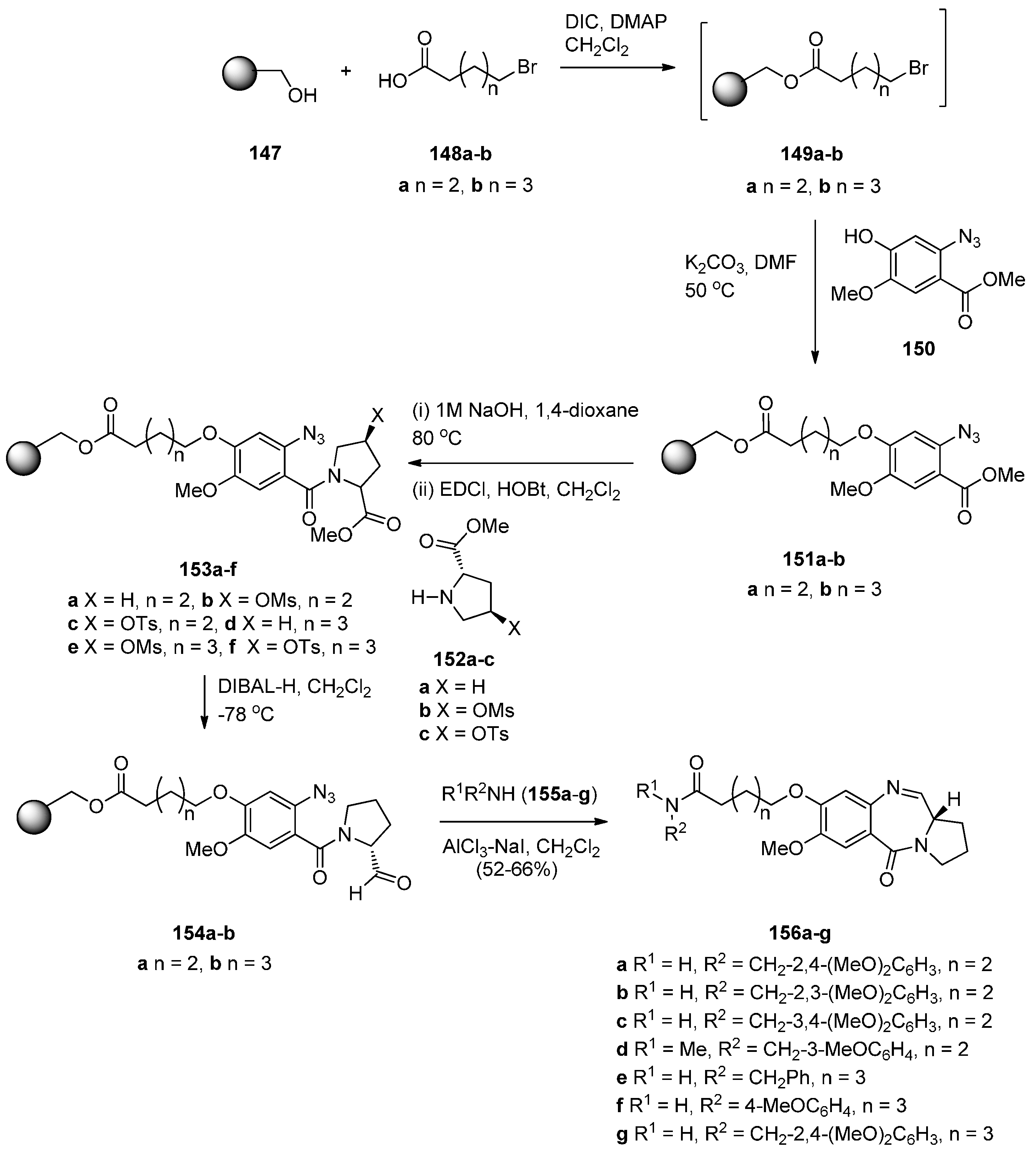
The use of N-(2-azidobenzoyl)pyrrolidine-2-carboxaldehydes to obtain PBD N10–C11 imines was reported independently by Eguchi and Molina [10] in 1995. They used the Staudinger reaction on the azido aldehyde to form an iminophosphorane which spontaneously cyclised via an aza-Wittig reaction with the adjacent aldehyde group of the molecule. Kamal and co-workers introduced polymer-supported reagents for this methodology and also reported various methods of reducing the nitro group of N-(2-azidobenzoyl)pyrrolidine-2-carboxaldehydes to an amino group followed by cyclisation to the PBDs [10]. The latter method of cyclisation combined with the use of a polymer support was recently published by Kamal et al. [67] who prepared a library of tricyclic PBDs 156a–g (Scheme 26). The synthetic approach used was first to couple the Wang resin 147 to the bromo-acids 148a,b and without isolating resulting resin-bound esters 149a, their reaction with methyl 2-azido-4-hydroxy-5-methoxybenzoate (150) [68] to provide the desired polymer supported ethers 151a,b. The aromatic ester group of ethers 151a,b was hydrolysed and subsequently coupled with substituted L-proline esters 152a–c to provide the key resin-bound intermediates 153a–f. The resin-bound azido esters 153a,b were selectively reduced with diisobutylaluminium hydride (DIBAL-H) to their corresponding resin-bound azido-aldehydes 154a,b and then submitted to a one-pot reaction of aluminium trichloride and sodium iodide assisted cleavage of the resin to the free acid, coupling with the added excess amount of amines 155a–g and concomitant tandem selective azido-reductive cyclocondensation with the aldehyde group, to afford the desired final PBD conjugates 156a–g, in 52%–66% yields. The synthetic method just described constitutes a novel route to C8–O-substituted PBD N10–C11 imine conjugates, employing the aluminium trichloride and sodium iodide system to execute four independent reactions under one roof, resin cleavage, amino and carboxylic acid coupling to amide, reduction of azide to amine and finally intramolecular condensation of amine and aldehyde to imine.
Cyclisation of N-(2-Azidobenzoyl)pyrrolidine-2-carboxaldehydes
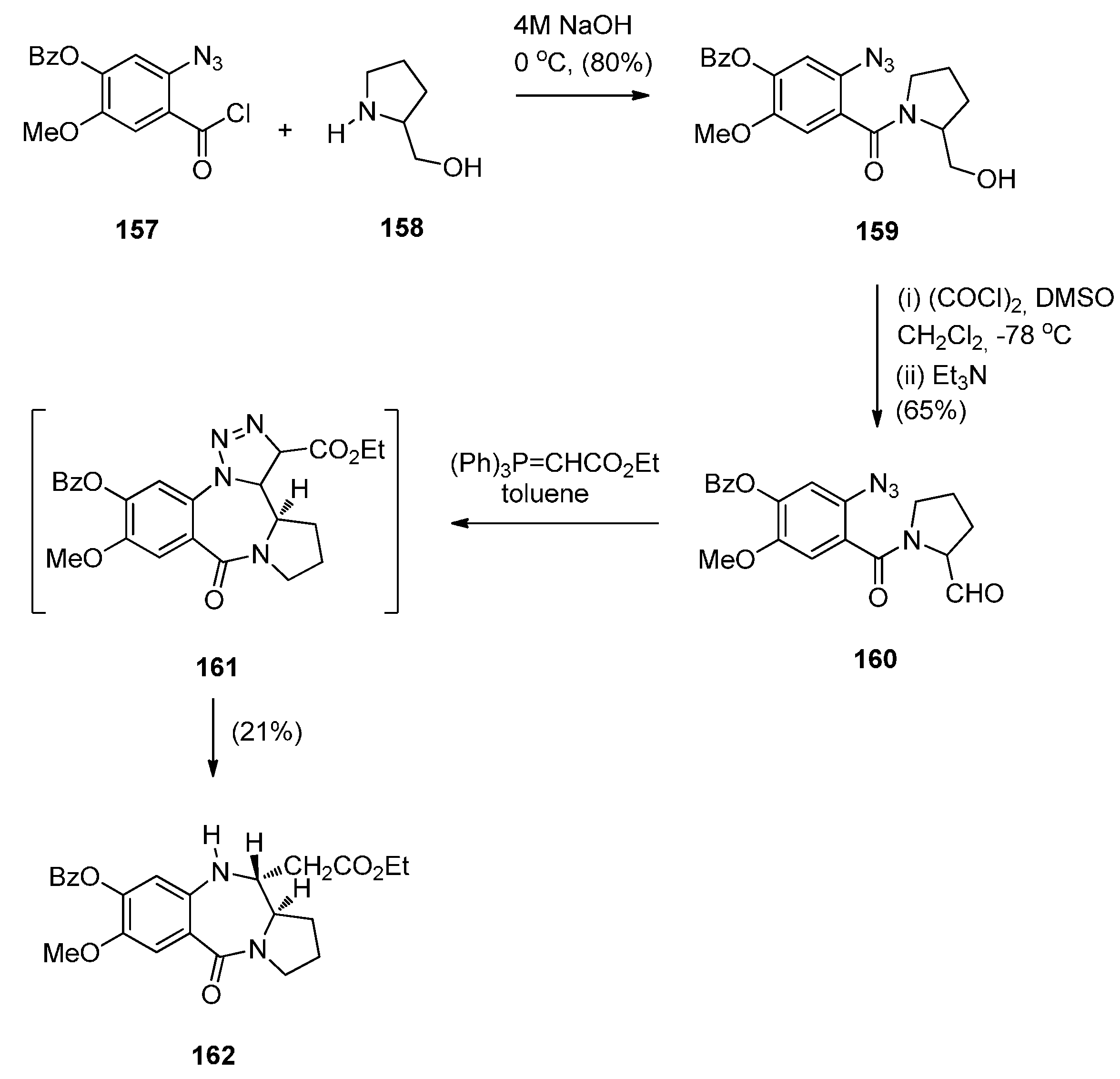
A rather intriguing but very low yielding approach to a PBD 5-one was described by Hemming et al. [69]. In the first step, the reaction involves acyl substitution between 2-azidobenzoyl chloride 157 and (S)-prolinol 158 to furnish amide 159 (Scheme 27) whose hydroxymethyl group was oxidised to afford, aldehyde 160 as a mixture of rotamers. Wittig reaction of this aldehyde with (carbethoxymethylene)triphenylphosphorane gave the corresponding α,β-unsaturated ester that underwent spontaneous intramolecular 1,3-dipolar cycloaddition, to give PBD 162 as a single diastereoisomer, in 21% yield. The authors assume that PBD 162 arises as a result of a free-radical based loss of nitrogen from intermediate triazoline 161 followed by hydrogen abstraction from the toluene solvent. Although the present synthetic route constitutes a novel method of preparing 11-ethoxycarbonylmethyl PBD 5-ones, the author has presented only one example and the route suffers from a very low yield in the final step.
Scheme 26.
Synthesis of C8-O-substituted PBD conjugates 156a–g.
Scheme 27.
Synthesis of PBD 162.

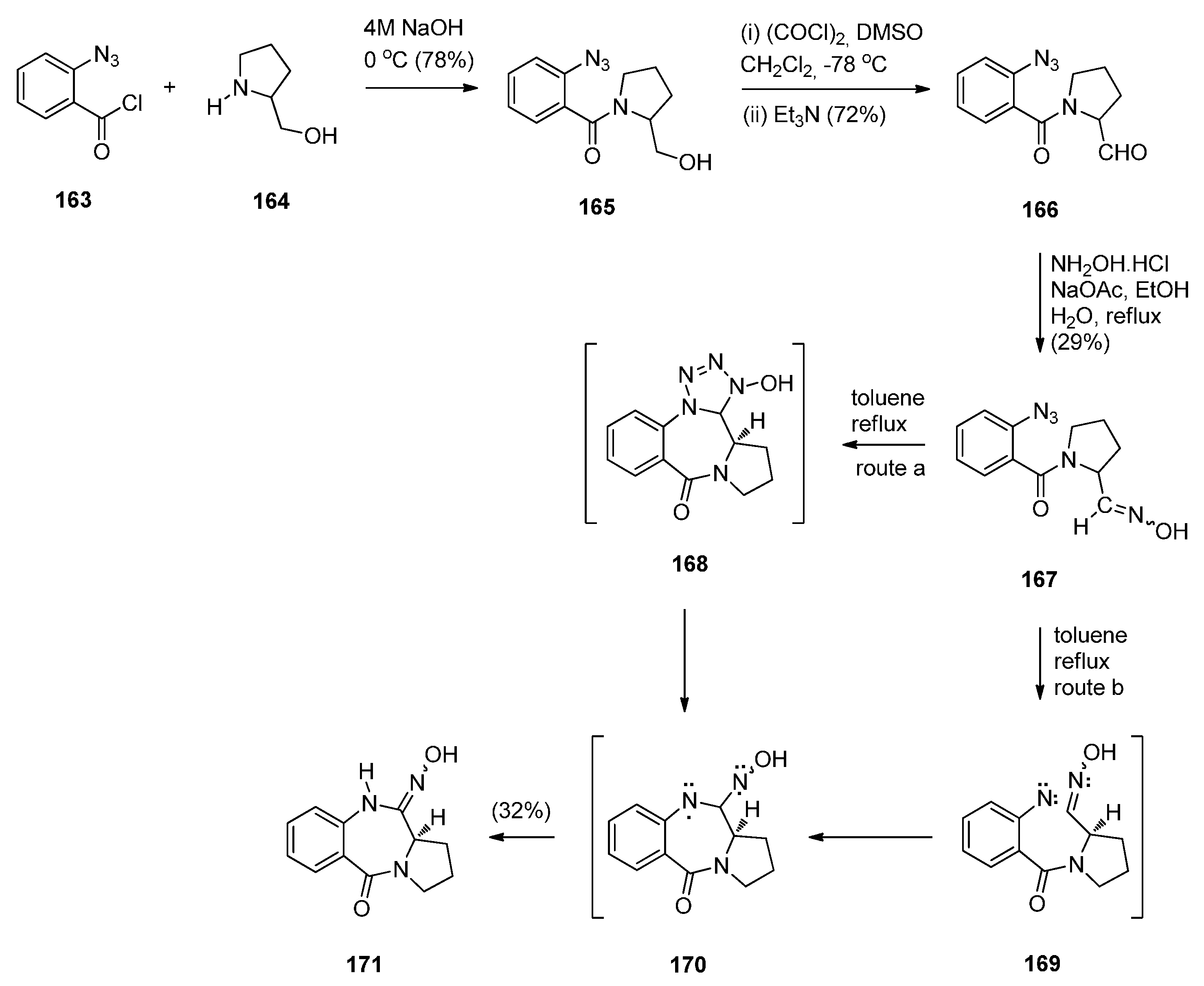
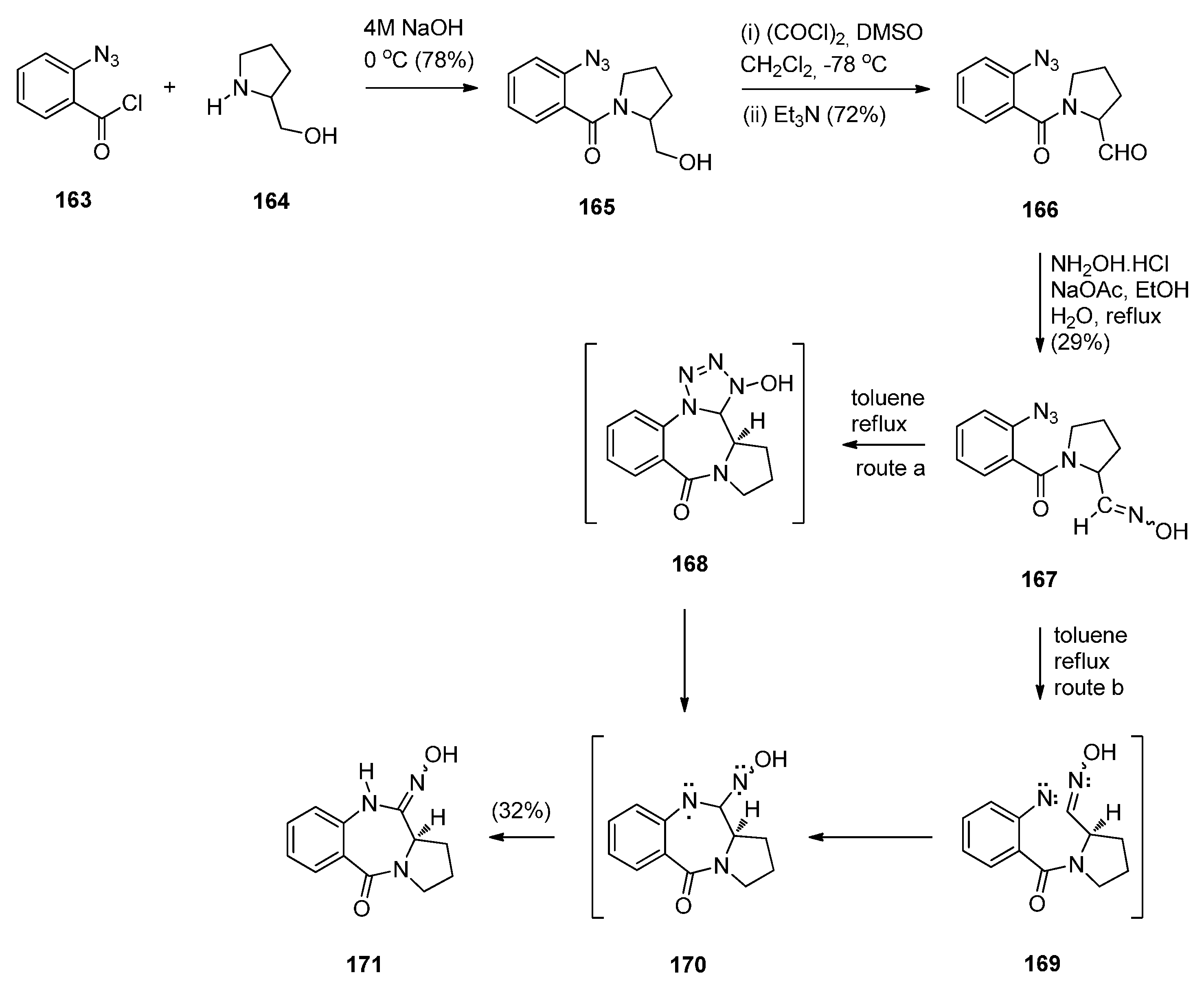
In the same year, Hemming et al. [70] reported the synthesis of azido-oxime 167 in order to study the potential of the azide to oxime intramolecular cycloaddition (Scheme 28). Acyl substitution of 2-azidobenzoyl chloride 163 by (S)-prolinol 164 afforded amide 165 that was subjected to Swern oxidation to yield azido-aldehyde 166. Although up to this step the yields of products were over 70%, the oxime 167 was obtained in only 29% yield and PBD 171 was isolated in 30% yield. The formation of this product is consistent with either intramolecular 1,3-dipolar cycloaddition followed by nitrogen extrusion and diradical rearrangement (route a) or nitrogen extrusion followed by nitrene insertion and diradical rearrangement (route b). This synthetic route leading to a N10–OH PBD 11-oxime is another novel preparation of a PBD 5-one although only one product in low yield is described by the authors.
Scheme 28.
Synthesis of PBD 171.
2.1.2. Pyrrolo[2,1-c][1,4]benzodiazepine-5,11-Diones (Dilactams) with a Non-Aromatic Pyrrole Ring
The relatively strong interest in synthesing PBD dilactams stems from the fact that although they contain a non-electrophilic amidic functionality at the N10–C11 region, they are known to interact with DNA predominantly by hydrogen bonding [10]. There are five most commonly used methods to obtain these compounds, reductive cyclisation of alkyl N-(2-azido or nitrobenzoyl)-pyrrolidine-2-carboxylates, cyclocondensation of isatoic anhydrides with substituted prolines, and cyclisation of 1-benzoyl-N-methoxypyrrolidine-2-carboxamides and of N-(2-trifluoromethylsulfonyl-oxybenzoyl)-pyrrolidine-2-carboxylic esters [10]. Although the most prolific of these methods is reductive cyclisation of alkyl N-(2-nitrobenzoyl)pyrrolidine-2-carboxylates, the need in most cases for acid-catalysed cyclisation of the intermediate alkyl N-(2-aminobenzoyl)pyrrolidine-2-carboxylates can be avoided when methyl N-(2-azidobenzoyl)pyrrolidine-2-carboxylates are used instead. These azido ester reductive cyclisations leading to PBD lactams have been pioneered by Kamal and co-workers [10] who recently have reported another application of this method (Section “Reductive Cyclisation of Methyl N-(2-Azidobenzoyl)pyrrolidine-2-Carboxylates”). Another report leading to PBD dilactams uses the isatoic anhydride route (Section “Reaction of isatoic anhydrides with d-prolines”).
Reductive Cyclisation of Methyl N-(2-Azidobenzoyl)pyrrolidine-2-Carboxylates
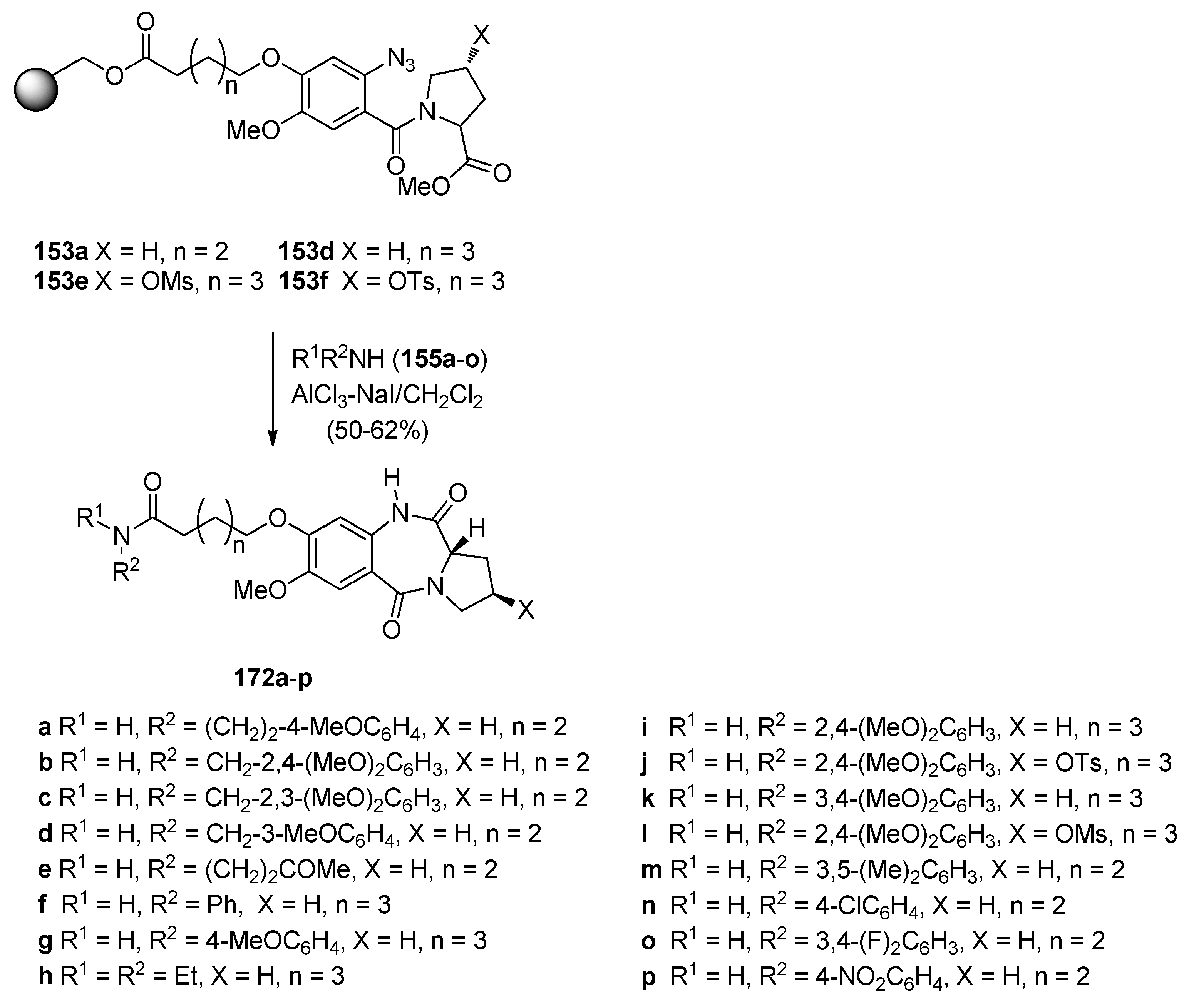
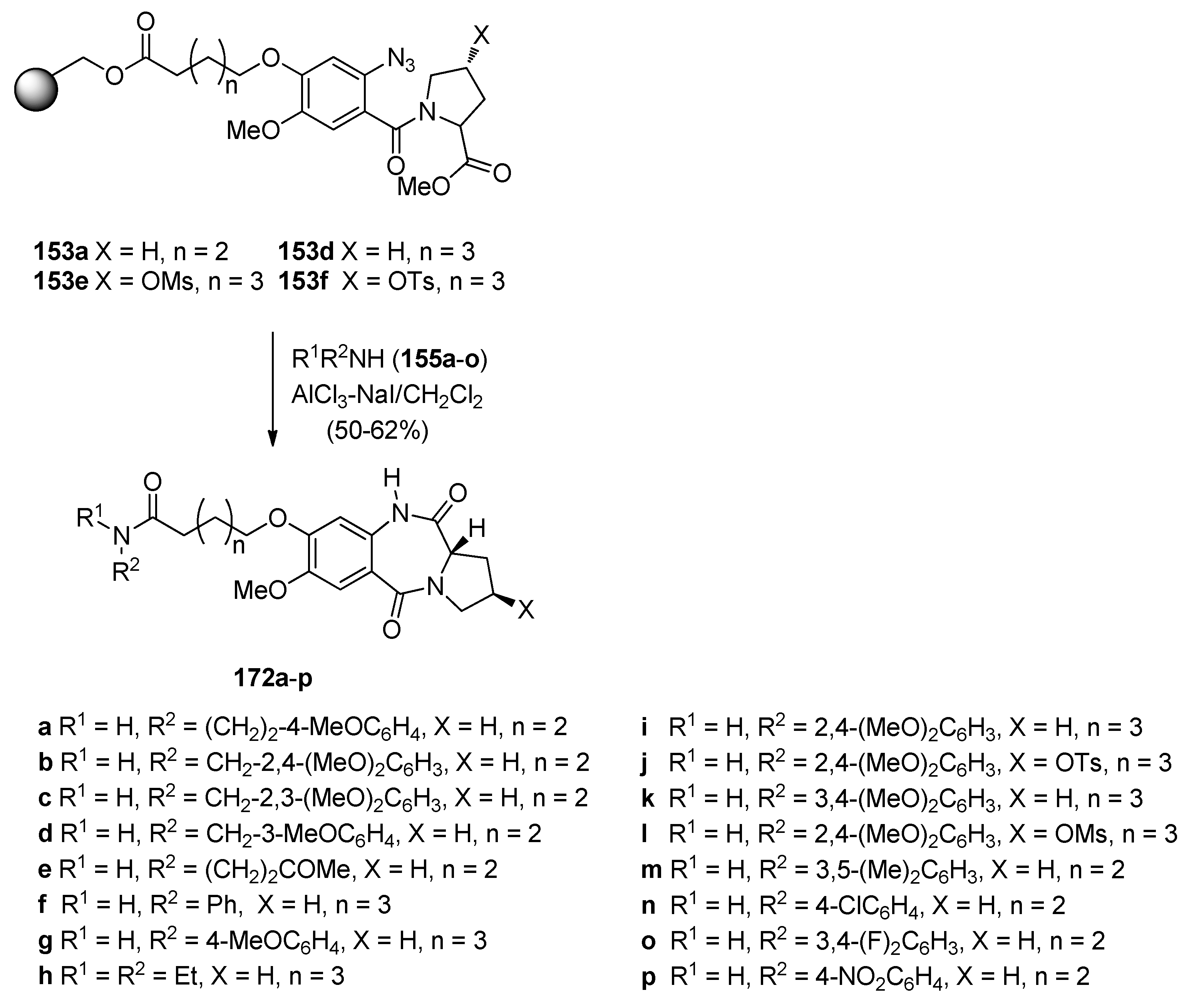
By picking two of the key resin-bound methyl N-(2-azidobenzoyl)pyrrolidine-2-carboxylates 153a,d described earlier (Section “Reductive cyclisation of N-(2-azidobenzoyl)pyrrolidine-2-carboxaldehydes”), Kamal et al. [67] used excess amines 155a–o and applied the same cyclisation procedure (Scheme 29) as they had done for the cyclisation of N-(2-azidobenzoyl)pyrrolidine-2-carboxaldehydes 154a,b (Section “Reductive Cyclisation of N-(2-azidobenzoyl)pyrrolidine-2-Carboxaldehydes”) and obtained the desired C8-substituted PBD-5,11-diones 172a–p, in 50%–62% yields. The yields of this cyclisation reaction are much lower than the yields (80%–99%) of PBD lactams obtained from corresponding non-resin bound methyl N-(2-azidobenzoyl)pyrrolidine-2-carboxylates using a variety of reagents such as, hexamethyldisilathiane in methanol, trimethylsilyl chloride with sodium iodide in acetonitrile, ferrous sulfate heptahydrate in aqueous ammonia solution ferric chloride with sodium iodide, zinc and ammonium formate in methanol, aqueous hydriodic acid, sodium iodide in acetic acid, boron trifluoride diethyl etherate with ethanethiol or sodium iodide, and, boride in acidic methanolic media under microwave radiation [10].
Scheme 29.
Synthesis of C8-O-substituted PBD conjugates 172a–p.
Reaction of Isatoic Anhydrides with d-Prolines
The advantage of using isatoic anhydrides in the synthesis of PBD dilactam scaffolds is their convenient preparation, their high yielding condensation with L-proline, and also with trans-4-hydroxy-l-proline and l-glutamic acid, and the ease of purifying the PBD products. Condensation procedures typically involve heating with dimethyl sulfoxide (DMSO) at 100–120 °C, refluxing in N,N-dimethylformamide (DMF), heating without solvent at 150 °C and without solvent under microwave radiation where reaction times of 3 min have been recorded.
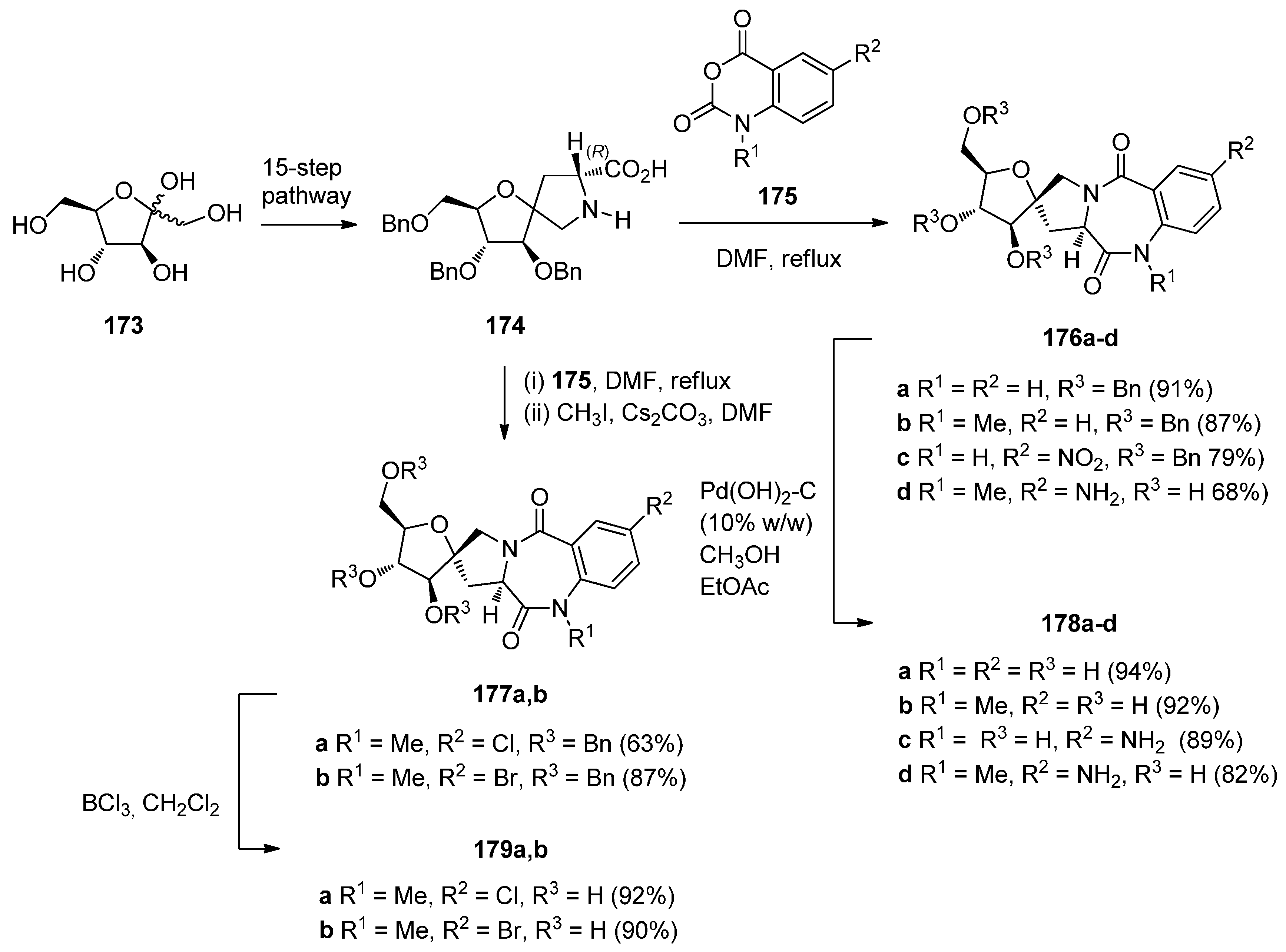
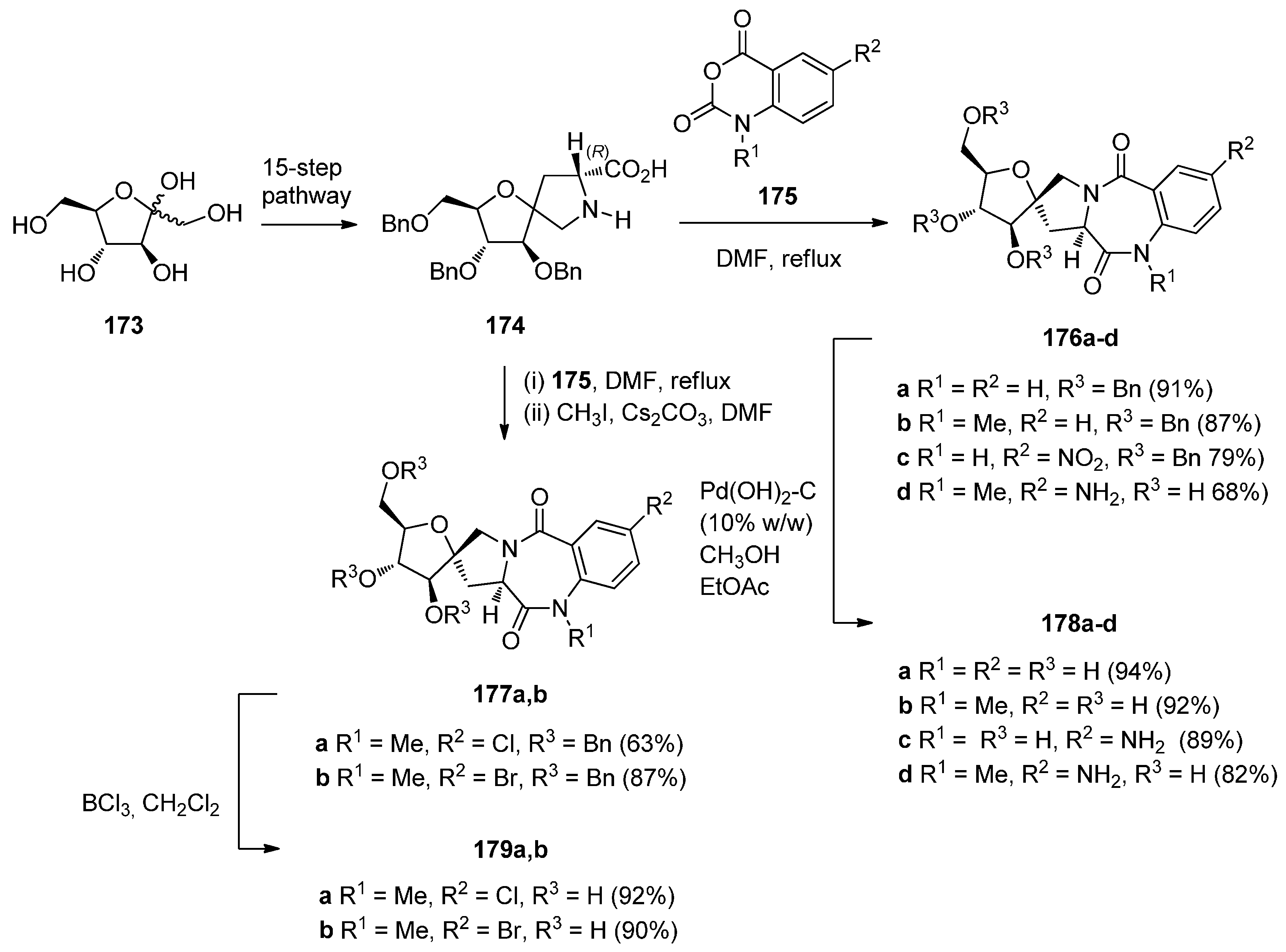
Araújo et al. [71] have applied the isatoic anhydride route to synthesise a library of enantiomeric and conformationaly constrained PBDs dilactams spiro-linked to d- or l-fructose rings 176a–d, 177a,b, 178a–d and 179a,b (Scheme 30) which were then evaluated as GABAA receptor ligands. The starting material for this synthetic route is the bicyclic spiro d-proline analogue 174 prepared through a 15-step pathway from d-fructose 173 as described earlier [72]. Thus, d-proline analogue 174 was condensed with isatoic anhydrides 175 to afford PBDs 176a–d with (11aR) configuration. Hydrogenolysis of PBDs 176a–d with palladium (II) hydroxide on carbon resulted both in debenzylation and reduction of the nitro group, yielding the sugar-based PBDs 178a–d. The synthesis of D-fructose-based PBDs 177a,b was achieved by condensation of compound 174 with 5-chloro- or 5-bromoisatoic anhydride derivatives of 175, followed by N-methylation. By using boron trichloride in dichloromethane debenzylation of PBDs 177a,b occurred without removal of the halogen atoms to give PBDs 179a,b. The yields of the PBD dilactams, obtained from condensing isatoic anhydrides and d- or l-prolines in boiling DMF are in the range 63%–91% whereas corresponding condensations in DMSO at around 110 °C or heating without solvent alone or under microwave radiation, are rarely under 80% [10].
Scheme 30.
Synthesis of spiro PBD derivatives 176a–d, 177a,b, 178a–d and 179a,b.
2.1.3. Pyrrolo[2,1-c][1,4]benzodiazepine-11-Ones with a Non-Aromatic Pyrrole Ring
Cyclodehydration of N-[2-(Aryl or Heteroaryl)hydroxymethyl]-l-Prolinamides
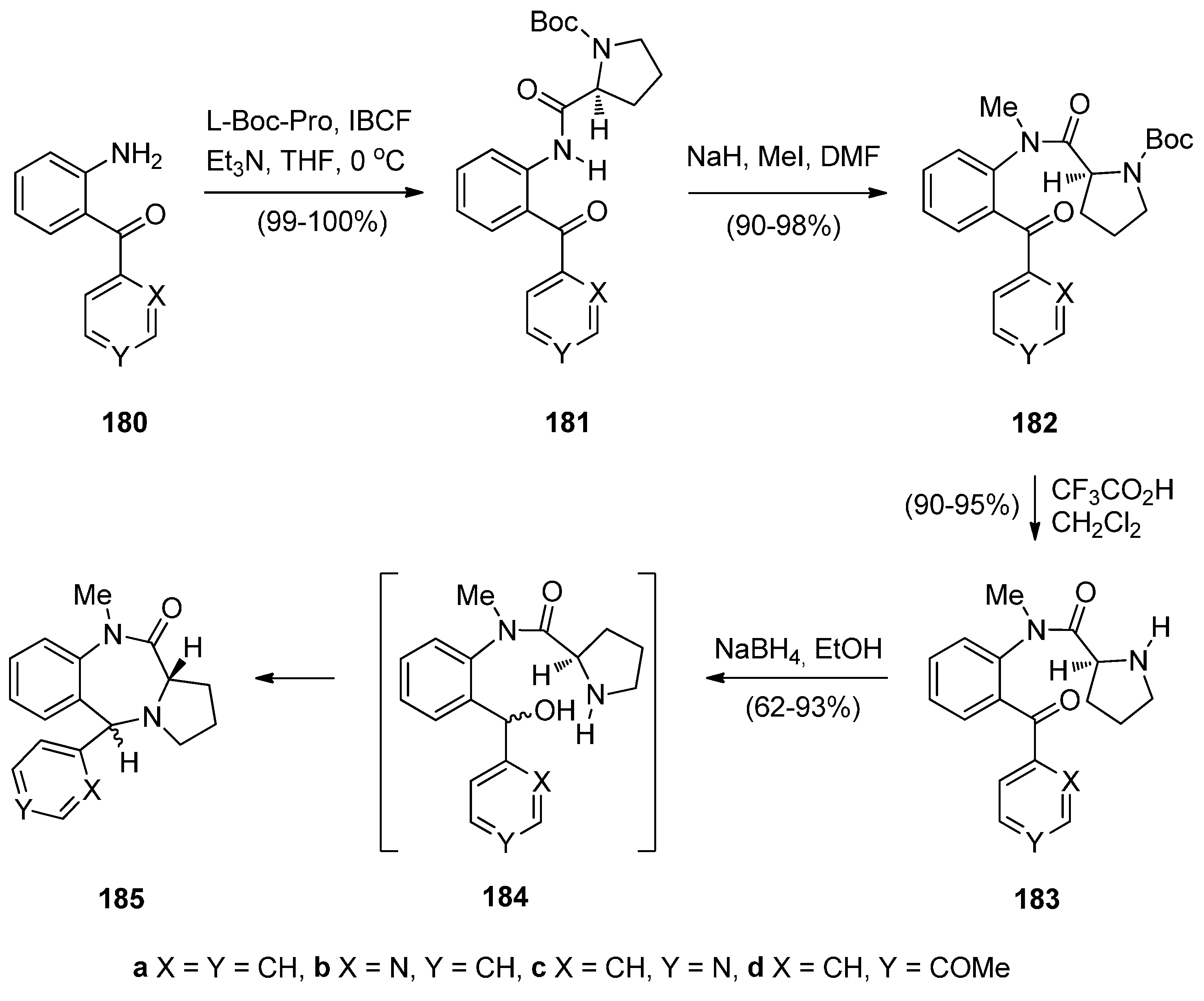
Legerén and Domínguez [73] combined the structural features of 5-aryl-1,4-benzodiazepin-2-ones and pyrrolo[2,1-c][1,4]benzodiazepin-11-ones, both biologically versatile molecular families, and developed a new synthetic approach towards novel 5-arylpyrrolo[2,1-c][1,4]benzodiazepin-11-ones 185a–d (Scheme 31) The starting materials for this synthesis, 2-aminophenylmethanones 180a–d, were added to solution of l-Boc-proline and triethylamine that had been treated with isobutyl chloroformate (IBCF), to afford amides 181a–d in virtually quantitative yield. Tertiary amides 182a–d were prepared in excellent yields by methylation of amides 181a–d which were then deprotected with trifluoroacetic acid (TFA) to amides 183. HPLC of compounds 182a and 183a on chiral columns confirmed that the stereochemical integrity of the proline stereocentre is intact. Compounds 183a–d were reduced to produce PBDs 185a–d via cyclodehydration of the intermediate amidobenzhydrols 184a–d. PBDs 185a–d were isolated in the corresponding 63, 93, 62 and 68% yields and trans/cis diastereoisomer ratios 75/25, 90/10, 95/05 and 80/20, respectively.
Scheme 31.
Synthesis of PBD derivatives 185a–d.

2.1.4. Pyrrolo[2,1-c][1,4]benzodiazepines with an Aromatic Pyrrole Ring
Cyclisation of N-(2-Aminobenzoyl)-1H-Pyrroles with Aldehydes.
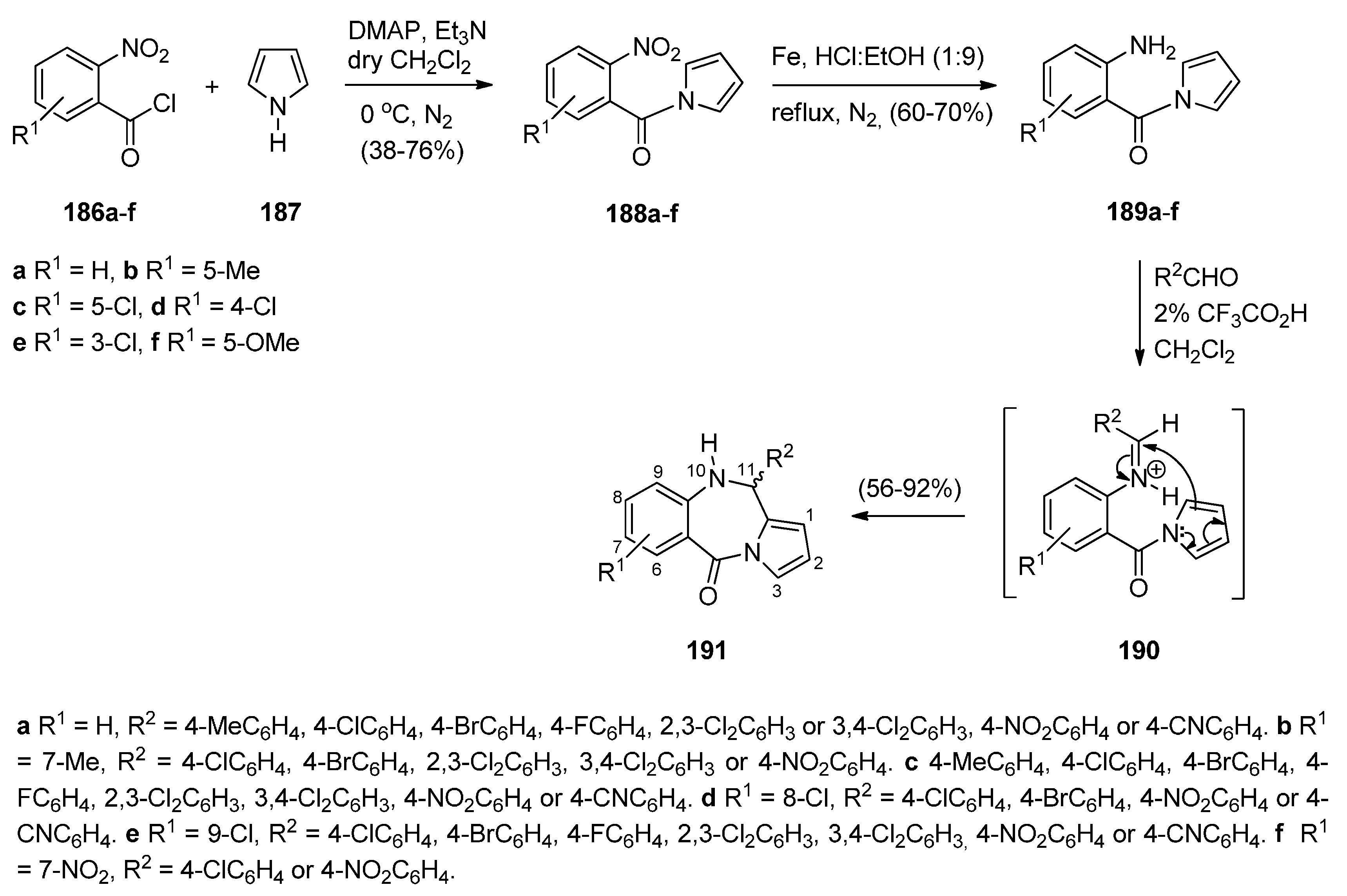
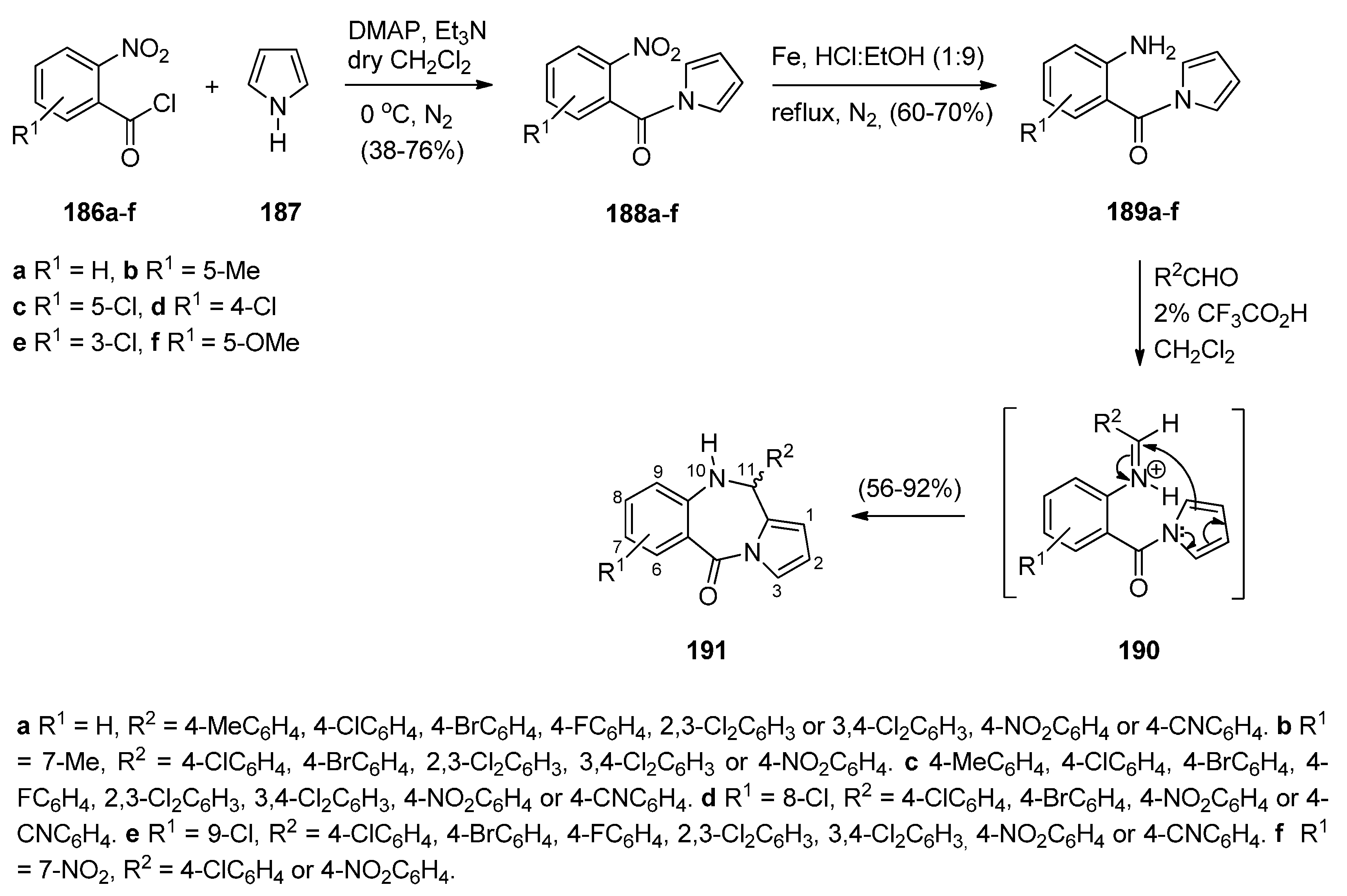
Kundu and co-workers [74] synthesized a large number of PBDs 191a–f (Scheme 32) with an aromatic pyrrole ring which are analogues of naturally occurring sibiromycin [75] and limazepine D [8]. In the first step of the synthetic route towards these compounds, 2-nitrobenzoyl chlorides 186a–f reacted with 1H-pyrrole 187 to afford (2-nitrophenyl)pyrrol-1-ylmethanones 188a–f. Reduction of the latter gave anilines 189a–f which underwent cationic π-(7-endo) cyclisation with a variety of aromatic aldehydes and 2% trifluoroacetic acid as catalyst to afford PBDs 191, with yields ranging from 56%–92%. In these reactions the intermediate electrophilic cationic iminium species is assumed to be intermediate 190.
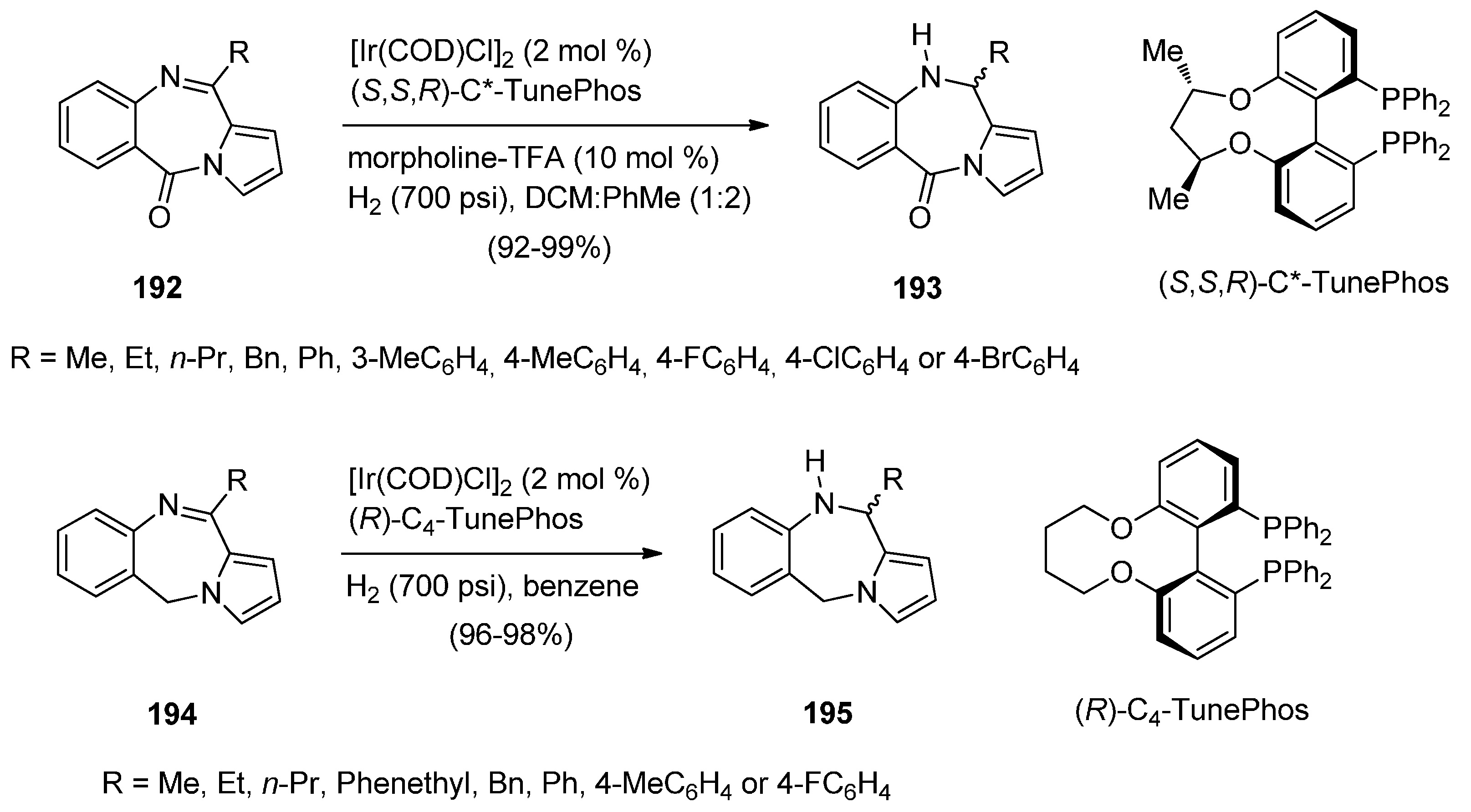
A novel and highly efficient Ir-catalysed asymmetric hydrogenation method for reducing the imine moiety contained in the seven-membered ring of 11-(alkyl or aryl)-5H-pyrrolo[2,1-c][1,4]-benzodiazepin-5-ones 193 and 11-(alkyl or aryl)-5H-pyrrolo[2,1-c][1,4]benzodiazepine 195 was developed by Zhou and co-workers [76] (Scheme 33). PBDs 193 and 195 were synthesised according to the procedures published by Kundu and co-workers [74] and Molinari et al. [77]. [Ir(COD)Cl]2 and (S,S,R)-C3*-TunePhos in a mixture of dichloromethane and toluene were added to the appropriate PBDs 192 and morpholine-trifluoroacetic acid and then hydrogenated at 700 psi of hydrogen to afford high yields of (+)-11-(alkyl or aryl)-10,11-dihydro-5H-PBDs 193 with 90%–96% ee. Similarly, but this time using [Ir(COD)Cl]2 and (R)-C4-TunePhos in benzene, the appropriate PBDs 194 were hydrogenated at 700 psi of hydrogen to afford high yields of (−)-11-(alkyl or aryl)-10,11-dihydro-5H-PBDs 195 with 82%–96% ee.
Scheme 32.
Synthesis of PBD derivatives 191a–f.
Scheme 33.
Synthesis of PBD derivatives 193 and 195.

Cyclisation of 2-(N-Phthalimido)-N-[2-(1H-pyrrol-1-yl-methyl)phenyl]-Acetamide
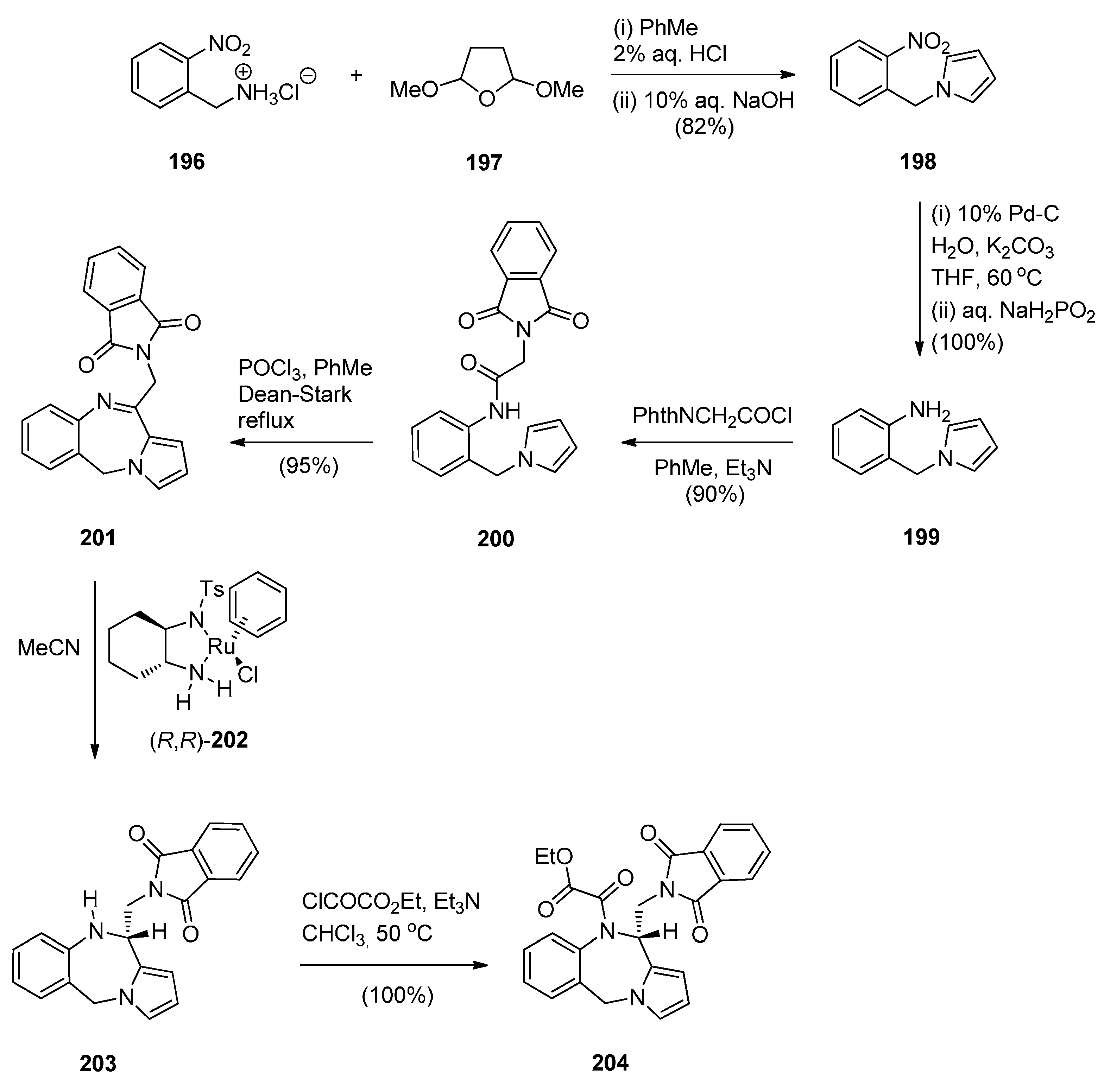
Three pyrrolo[2,1-c][1,4]benzodiazepines 201, 203 and 204 were synthesised by Czarnocki et al. [78] en route to (2-methyl-1,3,4,14b-tetrahydro-2H,10H-pyrazino[1,2-a]pyrrolo[2,1-c][1,4]benzodiazepine (aptazepine), a potent α2-adrenoreceptor blocking agent. The synthetic pathway that we adopted is presented in Scheme 34 where commercially available 2-nitrobenzylamine hydrochloride (196) is condensed with 2,5-dimethoxy-tetrahydrofuran to 1-(2-nitrobenzyl)-1H-pyrrole (198) which was reduced and subsequently acylated with N-phthalylglycyl chloride to afford amide 200. Conversion of this amide into the prochiral endocyclic imine 201 by a Bischler-Napieralski cyclisation and then asymmetric hydrogen-transfer using catalyst 202, afforded PBD (S)-203 with superior asymmetric induction (63% ee) and higher chemical yield (60%). Finally, amine 203 was reacted with ethyl chloro(oxo)acetate to afford amide (S)-204 in quantitative yield. All the steps in this synthetic route gave yields that were over 80%.
Scheme 34.
Synthesis of PBD derivatives 201, 203 and 204.

2.1.5. Pyrrolo[2,1-c][1,4]benzodiazepine-3-ones with a Non-Aromatic Pyrrole Ring
Reductive Cyclisation of in Situ Generated 1-(2-aminobenzyl)-5-aroylpyrrolidin-2-ones
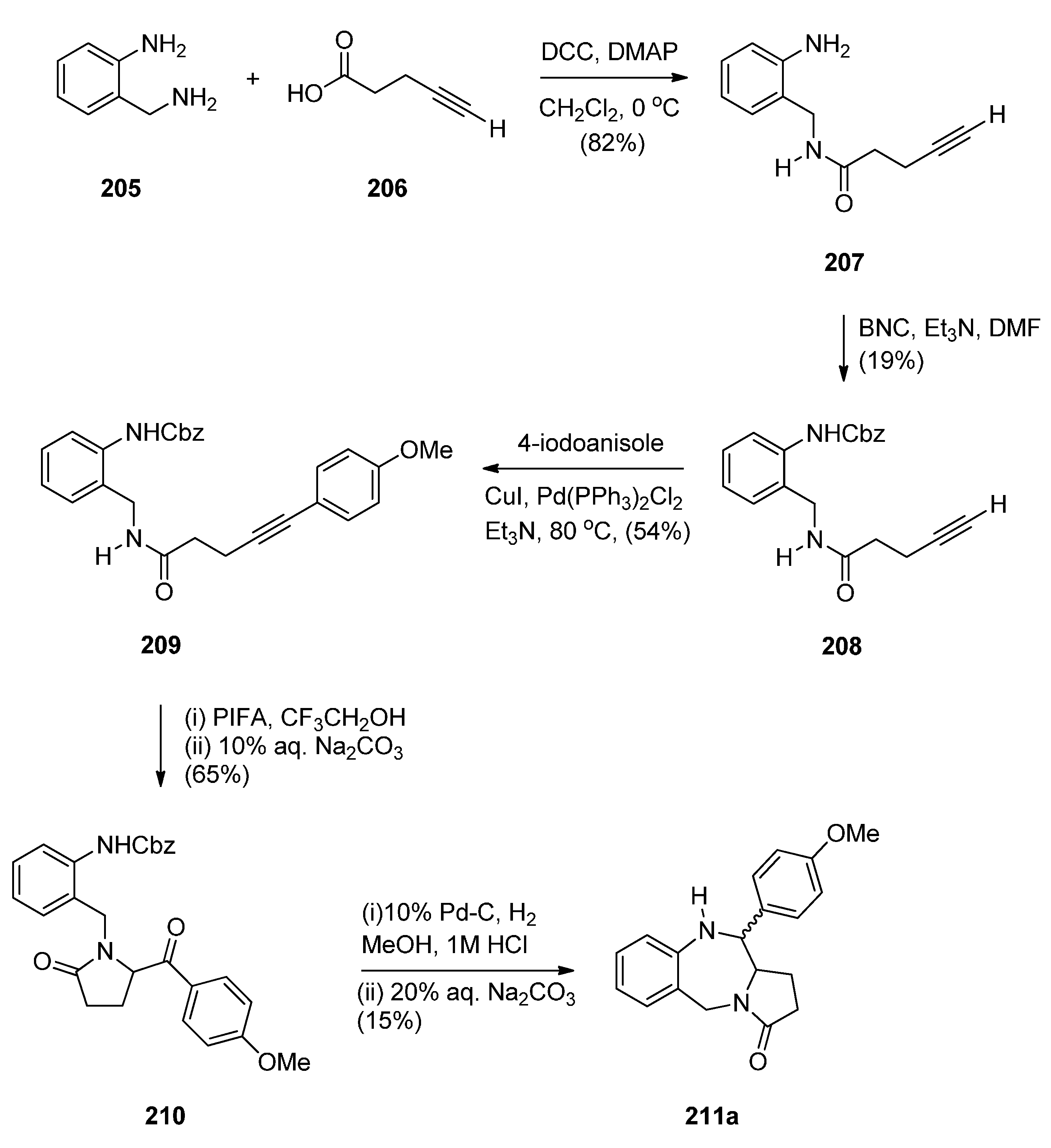
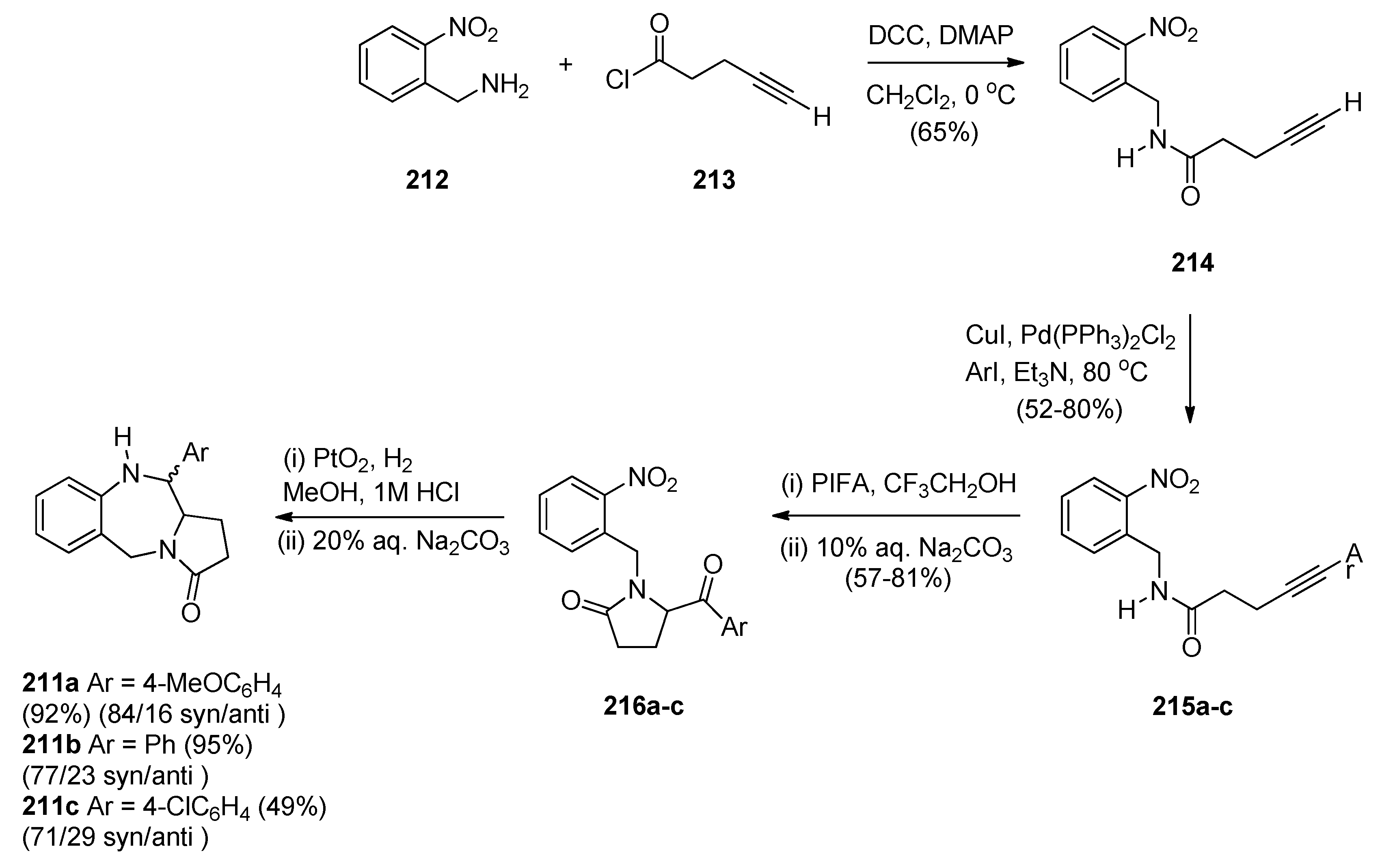
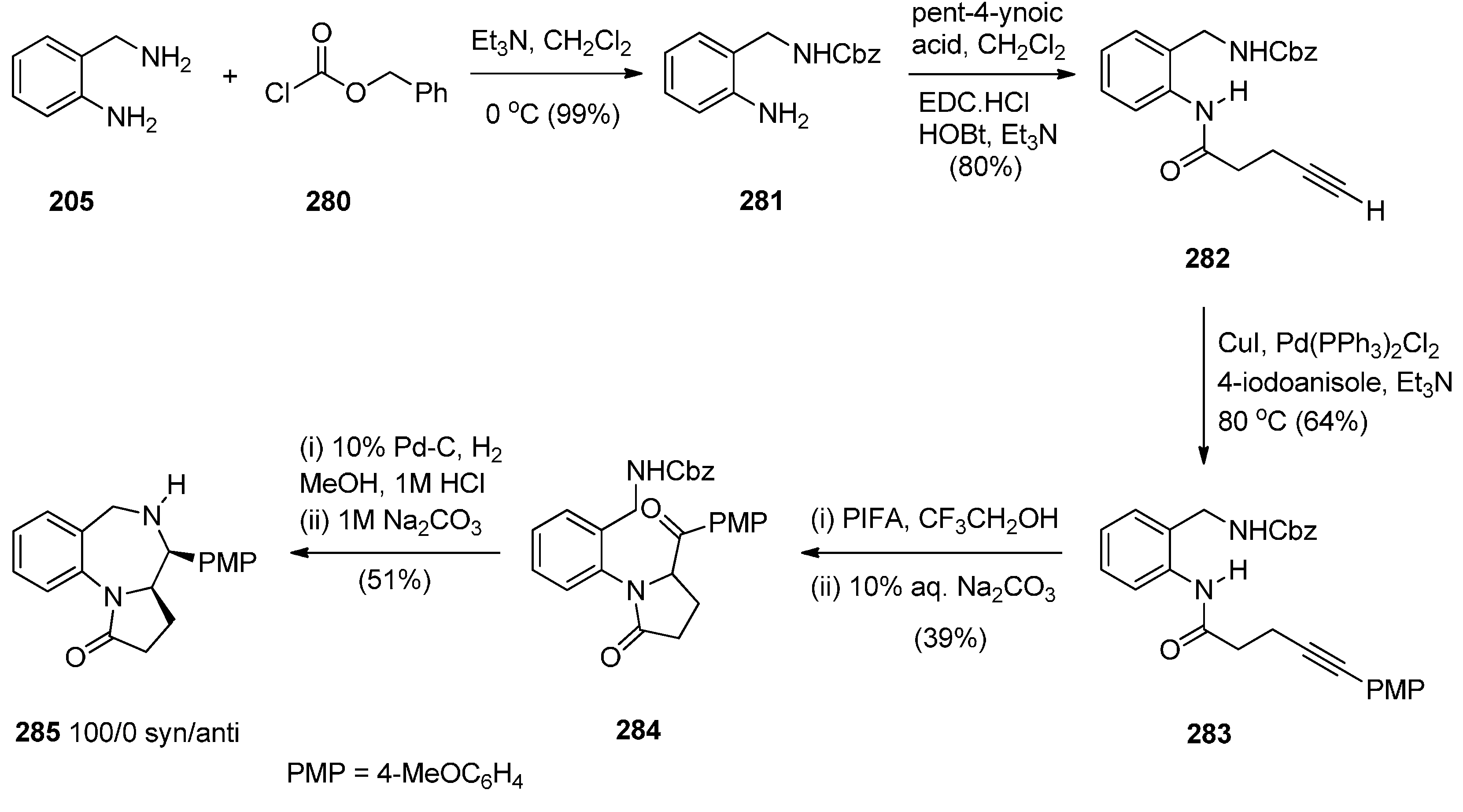
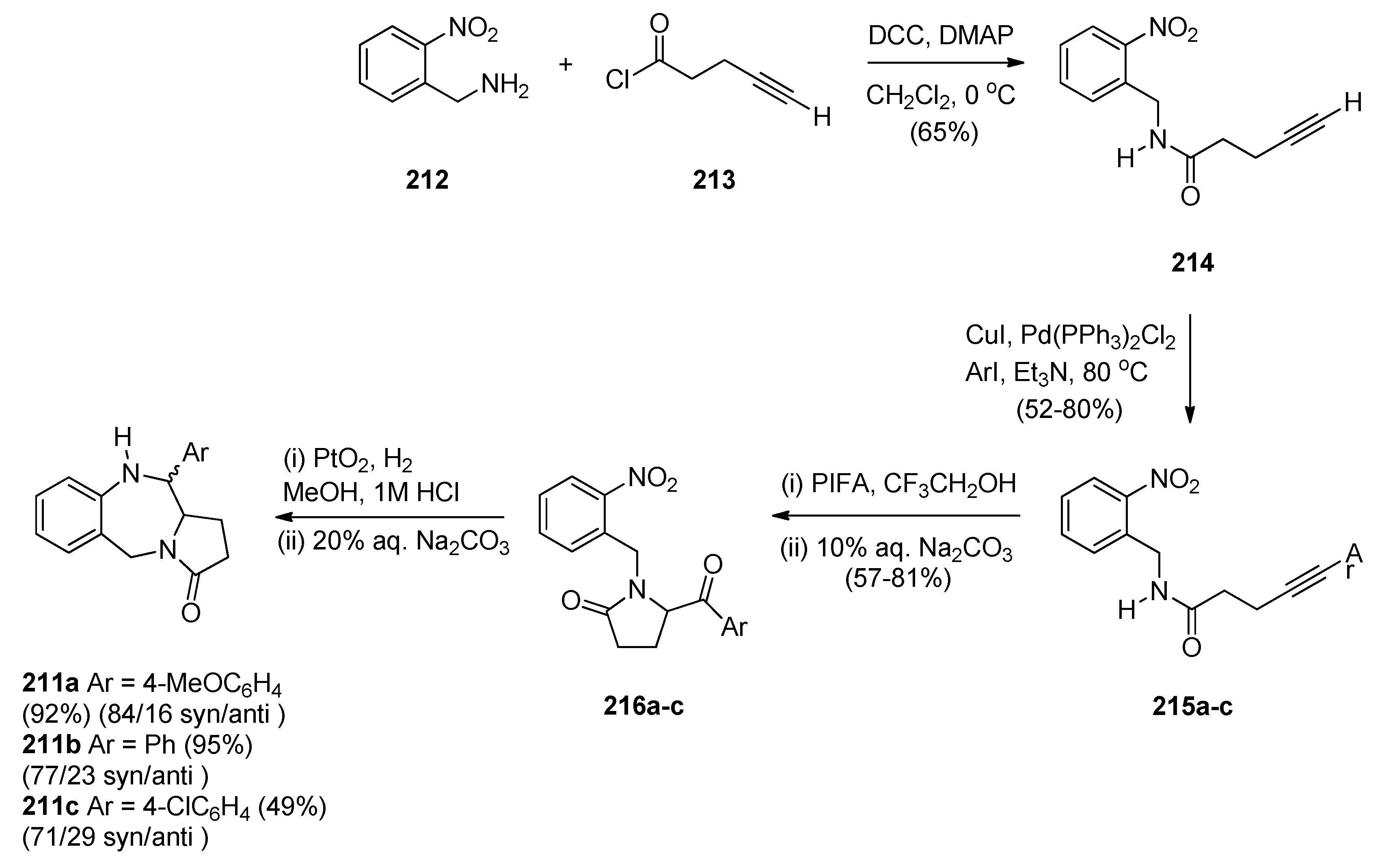
Domínguez and co-workers [79] synthesised PBD 211a by a sequence of reactions starting from 2-(aminomethyl)aniline (205) (Scheme 35). The key step towards the synthesis of PBD 211a is reductive cyclisation of amino-ketone 210 derived from transforming 205 with pent-4-ynoic acid (206) into amide 207, protecting as carboxybenzyl derivative 208, performing a Sonogashira coupling with 4-iodoanisole to give alkyne 209 and then [bis(trifluoroacetoxy)iodo]benzene (PIFA) mediated cyclisation to afford the pyrrolidinone 210, in good yield. Catalytic hydrogenation of 210 in acidic methanol produced PBD 211a in very low yield and poor diastereoselectivity (67/33, syn/anti), through a combination of three consecutive single processes, deprotection, condensation with the carbonyl group, and reduction of the resultant imine.
The authors considered the above synthetic route to PBD 211a quite unsatisfactory since it produced a low overall yield and poor diastereoselectivity in the final step. They therefore devised a new protection-free synthetic alternative to this compound and to two other derivatives 211b,c (Scheme 36). The synthesis started from 2-(nitrobenzyl)amine (212) and followed a route similar to that depicted in Scheme 35. Thus, after subjecting 212 to successive steps of amidation that gave 214, Sonogashira coupling that gave 215a and PIFA-mediated intramolecular cyclisation that gave pyrrolidinone 216a, the final reductive cyclisation step of 216a into PBD 211a was optimized by experimenting with different catalysts (10% by weight of PtO2, Ra-Ni, Pd(OH)2 or Pd-black, in methanol). The best conditions required the use of Adam’s catalyst (PtO2) under a hydrogen atmosphere at 70 psi. PBD 211a was obtained in excellent yield with 84/16 syn/anti stereoselectivity. PBDs 216b,c were prepared in a similar manner. Similar reductive cyclisation of 216b,c into the corresponding PBDs afforded 211b in excellent yield and 211c in moderate yield while both compounds showed good overall diastereoselectivities.
Scheme 35.
Synthesis of PBD derivative 211a.

Scheme 36.
Synthesis of PBD derivatives 211a–c.
2.2. Pyrrolo[1,2-a][1,4]benzodiazepines
A large number of [1,2-a] PBDs have been synthesised from the condensation of 1-(2-aminomethylphenyl)pyrroles with carbonyl compounds to provide non-isolable imines that undergo an intramolecular Mannich reaction, in acidic conditions, to form the diazepine ring (Section “Cyclisation of 1-(2-aminomethylphenyl)pyrrole with Carbonyl Compounds or a Hemiacetal”). Another frequently used method of synthesising [1,2-a] PBDs is reacting 1-(2-aminomethyl-phenyl)pyrroles with ethyl formate, carboxylic acid chlorides or anhydrides to form amides that are subjected to Bischler-Napieralski reaction conditions (Section “Cyclisation of Amide, Urea and Thiourea Derivatives of 1-(2-aminomethylphenyl)pyrrole”)
2.2.1. Pyrrolo[1,2-a][1,4]benzodiazepines with an Aromatic Pyrrole Ring
The most characteristic features of pyrrolo[1,2-a][1,4]benzodiazepines in this category are: (i) all PBDs have an aromatic pyrrole ring; (ii) no carbonyl groups on the diazepine ring and the PBD N5 is a secondary amine; (iii) no carbonyl groups on the diazepine ring and the PBD C4–N5 is an imine; (iv) no carbonyl groups on the diazepine ring and the PBD has a thiocarbonyl group at C4 as a C4–C5 thiolactam; (v) no carbonyl groups on the diazepine ring and the PBD N5–C6 is an imine or (vi) carbonyl group on diazepine ring which is an N5–C6 lactam of the PBD.
Cyclisation of 1-(2-Aminomethylphenyl)pyrrole with Carbonyl Compounds or a Hemiacetal
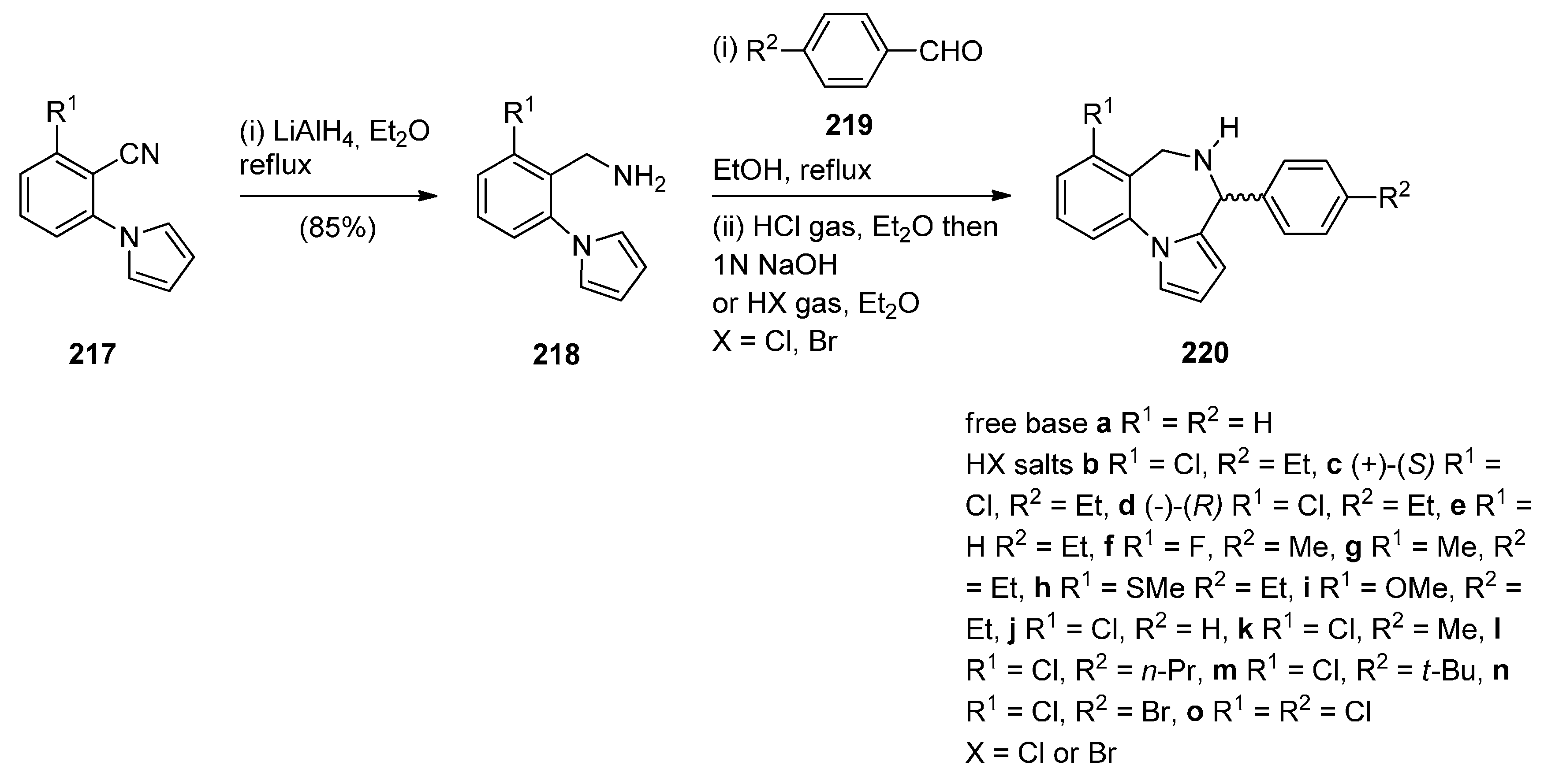
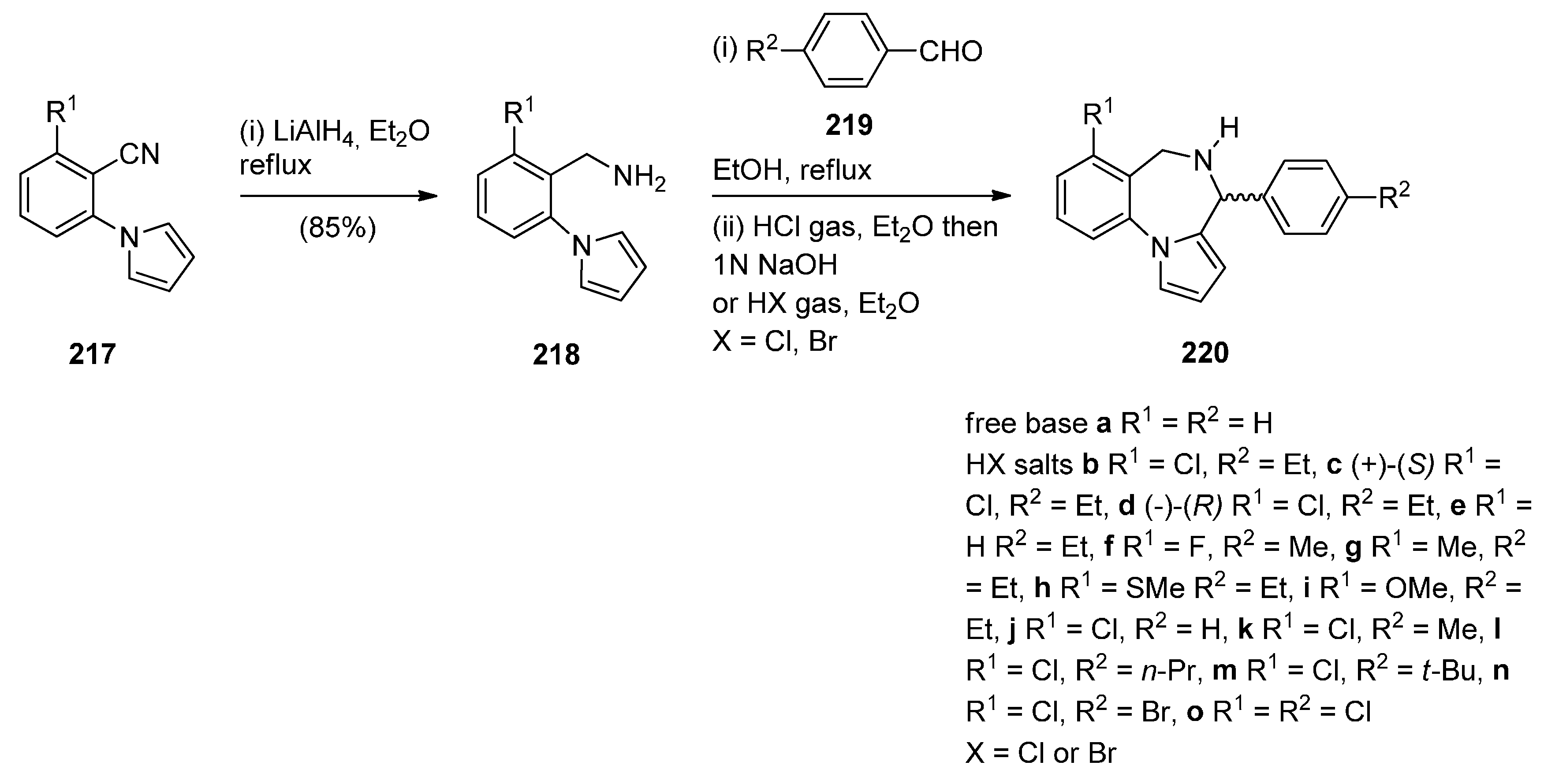
The first synthesis of the pyrrolo[1,2-a][1,4]benzodiazepine ring system was reported by Cheeseman and Rafiq [2]. 1-(2-Cyanophenyl)pyrrole (217a, Scheme 37) was prepared by heating under reflux 2-aminobenzonitrile with 2,5-diethoxytetrahydrofuran in acetic acid via a modified Hantzsch pyrrole synthesis, was reduced to primary amine 218 with lithium aluminium hydride (LiAlH4). Condensation of amine 218 with benzaldehyde 219a formed the corresponding imine which underwent a spontaneous intramolecular Mannich reaction to afford a racemic mixture of PBD 220a. Meerpoel et al. [44] use this methodology in the search for novel anti-fungal agents and synthesised racemic PBD salts 220b-o. Although in the former reaction the intramolecular Mannich step did not require the addition of a dry acid catalyst, in the latter reactions the starting materials were consumed faster during both the imine formation and the intramolecular step, by the addition of dry hydrochloric or hydrobromic acid in diethyl ether. Furthermore, PBD 220b was separated into its (+)-(S) enantiomer 220c and (−)-(R) enantiomer 220d using chiral HPLC. The absolute configuration of these compounds was found by vibrational circular dichroism (VCD)-FTIR measurement and cancellation.
Scheme 37.
Synthesis of PBD derivatives 220a–o.
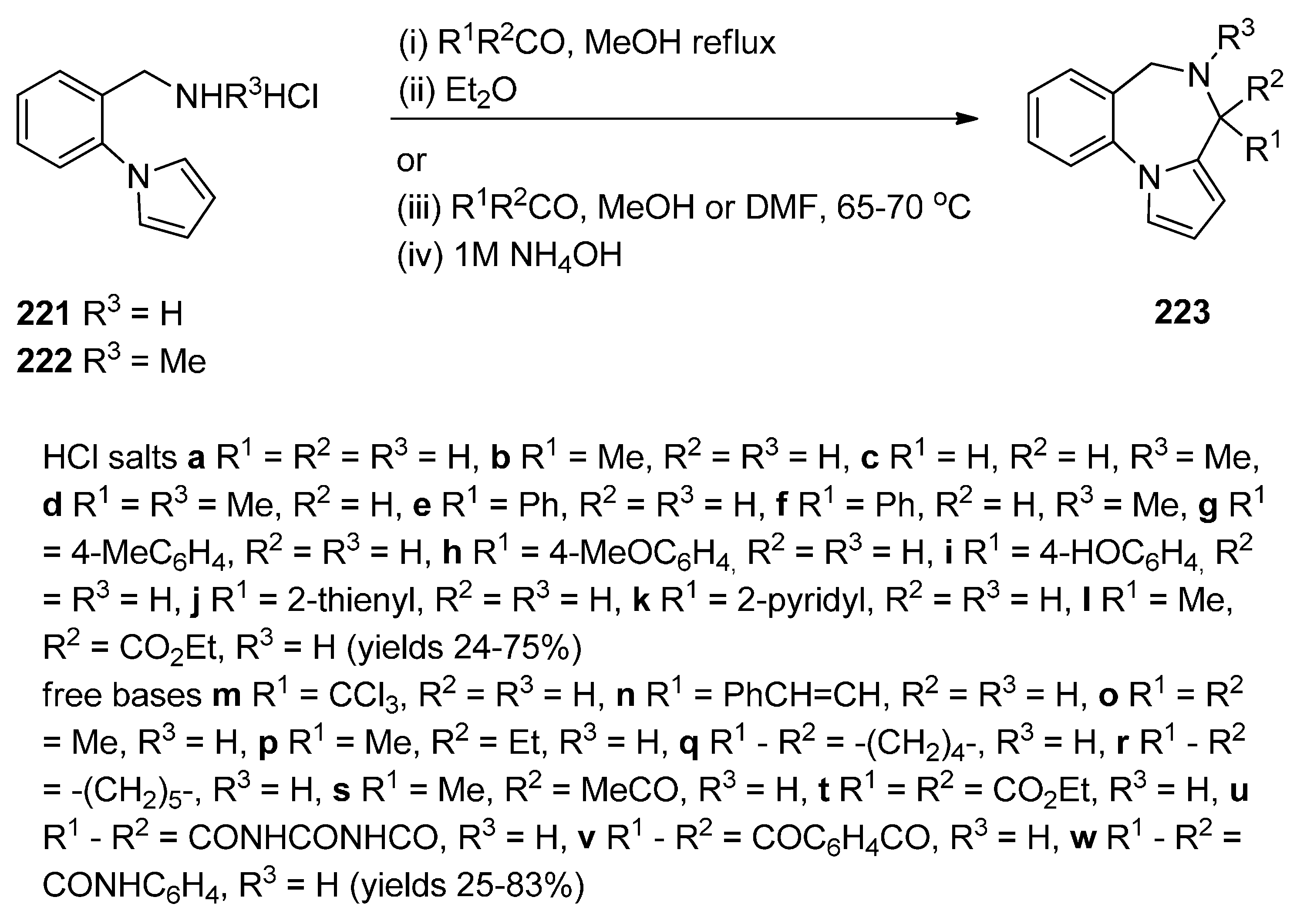
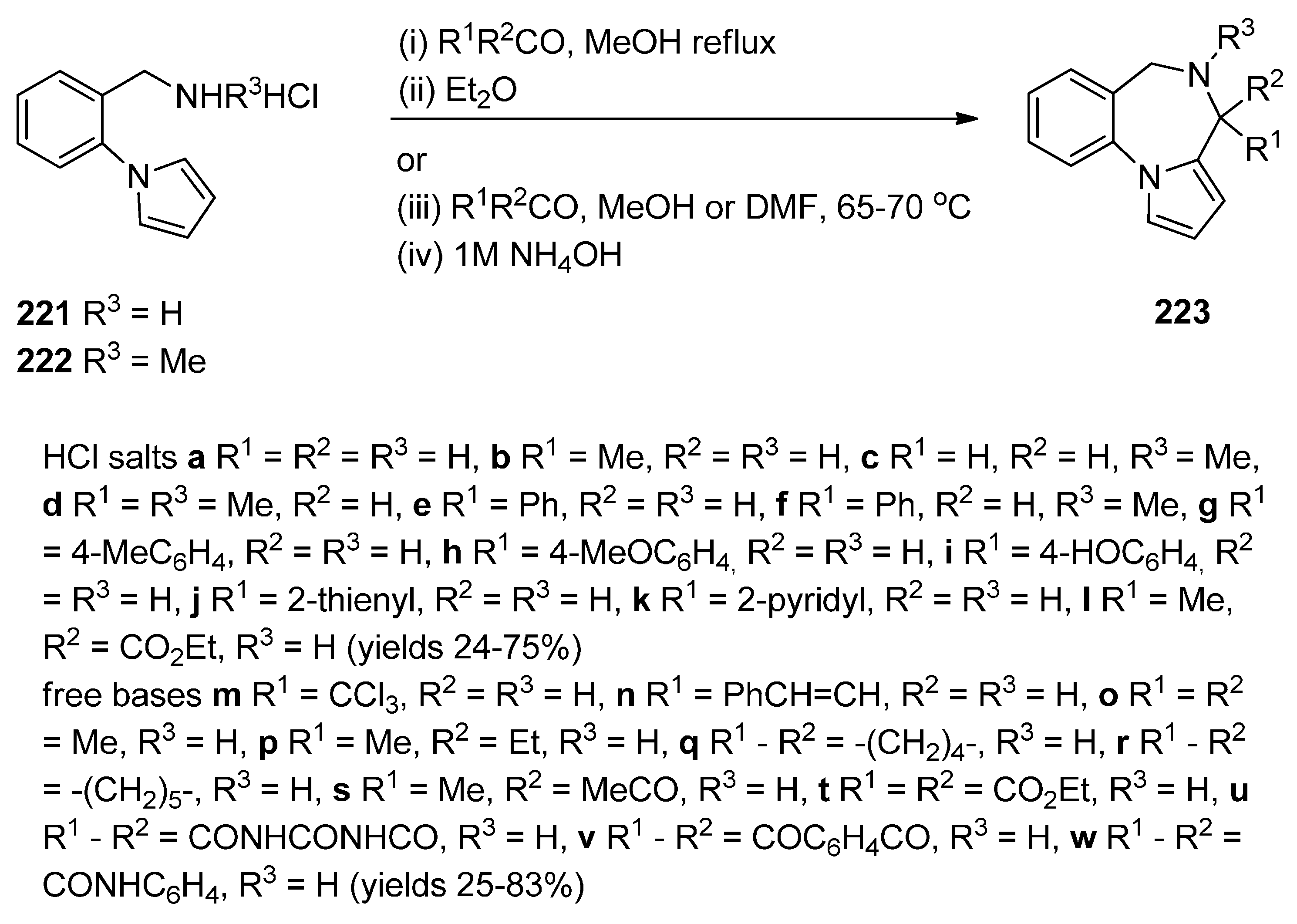
Raines et al. [80] expanded on the initial work of Cheeseman and Rafiq [2] by reacting the aminomethyl hydrochloride 221 and 222 (Scheme 38) with appropriate carbonyl compounds in boiling methanol and then precipitating the PBDs 223a–l, in poor to very good yields, by addition of diethyl ether. The carbonyl compounds used were aliphatic and aromatic aldehydes whereas one carbonyl compound was ethyl 2-oxopropanoate that yielded PBD 223l. Cheeseman and Greenberg [81] continued their line of work [2] and also used the cyclocondensation of aminomethyl hydrochloride 221 (Scheme 38) with a variety of carbonyl compounds in either methanol or DMF at 65–70 °C followed by basification with ammonium hydroxide, in order to release the PBD cyclic secondary amines 223m–w, in yields comparable to those obtained for compounds 223a–l.
Scheme 38.
Synthesis of PBD derivatives 223a–w.
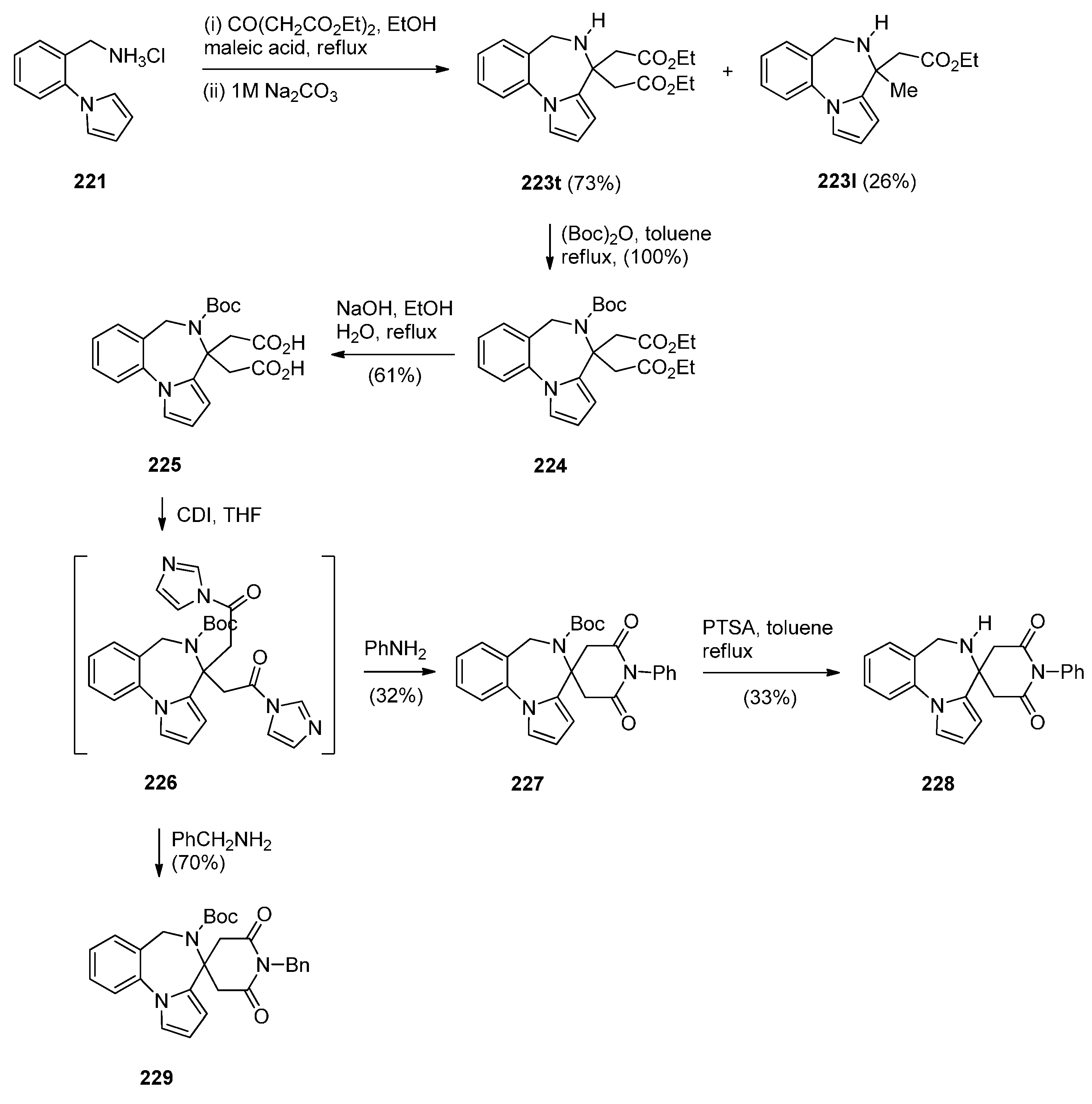
Artico and co-workers [82] became interested in PBDs due to their ongoing study on buspirone-like spiro derivatives and their desire to explore the usefulness 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepine-4,4,-diacetic acid diethyl ester (223t, Scheme 39) as a scaffold for the synthesis of polycyclic nitrogen compounds of potential pharmacological interest. The target spirodioxopiperidine-PBDs 227–229 were synthesised from the PBD diester 223t. This compound was formed together with PBD ester 223l from the reaction of aminomethyl hydrochloride 221 with diethyl 1,3-acetonedicarboxylate. Reaction of PBD 223t with di-tert-butyl dicarbonate (Boc2O) afforded Boc protected derivative 224. The latter compound was hydrolysed to the PBD diacetic acid 225 and then reacted with 1,1′-carbonyldiimidazole (CDI) to form the intermediate PBD diamide 226. Addition of aniline to the latter produced spirodioxopiperidine-PBD 227. The benzyl derivative 229 was obtained similarly from intermediate 226 by adding benzylamine in the place of aniline. Deprotection of the Boc group of 227 yielded spirodioxopiperidine-PBD 228.
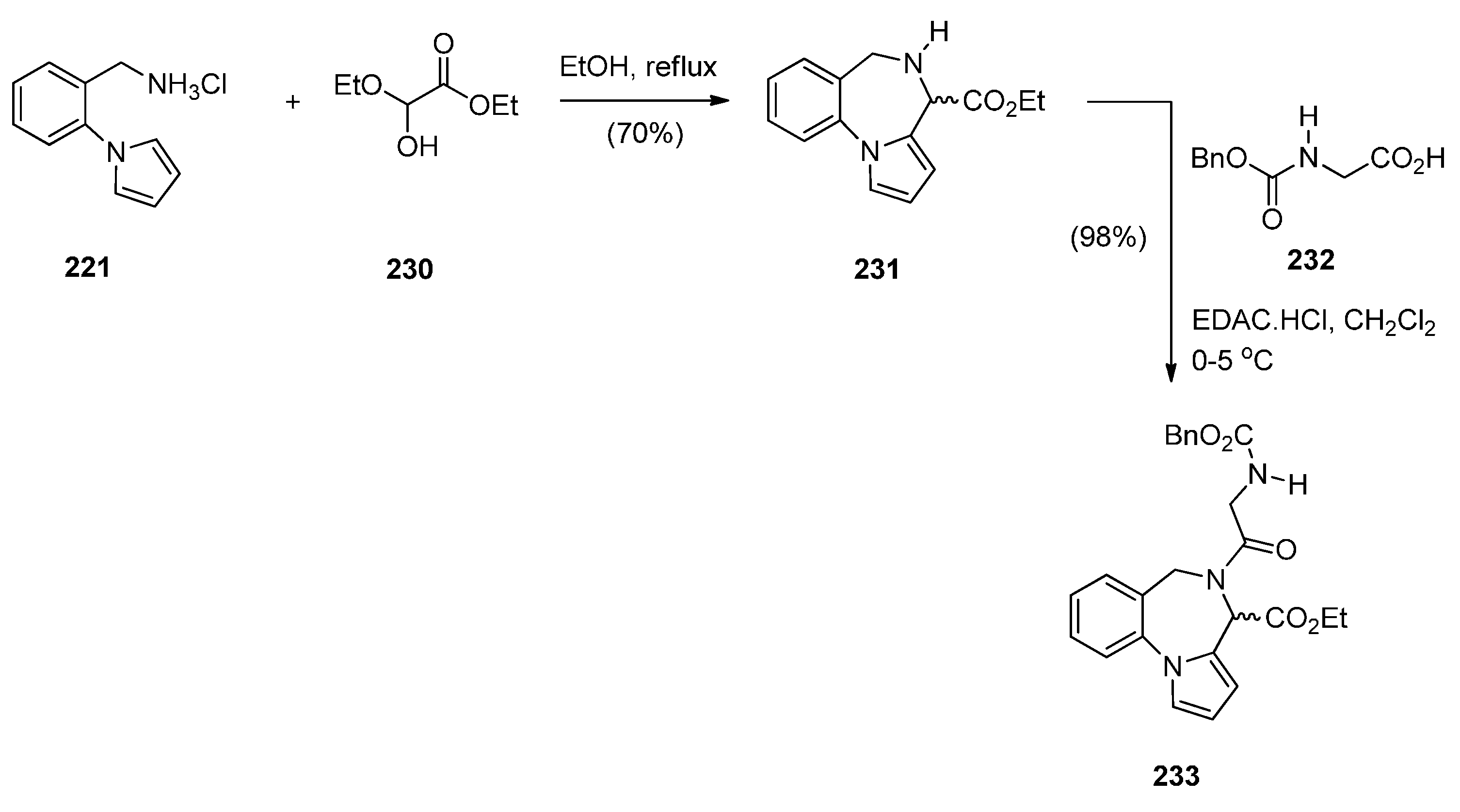
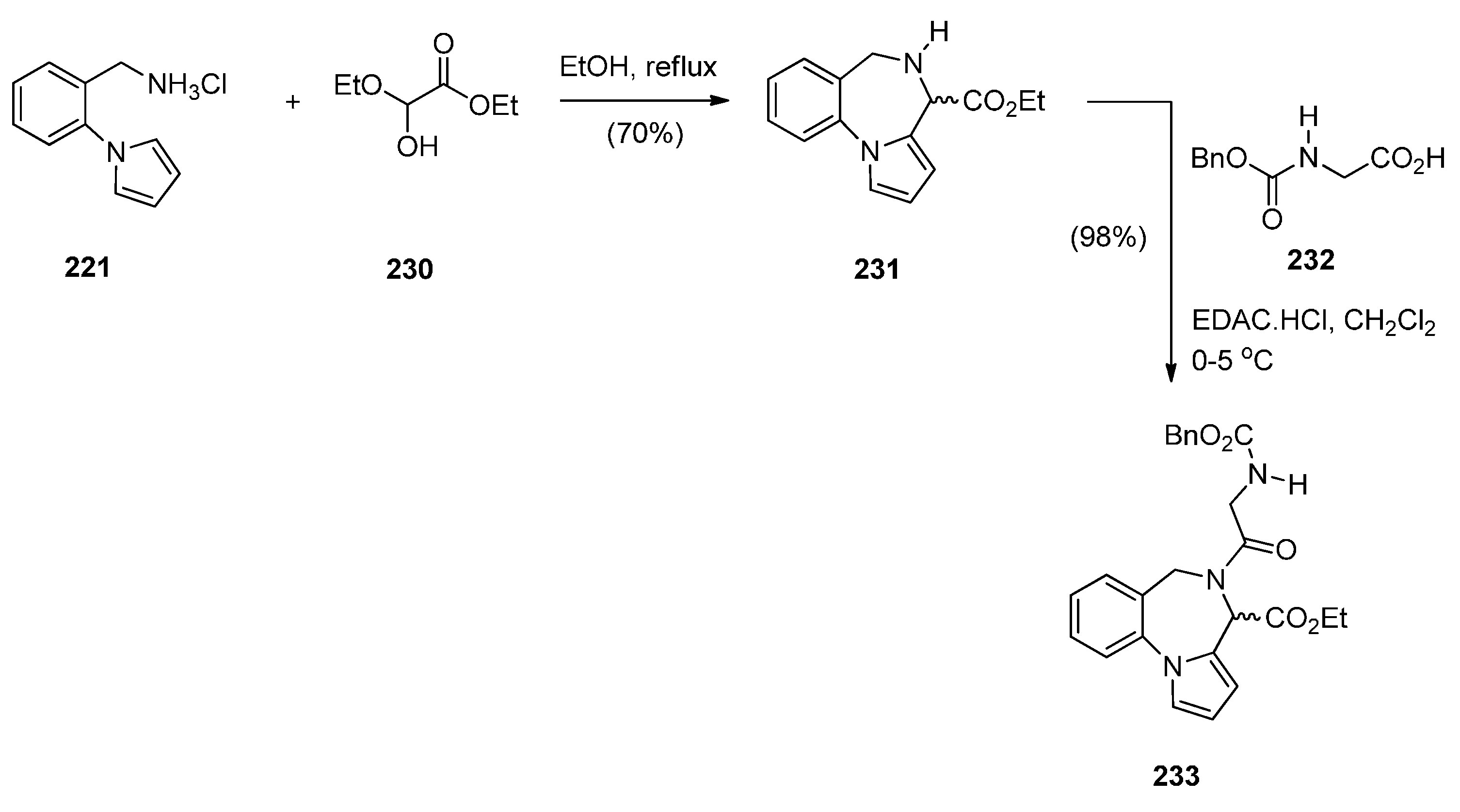
Massa, Corelli and co-workers [83] in search of new central nervous system agents with a pyrrole moiety and in particular pyrazino[2,1-c]pyrrolo[1,2-a][1,4]benzodiazepines, found out that the best route to synthesise these compounds is via PBDs 231 and 233 (Scheme 40). Therefore, Pictet-Spengler condensation of amine hydrochloride 221 with ethyl 2-ethoxy-2-hydroxyacetate (230) provided racemic PBD 231. The secondary amino group of 231 was derivatised by reaction with benzyloxycarbonylglycine 232 in the presence of l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDAC.HCl) to provide racemic PBD 233, in excellent yield. The methyl ester of 231 was prepared analogously [46].
Scheme 39.
Synthesis of PBD derivatives 223–229.

Scheme 40.
Synthesis of PBD derivatives 231 and 233.
Cyclisation of Amide, Urea And Thiourea Derivatives of 1-(2-Aminomethylphenyl)pyrrole
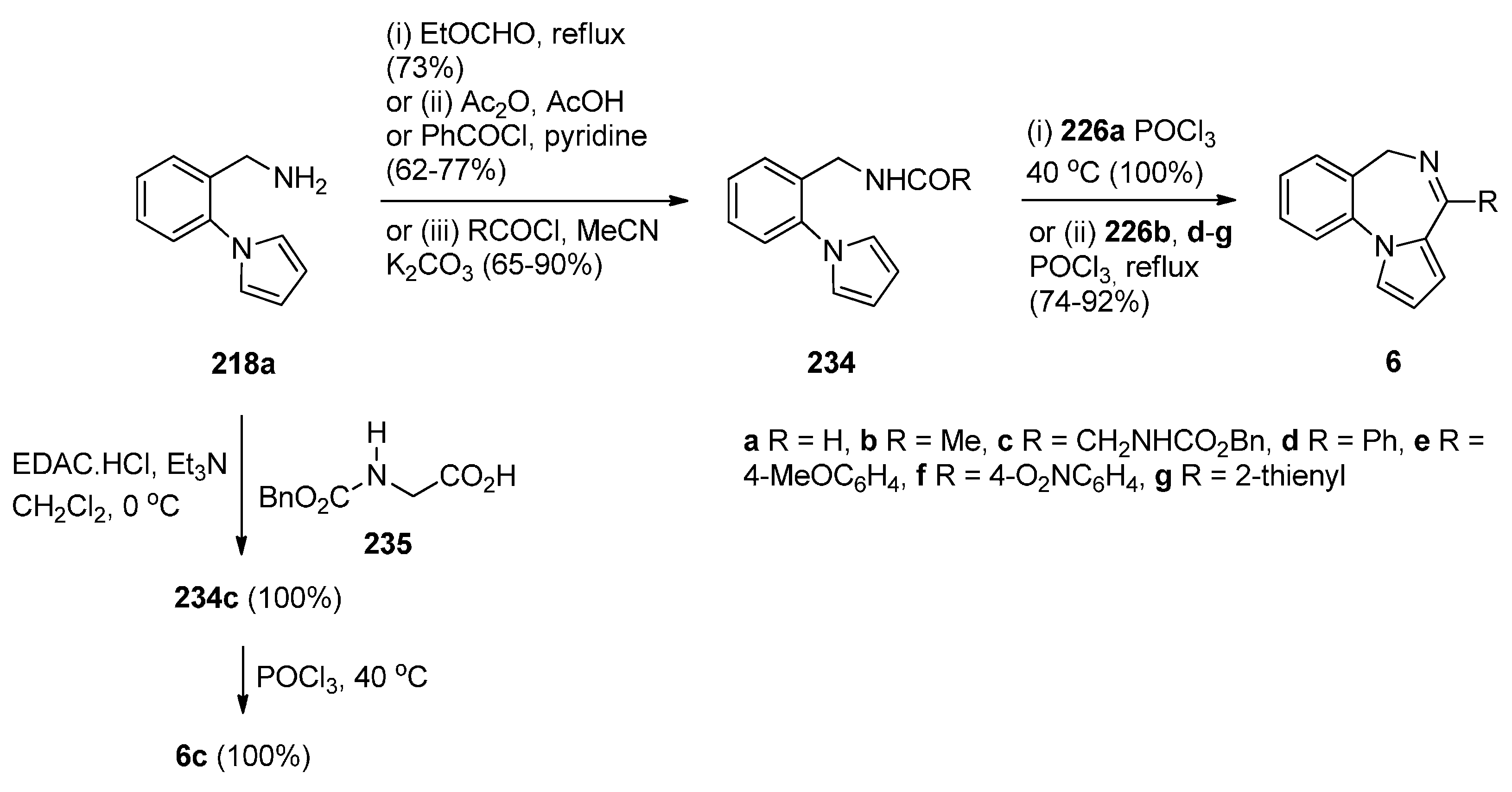
Cheeseman and Rafiq [2] were the first to report the application of the Bischler–Napieralski reaction on amidomethyl derivatives 234b,d to produce the corresponding PBD C4–N5 imines 6b,d (Scheme 41). Amides 234b,d were derived from aminomethyl derivative 218a by reaction with acetic anhydride and benzoyl chloride, respectively. En route to the synthesis of tetracyclic nitrogen heterocycles as potential pharmacophoric structures for drug design, Massa et al. [84] prepared PBDs 6a,c (Scheme 41). Both compounds were prepared from amine 218a. The parent PBD 6a required heating the amine with ethyl formate to give formamide 234c and then cyclisation with phosphoryl chloride at 40 °C while PBD 6c was prepared by coupling the amine with protected glycine 235 in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDAC.HCl) to produce amide 234c and then cyclisation using similar reaction conditions. In two recent publications Voskressensky et al. [85,86] used the same methodology to synthesise novel PBDs 6b,d–g from amine 218a via cyclisation of amides 234b,d–g. The yields of the Bischler–Napieralski cyclisation step are noted to be from 74% with up to quantitative yield. Quantitative yields were obtained only when the precursors contained either N-methylformamide or N-methylacetamide groups and the reactions were heated in phosphoryl chloride at 40 °C. Heating under reflux in phosphoryl chloride is by far most frequently used for precursors containing N-methylbenzamide, N-methylarylamide and N-methylthiophene-2-carboxamide groups.
Scheme 41.
Synthesis of PBD derivatives 6a–g.
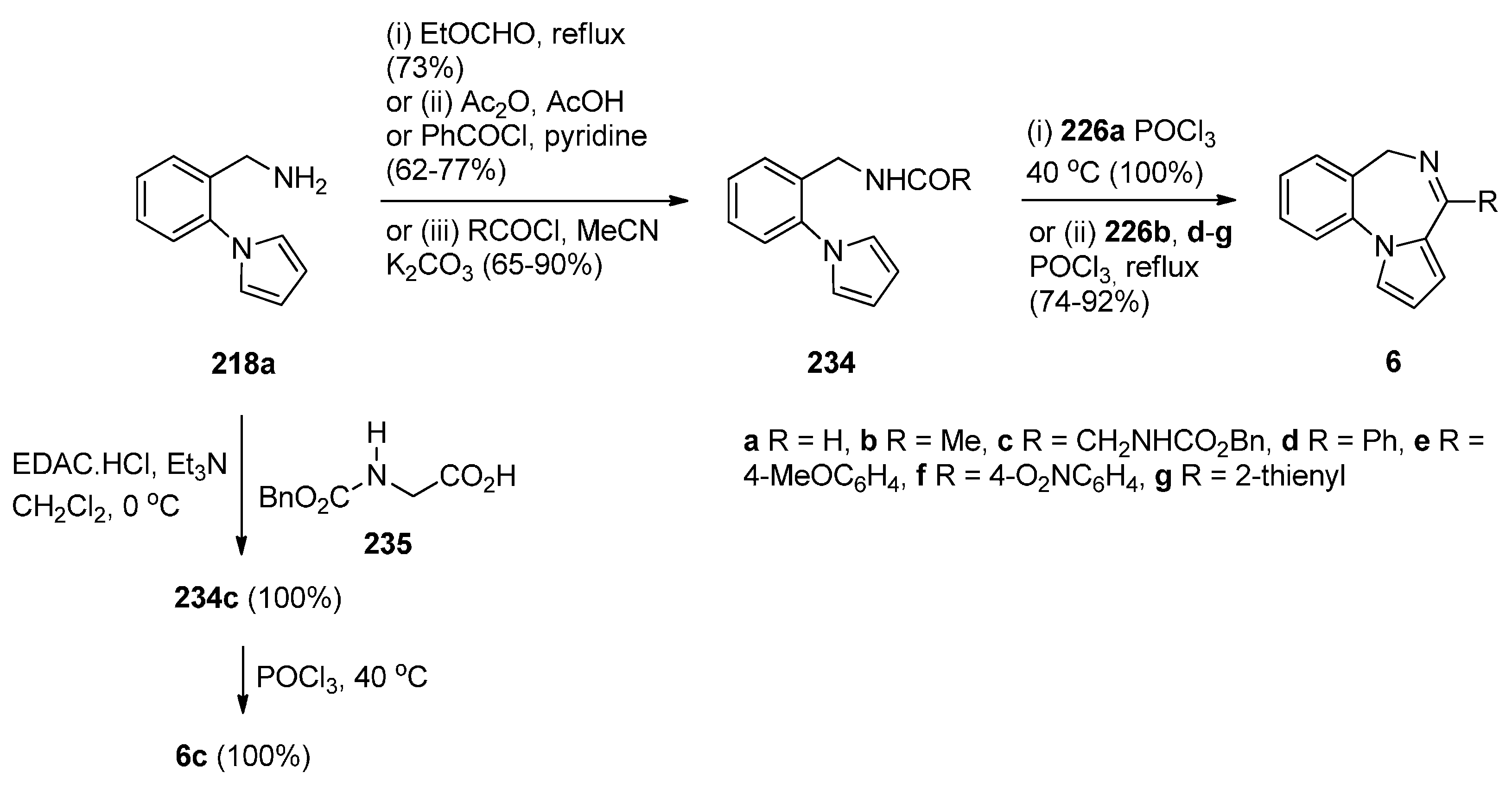
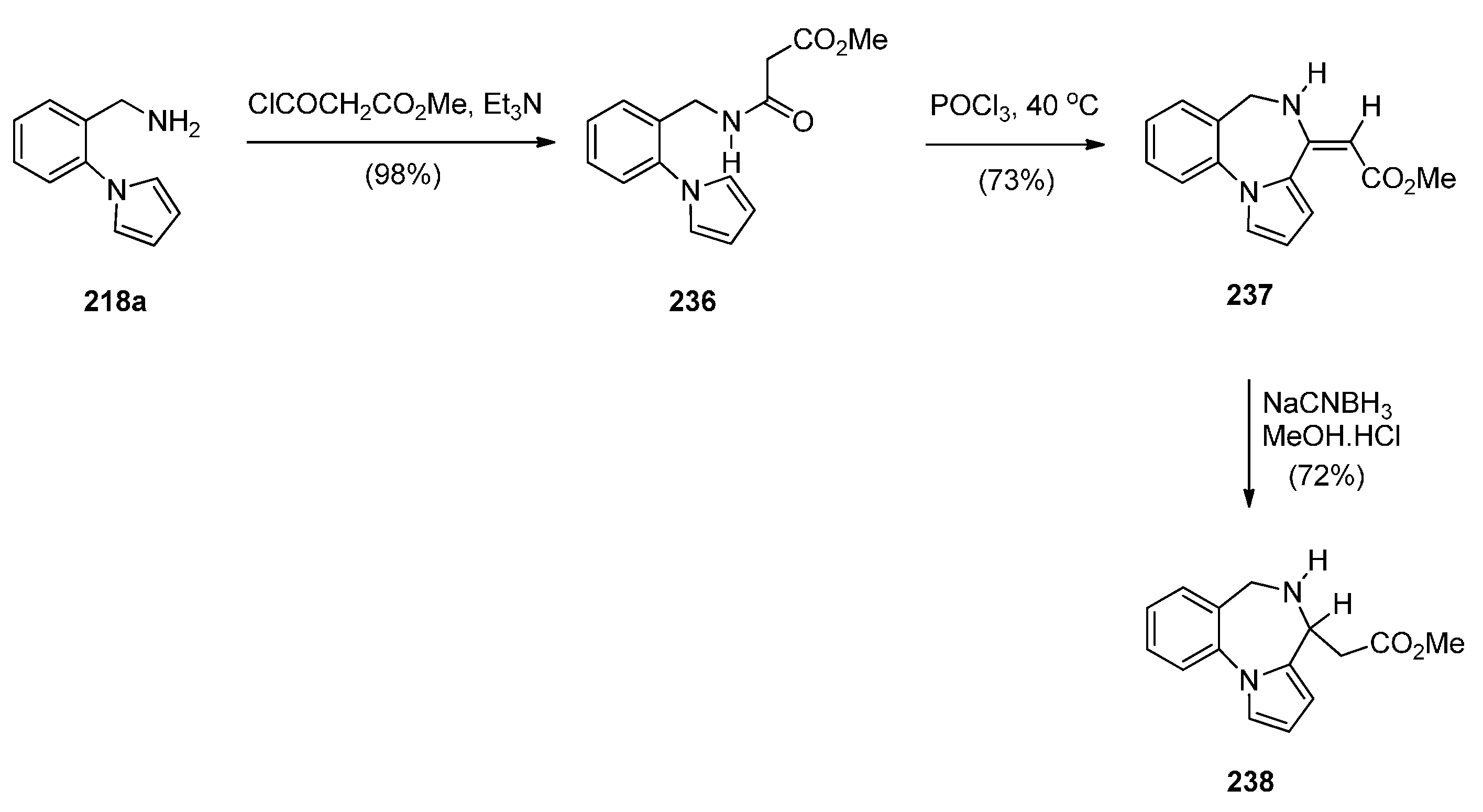
In the Section “Cyclisation of 1-(2-Aminomethylphenyl)pyrrole with Carbonyl Compounds or a Hemiacetal”, Artico and co-workers [82] synthesized the 4-methyl ethyl ester derivative of 238 (i.e., 223l) by an intramolecular Mannich reaction (Scheme 38). In the same paper he also presented a convenient synthetic route to PBD methyl ester 238, unsubstituted at position 4 (Scheme 42). Following the isolation of acetamidopyrrole 236 in high yield from the reaction between and methyl malonyl chloride, the Bischler–Napieralski reaction provided PBD enamine 237, after tautomerisation of the initially formed PBD C4–N5 imine. Compound 237 was then reduced with sodium cyanoborohydride to afford PBD 238.
Scheme 42.
Synthesis of PBD derivatives 237 and 238.
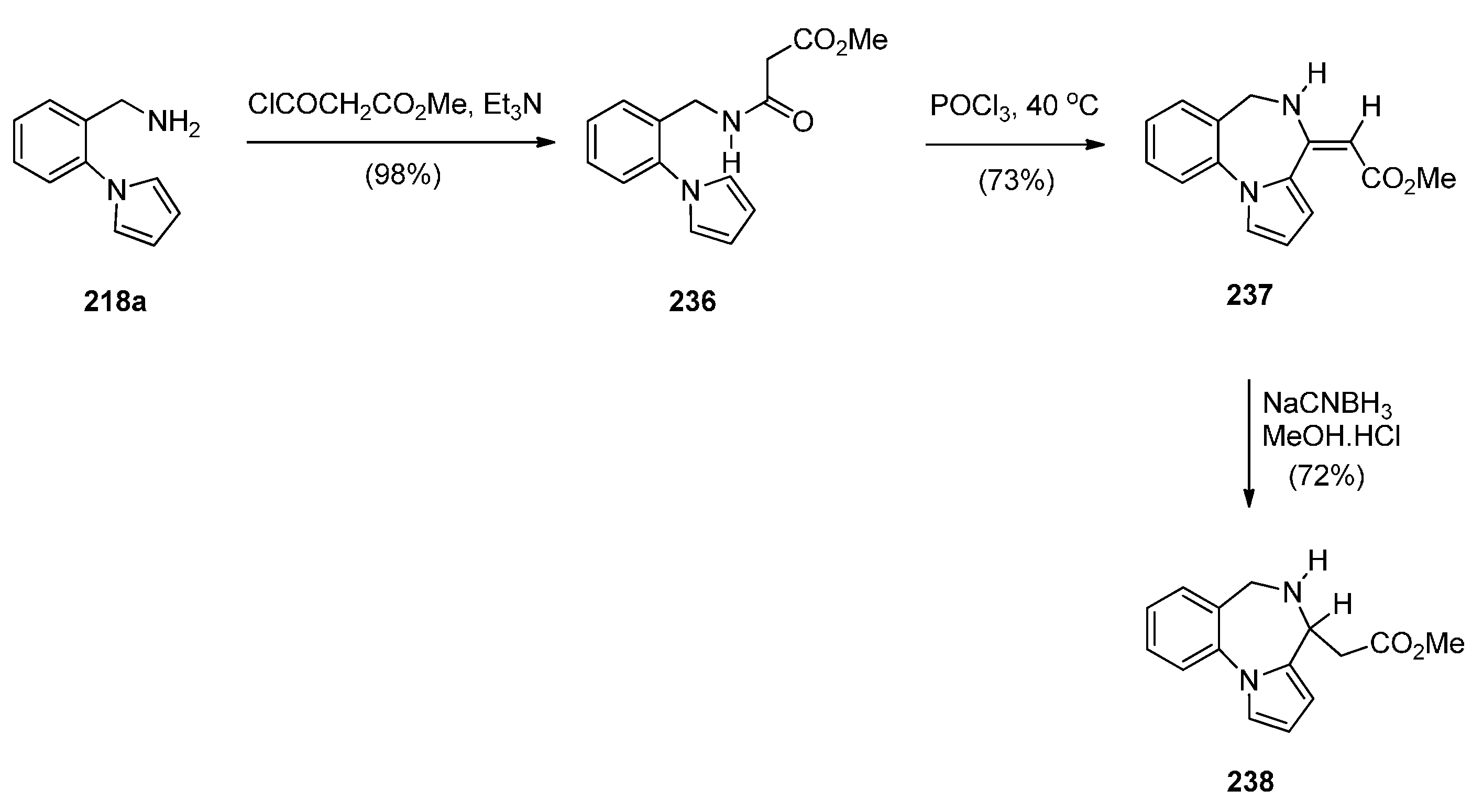
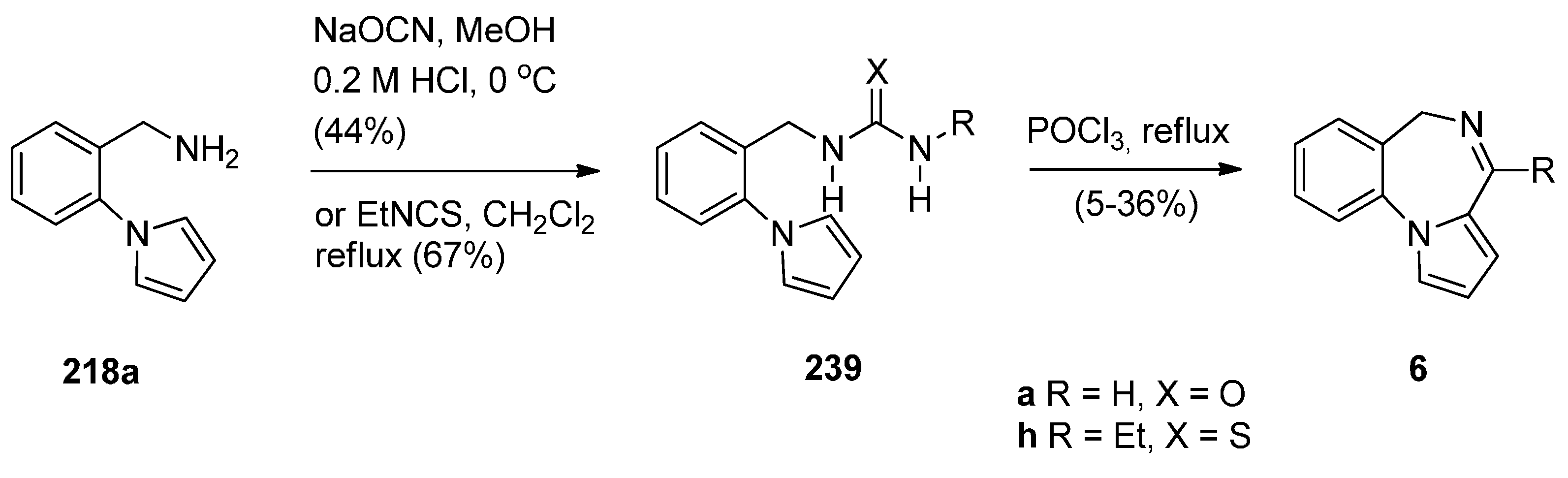
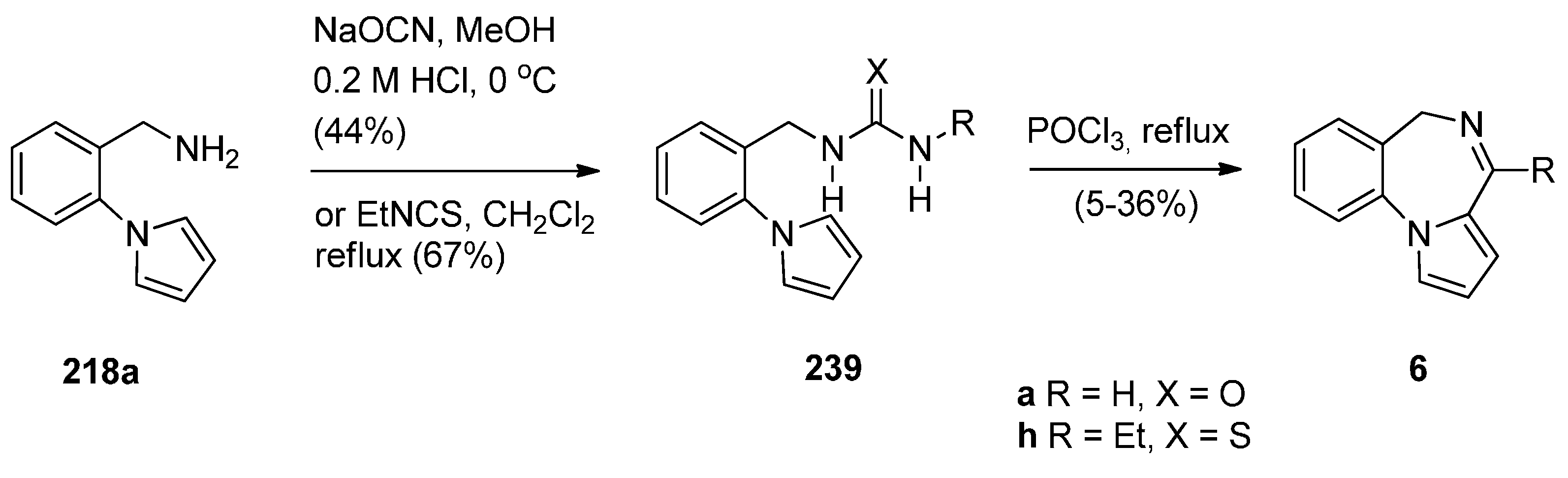
Another method of synthesizing [1,2-a] PBD C4–N5 imines with an aromatic pyrrole ring is using Bischler–Napieralski reaction conditions on precursors such as 239 (Scheme 43) containing, instead of an amide function group, a urea or thiourea group. Duceppe and Gauthier [87] used this method to synthesize PBDs 6a,h during an effort to prepare tricyclic compounds incorporating a pyrrole ring that could be of potential pharmacological interest. PBDs 6a,h were prepared from amine 218a in two steps by reaction of with sodium cyanate to give urea 239a and reaction of 218a with ethyl isothiocyanate to give thiourea 239h. Heating 239a,h with phosphoryl chloride provided parent PBD 6a in only 5% yield and PBD 6h in 36% yield.
Scheme 43.
Synthesis of PBD derivatives 6a,h.
Cyclisation of the Isothiocyanate Derivative of 1-(2-Aminomethyl-phenyl)pyrrole
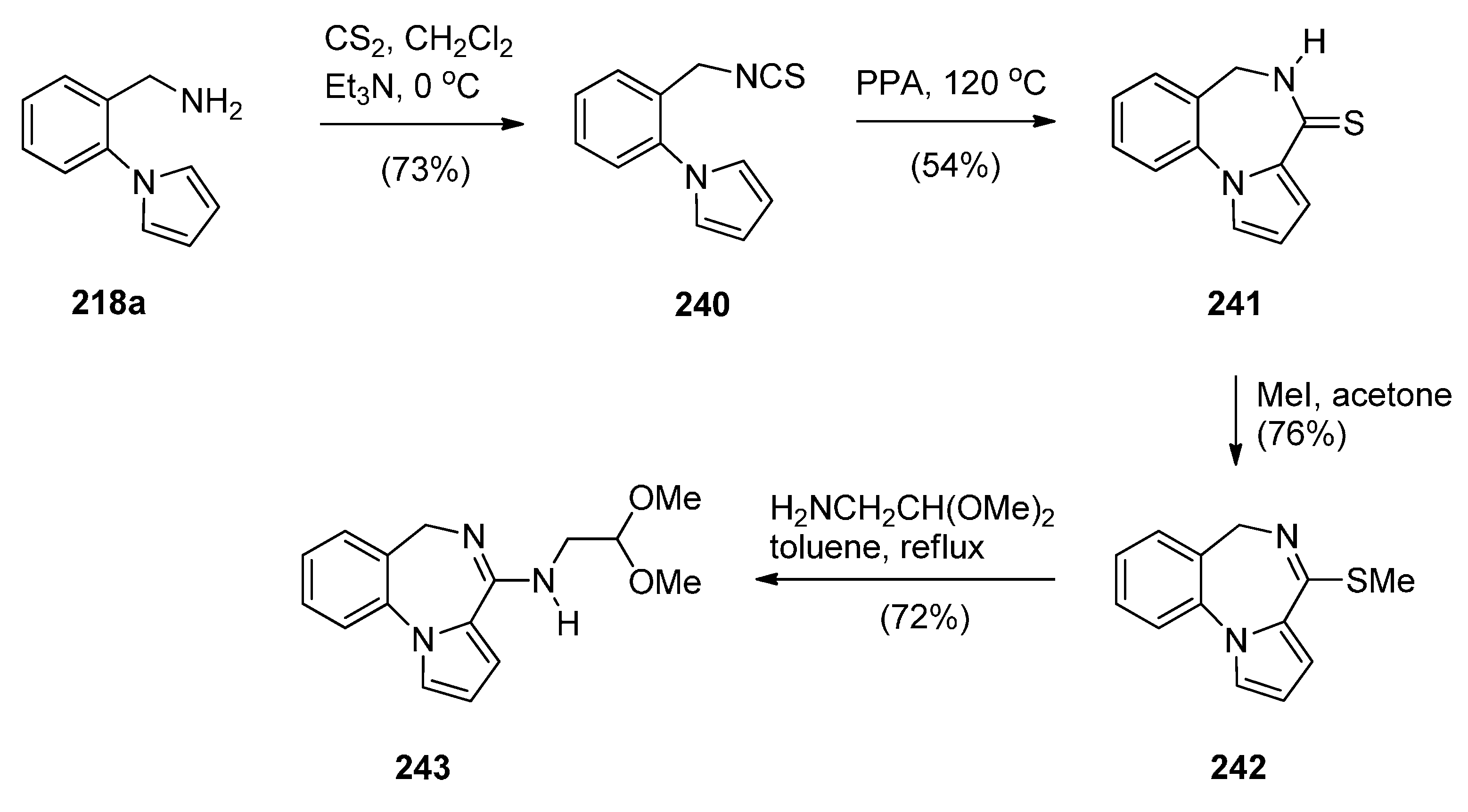
Duceppe and Gauthier [87] investigated yet another route to [1,2-a] PBD C4–N5 imines and this time targeted compound 242 (Scheme 44) whose 4-methylthio group was substituted by a primary amine, thus showing its potential in building a library of C4 substituted derivatives for biological evaluation. In the first step towards PBD 242 amine 218a reacted with carbon disulfide followed by subsequent treatment of the resulting isothiocyanate 240 with polyphosphoric acid (PPA) at 120 °C. Thiolactam 241 was S-methylated with methyl iodide to afford C4–methylthio PBD 242 which was then reacted with 2-aminoacetaldehyde dimethylacetal to furnish amidoketal PBD 243. The cyclisation of isothiocyanate 240 in polyphosphoric acid at 120 °C provided PBD thiolactam 241 in a mediocre 54% yield whereas the Bischler–Napieralski cyclisations of amides 234 and 236 (Scheme 41 and Scheme 42) provided PBDs 6 and 237, in yields of 73%–100%.
Scheme 44.
Synthesis of PBD derivatives 241–243.

Cyclocondensation of 2-Aminomethyl-1-(2-aroyl)pyrrole Derivatives
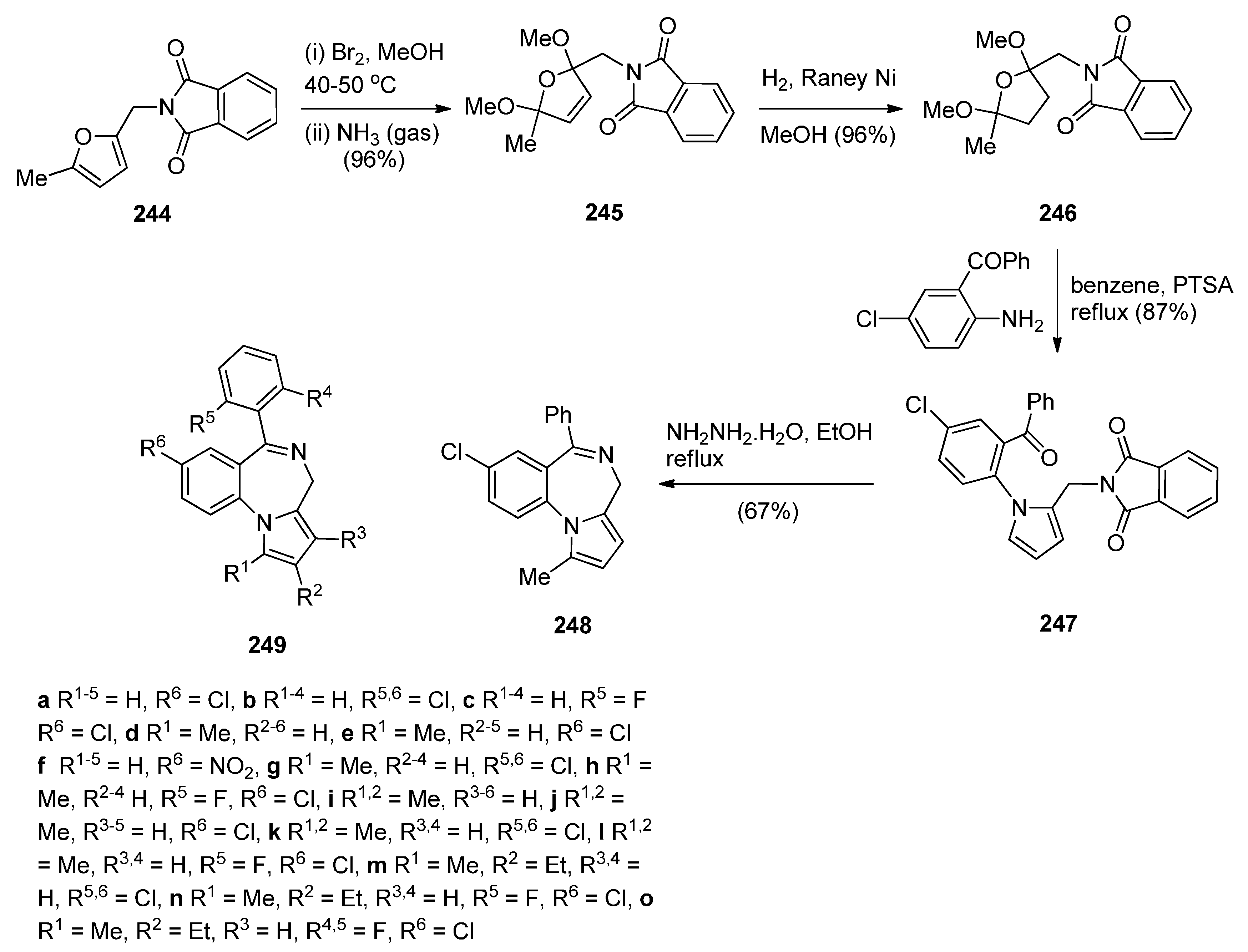
Hara and co-workers [88] described a convenient high yielding synthesis of PBD N5–C6 imine 248 in four steps from 2-methyl-5-phthalimidomethylfuran 244 (Scheme 45). Bromination at the α-positions of the furan ring of compound 244 followed by substitution from methanol yielded 2,5-dimethoxydihydrofuran 245. Compound 245 was obtained as a near 1:1 mixture of trans/cis isomers that were separated by fractional crystallisation. Trans-isomer 245 was then reduced by catalytic hydrogenation to the tetrahydrofuran 246. Next, compound 246 was condensed with 2-amino-5-chlorobenzophenone, to afford pyrrolylbenzophenone 247. The latter was heated with hydrazine hydrate causing cleavage of the phthaloyl group and cyclocondensation of the resulting intermediate aminoketone to yield PBD 248. Hara et al. [45] continued to use this synthetic methodology and one year later presented the synthesis of fifteen more examples of this tricyclic ring, that is PBDs 249a–o.
Scheme 45.
Synthesis of PBD derivatives 248 and 249a–o.
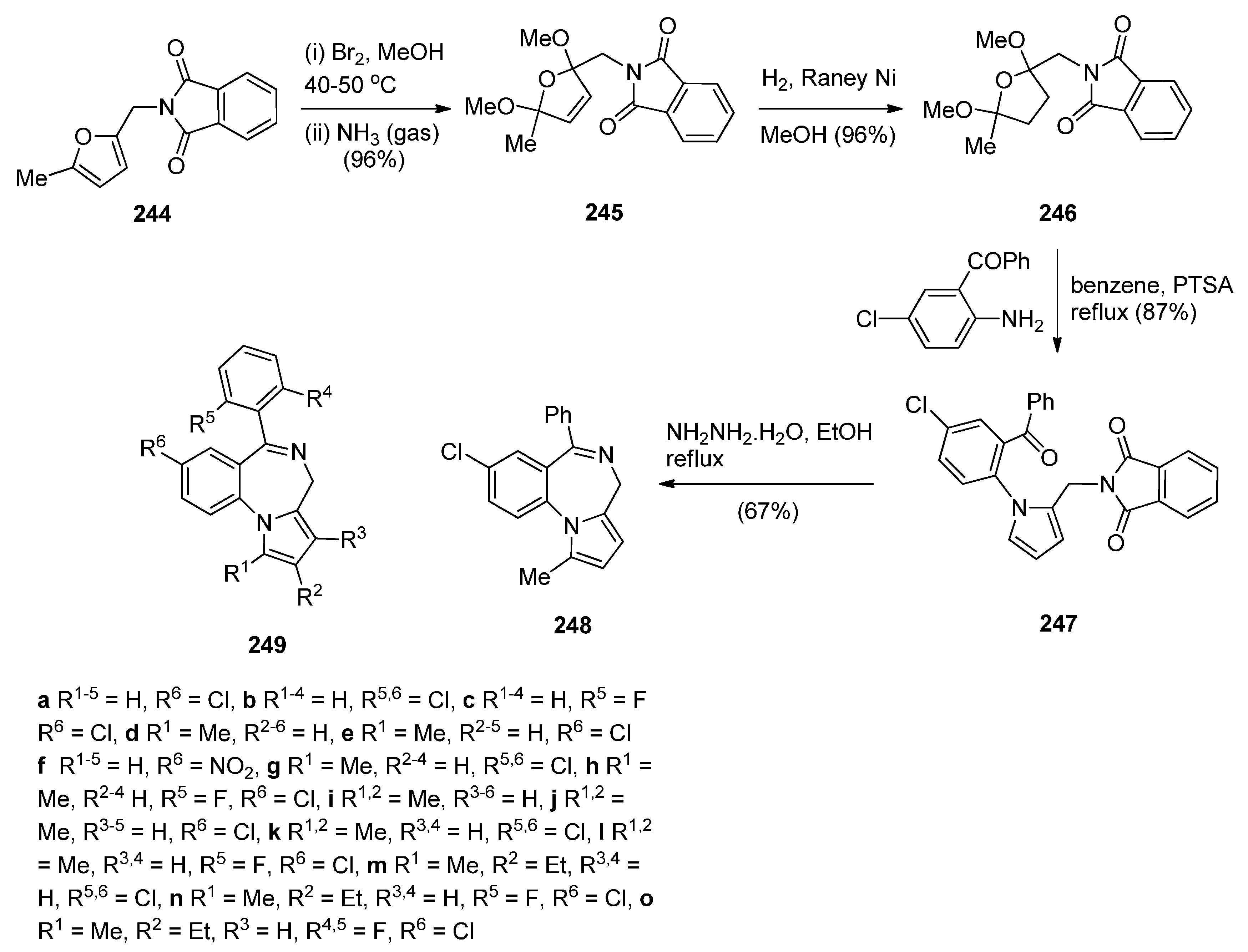
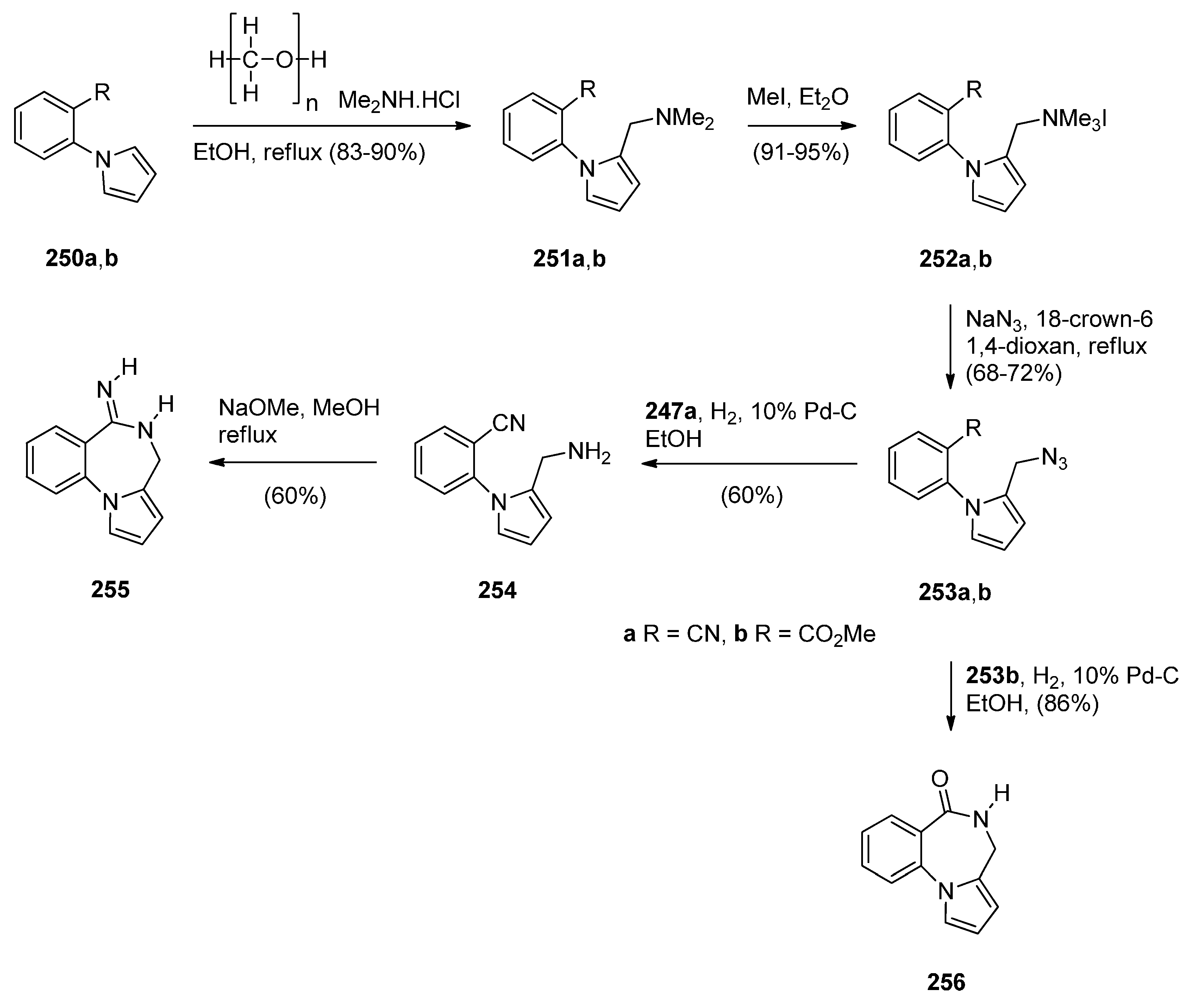
Within the framework of their scientific interests regarding the synthesis of nitrogen containing tricyclic compounds of potential pharmaceutical value, Korakas and Varvounis [89,90] presented the preparation of PBDs 255 and 256 from 1-arylpyrroles 250a,b (Scheme 46). In the first step, pyrroles 250a,b were subjected to Mannich reaction conditions to afford the respective dimethylamino-methylpyrroles 251a,b which were treated with methyl iodide to produce the corresponding quaternary ammonium salts 252a,b. Displacement of triethylamine from quaternary salts 252a,b by azide anion afforded azides 253a,b that were reduced by catalytic hydrogenation to afford, in the case of nitrile 253a, aminomethylpyrrole 254 whereas in the case of ester 253b, intermediate 2-aminomethyl-1-(2-methoxycarbonylphenyl)pyrrole reacted further by intramolecular acyl substitution, to yield PBD 256. Upon heating aminonitrile 254 in methanolic sodium methoxide, intramolecular nucleophilic addition occurred to produce PBD 6-one 255.
Scheme 46.
Synthesis of PBD derivatives 255 and 256.
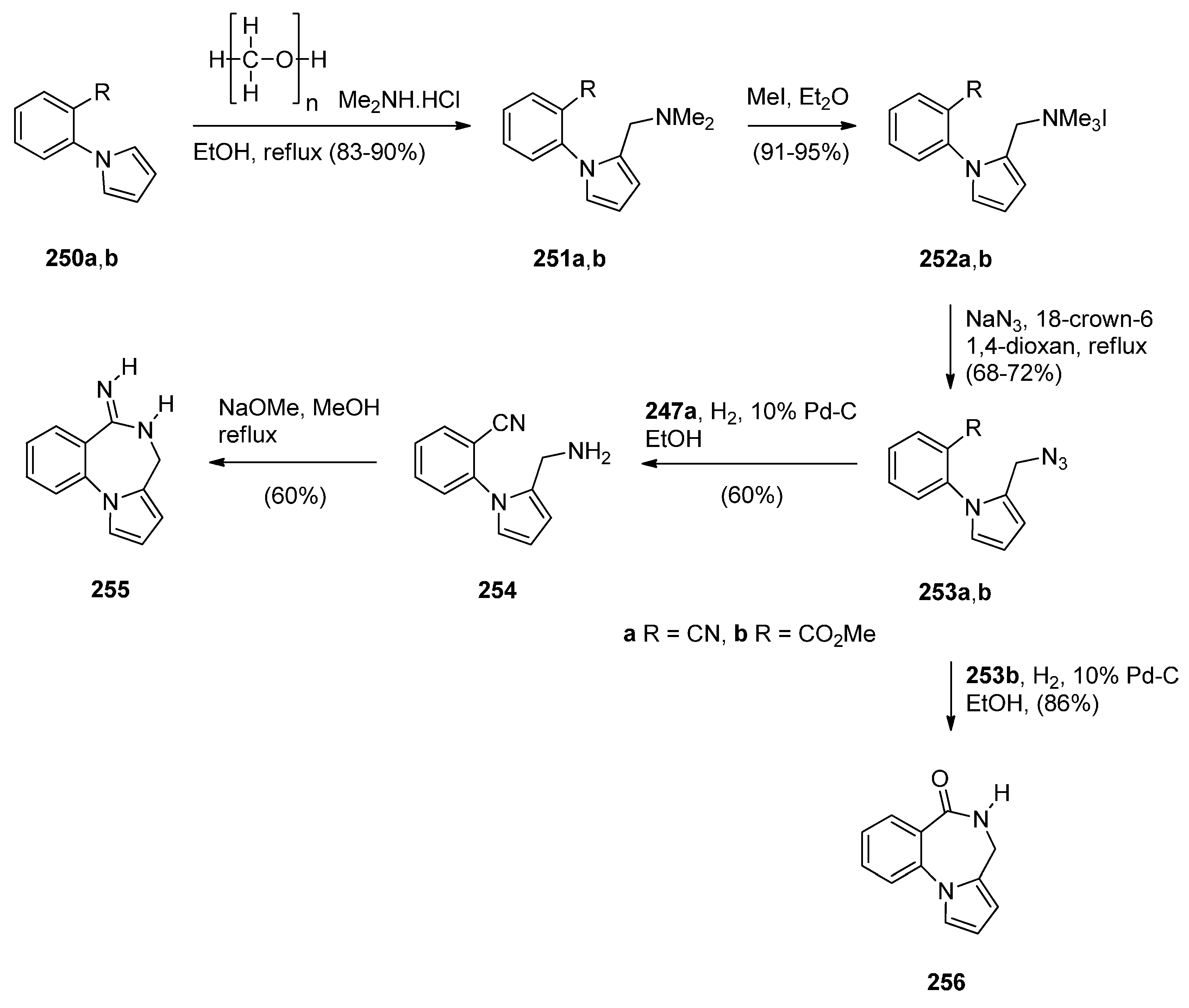
Cyclocondensation of N-(2,5-Dicarbonylalkyl)-2-nitrobenzamide Precursors Then Formation of Pyrrole Ring
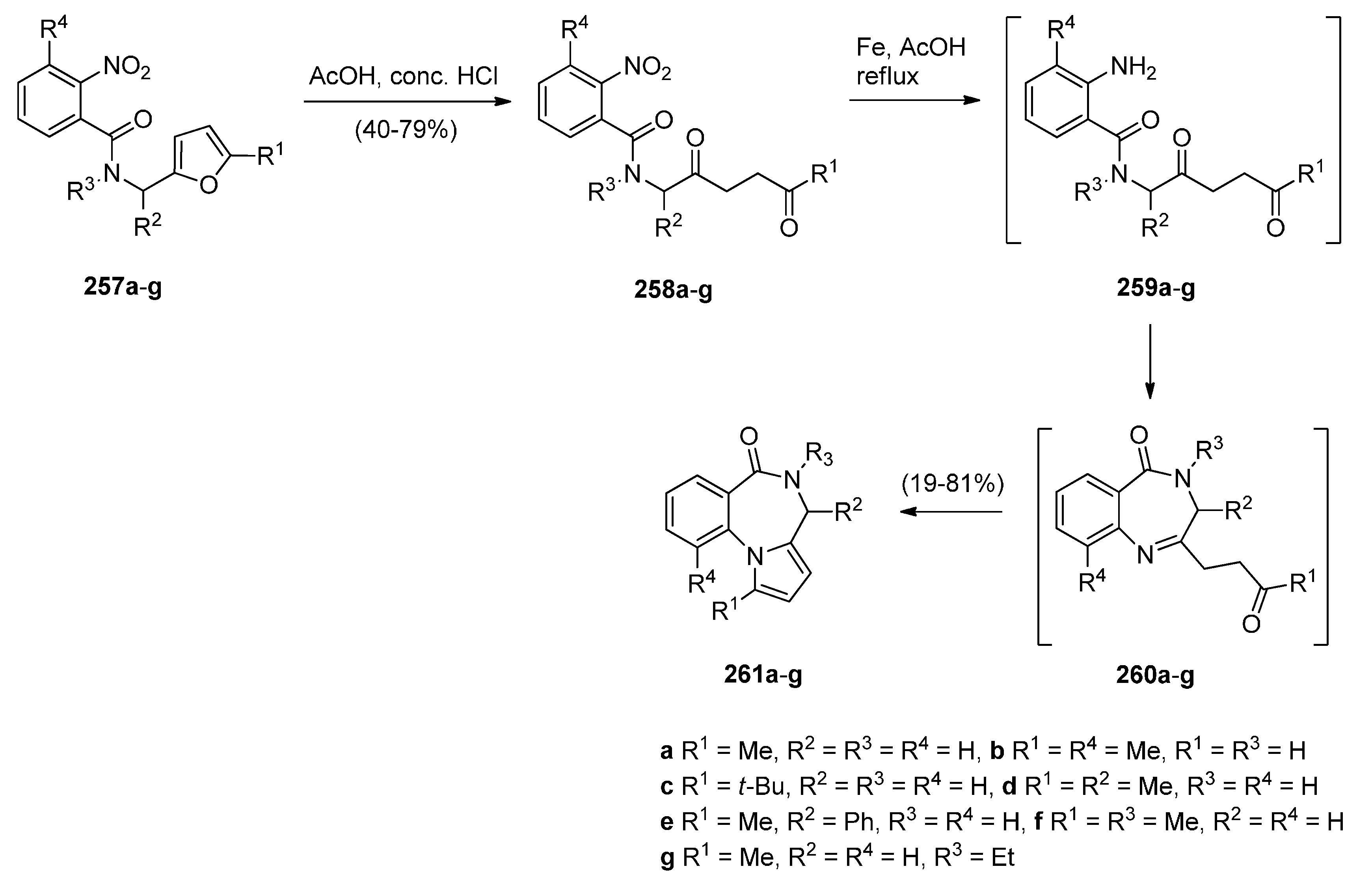
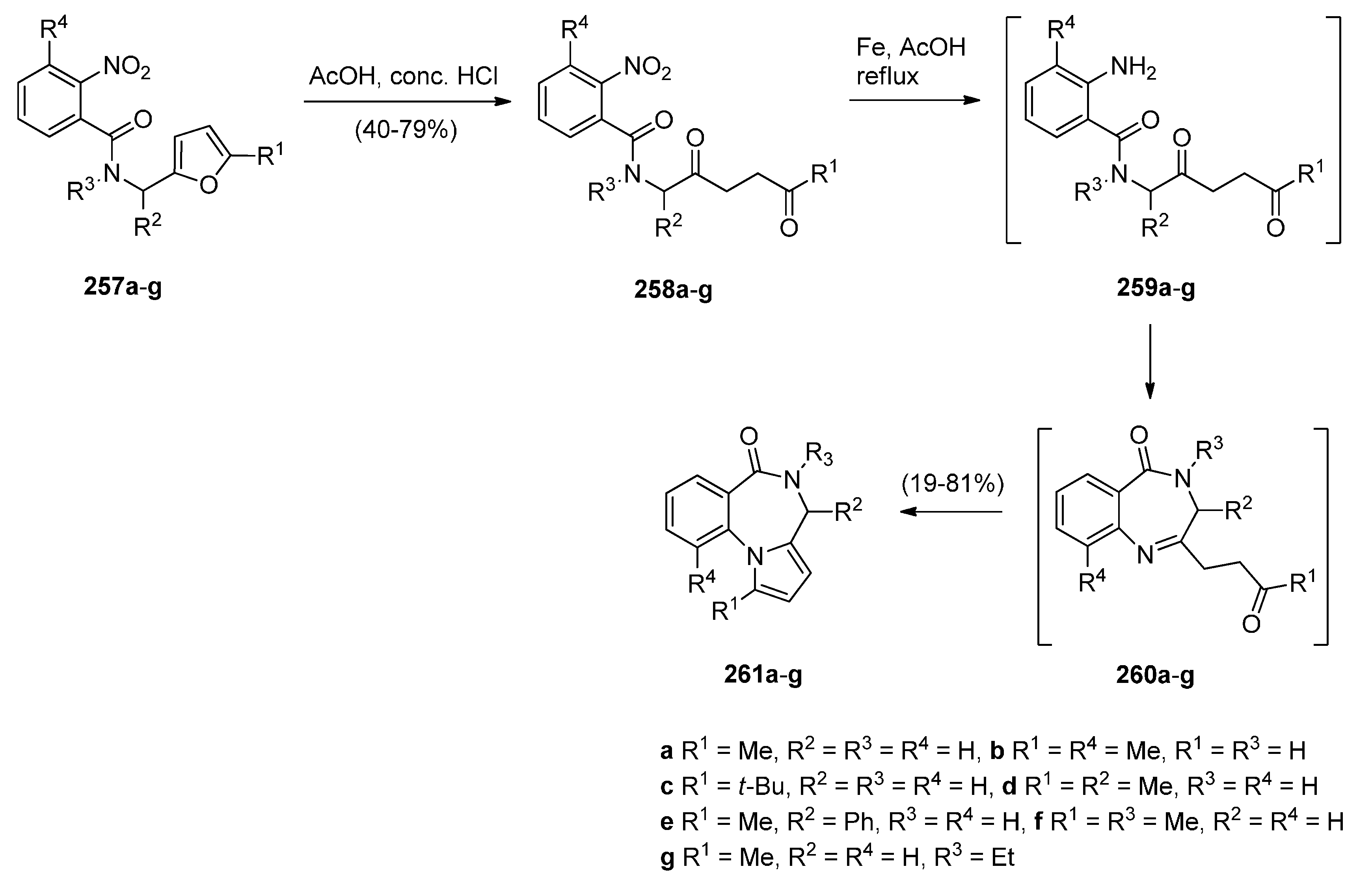
Based on the importance of pyrrolo[1,2-a][1,4]benzodiazepines as therapeutic molecules, Butin et al. [91] implemented a new method to synthesise PBDs 261a–g (Scheme 47), that consists of acid-catalyzed furan ring opening of N-(furfuryl)-2-nitrobenzamides 257a–g followed by a one pot reduction of the nitro group to amino and subsequent cyclisation with concomitant formation of the diazepine and pyrrole rings. Hence, at first the efficiency of the furan ring-opening reaction was tested with various acids and it was found that furans 257a–g treated with a mixture of glacial acetic acid and concentrated hydrochloric acid afforded nitrodiketones 261a–g in yields ranging from 40%–79%. Reduction of these compounds in glacial acetic with iron powder produced non-isolable amines 259a–g that cyclocondensed spontaneously to afford intermediate benzodiazepines 260a–g which underwent another cyclocondensation to afford PBDs 261a–g. With the exception of 261c, obtained in only 19% yield due to steric hinderance from the t-Bu group, PBDs 261a,b,d–g were isolated in very good yields.
2.2.2. Pyrrolo[1,2-a][1,4]benzodiazepines with a Non-Aromatic Pyrrole Ring
The most characteristic features of [1,2-a] PBDs in this category are: (i) non-aromatic pyrrole ring in all PBDs; (ii) one carbonyl group on the diazepine ring either at positions 4 or 6 of the PBD, N atoms are at position 5 are substituted part of either C4–N5 lactams or N5–C6 lactams, or; (iii) no carbonyl groups on the diazepine ring and the PBD N5 is a secondary amine.
Scheme 47.
Synthesis of PBD derivatives 261a–g.
Cyclisation of a 2-Fluoro-N-(pyrrolidin-2-ylmethyl)benzamide Precursor
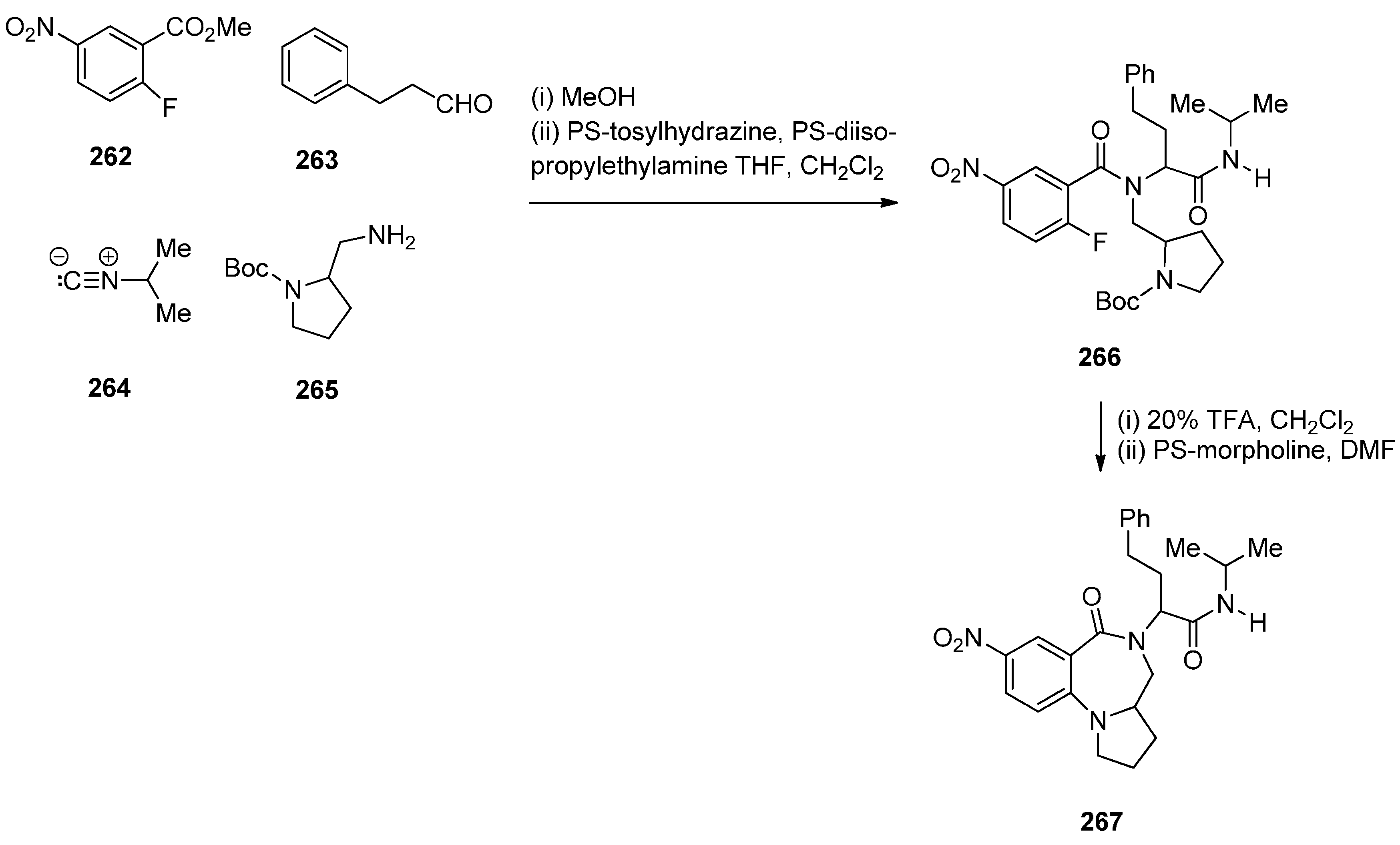
Tempest, Hulme and co-workers [92] developed a modified Ugi-4-component reaction to synthesise a series of biologically useful polycyclic heterocyclic cores. The synthesis of PBDs 267 (Scheme 48) by this method involves reacting 2-fluoro-5-nitrobenzoic acid (262) with 3-phenyl-propanal (263), isopropyl isocyanide (264) and tert-butyl 2-(aminomethyl)pyrrolidine-1-carboxylate (265), followed by the addition of polymer-bound PS-tosylhydrazine and PS-diisopropylethylamine, to afford Ugi product 266, in good yield. Boc-deprotection of 266 by 20% trifluoroacetic acid (TFA) in dichloromethane and intramolecular SNAr by subsequent addition of a proton scavenger such as polymer-bound PS-morpholine in DMF, produced PBD 267, in very good yield.
Scheme 48.
Synthesis of PBD 267.

Rearrangement of Intermediate 3,4′-Dioxospiro[pyrrolidine-1,1′-quinazoline]ylides
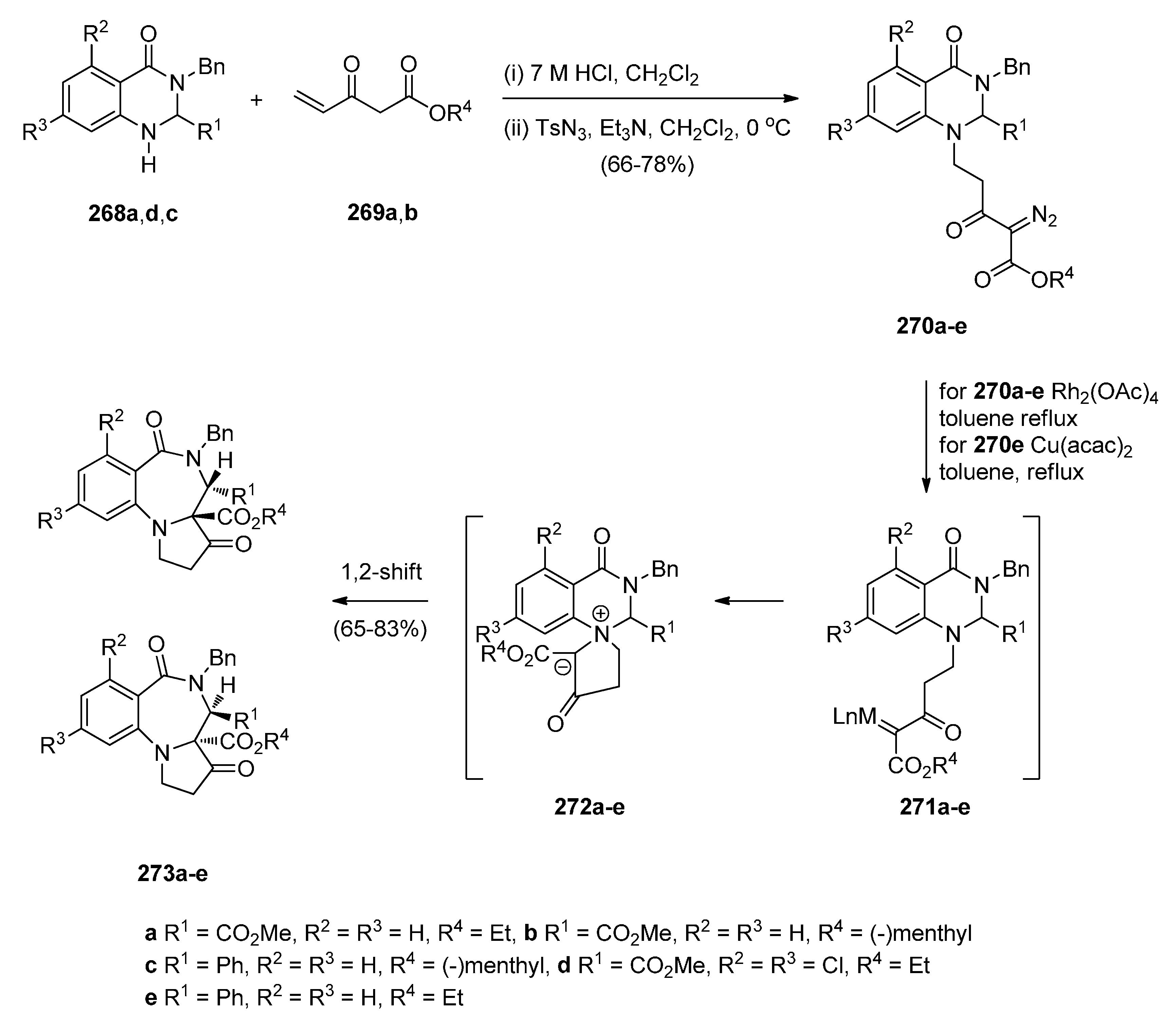
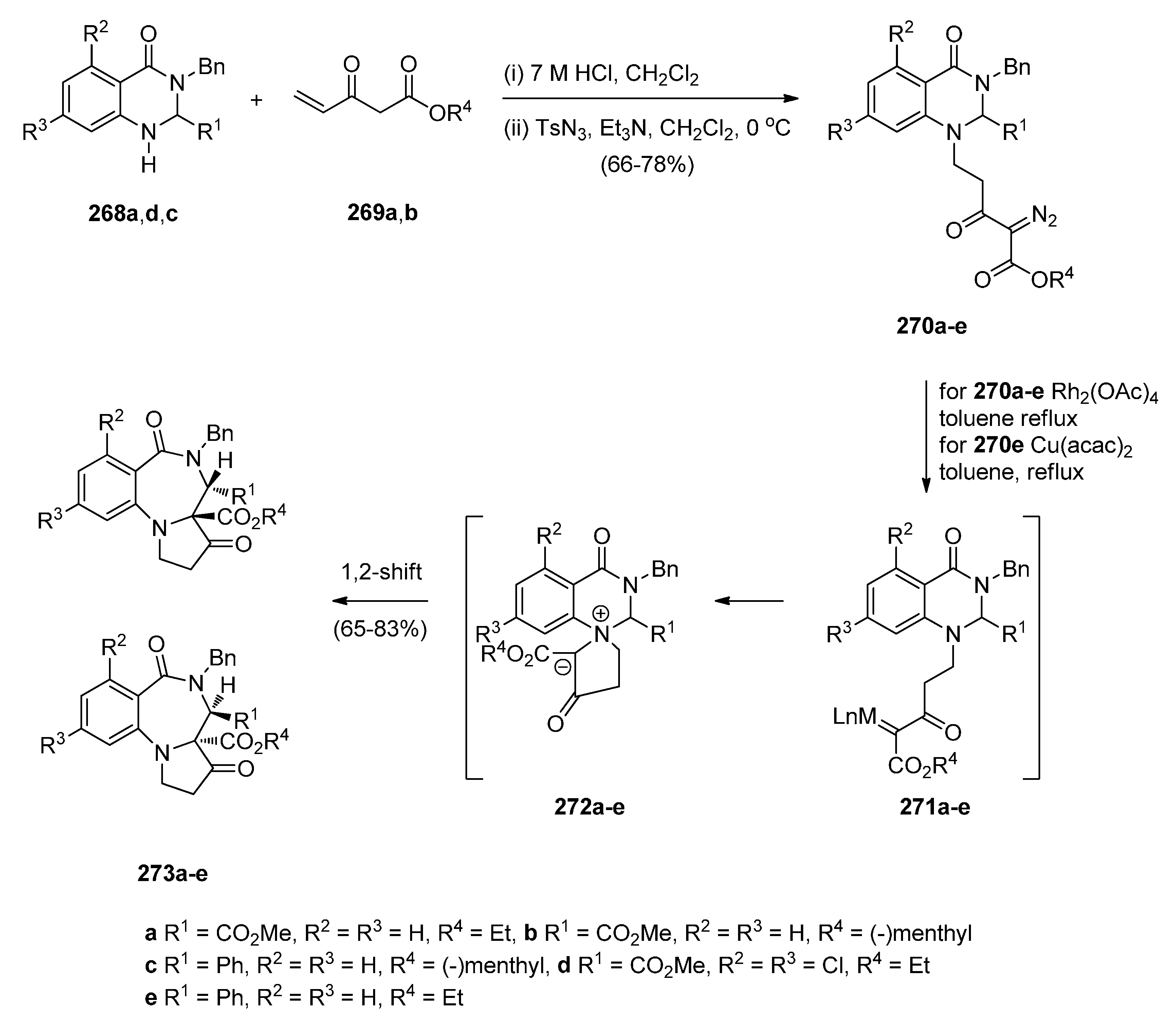
Continuing their work on the synthesis of enantiopure bicyclic and tricyclic alkaloids, Saba and co-workers [93] describe the synthesis of PBDs 273a–e (Scheme 49) by using the metallo carbenoid/spiro-[6,5]-ammonium ylide/Stevens[1,2]-shift with a ring-expansion sequence. The reaction sequence starts by the conjugate addition of (±)-methyl 4-oxo-1,2,3,4-tetrahydroquinazoline-2-carboxylates 268a,d or (±)-3-benzyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-one 268c, to the appropriate ethyl-3-ketopent-4-enoate 269a or (–)-menthyl-3-ketopent-4-enoate dichloromethane 269b in dichloromethane containing hydrochloric acid, followed by diazo group transfer reaction with tosyl azide. The corresponding diazo compounds 270a–e were isolated in good yields. Rhodium(II) acetate dimer [Rh2(OAc)4] catalyzed diazo-decomposition of 270a–d was performed in refluxing toluene and it took forty minutes to give the PBDs 273a–d as the sole products and in very good yields. For 270e the reaction worked equally well when copper(II) acetylacetonate [Cu(acac)2] was used as catalyst, requiring only a few minutes boiling in toluene. PBD 273e was isolated in 80% yield. It is expected that intermediate spiro-[6,5]-ammonium ylides 272, that are formed via the nitrogen trapping of the metallo carbenoid group correctly situated on the pendant of intermediate 271, undergo sigmatropic rearrangement to furnish the PBDs 273a–d. Compounds 273a,d and e were isolated as single diastereoisomers with trans configuration the substituents at the C-3a and C-4 positions whereas the chiral (–)-menthyl substituted derivatives 273b and 273c were obtained as a 1:1 mixture of trans diastereomers that could not be separated by column chromatography.
Scheme 49.
Synthesis of PBD derivatives 273a–e.
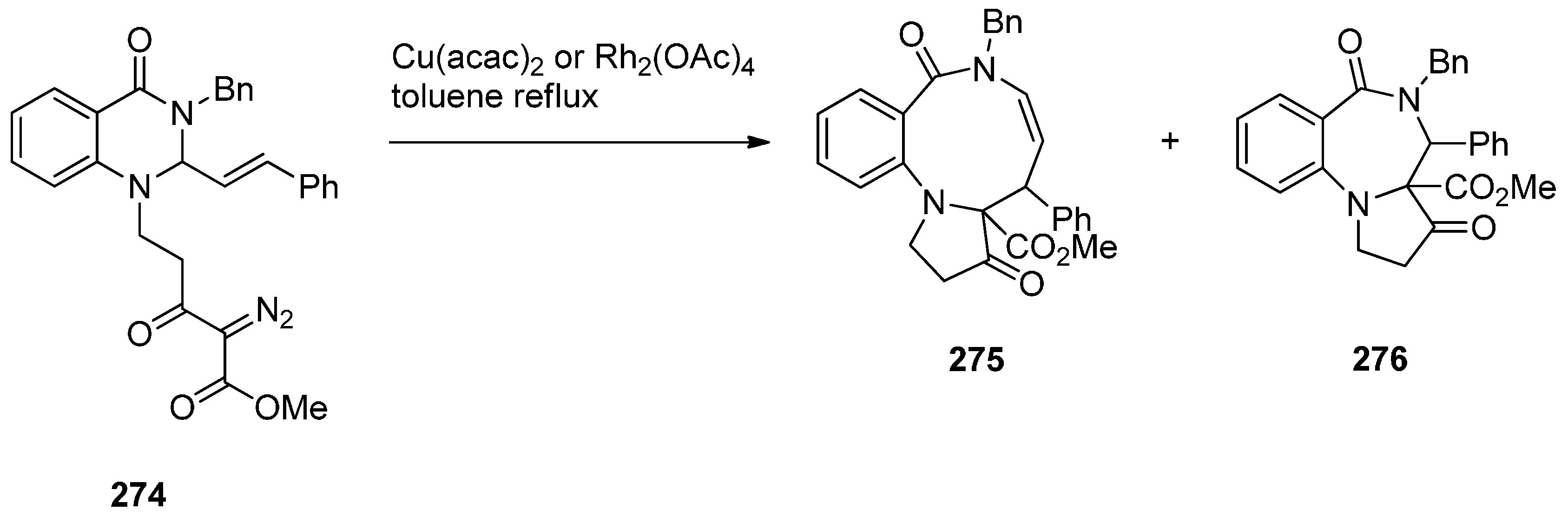
Two years later Saba and co-workers [94] prepared diazoquinazolinone 274, using similar methodology as explained above (Scheme 49), to find out that the copper(II) acetylacetonate [Cu(acac)2] catalysed diazo decomposition of 274 (Scheme 50) gave a mixture of the pyrrolobenzodiazonine 275 and PBD 276, in a 81:19 ratio. The same reaction using rhodium(II) acetate dimer [Rh2(OAc)4] afforded a mixture of 275 and PBD 276 in a 54:46 ratio. In both reactions the decompositions gave quantitative overall yields.
Scheme 50.
Synthesis of PBD 276.

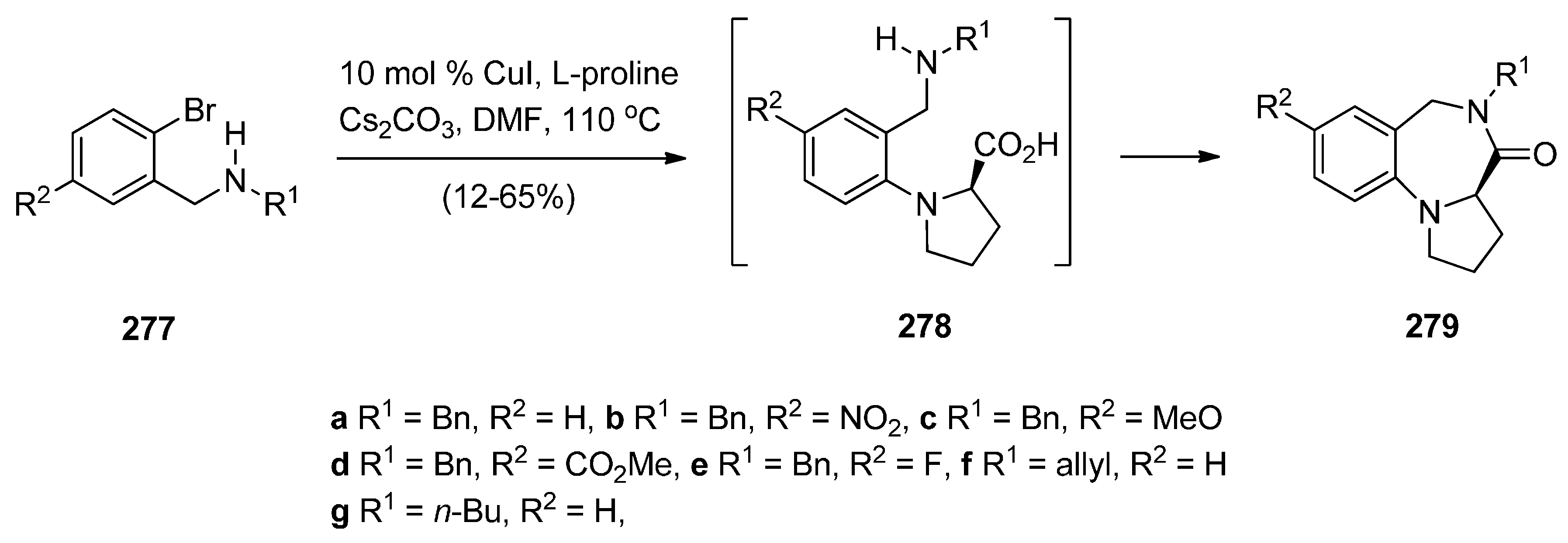
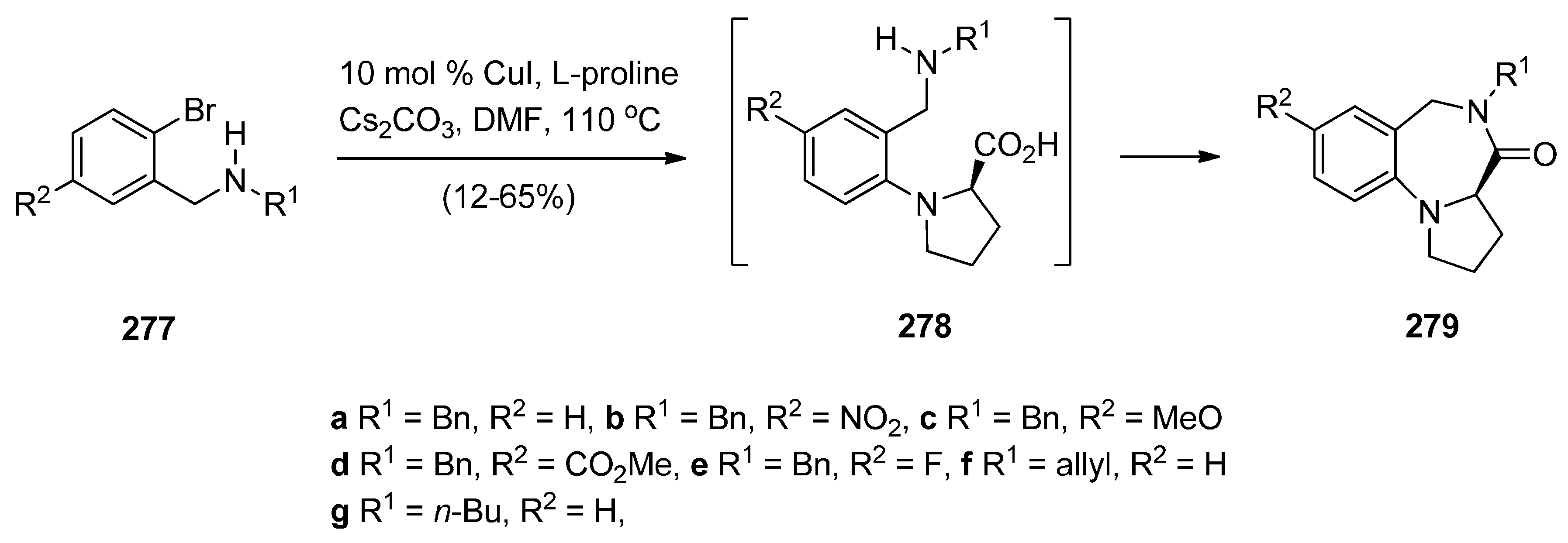
There are numerous substituted 1,4-benzodiazepin-3-ones that exhibit useful biological activity. Gao, Ma and co-workers [95] have recently shown interest in the chemistry of amino acid promoted copper iodide-catalysed C–N bond formation and used this method to synthesise PBDs 279a–g (Scheme 51). Thus, of N-(benzyl, allyl or n-butyl)-2-bromobenzylamines 277a–g were coupled with l-proline using copper iodide and cesium carbonate to give first the intermediate coupling products 278a–g which underwent intramolecular coupling to yield PBDs 279a–g. It was found the electronic-rich substrate 277c provided low yield (12%) of PBD 279c.
Scheme 51.
Synthesis of PBD derivatives 249a–g.
Reductive Cyclisation of 1-[2-(Aminomethyl)phenyl]-5-(4-methoxybenzoyl)pyrrolidin-2-one
Domínguez and co-workers [79], (Section “Reductive cyclisation of in situ generated 1-(2-aminobenzyl)-5-aroylpyrrolidin-2-ones”), have also reported the synthesis of 4-(4-methoxyphenyl) PBD 1-one 285 (Scheme 52). The synthetic route starts by protecting selectively 2-(aminomethyl)aniline (205) as carbamate 281. Coupling 281 with pent-4-ynoic acid using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC.HCl) and N-hydroxybenzotriazole (HOBt) afforded amide 282. Next, Sonogashira coupling between alkyne group of 282 and 4-iodoanisole under standard reaction conditions, rendered 4-pentyamide 283. Formation of the pyrrolidinone ring of 284 required (bis(trifluoroacetoxy)iodo)benzene (PIFA)-mediated cyclisation of 283 followed by basic hydrolysis. Catalytic hydrogenation of 284 with 10% palladium-on-carbon in acidic media caused successive deprotection, cyclodehydration of the ensuing aminoketone and reduction of the resulting cyclic imine to give the syn-diastereomer 285, exclusively. The overall yield of PBD 285 is 10%.
Scheme 52.
Synthesis of PBD 285.

2.3. Pyrrolo[1,2-d][1,4]benzodiazepines
2.3.1. Pyrrolo[1,2-d][1,4]benzodiazepines with a Non-Aromatic Pyrrole Ring
There are only five examples of [1,2-d] PBDs where: (i) all have a saturated pyrrole ring with a carbonyl group adjacent the N atom; (ii) two compounds have a carbonyl group on the diazepine ring which forms a C6–N7 lactam of the PBD and (iii) three compounds are without a carbonyl group on the diazepine ring and the PBD has an N7 tertiary amine.
Rearrangement of Pyrrolo[2,1-a]isoquinolinediones
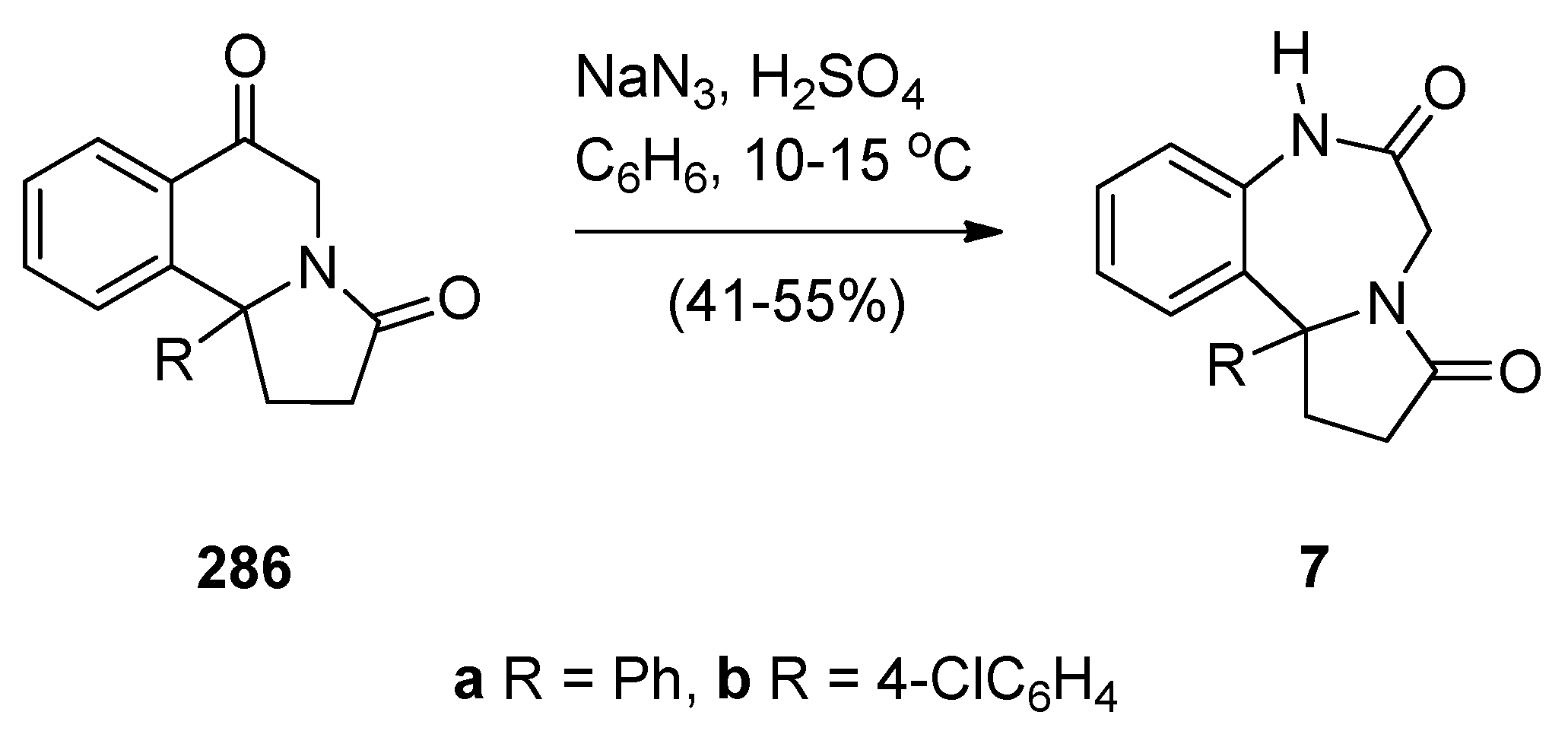
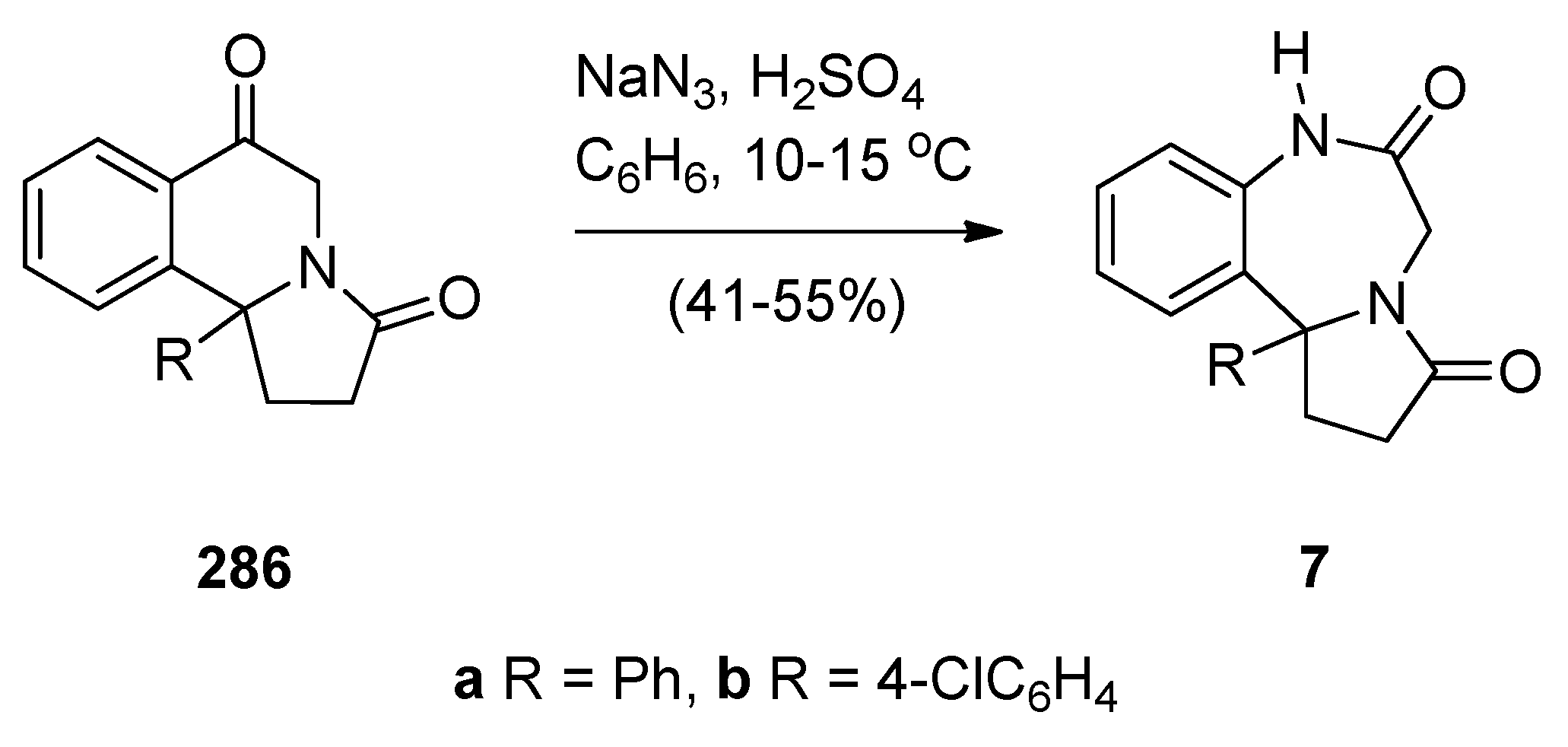
The first two examples of [1,2-d] PBDs 7a,b (Scheme 53) were reported in 1977 by Saito and co-workers [3]. Dihydropyrrolo[2,1-a]isoquinolinediones 286a,b were subjected to Schmidt reaction conditions, namely sodium azide and sulphuric acid in benzene at low temperature to yield PBDs 7a,b in moderate yields.
Scheme 53.
Synthesis of PBD derivatives 7a,b.
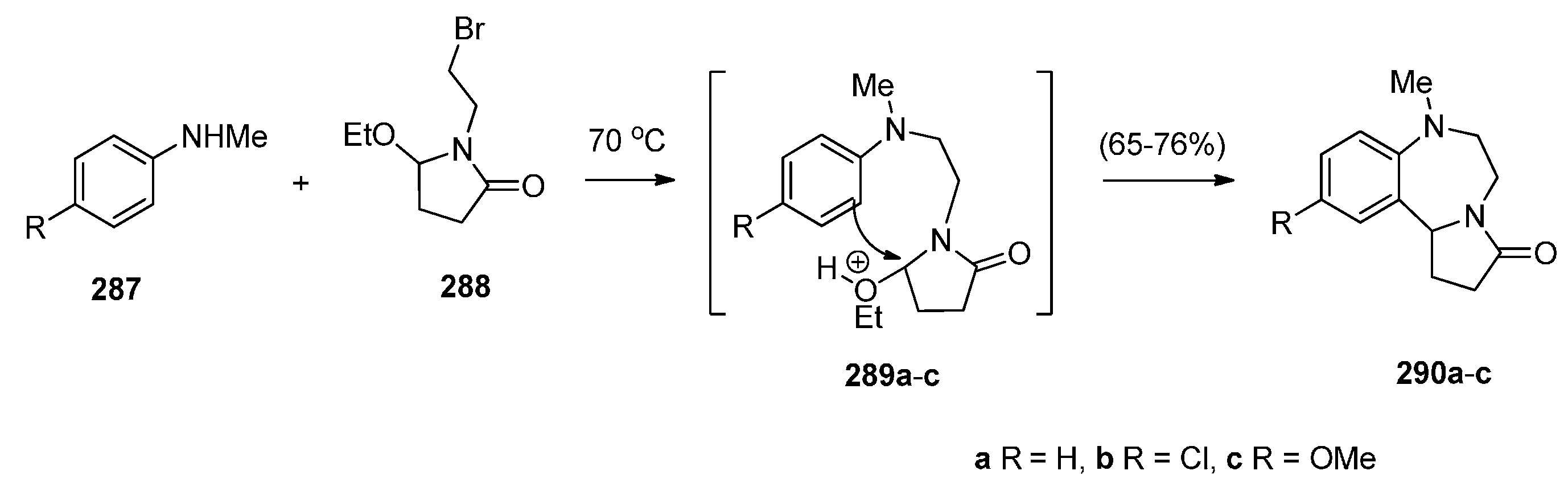
Cyclisation of 1-(2-Anilinoethyl)-5-ethoxypyrrolidin-2-ones
Kraus and Yue [96] became interested in synthesizing PBDs 290a–c (Scheme 54) due to their work on intermolecular aminoalkylations on aromatic rings. They used as starting material 1-(2-bromoethyl)-5-ethoxypyrrolidin-2-one 288 which was heated with the appropriate anilines 287a–c at 70 °C without solvent to give the non-isolable alkylated anilines 289a–c, that underwent spontaneous intramolecular substitution of the protonated ethoxy group by the aniline C-2, to afford PBDs 290a–c in very good yields.
Scheme 54.
Synthesis of PBD derivatives 290a–c.

2.3.2. Pyrrolo[1,2-d][1,4]benzodiazepines with an Aromatic Pyrrole Ring
There are thirty-eight examples of [1,2-d] PBDs where: (i) all PBDs have an aromatic pyrrole ring, (ii) thirty-three compounds have a carbonyl group on the diazepine ring which is an C6-N7 lactam of the PBD and (iii) five compounds have a carbonyl group on the diazepine ring which is a C6-N7 N-substituted lactam of the PBD.
Reductive Cyclisation of [2-Methyl-5-(2-nitrophenyl)pyrrol-1-yl]acetic Acids
The synthesis of PBDs 295c–e from diketones 291a,b has been reported by Aiello et al. [97] (Scheme 55). Thus, 3-acetyl-1-(2-nitrophenyl)pentane-1,4-dione 291a or ethyl 2-acetyl-4-(2-nitro-phenyl)-4-oxo-butanoate 291a,b were reacted with ammonium acetate on one hand and either glycine or alanine in the other, to produce 1-(2-nitrophenyl)pyrroles 292a,b and 294c–e, respectively. Reduction of pyrroles 292a,b by catalytic hydrogenation afforded amino pyrroles 293a,b. It was found that cyclisation of 293a,b with bromoacetyl bromide was best performed with triethylamine in dichloromethane at low temperature but even after column chromatography PBDs 295c,e were isolated in less than 10% yield. This problem was overcome by the shorter and more efficient route whereby the nitro acids 294c–e were submitted to catalytic hydrogenation which caused reduction of the nitro group to amino and spontaneous cyclocondensation to yield PBDs 295c–e, in very good yields. The by-products of the reaction between amino pyrroles 293a,b and bromoacetyl bromide were identified in a later publication [98].
Scheme 55.
Synthesis of PBD derivatives 295c–e.
Cyclisation of N-(2-Carbonylphenyl)glycinamide Precursors Then Formation of Pyrrole Ring
Lucca and Otto [48] became aware from studies conducted by the Janssen Pharmaceutical Company that certain benzodiazepine derivatives synthesized for a peptidomimetics program could have antiviral properties against HIV-I. One of these compounds was PBD 301b and in this context a large number of analogues 301a–m were synthesized and tested. Selected syntheses are presented for PBDs 301b,c,d,g,j,k in Scheme 56. The first step of the synthesis utilizes an excess of Grignard reagent 297 on anthranilonitrile 296 to give aminoketone 298 which is then coupled with either N-α-Boc-d-aspartic acid β-benzyl ester or Boc-O-benzyl-l-tyrosine in the presence of isobutyl chloroformate and N-methylmorpholine (NMM), to provide amides 299a,b. Next, N-Boc deprotection and cyclocondensation produces benzodiazepines 300a,b which upon heating in an aqueous solution of oxalic acid caused cyclisation into PBDs 301b,g. Debenzylation by catalytic hydrogenation yielded PBD carboxylic acids 301c,d. Furthermore, PBD 301g was N-alkylated to PBD 301j and by deprotecting both benzyl groups of this compound with trimethylsilyl iodide (TMSI), diphenolic PBD 301k was obtained. The authors refer to the use of standard literature procedures and do not report the yields of the synthesized compounds. The results of the antiviral activity tests of compounds 301a–m showed that compound 301b is the most active with an IC90 of 0.29 μg/mL.
Scheme 56.
Synthesis of PBD derivatives 248 and 249a–o.
Butin et al. [99] applied the same principle of an earlier method of synthesizing pyrrolo[1,2-a][1,4]benzodiazepines [91] from furfuryl derivatives in order to synthesize pyrrolo[1,2-d][1,4]benzodiazepines 306a–f (Scheme 57). The starting materials for this synthesis are the 2-(2-aminoaryl)furans 302a–f which were reacted with 2-(phthalimido)acetyl chloride in benzene to afford amides 303a–f. Hydrazinolysis of the phthalamido ring of 303a–f produced the corresponding amines 304a–f which were heated in a mixture of acetic acid and hydrochloric acid causing ring opening of the furan ring to afford the intermediate crude aminodiketones 305a–f. These were heated in glacial acetic acid causing two consecutive cyclodehydrations to afford PBDs 306a–f in low yields. One reason for the low yields of the PBDs is incomplete conversion of furans 304 into intermediate diketones 305 which is possibly due to the conjugation between furan and benzene rings.
Scheme 57.
Synthesis of PBD derivatives 306a–f.
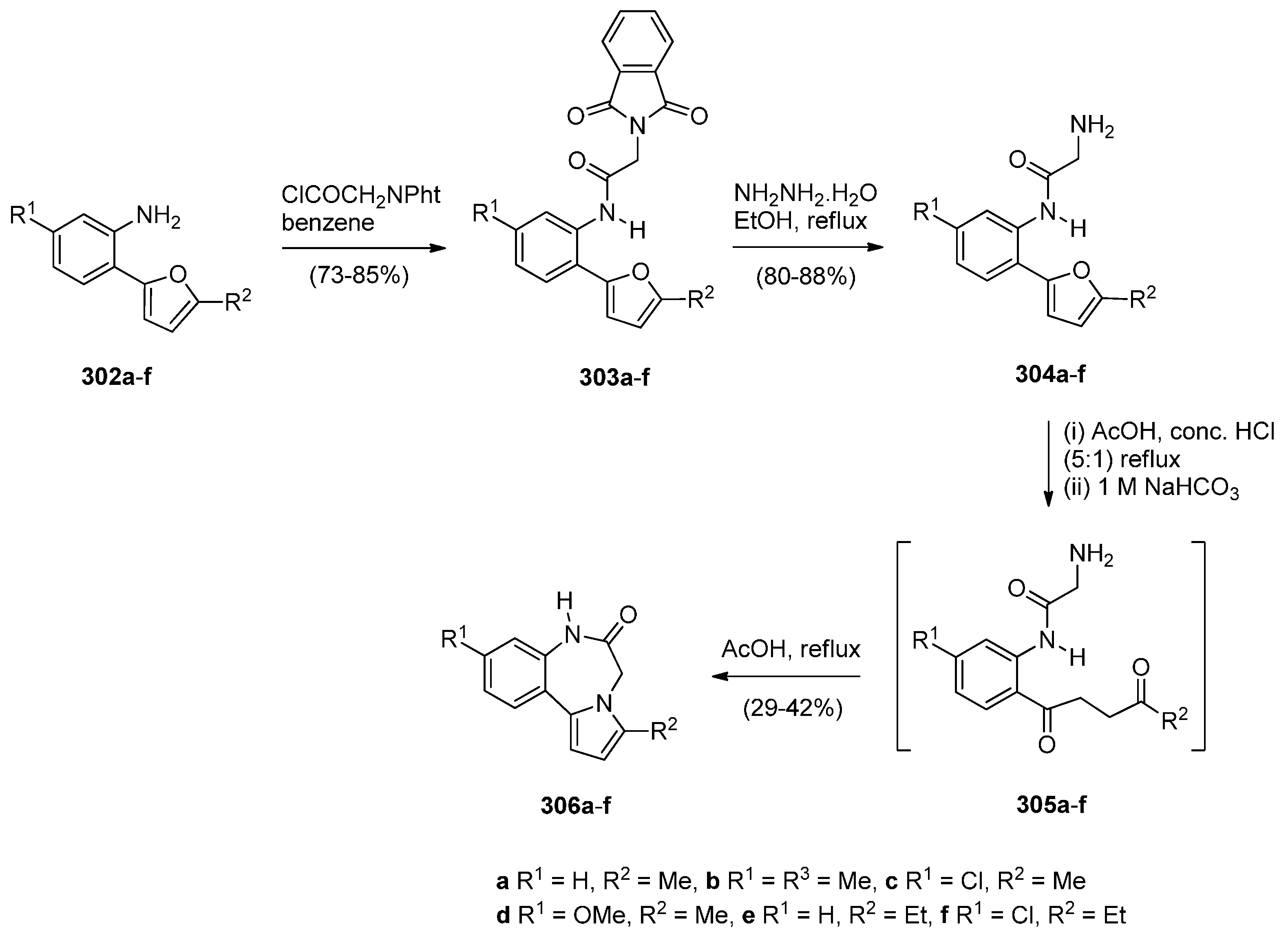
1,4-Diazepin-2-ones are considered important synthetic targets because they have shown therapeutic potential against inflammation and autoimmune diseases and have served as receptor ligands and enzyme inhibitors. They also have the ability to mimic natural γ- and β-turn peptide secondary structure. In order to investigate novel diazepinone analogues as potential γ-turn mimics, Dörr and Lubell [100] synthesized PBDs 312a–g and 314a–g (Scheme 58) and examined them by X-ray diffraction since X-ray structural analysis of certain diazepin-2-ones has revealed that their amino acid components adopt dihedral angle values similar to those of the central residue in a γ-turn. The starting point for the synthesis of these PBDs is 1-(2-aminophenyl)pent-4-en-1-one (307) which was coupled to Boc-l-α-amino acids 308a–g using N,N′-dicyclohexylcarbodiimide (DCC), and 4-dimethyl-aminopyridine (DMAP) while the addition of N-hydroxybenzotriazole (HOBt) prevented racemisation so that amides 309a–g were obtained, after chromatography, in 67%–97% yields and >93% enantiomeric excess. Two different routes to PBDs 312a–g have been presented. The first route involves removal of the Boc group from ketones 309a–g, cyclocondensation of the formed aminoketones and free-basing of the cyclic imine hydrochlorides, to afford benzodiazepinones 310a–g. Ozonolysis of these compounds and subsequent reduction of the formed ozonide with excess dimethyl sulphide, provided (S)-PBDs 312a–g. The second route to these compounds requires oxidative cleavage of olefins 309a–g to provide 1,4-ketoaldehydes 311a–g that underwent microwave irradiation causing cleavage of the Boc group followed by two consecutive cyclocondensations to afford (S)-PBDs 312a–g, in slightly better yields than the first method. The 3-methyl analogues of the latter compounds, (S)-PBDs 314a–g, were prepared from 1,4-diones 313a–g using the same principle of forming first the diazepine ring followed by the pyrrole ring but under different reaction conditions. Thus, trifluoroacetic acid (TFA) was applied to remove the Boc group of compounds 313a–g, the resulting ammonium trifluoroacetates were free-based using Amberlyst A-21 ion-exchange resin while the non-isolable aminoketones that were produced, underwent double cyclocondensation with p-toluenesulfonic acid as catalyst, to provide (S)-PBDs 314a–g. Next, the Tsuji-Wacker oxidation was applied to convert the terminal ethene group of compounds 309a–g to the ethanal group of compounds 313a–g. Comparing the two double cyclisations of diketones 311a–g and 313a–g leading to the respective PBDs 312a–g and 314a–g, it seems that the use of microwave irradiation at 150 °C is more efficient without the need to use solvents and reagents.
Scheme 58.
Synthesis of PBD derivatives 312a–g and 314a–g.

3. Conclusions
The interest in synthesising pyrrolo[1,4]benzodiazepines is mainly due to the ability of pyrrolo[2,1-c][1,4]benzodiazepines to interact with DNA. The molecules fit perfectly in the minor groove of DNA because of their right-handed helical conformation adopted from the (S)-configuration at C11a and interact with a selectivity for 5′-purine-G-purine sequences forming a covalent bond with guanine residues. The synthesis of these molecules extends from monomeric analogues of the natural products to dimeric adducts and recently to trimeric adducts. The evaluation of the antitumour activity of these compounds is dominated by in vitro cytotoxicity against tumour cell lines, DNA thermal denaturation experiments and DNA footprinting assays used to measure the DNA-binding affinity and establish the sequence selectivity. Although pyrrolo[1,2-a][1,4]benzodiazepines and pyrrolo[1,2-d][1,4]benzodiazepines have been very much less studied, they possess a wide range of useful biological activities.
Acknowledgments
The results presented by the author in the current review materialized due to the financial support by the Research Committee of the University of Ioannina.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Leimgruber, W.; Batcho, A.D.; Czajkowski, R.C. Total synthesis of anthramycin. J. Am. Chem. Soc. 1968, 90, 5641–5643. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, G.W.H.; Rafiq, M. Further cyclisation reactions of 1-arylpyrroles. J. Chem. Soc. C 1971. [Google Scholar] [CrossRef]
- Yamawaki, Y.; Watanabe, M.; Yamamura, S.; Saito, S. Studies on synthetic drugs. II. Syntheses of pyrrolo[1,2-d][1,4]benzodiazepine derivatives. Yakugaku Zasshi 1977, 97, 135–142. [Google Scholar]
- Leimgruber, W.; Stefanovic, V.; Shenker, F.; Karr, A.; Berger, J. Isolation and characterization of anthramycin, a new antitumour antibiotic. J. Am. Chem. Soc. 1965, 87, 5791–5793. [Google Scholar] [CrossRef] [PubMed]
- Thurston, D.E. Advances in the study of pyrrolo[2,1-c][1,4]benzodiazepine (PBD) antitumour antibiotics. In Molecular Aspects of Anticancer Drug-DNA Interactions; Neidle, S., Waring, M.J., Eds.; The Macmillan Press Ltd.: London, UK, 1993; pp. 54–88. [Google Scholar]
- Arora, S.K. Structural investigations of mode of action of drugs. II. Molecular structure of anthramycin methyl ether monohydrate. Acta Crystallogr. B 1979, 35, 2945–2948. [Google Scholar] [CrossRef]
- Hurley, L.H.; Reck, T.; Thurston, D.E.; Langley, D.R.; Holden, K.G.; Hertzberg, R.P.; Hoover, J.R.E.; Gallagher, G., Jr.; Faucette, L.F. Pyrrolo[1,4]benzodiazepine antitumour antibiotics: Relationship of DNA alkylation and sequence specificity to the biological activity of natural and synthetic compounds. Chem. Res. Toxicol. 1988, 1, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Fotso, S.; Zabriskie, T.M.; Proteau, P.J.; Flatt, P.M.; Santosa, D.A.; Mahmud, T. Limazepines A–F, pyrrolo[1,4]benzodiazepine antibiotics from an Indonesian Micrococcus sp. J. Nat. Prod. 2009, 72, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, S.; Yamamoto, Y.; Hayashi, M.; Komiyama, K.; Ishibashi, M. Cycloanthranilylproline-Derived Constituents from a Myxomycete Fuligo candida. Chem. Pharm. Bull. 2004, 52, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-P.; Yu, H.-S.; Sung, P.-J.; Tsai, F.-Y.; Shen, Y.-K.; Long-Sen Chang, L.-S.; Wang, J.-J. DC-81-Indole Conjugate Agent Induces Mitochondria Mediated Apoptosis in Human Melanoma A375 Cells. Chem. Res. Toxicol. 2007, 20, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Thurston, D.E.; Bose, D.S. Synthesis of DNA interactive pyrrolo[2,1-c][1,4]benzodiazepines. Chem. Rev. 1994, 94, 433–465. [Google Scholar] [CrossRef]
- Antonow, D.; Thurston, D.E. Synthesis of DNA-interactive pyrrolo[2,1-c][1,4]benzodiazepines (PBDs). Chem. Rev. 2011, 111, 2815–2864. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, B. Biosynthesis, synthesis, and biological activities of pyrrolobenzodiazepines. Med. Res. Rev. 2012, 32, 254–293. [Google Scholar] [CrossRef] [PubMed]
- Kaliszczak, M.; Antonow, D.; Patel, K.I.; Howard, P.; Jodrell, D.I.; Thurston, D.E.; Guichard, S.M. Optimization of the antitumor activity of sequence-specific pyrrolobenzodiazepine derivatives based on their affinity for ABC transporters. Am. Assoc. Pharm. Sci. J. 2010, 12, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Vassoler, H.; James, C.H.; Thurston, D.E. DNA sequence preference and adduct orientation of pyrrolo[2,1-c][1,4]benzodiazepine antitumor agents. ACS Med. Chem. Lett. 2010, 1, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Lee, W.; Kris, M.G.; Miller, V.A.; Krug, L.M.; Azzoli, C.G.; Senturk, E.; Calcutt, M.W.; Rizvi, N.A. A phase I trial of SJG-136 (NSC#694501) in advanced solid tumours. Cancer Chemother. Pharmacol. 2010, 65, 833–838. [Google Scholar] [PubMed]
- Hartley, J.A.; Hamaguchi1, A.; Coffils, M.; Martin, C.R.H.; Suggitt, M.; Chen, Z.; Gregson, S.J.; Masterson, L.A.; Tiberghien, A.C.; Hartley, J.M.; et al. SG2285, a novel C2-aryl-substituted pyrrolobenzodiazepine dimer pro-drug that cross-links DNA and exerts highly potent antitumour activity. Cancer Res. 2010, 70, 6849–6858. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.M.; Buhrow, S.A.; Kuffel, M.J.; Jia, L.; Spanswick, V.J.; Hartley, J.A.; Thurston, D.E.; Tomaszewski, J.E.; Ames, M.M. Pharmacokinetics, pharmacodynamics and metabolism of the dimeric pyrrolobenzodiazepine SJG-136 in rats. Cancer Chemother. Pharmacol. 2011, 68, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Rosado, H.; Rahman, K.M.; Feuerbaum, E.-A.; Hinds, J.; Thurston, D.E.; Taylor, P.W. The minor groove-binding agent ELB-21 forms multiple interstrand and intrastrand covalent cross-links with duplex DNA and displays potent bactericidal activity against methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2011, 66, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; James, C.H.; Bui, T.T.T.; Drake, A.F.; Thurston, D.E. Observation of a single-stranded DNA/pyrrolobenzodiazepine adduct. J. Am. Chem. Soc. 2011, 133, 19376–19385. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; James, C.H.; Thurston, D.E. Effect of base sequence on the DNA cross-linking properties of pyrrolobenzodiazepine (PBD) dimers. Nucleic Acids Res. 2011, 39, 5800–5812. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; James, C.H.; Thurston, D.E. Observation of the reversibility of a covalent pyrrolobenzodiazepine (PBD) DNA adduct by HPLC/MS and CD spectroscopy. Org. Biomol. Chem. 2011, 9, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Li, W.; Khullar, A.; Reixach, N.; Gerratana, B. Mutasynthesis of a potent anticancer sibiromycin analogue. ACS Chem. Biol. 2012, 7, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A.; Hochhauser, D. Small molecule drugs—Optimizing DNA damaging agent-based therapeutics. Curr. Opin. Pharm. 2012, 12, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A.; Hamaguchi, A.; Suggitt, M.; Gregson, S.J.; Thurston, D.E.; Howard, P.W. DNA interstrand cross-linking and in vivo antitumor activity of the extended pyrrolo[2,1-c][1,4]benzodiazepine dimer SG2057. Investig. New Drugs 2012, 30, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Rosado, H.; Moreira1, J.B.; Feuerbaum, E.-A.; Fox, K.R.; Stecher, E.; Howard, P.W.; Gregson, S.J.; James, C.H.; de la Fuente, M.; et al. Antistaphylococcal activity of DNA-interactive pyrrolobenzodiazepine (PBD) dimers and PBD-biaryl conjugates. J. Antimicrob. Chemother. 2012, 67, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Thurston, D.E.; Vassoler, H.; Jackson, P.J.M.; James, C.H.; Rahman, K.M. Effect of hairpin loop structure on reactivity, sequence preference and adduct orientation of a DNA-interactive pyrrolo[2,1-c][1,4]benzodiazepine (PBD) antitumour agent. Org. Biomol. Chem. 2015, 13, 4031–4040. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Burke, P.J.; Lyon, R.P.; Meyer, D.W.; Sussman, D.; Anderson, M.; Hunter, J.H.; Leiske, C.I.; Miyamoto, J.B.; Nicholas, N.D.; et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjug. Chem. 2013, 24, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.S.K.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, S.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.A.; Thomas, H.; Pillow, T.H.; Aristoff, P. Antibody-drug Conjugates for the treatment of cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. Antibody-Drug Conjugates Delivering DNA Cytotoxics. In Cancer Drug Design and Discovery, 2nd ed.; Neidle, S., Ed.; Academic Press: London, UK, 2014; pp. 479–490. [Google Scholar]
- Rahman, K.M.; Corcoran, D.B.; Bui, T.T.T.; Jackson, P.J.M.; David, E.; Thurston, D.E. Pyrrolobenzodiazepines (PBDs) do not bind to DNA G-quadruplexes. PLoS ONE 2014, 9, e105021. [Google Scholar] [CrossRef] [PubMed]
- Schneditz, G.; Rentner, J.; Roier, S.; Pletz, J.; Herzog, K.A.T.; Troeger, R.B.H.; Schild, S.; Weber, H.; Breinbauer, R.; Gorkiewicz, G.; et al. Enterotoxicity of a nonribosomal peptide causes antibiotic-associated colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13181–13186. [Google Scholar] [CrossRef] [PubMed]
- Rettig, M.; Langel, W.; Kamal, A.; Weisz, K. NMR structural studies on the covalent DNA binding of a pyrrolobenzodiazepine–naphthalimide conjugate. Org. Biomol. Chem. 2010, 8, 3179–3187. [Google Scholar] [CrossRef] [PubMed]
- Hopton, S.R.; Thompson, A.S. Nuclear Magnetic Resonance Solution Structures of Inter- and Intrastrand Adducts of DNA Cross-Linker SJG-136. Biochemistry 2011, 50, 4720–4732. [Google Scholar] [CrossRef] [PubMed]
- Raju, G.; Srinivas, R.; Reddy, V.S.; Idris, M.M.; Kamal, A.; Nagesh, N. Interaction of pyrrolobenzodiazepine (PBD) ligands with parallel intermolecular G-quadruplex complex using spectroscopy and ESI-MS. PLoS ONE 2012, 7, e35920. [Google Scholar] [CrossRef]
- Jackson, P.J.M.; James, C.H.; Jenkins, T.C.; Rahman, K.M.; David, E.; Thurston, D.E. Computational studies support the role of the C7-sibirosamine sugar of the pyrrolobenzodiazepine (PBD) sibiromycin in transcription factor inhibition. ACS Chem. Biol. 2014, 9, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Jang, J.-H.; Choo, S.-J.; Kim, S.-O.; Kim, J.W.; Ko, S.-K.; Soung, N.-K.; Lee, J.-S.; Kim, C.J.; Oh, H.; et al. Boseongazepines A–C, pyrrolobenzodiazepine derivatives from a Streptomyces sp. 11A057. Bioorg. Med. Chem. Lett. 2014, 24, 1802–1804. [Google Scholar] [CrossRef] [PubMed]
- Corelli, F.; Massa, S.; Stefancich, G.; Ortenzi, G.; Artico, M.; Pantaleoni, G.C.; Palumbo, G.; Fanini, D.; Giorgi, R. Benzodiazepines with Both Sedative and Analgesic Activities. Eur. J. Med. Chem 1986, 21, 445–449. [Google Scholar]
- Mai, A.; di Santo, R.; Massa, S.; Artico, M.; Pantaleoni, G.C.; Giorgi, R.; Coppolino, M.F.; Barracchini, A. Pyrrolobenzodiazepines with antinociceptive activity: Synthesis and pharmacological activities. Eur. J. Med. Chem. 1995, 30, 593–601. [Google Scholar] [CrossRef]
- Massa, S.; Artico, M.; Mai, A.; Corelli, F.; Pantaleoni, G.C.; Giorgi, R.; Ottaviani, D.; Cagnotto, A. Pyrrolobenzodiazepine and related systems. I. Synthesis and pharmacological evaluation of new 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepine derivatives. Farmaco 1990, 45, 1265–1281. [Google Scholar] [PubMed]
- Meerpoel, L.; van Gestel, J.; van Gerven, F.; Woestenborghs, F.; Marichal, P.; Sipido, V.; Terence, G.; Nash, R.; Corens, D.; Richards, R.D. Pyrrolo[1,2-a][1,4]benzodiazepine: A novel class of non-azole anti-dermatophyte anti-fungal agents. Bioorg. Med. Chem. Lett. 2005, 15, 3453–3458. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kayama, Y.; Mori, T.; Itoh, K.; Fujimori, H.; Sunami, T.; Hashimoto, Y.; Ishimoto, S. Diazepines. 5. Synthesis and Biological Action of 6-Phenyl-4H-pyrrolo[1,2-a][1,4]benzodiazepines. J. Med. Chem. 1978, 21, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.; Corelli, F.; Artico, M.; Mai, A.; Silvestri, R.; Pantaleoni, G.C.; Palumbo, G.; Fanini, D.; Giorgi, R. 5-Aroyl-5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepin-4-carboxylic acids: Synthesis and analgesic and neurobehavioral activity. Farmaco 1989, 44, 109–123. [Google Scholar] [PubMed]
- Massa, S.; Artico, M.; Mai, A.; Corelli, F.; Botta, M.; Tafi, A.; Pantaleoni, G.C.; Giorgi, R.; Coppolino, M.F.; Cagnotto, A.; et al. Pyrrolobenzodiazepines and related systems. 2. Synthesis and biological properties of isonoraptazepine derivatives. J. Med. Chem. 1992, 35, 4533–4541. [Google Scholar] [CrossRef] [PubMed]
- De Lucca, G.V.; Otto, M.J. Synthesis and anti-HIV activity of pyrrolo[1,2-d][1,4]benzodiazepine-6-ones. Bioorg. Med. Chem. Lett. 1992, 2, 1639–1644. [Google Scholar] [CrossRef]
- Kamal, A.; Rajender; Reddy, R.; Reddy, M.K.; Balakishan, G.; Shaik, T.B.; Chourasia, M.; Sastry, G.N. Remarkable enhancement in the DNA-binding ability of C2-fluoro substituted pyrrolo[2,1-c]-[1,4]benzodiazepines and their anticancer potential. Bioorg. Med. Chem. 2009, 17, 1557–1572. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Reddy, M.K.; Ramaiah, M.J.; Reddy, R.J.S.; Srikanth, Y.V.V.; Dastagiri, D.; Bharathi, E.V.; Pushpavalli, S.N.; Sarma, P.; Pal-Bhadra, M. Synthesis and biological evaluation of estradiol linked pyrrolo[2,1-c][1,4]benzodiazepine (PBD) conjugates as potential anticancer agents. Bioorg. Med. Chem. 2011, 19, 2565–2581. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Reddy, M.K.; Ramaiah, M.J.; Srikanth, Y.V.V.; Reddy, R.V.S.; Kumar, G.B.; Pushpavalli, S.N.; Bag, I.; Juvekar, A.; Sen, S.; et al. Synthesis of aryl-substituted naphthalene-linked pyrrolobenzodiazepine conjugates as potential anticancer agents with apoptosis-inducing ability. ChemMedChem 2011, 6, 1665–1679. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Prabhakar, S.; Ramaiah, M.J.; Reddy, P.V.; Reddy, C.R.; Mallareddy, A.; Shankaraiah, N.; Reddy, T.L.M.; Pushpavalli, S.N.; Pal-Bhadra, M. Synthesis and anticancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers. Eur. J. Med. Chem. 2011, 46, 3820–3831. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Shetti, R.V.; Ramaiah, M.J.; Swapna, P.; Reddy, K.S.; Mallareddy, A.; Rao, M.P.N.; Chourasia, M.; Sastry, G.N.; Juvekar, A.; et al. Carbazole-pyrrolo[2,1-c][1,4]benzodiazepine conjugates: Design, synthesis, and biological evaluation. Med. Chem. Commun. 2011, 2, 780–788. [Google Scholar] [CrossRef]
- Thurston, D.E.; Murty, V.S.; Langley, D.R.; Jones, G.B. O-Debenzylation of a pyrrolo[2,1-c][1,4]benzodiazepine in the presence of a carbinolamine functionality: Synthesis of DC-81. Synthesis 1990, 1, 81–84. [Google Scholar] [CrossRef]
- Kamal, A.; Ramakrishna, G.; Nayak, V.L.; Raju, P.; Rao, A.V.S.; Viswanath, A.; Vishnuvardhan, M.V.P.S.; Ramakrishna, S.; Srinivas, G. Design and synthesis of benzo[c,d]indolone-pyrrolobenzodiazepine conjugates as potential anticancer agents. Bioorg. Med. Chem. 2012, 20, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Srikanth, Y.V.V.; Ramaiah, M.J.; Khan, M.N.A.; Reddy, M.K.; Ashraf, Md.; Lavanya, A.; Pushpavalli, S.N.; Pal-Bhadra, M. Synthesis, anticancer activity and apoptosis inducing ability of bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine conjugates. Bioorg. Med. Chem. Lett. 2012, 22, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Ramakrishna, G.; Ramaiah, M.J.; Viswanath, A.; Ayinampudi, V.S.R.; Bagul, C.; Mukhopadyay, D.; Pushpavalli, S.N.; Pal-Bhadra, M. Design, synthesis and biological evaluation of imidazo[1,5-a]pyridine–PBD conjugates as potential DNA-directed alkylating agents. Med. Chem. Commun. 2013, 4, 697–703. [Google Scholar] [CrossRef]
- Kamal, A.; Sreekanth, K.; Shankaraiah, N.; Sathish, M.; Nekkanti, S.; Srinivasulu, V. Dithiocarbamate/piperazine bridged pyrrolobenzodiazepines as DNA-minor groove binders: Synthesis, DNA-binding affinity and cytotoxic activity. Bioorg. Chem. 2015, 59, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.S.; Idrees, M.; Todewale, I.K.; Jakka, N.M.; Rao, J.V. Hybrids of privileged structures benzothiazoles and pyrrolo[2,1-c][1,4]benzodiazepin-5-one, and diversity-oriented synthesis of benzothiazoles. Eur. J. Med. Chem. 2012, 50, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Langley, D.R.; Thurston, D.E. A versatile and efficient synthesis of carbinolamine-containing pyrrolo[1,4]benzodiazepines via the cyclisation of N-(2-aminobenzoyl)-pyrrolidine-2-carboxaldehyde diethyl thioacetals: total synthesis of prothracarcin. J. Org. Chem. 1987, 52, 91–97. [Google Scholar] [CrossRef]
- Wells, G.; Martin, C.R.H.; Howard, P.W.; Sands, Z.A.; Laughton, C.A.; Tiberghien, A.; Woo, C.K.; Masterson, L.A.; Stephenson, M.J.; Hartley, J.A.; et al. Design, synthesis, and biophysical and biological evaluation of a series of pyrrolobenzodiazepine-poly(N-methylpyrrole) conjugates. J. Med. Chem. 2006, 49, 5442–5461. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, M.; Kluza, J.; Wells, G.; O’Hare, C.C.; Forni, C.; Mantovani, R.; Howard, P.W.; Morris, P.; Thurston, D.E.; Hartley, J.A.; et al. Inhibition of DNA binding of the NF-Y transcription factor by the pyrrolobenzodiazepine-polyamide conjugate GWL-78. Mol. Cancer Ther. 2008, 7, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Brucoli, F.; Hawkins, R.M.; James, C.H.; Wells, G.; Jenkins, T.C.; Ellis, T.; Hartley, J.A.; Howard, P.W.; Thurston, D.E. Novel C8-linked pyrrolobenzodiazepine (PBD)–heterocycle conjugates that recognize DNA sequences containing an inverted CCAAT box. Bioorg. Med. Chem. Lett. 2011, 21, 3780–3783. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Jackson, P.J.M.; James, C.H.; Basu, B.P.; Hartley, J.A.; de la Fuente, M.; Schatzlein, A.; Robson, M.; Pedley, R.B.; Pepper, C.; et al. GC-targeted C8-linked pyrrolobenzodiazepine-biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J. Med. Chem. 2013, 56, 2911–2935. [Google Scholar] [CrossRef] [PubMed]
- Brucoli, F.; Hawkins, R.M.; James, C.H.; Jackson, P.J.M.; Wells, G.; Jenkins, T.C.; Ellis, T.; Kotecha, M.; Hochhauser, D.; Hartley, J.A.; et al. An extended pyrrolobenzodiazepine–polyamide conjugate with selectivity for a DNA sequence containing the ICB2 transcription factor binding site. J. Med. Chem. 2013, 56, 6339–6351. [Google Scholar] [CrossRef] [PubMed]
- Kolakowski, R.V.; Young, T.D.; Howard, P.W.; Jeffrey, S.C.; Senter, P.D. Synthesis of a C2-arylpyrrolo[2,1-c][1,4]benzodiazepine monomer enabling the convergent construction of symmetrical and non-symmetrical dimeric analogs. Tetrahedron Lett. 2015, 56, 4512–4515. [Google Scholar] [CrossRef]
- Kamal, A.; Prabhakar, S.; Shankaraiah, N.; Markandey, N.; Reddy, P.V.; Srinivasulu, V.; Sathish, M. AlCl3–NaI assisted cleavage of polymer-bound esters with concomitant amine coupling and azido-reductive cyclisation: Synthesis of pyrrolobenzodiazepine derivatives. Tetrahedron Lett. 2013, 54, 4435–4441. [Google Scholar] [CrossRef]
- Kamal, A.; Shankaraiah, N.; Devaiah, V.; Reddy, K.L. Solid-phase synthesis of fused [2,1-b]-quinazolinone alkaloids. Tetrahedron Lett. 2006, 47, 9025–9028. [Google Scholar]
- Hemming, K.; Chambers, C.S.; Jamshaid, F.; O’Gorman, P.A. Intramolecular azide to alkene cycloadditions for the construction of pyrrolobenzodiazepines and azetidinobenzodiazepines. Molecules 2014, 19, 16737–16756. [Google Scholar] [CrossRef] [PubMed]
- Hemming, K.; Chambers, C.S.; Hamasharif, M.S.; João, H.; Khan, M.N.; Nilesh Patel, N.; Airley, R.; Day, S. Azide based routes to tetrazolo and oxadiazolo derivatives of pyrrolobenzodiazepines and pyrrolobenzothiadiazepines. Tetrahedron 2014, 70, 7306–7317. [Google Scholar] [CrossRef]
- Araújo, A.C.; Rauter, A.P.; Nicotra, F.; Airoldi, C.; Costa, B.; Cipolla, L. Sugar-based enantiomeric and conformationally constrained pyrrolo[2,1-c][1,4]benzodiazepines as potential GABAA ligands. J. Med. Chem. 2011, 54, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, L.; Redaelli, C.; Nicotra, F. Synthesis of a spiro d-proline analogue bearing d-fructose. Lett. Drug Des. Discov. 2005, 2, 291–293. [Google Scholar] [CrossRef]
- Legerén, L.; Domínguez, D. Synthesis of 5-arylpyrrolo[2,1-c][1,4]benzodiazepines under mild cyclodehydration conditions. Tetrahedron 2010, 66, 2718–2722. [Google Scholar] [CrossRef]
- Sharma, S.K.; Mandadapu, A.K.; Kumaresan, K.; Arora, A.; Gauniyal, H.M.; Kundu, B. Efficient synthesis of naturally occurring skeleton 5–7–6 tricyclic pyrrolo[2,1-c][1,4]benzodiazepin-5-one and its derivatives via cationic π-cyclisation. Synthesis 2010, 23, 4087–4095. [Google Scholar]
- Leber, J.D.; Hoover, J.R.E.; Holden, K.G.; Johnson, R.K.; Hecht, S.M. A revised structure of sibiromycin. J. Am. Chem. Soc. 1988, 110, 2992–2993. [Google Scholar] [CrossRef]
- Gao, K.; Wu, B.; Yu, C.-B.; Chen, Q.-A.; Ye, Z.-S.; Zhou, Y.G. Iridium catalyzed asymmetric hydrogenation of cyclic imines of benzodiazepinones and benzodiazepines. Org. Lett. 2012, 14, 3890–3893. [Google Scholar] [CrossRef] [PubMed]
- Molinari, A.J.; Trybulski, E.J.; Bagli, J.; Croce, S.; Considine, J.; Qi, J.; Ali, K.; Demaio, W.; Lihotz, L.; Cochran, D. Identification and synthesis of major metabolites of Vasopressin V2-receptor agonist WAY-151932, and antagonist, Lixivaptan®. Bioorg. Med. Chem. Lett. 2007, 17, 5796–5800. [Google Scholar] [CrossRef] [PubMed]
- Roszkowski, P.; Maurin, J.K.; Czarnocki, Z. First enantioselective synthesis of aptazepine. Synthesis 2012, 2, 241–246. [Google Scholar]
- Pardo, L.M.; Tellitu, I.; Esther Domínguez, E. A versatile PIFA-mediated approach to structurally diverse pyrrolo(benzo)diazepines from linear alkynylamides. Tetrahedron 2010, 66, 5811–5818. [Google Scholar] [CrossRef]
- Raines, S.; Chai, S.Y.; Palopoli, F.P. Mannich reactions. Synthesis of 4,5-dihydropyrrolo[l,2-a]quinoxalines, 2,3,4,5-tetrahydro-lH-pyrrolo[l,2-a][l,4]diazepines and 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepines. J. Heterocycl. Chem. 1976, 13, 711–716. [Google Scholar] [CrossRef]
- Cheeseman, G.W.H.; Greenberg, S.G. Synthesis of 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepines. J. Heterocycl. Chem. 1979, 16, 241–244. [Google Scholar] [CrossRef]
- Massa, S.; Mai, A.; di Santo, R.; Artico, M. 5,6-Dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepine-4,4-diacetic acid diethyl ester, an useful synthon for the synthesis of new polycyclic nitrogen systems of pharmacological interest. J. Heterocycl. Chem. 1993, 30, 897–903. [Google Scholar] [CrossRef]
- Massa, S.; Mai, A.; Artico, M.; Corelli, F.; Botta, M. Synthesis of 3b,4,6,7-tetrahydro-5H,9H-pyrazino[2,l-c]-pyrrolo[1,2-a][l,4]benzodiazepine, a valuable precursor of potential central nervous system agents. Tetrahedron 1989, 45, 2763–2772. [Google Scholar] [CrossRef]
- Massa, S.; di Santo, R.; Costi, R.; Artico, M. Research on nitrogen containing heterocyclic compounds. XX. Synthesis of 8H-Imidazo[5,1-c]pyrrolo[1,2-a][1,4]benzodiazepine and its 6-derivatives. J. Heterocycl. Chem. 1993, 30, 749–753. [Google Scholar] [CrossRef]
- Voskressensky, L.G.; Borisova, T.N.; Babakhanova, M.I.; Titov, A.A.; Chervyakova, T.M.; Novikov, R.A.; Butin, A.S.; Khrustalev, V.N.; Varlamov, A.V. Transformation of 4-substituted tetrahydropyrrolo-benzodiazepines in a three-component reaction with methyl propiolate and indole. Chem. Heterocycl. Compd. 2014, 49, 1785–1794. [Google Scholar] [CrossRef]
- Voskressensky, L.G.; Borisova, T.N.; Babakhanova, M.I.; Chervyakova, T.M.; Titov, A.A.; Butin, A.S.; Nevolina, T.A.; Khrustalev, V.N.; Varlamov, A.V. Synthesis of pyrrolo[1,2-a][1,6]benzodiazonines from pyrrolo[1,2-a][1,4]benzodiazepines and alkynes containing electron-acceptor substituents. Chem. Heterocycl. Compd. 2013, 49, 1024–1032. [Google Scholar] [CrossRef]
- Duceppe, J.S.; Gauthier, J. Synthesis of 5H-imidazo[2,1-c]pyrrolo[1,2-a][1,4]benzodiazepine. J. Heterocycl. Chem. 1984, 21, 1685–1687. [Google Scholar] [CrossRef]
- Kayama, Y.; Hara, T.; Itoh, K.; Sunami, T. Diazepines II. A new synthesis of 4H-pyrrolo[1,2-a][1,4]-benzodiazepine. J. Heterocycl. Chem. 1977, 14, 171–172. [Google Scholar] [CrossRef]
- Korakas, D.; Varvounis, G. A convenient synthesis of 2-aminomethyl-1-arylpyrroles. Synthesis 1994, 1994, 164–166. [Google Scholar] [CrossRef]
- Korakas, D.; Varvounis, G. Synthesis of 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepine and 10,11-dihydro-5H,12H-pyrrolo[2,1-c][1,4]benzodiazocine derivatives via cyclisation of 2-aminomethylpyrroles. J. Heterocycl. Chem. 1994, 31, 1317–1320. [Google Scholar] [CrossRef]
- Butin, A.V.; Nevolina, T.A.; Shcherbinin, V.A.; Trushkov, I.V.; Cheshkov, D.A.; Krapivin, G.D. Furan ring opening-pyrrole ring closure: A new synthetic route to aryl(heteroaryl)-annulated pyrrolo[1,2-a][1,4]diazepines. Org. Biomol. Chem. 2010, 8, 3316–3327. [Google Scholar] [CrossRef] [PubMed]
- Tempest, P.; Ma, V.; Kelly, M.G.; Jones, W.; Hulme, C. MCC/SNAr methodology. Part 1: Novel access to a range of heterocyclic cores. Tetrahedron Lett. 2001, 42, 4963–4968. [Google Scholar] [CrossRef]
- Mucedda, M.; Muroni, D.; Saba, A.; Manassero, C. Concise diastereospecific pyrrolo[1,2-a][1,4]-benzodiazepinone synthesis. Tetrahedron 2007, 63, 12232–12238. [Google Scholar] [CrossRef]
- Muroni, D.; Mucedda, M.; Saba, A. Efficient synthesis of 1-azatricyclic ring systems from anthranylamide. Heterocycles 2009, 78, 635–644. [Google Scholar]
- Wang, H.; Jiang, Y.; Gao, K.; Ma, D. Facile synthesis of 1,4-benzodiazepin-3-ones from o-bromobenzylamines and amino acids via a cascade coupling/condensation process. Tetrahedron 2009, 65, 8956–8960. [Google Scholar] [CrossRef]
- Kraus, G.A.; Yue, S. Amidoalkylation Reactions of Anilines. A direct synthesis of benzodiazepines. J. Org. Chem. 1983, 48, 2936–2937. [Google Scholar] [CrossRef]
- Aiello, E.; Dattolo, G.; Cirrincione, G.; Plescia, S.E.; Daidone, G. Polycondensed nitrogen heterocycles. VII. 5,6-Dihydro-7H-pyrrolo[1,2-d][1,4]benzodiazepin-6-ones. A novel series of annelated 1,4-benzodiazepines. J. Heterocycl. Chem. 1979, 16, 209–211. [Google Scholar] [CrossRef]
- Dattolo, G.; Cirrincione, G.; Aiello, E. Polycondensed nitrogen heterocycles. IX. 5,6-Dihydro-7H-pyrrolo[1,2-d][1,4]benzodiazepin-6-one. J. Heterocycl. Chem. 1980, 17, 701–703. [Google Scholar] [CrossRef]
- Nevolina, T.A.; Shcherbinin, V.A.; Serdyuk, O.V.; Butin, A.V. Furan ring opening-pyrrole ring closure: A new route to pyrrolo[1,2-d][1,4]benzodiazepin-6-ones. Synthesis 2011, 21, 3547–3551. [Google Scholar] [CrossRef]
- Dörr, A.A.; Lubell, W.D. γ-Turn mimicry with benzodiazepinones and pyrrolobenzodiazepinones synthesized from a common amino ketone intermediate. Org. Lett. 2015, 17, 3592–3595. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Varvounis, G. An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines. Molecules 2016, 21, 154. https://doi.org/10.3390/molecules21020154
AMA Style
Varvounis G. An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines. Molecules. 2016; 21(2):154. https://doi.org/10.3390/molecules21020154
Chicago/Turabian StyleVarvounis, George. 2016. "An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines" Molecules 21, no. 2: 154. https://doi.org/10.3390/molecules21020154