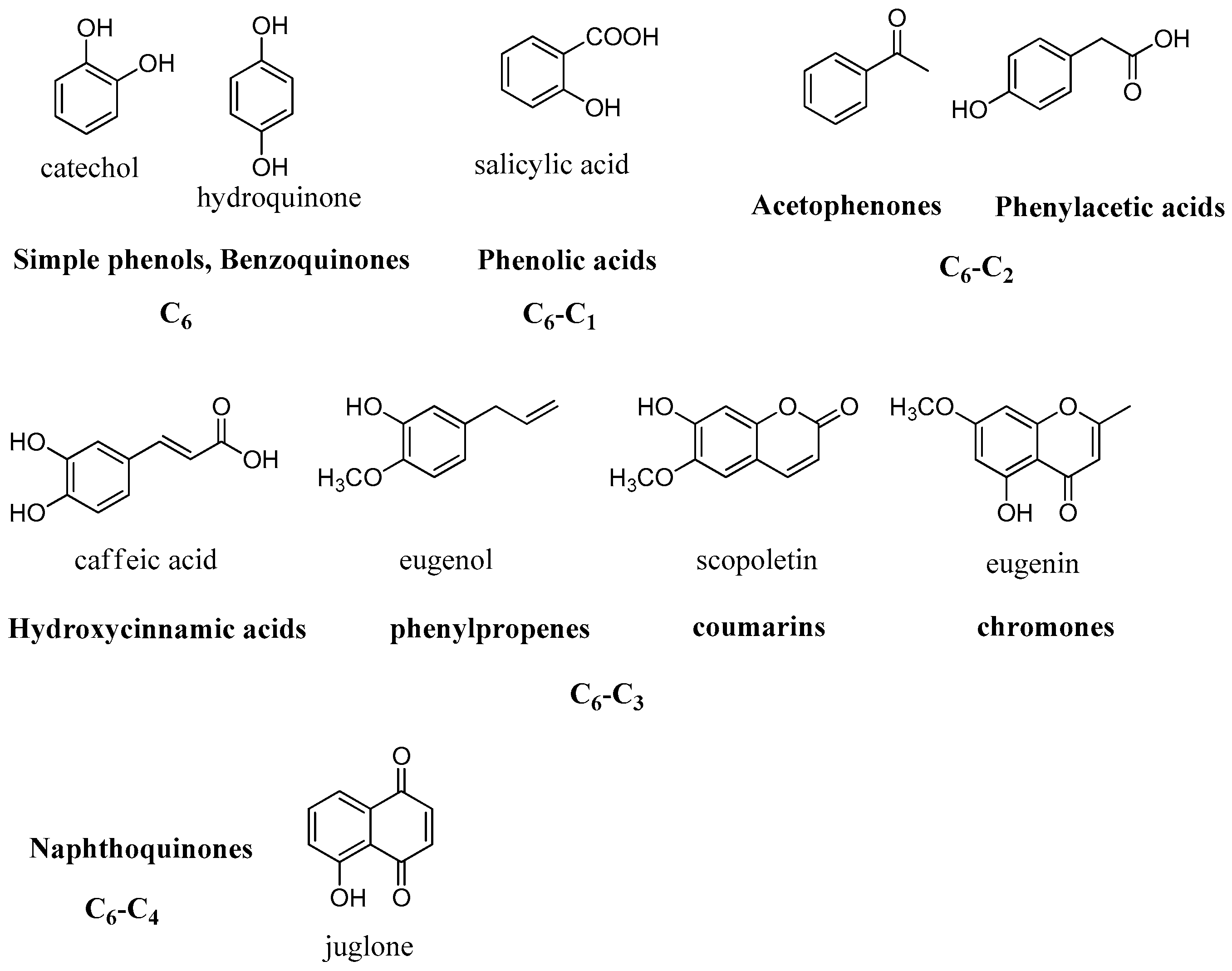
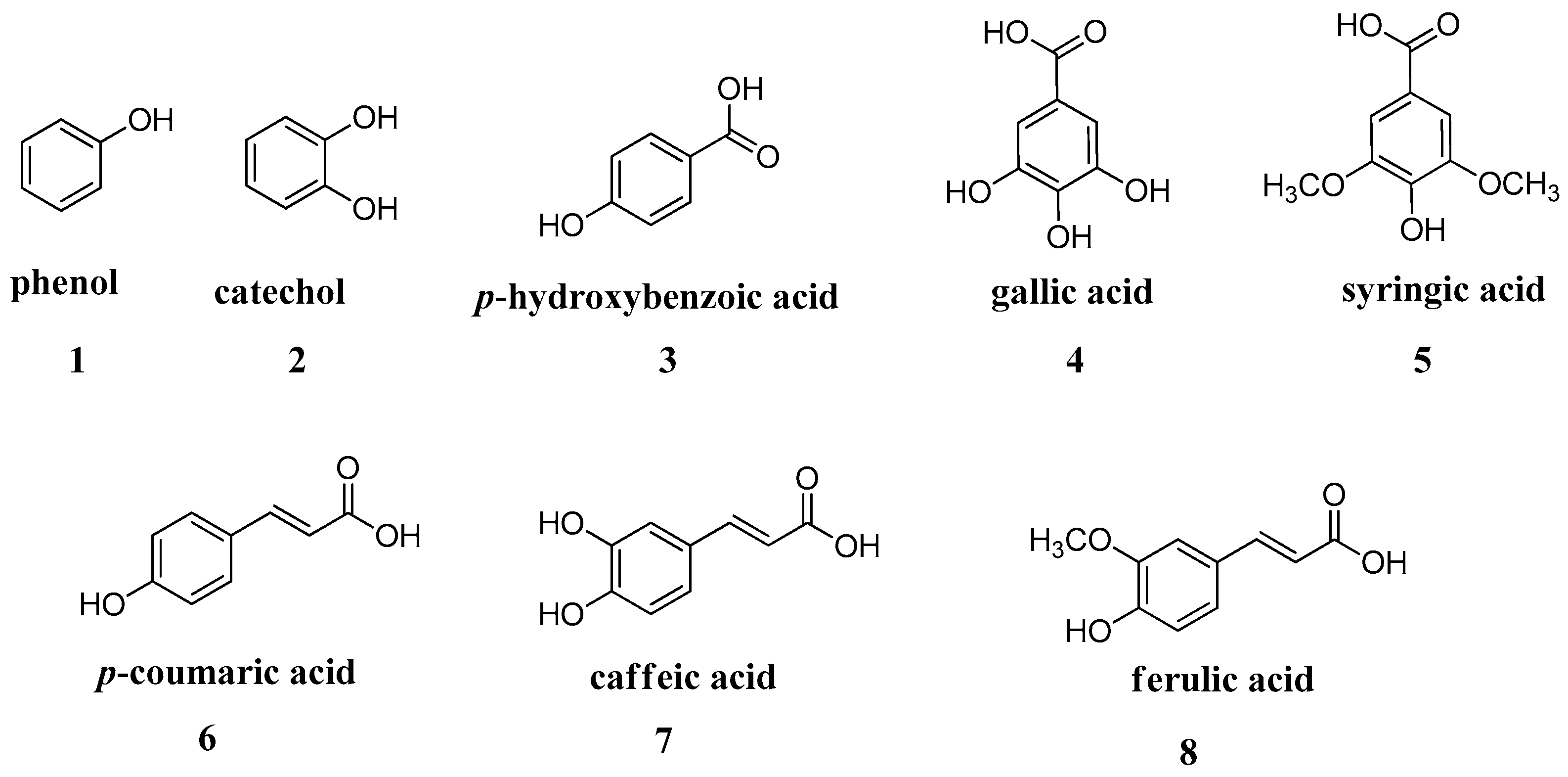
2.2. Simple Phenols
These findings opened the way for the exploration of other phenolic and polyphenolic natural products. In vitro kinetic studies of a series of phenols widely available on the market such as salicylic acid, resorcinol,
p-coumaric, caffeic, ferulic, gallic acids, etc., showed that these are affective CA inhibitors in the low micromolar range (
Figure 6) [
45,
46].
The presence of free hydroxyl groups at position 4’ in combination with the double bond in the phenylpropanoid skeleton, as in
p-coumaric acid, clearly enhanced the inhibitory activity when compared to the simple phenol moiety. Similar results were obtained in a further study with a small group of monophenolic constituents, where
trans-cinnamic (
14),
o-coumaric (
15) and chlorogenic (
16) acids were more effective than simple phenolic constituents such as benzoic acid (
9) [
45] (
Figure 7 and
Table 1).
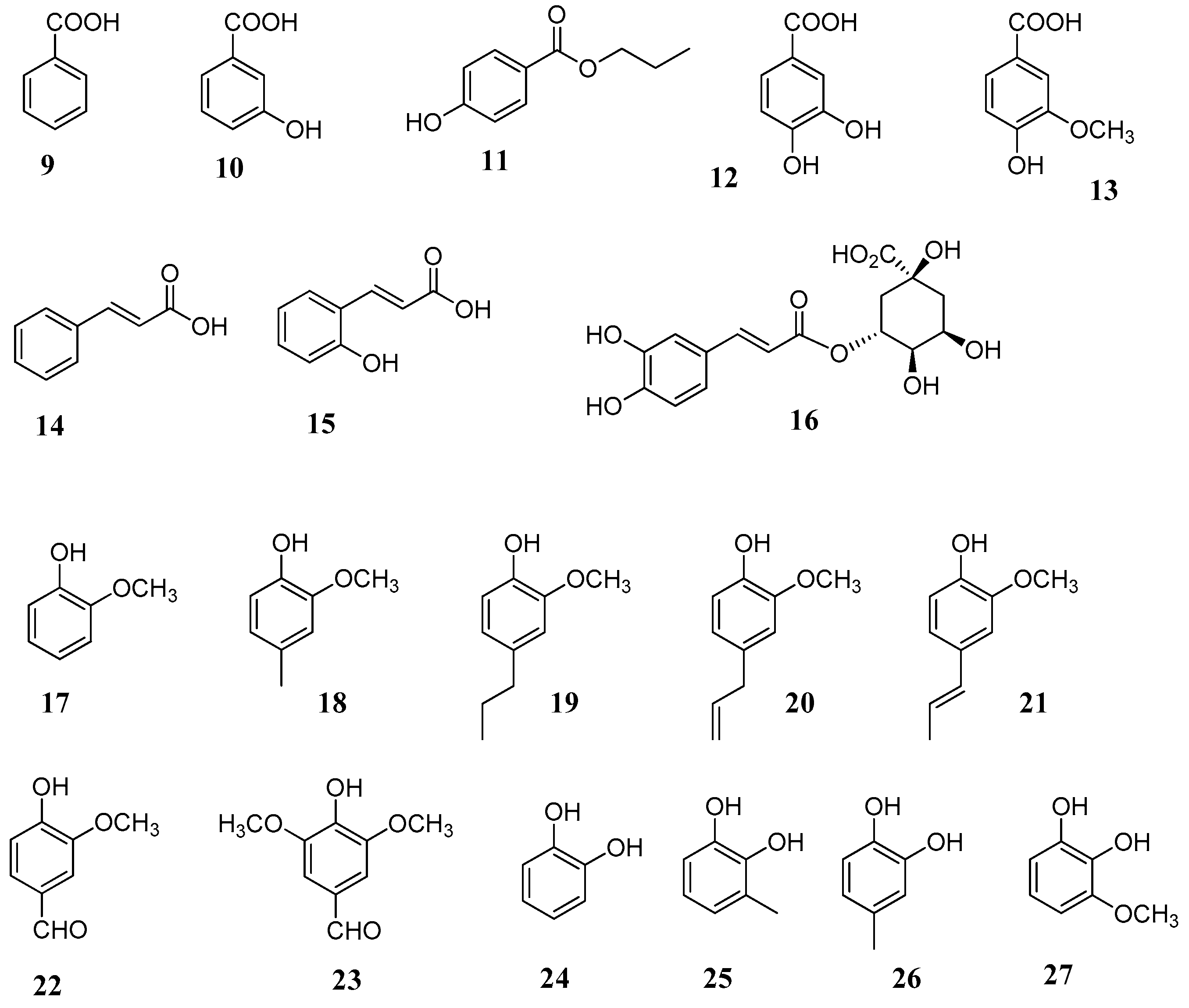
In another study several derivatives of guaiacol and catechol (
17–
27) were considered for their CA inhibitory activity against human CA I, II, IX and XII isoforms [
49]. Among the compounds tested in vitro were the volatile constituents eugenol (
20), isoeugenol (
21) and vanillin (
22), which are major components of many aromatic herbs and spices such as clove, basil, anise seed,
Vanilla sp., etc. All compounds showed micromolar inhibition potencies spanning between 2.2–10.92 μM with the exception of the catechol (
Table 2). Among the tested phenolic derivatives, compounds 4-methyl-catechol (
26) and 3-methoxycatechol (
27) showed potent activity as inhibitors of the tumour-associated transmembrane isoforms hCA IX and XII with K
I values in the submicromolar range.
Combination of the phenolic moiety with other functional groups is a common strategy to obtain products with enhanced activity. For example, in the so-called “sugar approach” the incorporation of a hydrophilic moiety of a sugar permits to the molecule to anchor in a different manner to the enzyme cavity. Furthermore, it increases its selectivity towards the membrane bound CA isoforms over the cytosolic ones [
50,
51]. This is desirable since selectivity against specific hCA isoforms might reduce side effects.
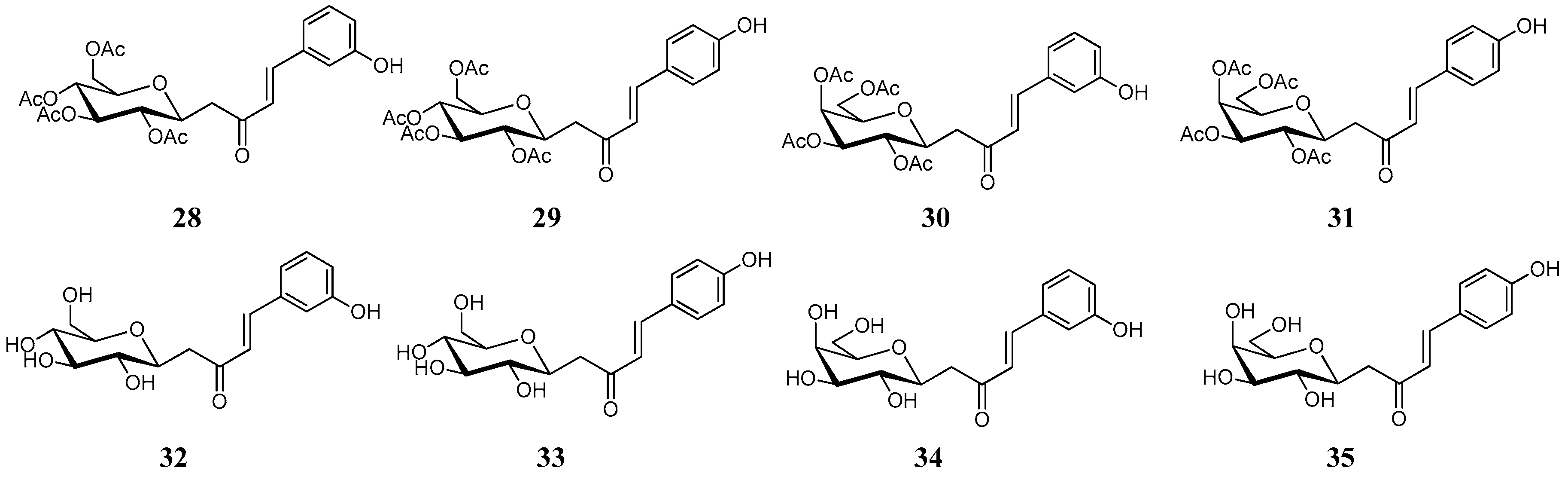
In this concept, a series of synthetic
C-cinnamoyl glycosides were synthetized and considered for the inhibition on twelve mammalian isoforms of carbonic anhydrase (
Figure 8) [
52]. These compounds combined two functional groups with known activity against the hCAs: the cinnamoyl and the glucose group. In order to avoid undesirable degradation, the
O-glycosidic bond was replaced by the more stable
C-glycosidic one. The
C-cinnamoyl glycosides (
28–
35) were generally effective CAs, with inhibition constants in the low micromolar range against CA I, II, IV, VA, VB, VI, VII, IX, XII, XIII, XIV and ineffective inhibitors of CA III (
Table 3).
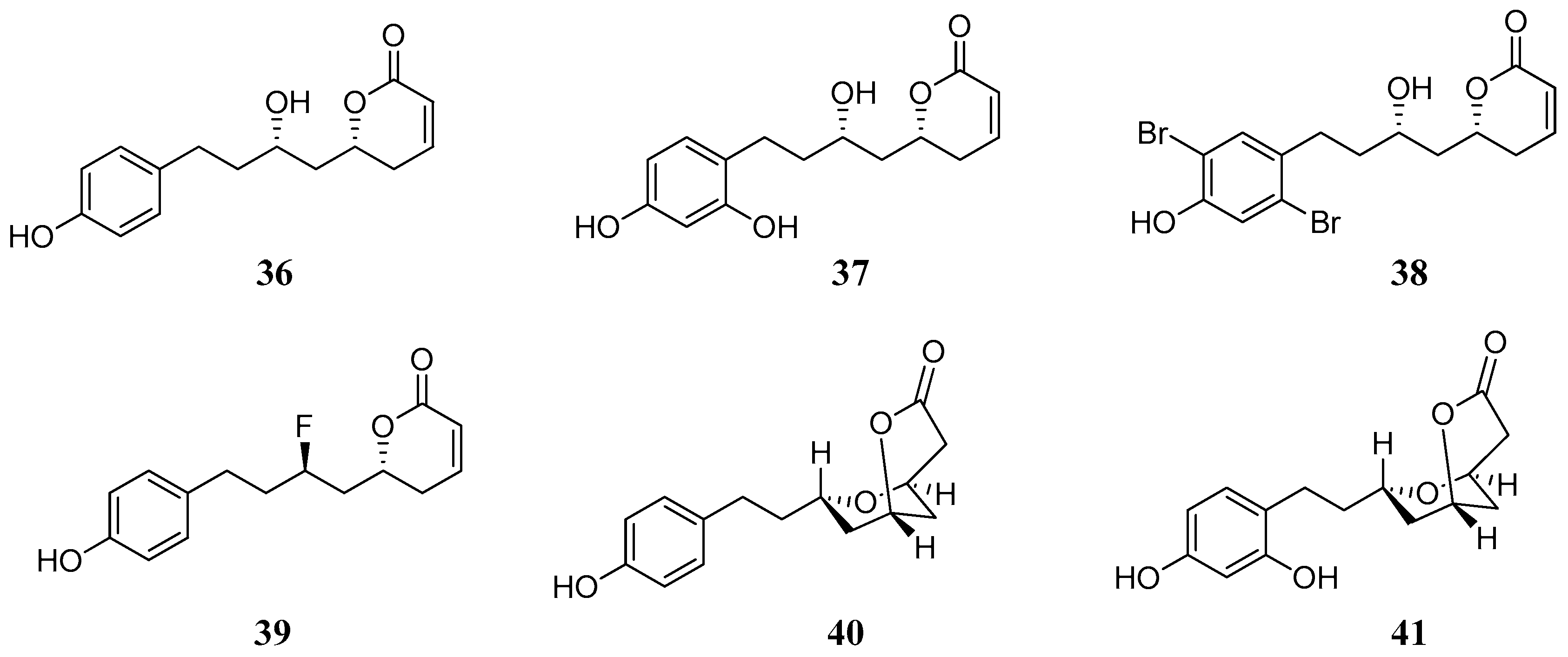
Another hybrid molecule recently investigated against several hCA isozymes [
53] is Dodoneine
36, one of the active principles of the medicinal plant
Agelanthus dodoneifolius (DC) Polhilland Wiens. The plant is used in traditional medicine for the treatment of hypertension. Structurally
36 incorporates both the phenol and lactone (α-pyrone) moieties. Dodoneine showed to be selective in inhibiting the hCA isoforms I, III, IV, XIII and XIV with K
Is in the range of 5.5–10.4 µM. CA I, III and XIII are cytosolic, CA IV is localized in plasma membrane and hCA XIV is a membrane bounded isozyme. Further in vitro and in vivo studies in rat aorta and vascular smooth muscle cells showed that hCA II, III, XIII and XIV were expressed in rat aorta while only the hCA isoforms III, XIII and XIV were expressed in smooth muscle cells. Thus it is reasonable to assume that dodoneine induced vasorelaxation by a dual mode of action including both block of the L-type calcium channels and inhibition of the hCA isoforms therein expressed [
54]. A series of synthetic analogues of dodoneine (
37–
41), reported in
Figure 9, were prepared and tested for their inhibition properties against the hCAs. Among them the bicyclic compound
40 which is normally found in the plant in small amounts and its hydroxylated derivative
41 and the fluorinated analogue
39 (
Table 4). Compound
39 inhibited all hCA isoforms, while
41 inhibited selectively hCA III (K
I = 5.1 nM) and mCA XIII (K
I = 340 nM).
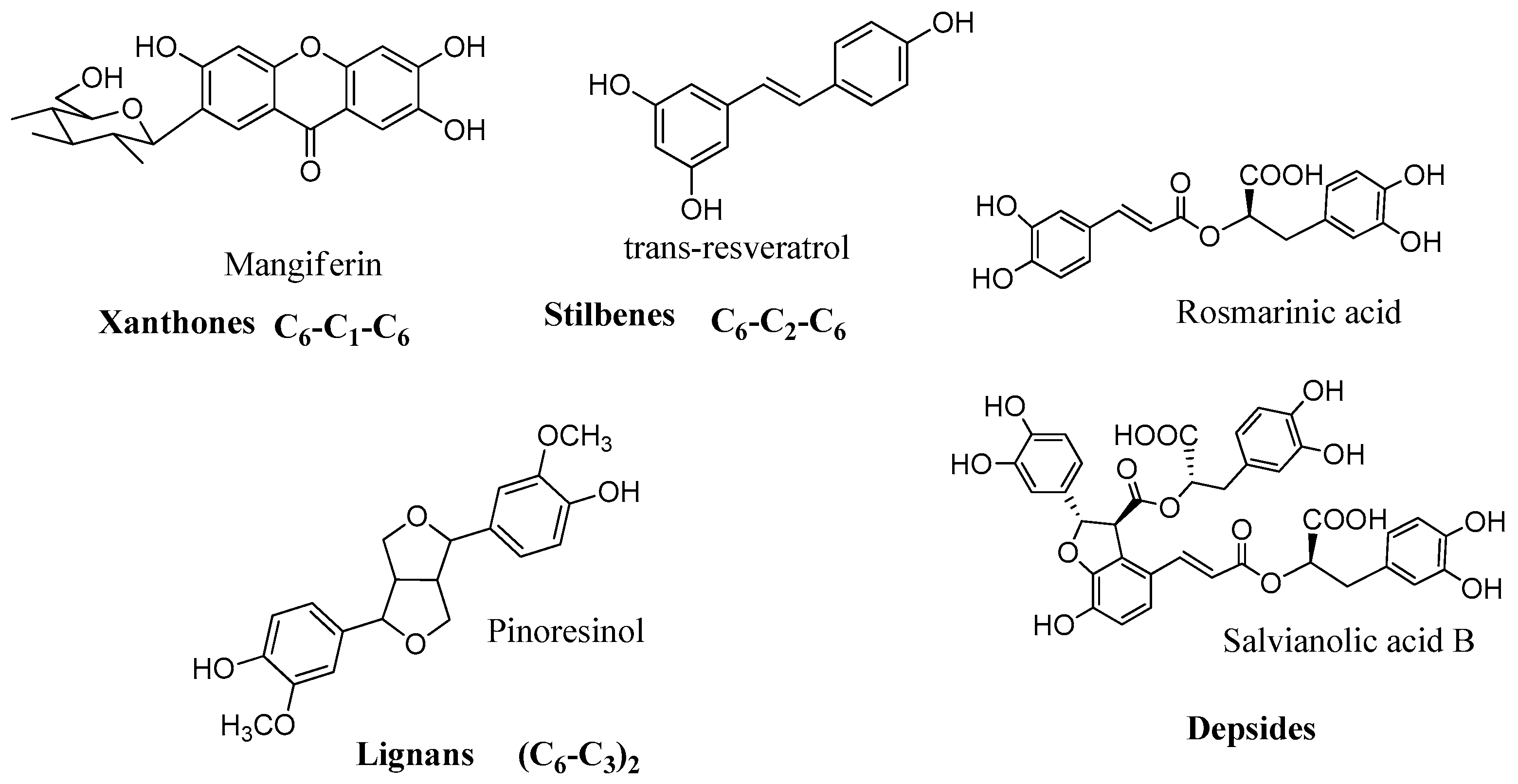
2.3. Polyphenols
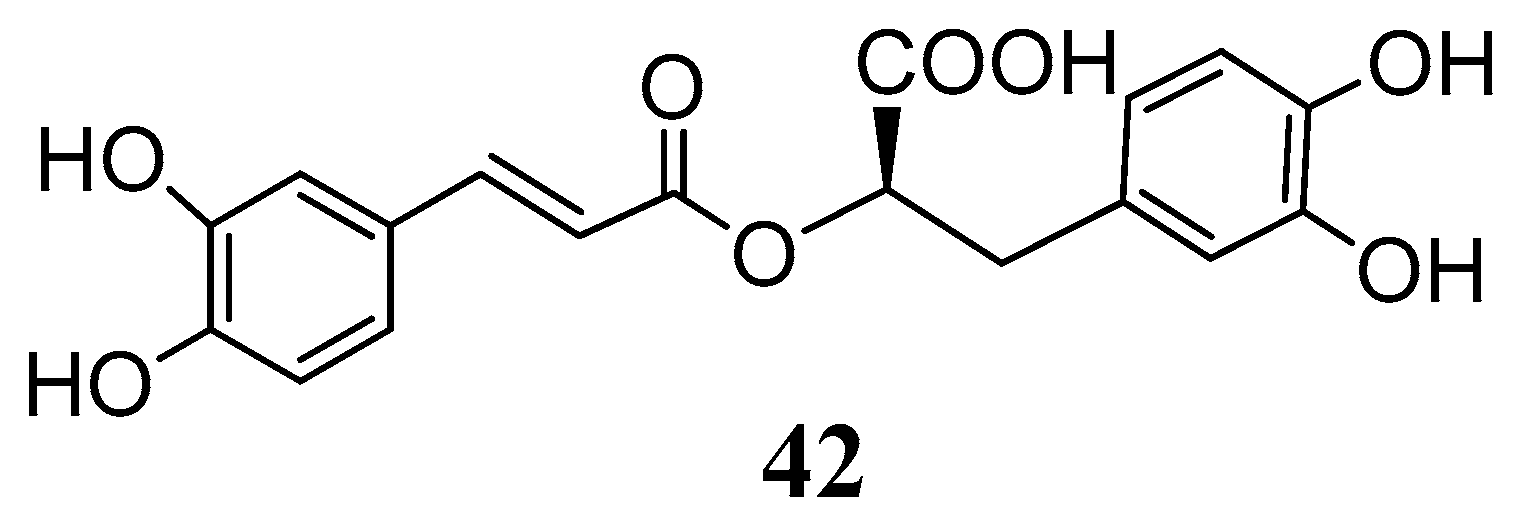
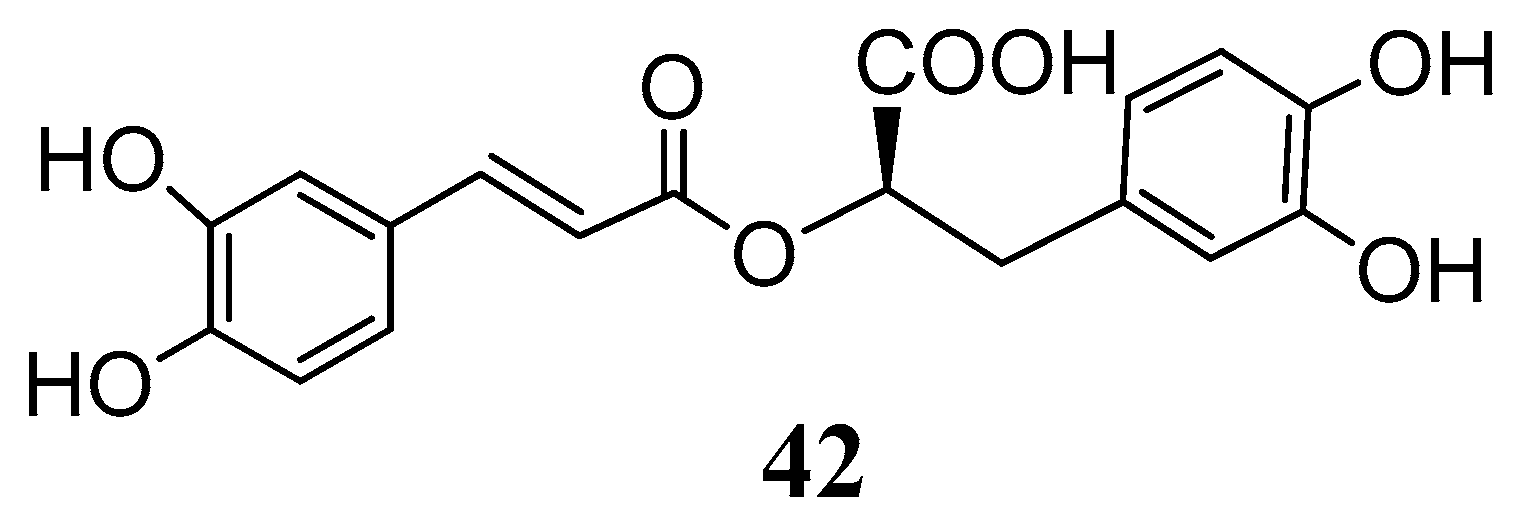
Recently rosmarinic acid (
42) was reported to be a potent inhibitor of the hCA I and II isoenzymes at low micromolar range with K
I values of 86.0 µM and 57.0 µM, respectively (
Figure 10) [
55]. Rosmarinic acid is a powerful antioxidant found in many aromatic, medicinal and culinary herbs of the Lamiaceae family, such as rosemary, sage, oregano and thyme [
56]. Chemically it belongs to the class of depsides, or the so-called labiataetannins [
41], which are obtained by condensation of caffeic acid units or by combination of caffeic acid with 3,4-dihydroxyphenyl lactic acid, to form dimers, trimers and tetramers.
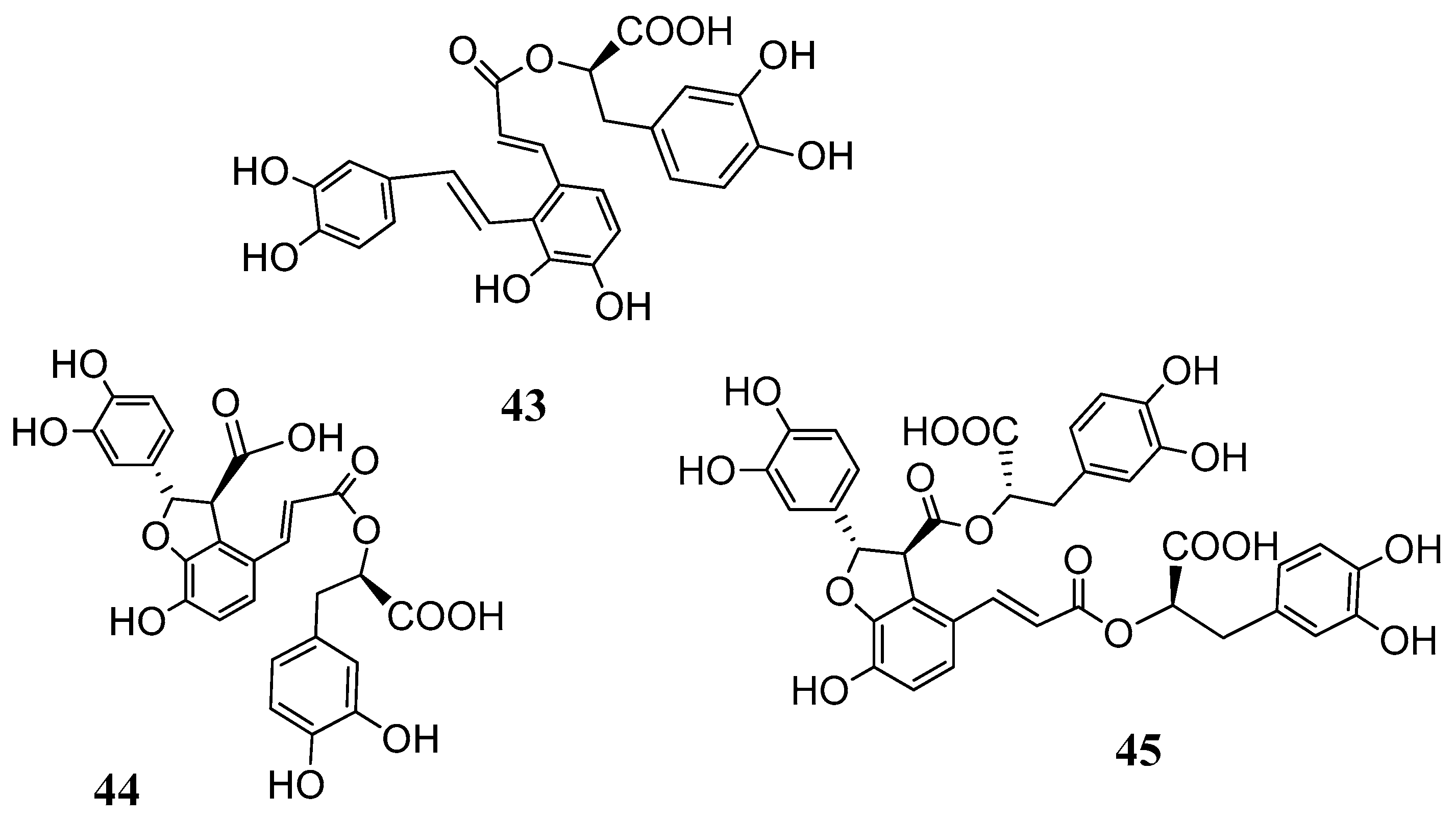
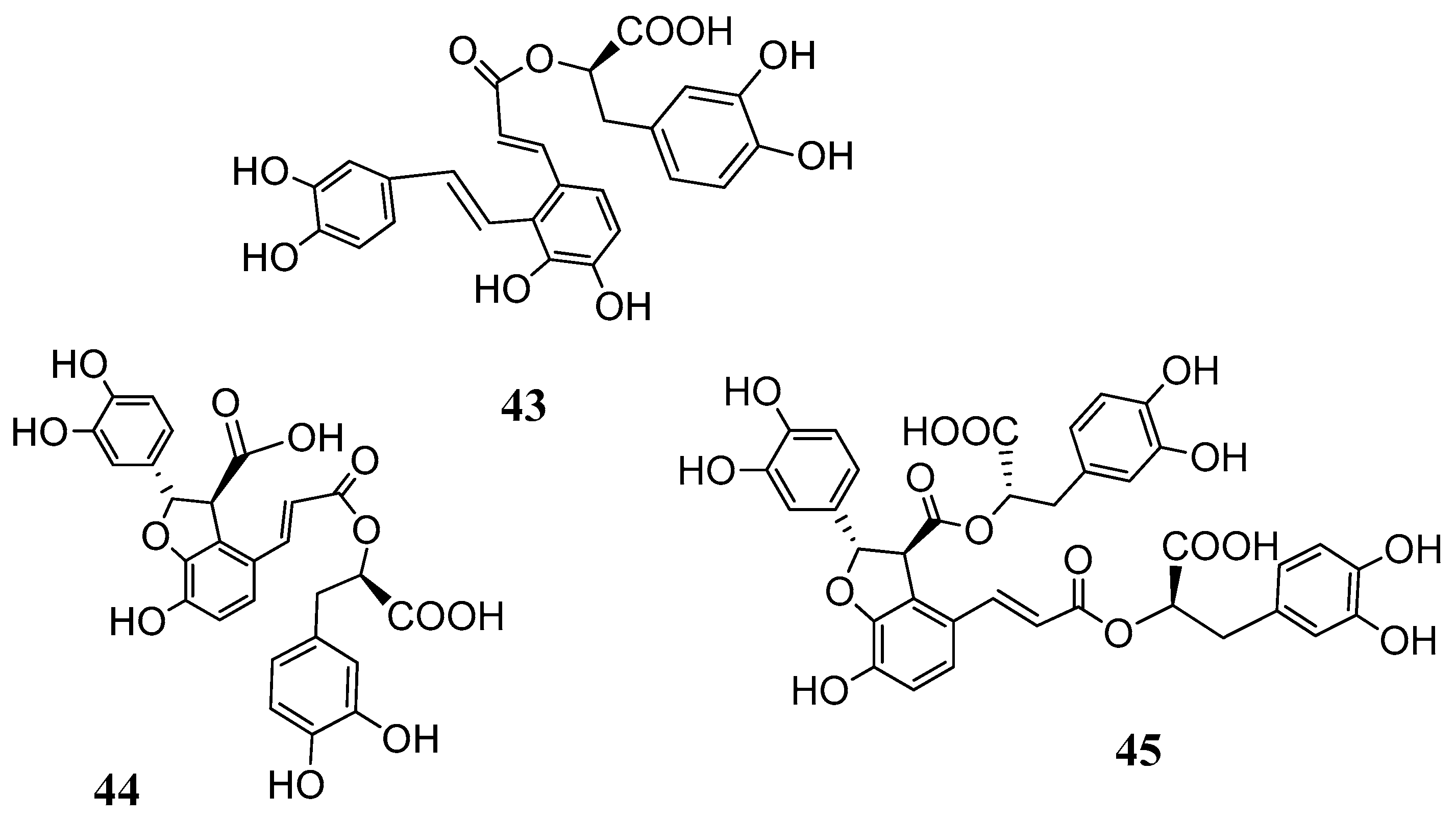
Based on these results further investigations were carried out with the salvianolic acids
43–
45 (
Figure 11) isolated from
Salvia miltiorrhiza (Lamiaceae) [
57,
58].
Salvia miltiorrhiza or Danshen is one of the most important herbal medicinal drugs in the Chinese Traditional Medicines (TCM) to treat bleeding disorders (e.g., menstrual bleeding) and blood stasis [
59]. Salvianolic acids are together with tanshinones (lipophilic, diterpenic quinones) the main active principles of the herbal drug. These hydrophilic derivatives, abundant in the roots of the plant, incorporate three to four units of caffeic/3,4-dihydroxyphenyl lactic acids. They have attracted interest due to their protective roles in CNS neuronal injuries and degeneration. The main mechanism involved in such processes is the decrease of reactive oxygen species (ROS) levels. Furthermore, there is evidence of the protective role of salvianolic acids in several cardiovascular diseases, osteoporosis, liver fibrosis and hepatic failure [
59,
60,
61].
The CAs inhibition studies reported in
Table 5 clearly showed that depsides
43–
45 were ineffective hCA I and weak hCA II inhibitors. On the other hand, they were inhibitors of the hCA IV with K
I values in the range of 62.2–102.1 nM and comparable to the standard drug acetazolamide
AAZ (74.0 nM). Even if a proper structure–activity-relationship (SAR) consideration is difficult to be drawn it is noteworthy that the flexible and more containing catechol moieties salvianolic acids A and B (
43 and
45) were better inhibitors when compared to the lithospermic acid
44. Furthermore, salvianolic acids A and B were medium and high nanomolar inhibitors (K
I 39.8 and 453.6 nM of the tumor associated CA XII. By contrast, lithospermic acid (
44) showed to be a very potent hCA XII inhibitor with a K
I value of 4.8 nM. The presence of ortho-hydroxyl moieties in these molecules, the possibility to release in situ caffeic acid or even the presence of free carboxyl groups are some of the structural characteristics that play an important role in the CA inhibitory activity. In any case such results put in evidence a new class of natural products which deserves further investigation in order to decipher a proper SAR correlation.
Another recent example is constituted by a series of phenolic acid esters (
Scheme 1) incorporating caffeic, ferulic, and
p-coumaric acid, and benzyl,
m/p-hydroxyphenethyl- as well as
p-hydroxy-phenethoxy-phenethyl moieties, which were investigated for their inhibitory effects against the mammalian isozymes of human (h) or murine (m) origin, hCA I–hCA XII, mCA XIII and hCA XIV [
62]. These enzymes were inhibited in the submicromolar range by many of these derivatives (with K
Is of 0.31–1.03 μM against hCA VA, VB, VI, VII, IX and XIV). The off-target, highly abundant isoforms hCA I and II, as well as hCA III, IV and XII were poorly inhibited by many of these esters, although the original phenolic acids were micromolar inhibitors. These phenols, like others investigated earlier, possess a CA inhibition mechanism distinct of the sulfonamides/sulfamates, clinically used drugs for the treatment of a multitude of pathologies, but with severe side effects due to hCA I/II inhibition.
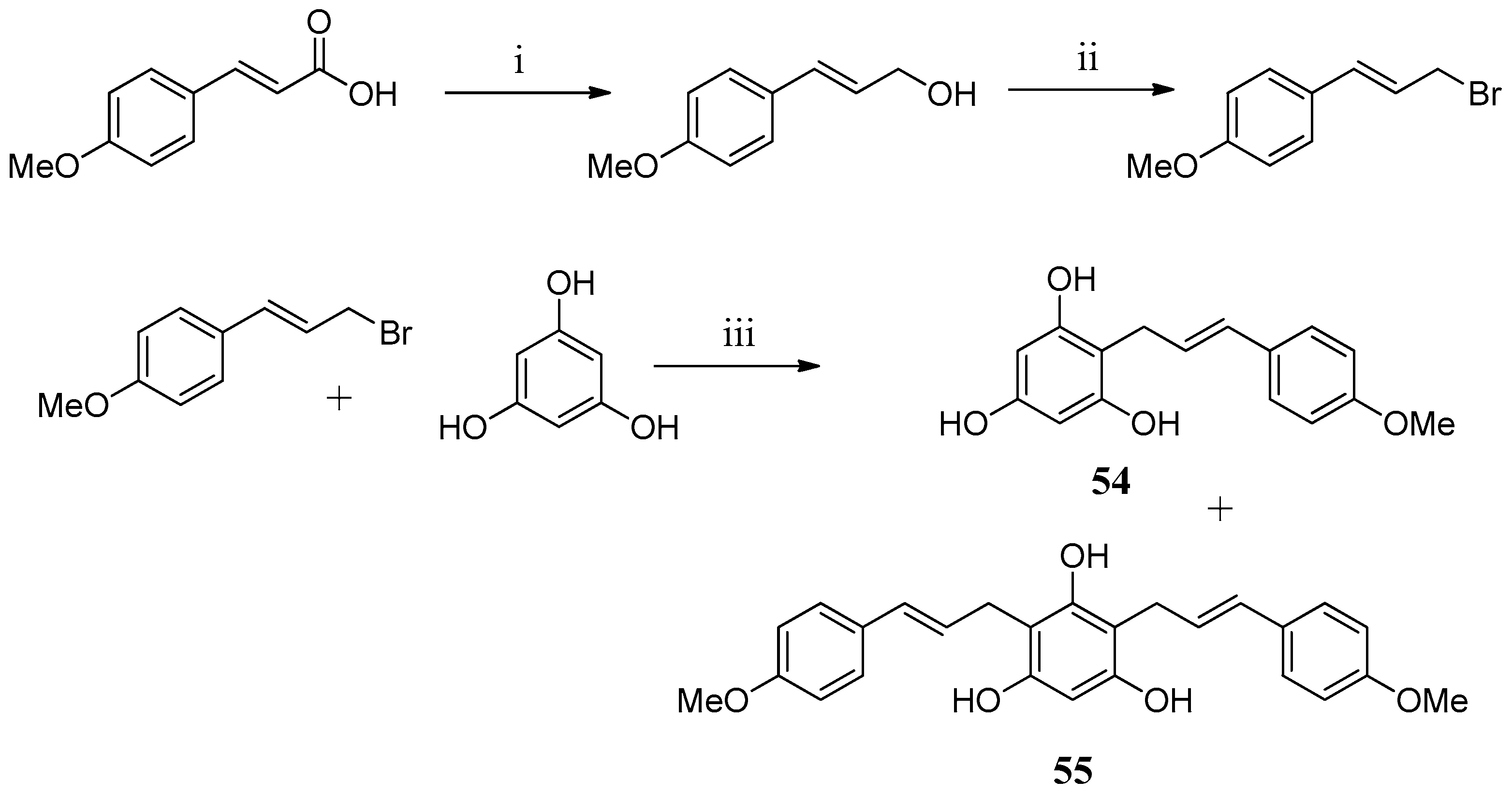
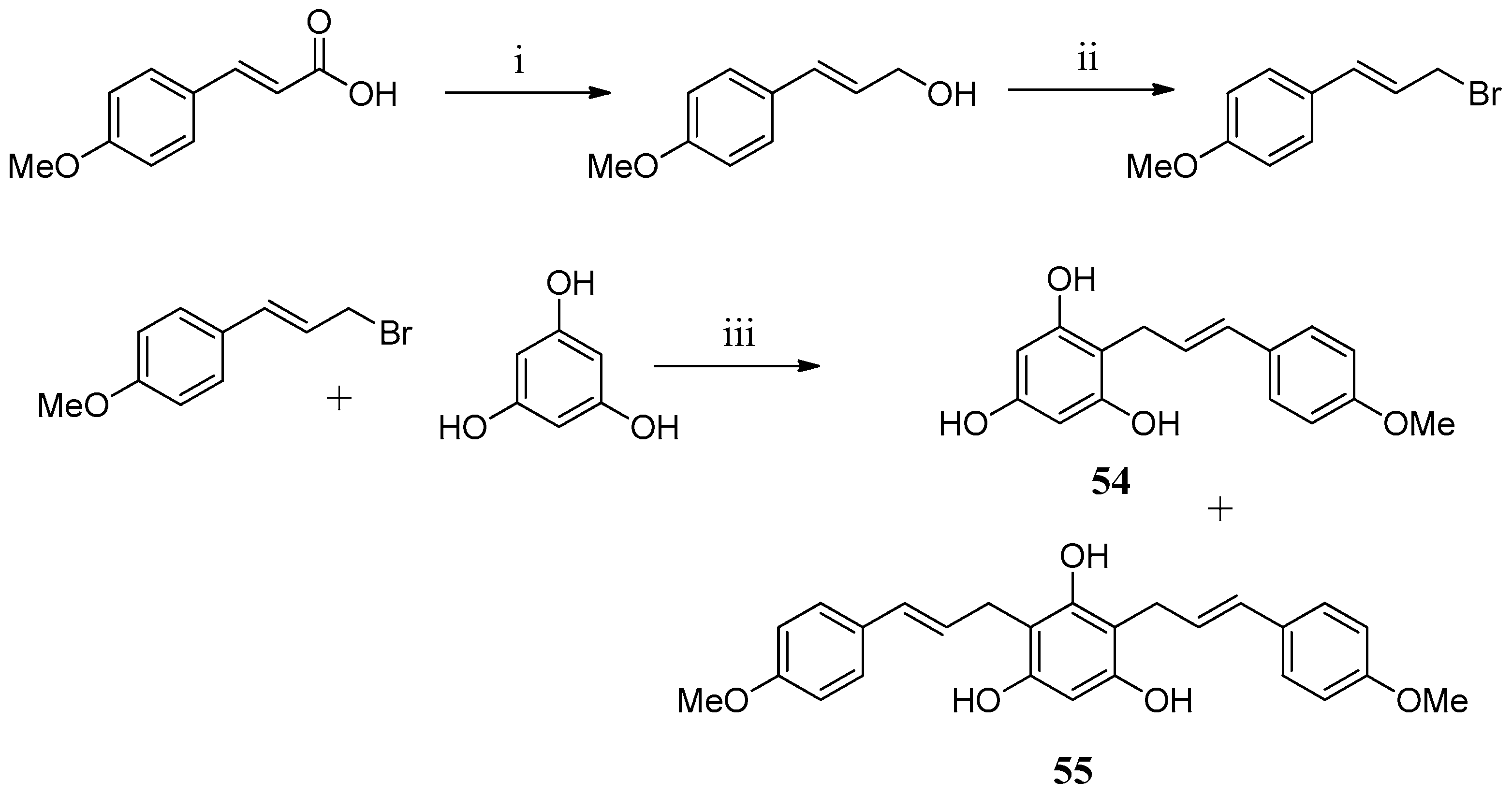
In an attempt to find new chemotypes, two floroglucinol derivatives
54,
55 were prepared and tested against hCA I and hCA II (
Scheme 2). Unfortunately, none of the compounds proved to be active [
63].
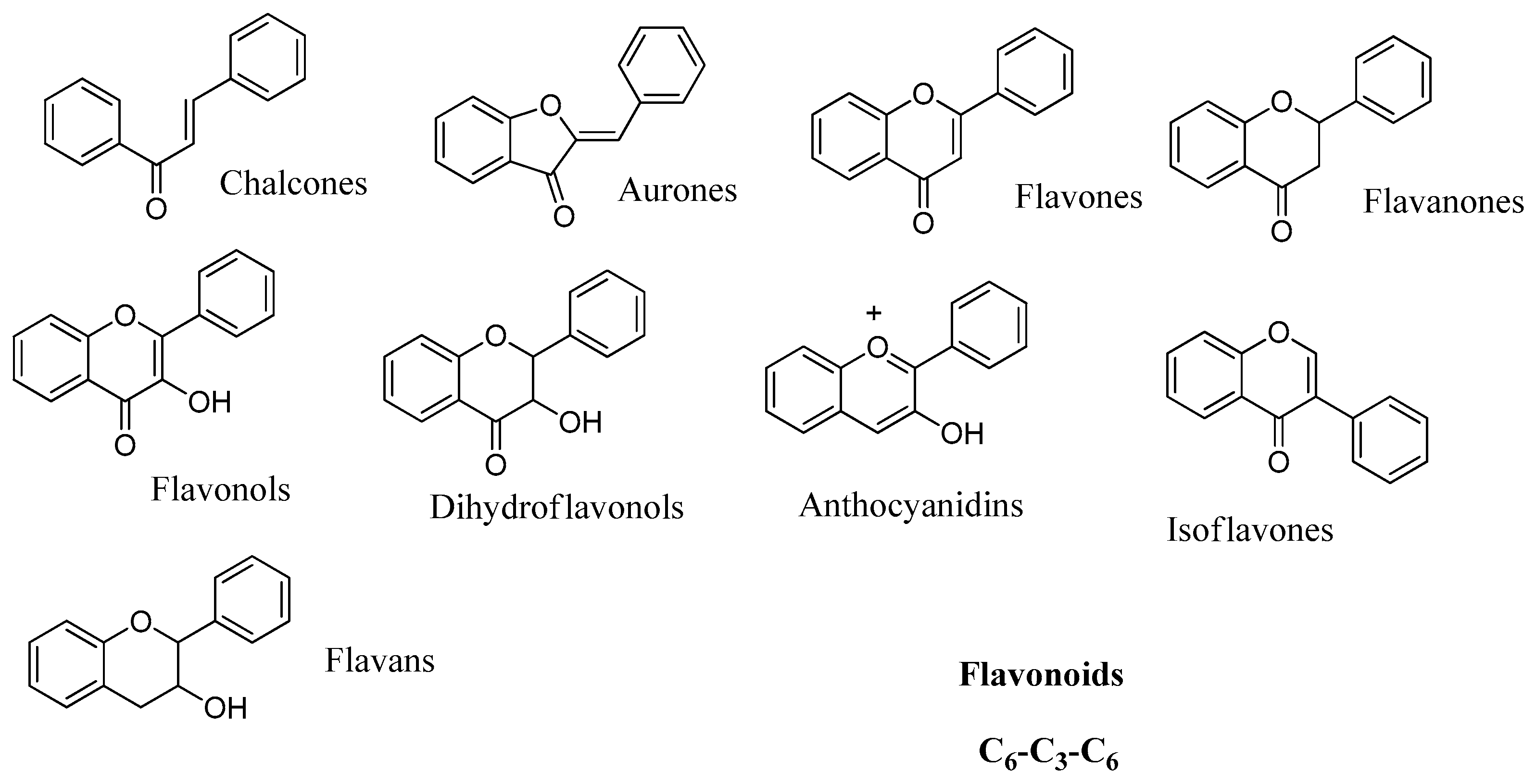
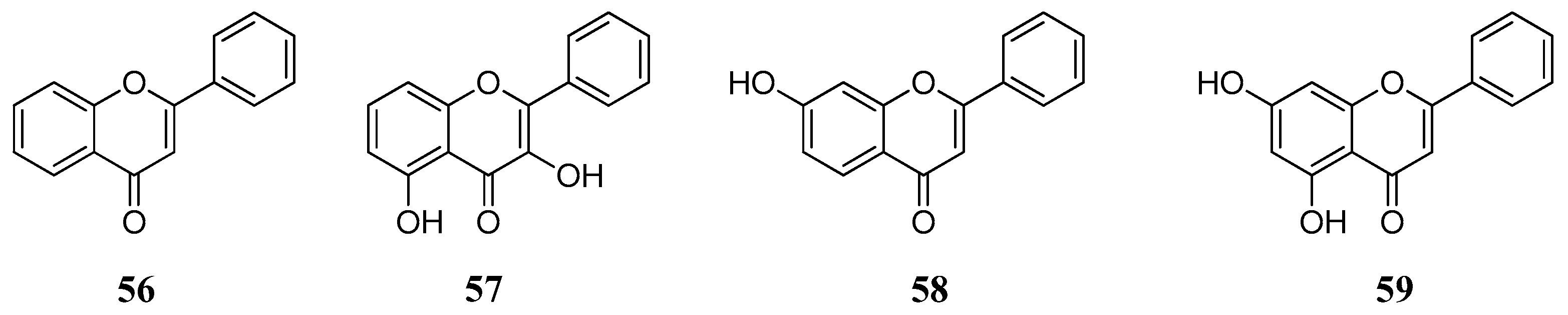
2.4. Flavonoids
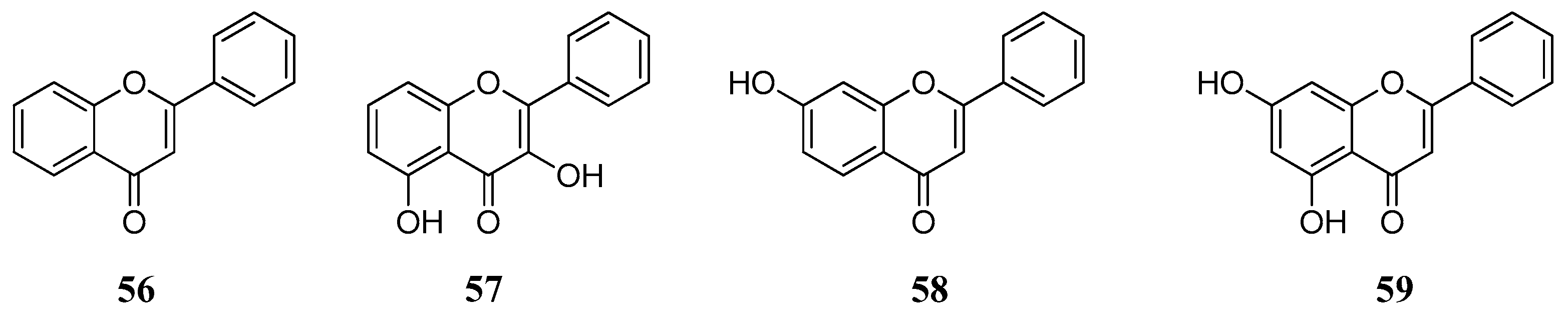
Data of the interaction of flavonoids with CAs indicate that these may be another category of CAIs of interest, which can be used as leads for generating more potent CAIs. Preliminary test of a small number of basic flavonoid structures
56–
59 (
Figure 12) against hCA I, II, IX and XII showed that these act as low micromolar inhibitors and their effect is independent of the incubation time [
64]. This fact indicates that flavonoids inhibit CAs probably through a mechanism similar to the one of phenol rather than hydrolysis of the chromenone as in the case of coumarins.
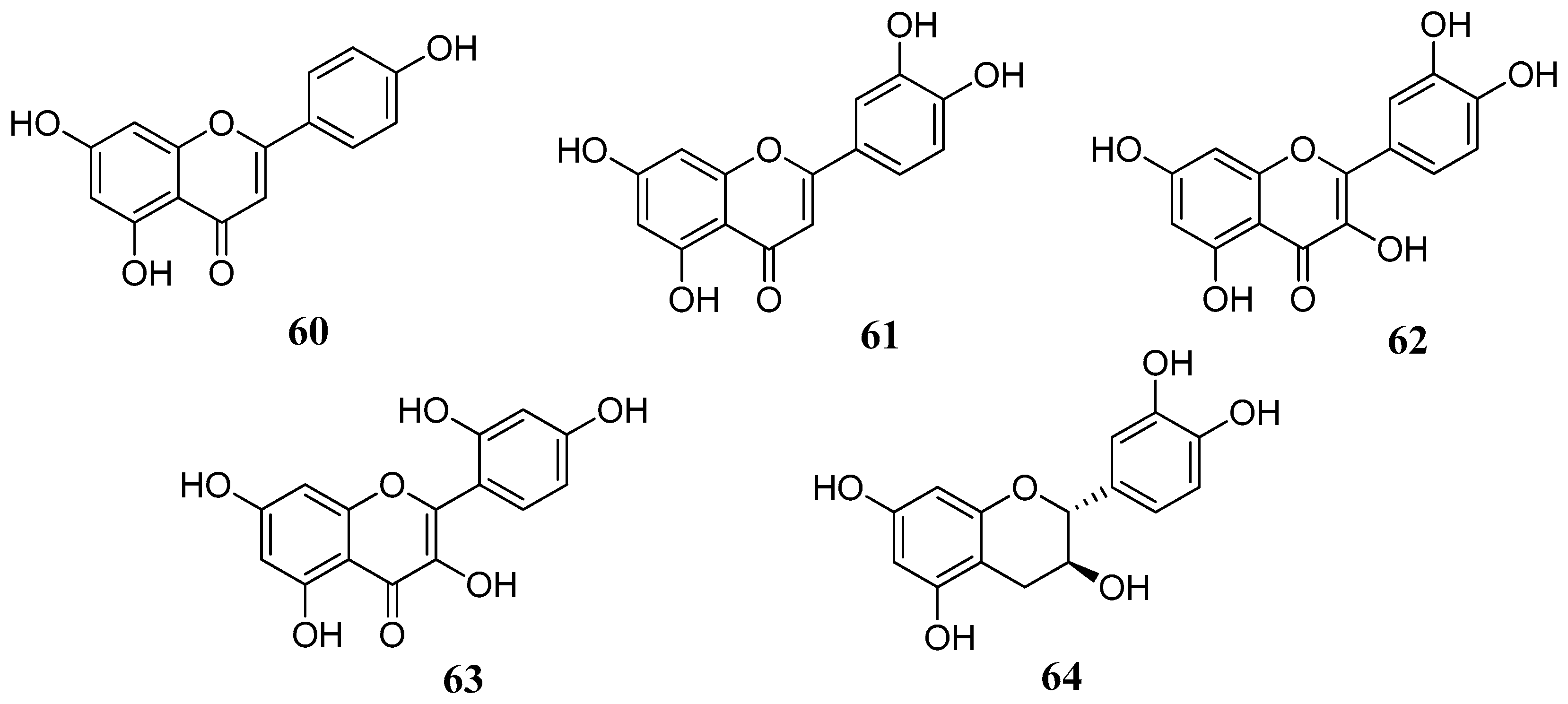
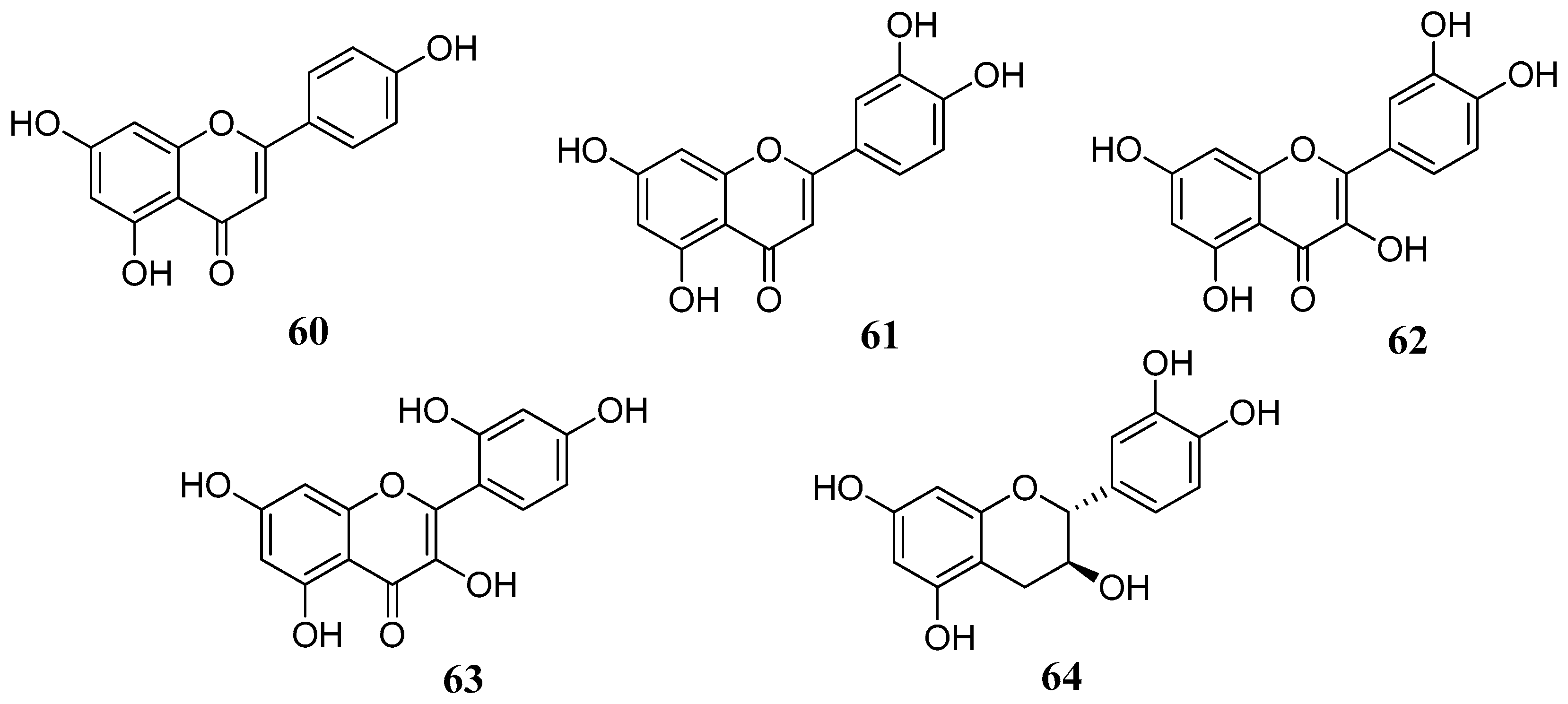
This hypothesis was further confirmed in several studies including flavonoids lacking the C-4 carbonyl group. A series of flavonoids
60–
64 with different substitution patterns were investigated for their inhibitory effects against four α-CA isozymes from human and bovine (hCA I, hCA II, bCA III, hCA IV) tissues: the flavones apigenin (
60) and luteolin (
61), the flavonols quercetin (
62) and morin (
63) and the flavan-3-ol, catechin (
64) (
Figure 13) [
65]. As it can be observed from
Table 6, flavonoids
60–
64 were quite effective, similar to the standards used. The four hCA isozymes showed quite diverse inhibition profiles with these compounds suggesting that the type of flavonoid and/or substitution pattern are important for the activity. As briefly outlined above, the C-4 carbonyl group is not so important for the inhibition, as shown by the example of catechin (
64). This is another indication that the inhibition mechanism does not proceed via hydrolysis (CA esterase activity). Instead, the presence of one or two neighboring hydroxyl groups is essential, as shown by the example of morin
67 which is the less potent compound compared to quercetin
62 or luteolin
61. The latter,
61 showed the most effective inhibitory activity with K
Is in the low micromolar range.
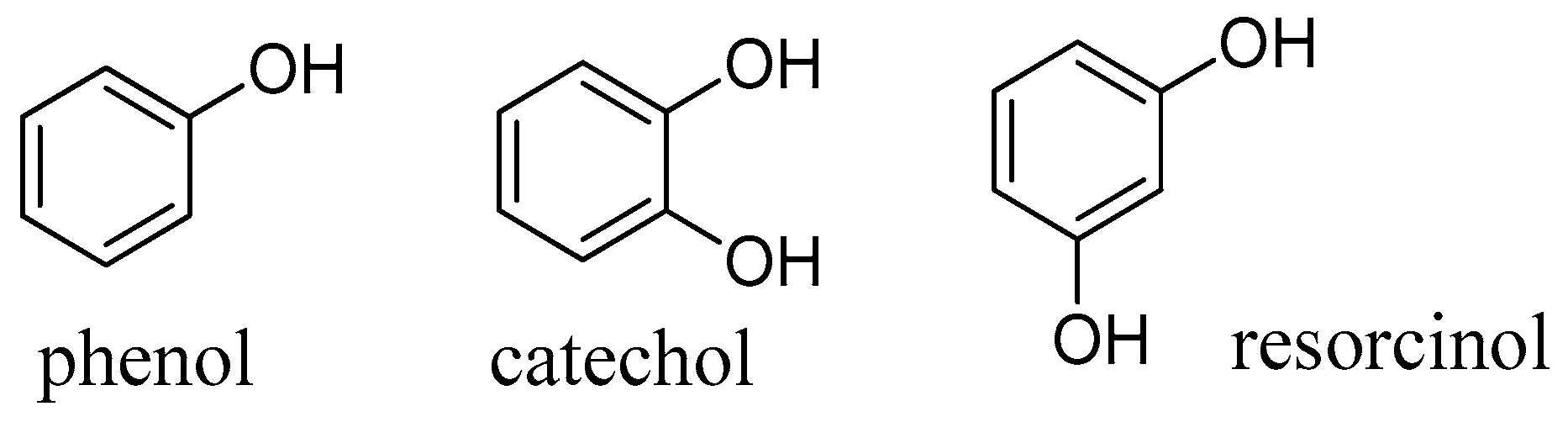
Comparison with the relative monophenols, catechol, phenol and resorcinol (
Figure 14), which were considerably less active, suggests that also the ring A of flavonoids may be involved in the inhibitory mechanism. Whether this is due to a different mechanism or simply stronger binding to the enzyme cavity (e.g. by further hydrogen bonds) is to be investigated in the future.
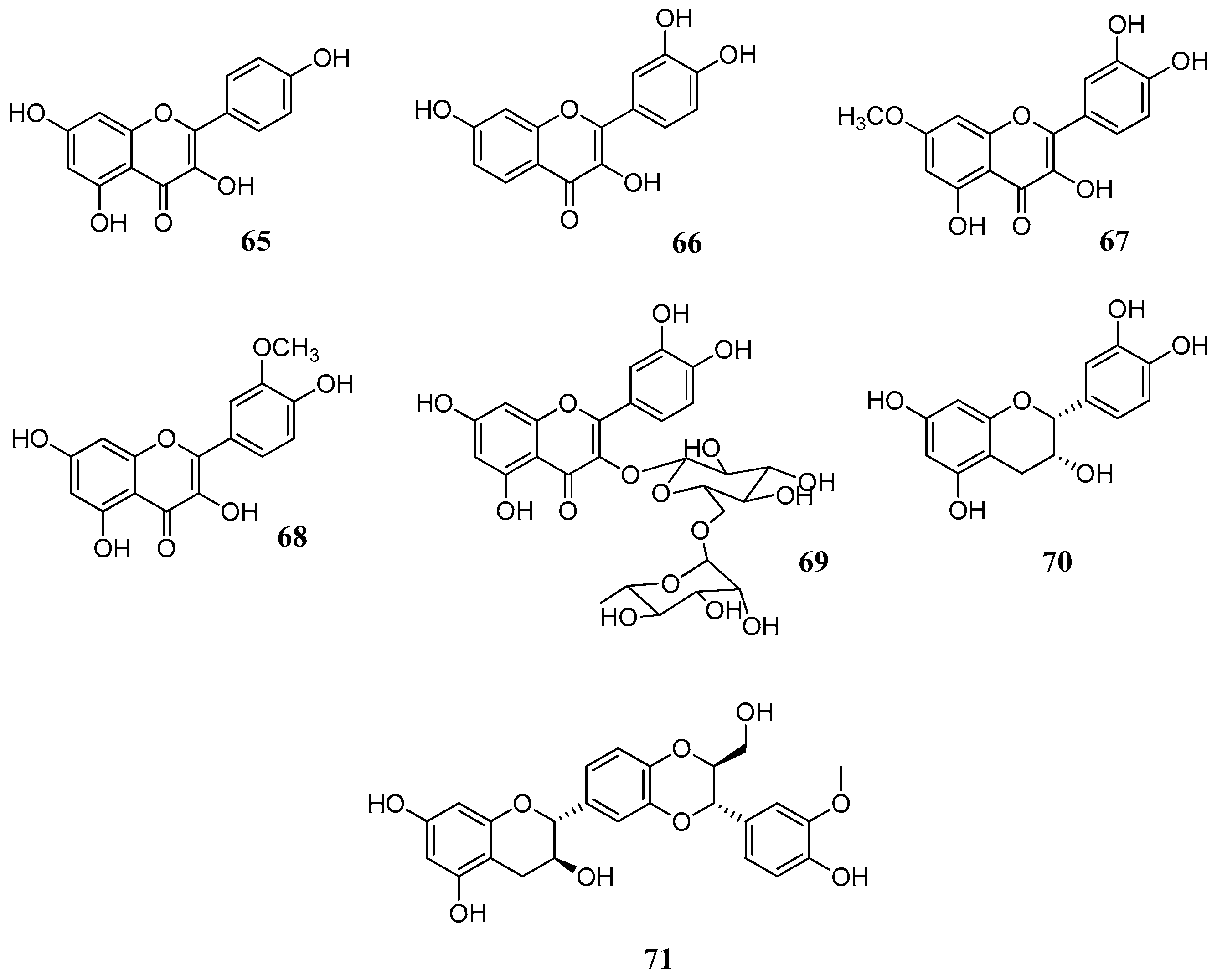
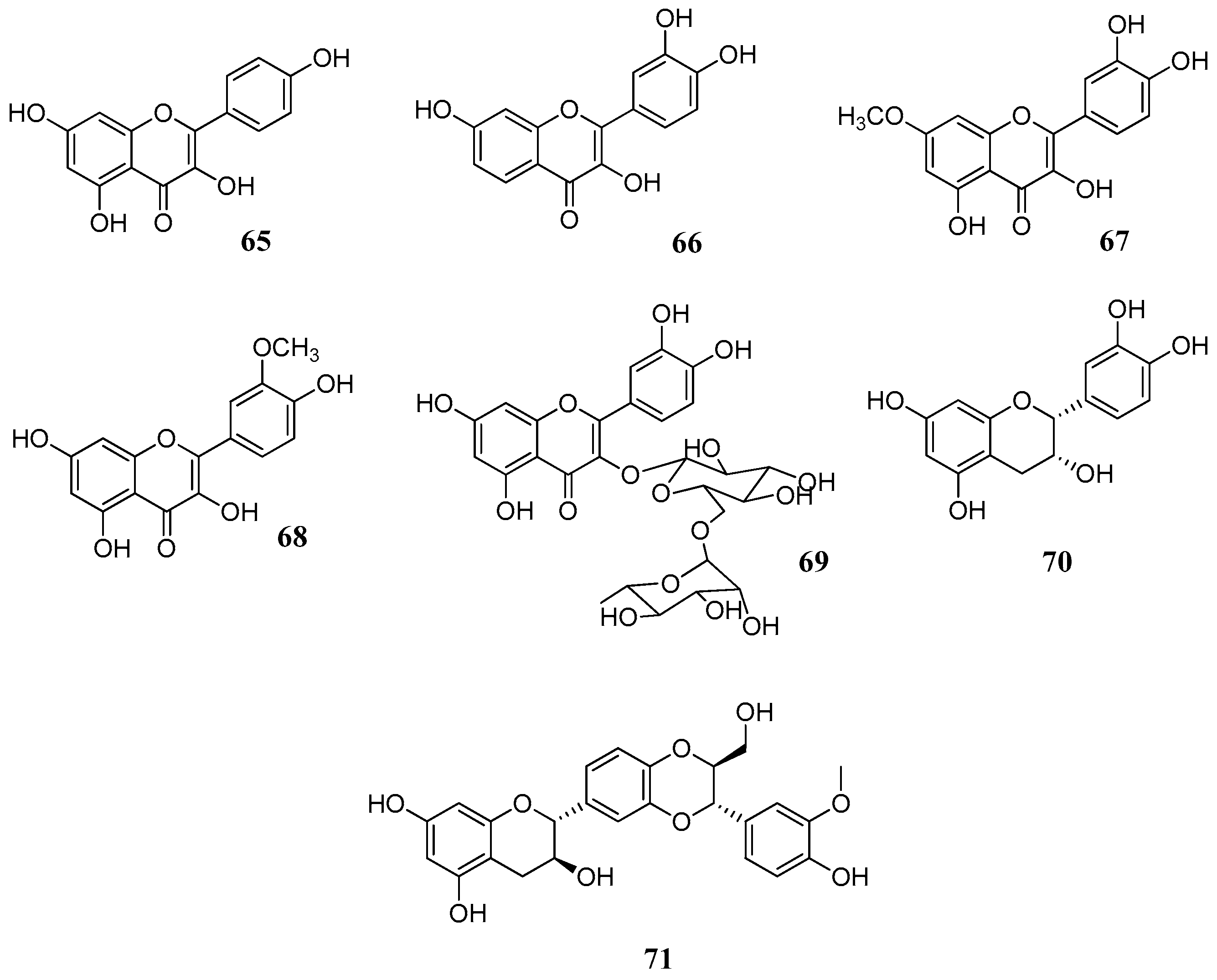
Two further studies [
46,
67] with several flavonoids
60,
62,
64–
71 (
Figure 15,
Table 7 and
Table 8) against human CA I, II, IV, VI and bovine CA III isoforms gave further evidence that the catechol moiety on C-2 enhances the activity, as demonstrated in the case of fisetin (
66), rhamnetin (
67) and rutin (
69). Although a comparison between results of
Table 7,
Table 8 and
Table 9 is not feasible due to the different enzymatic assays used and to different enzyme purities, it can be observed that catechin (
64) and epicatechin (
70) have similar activities despite different stereochemistry on C-3. Similarly, the hydroxyl group of C-3 seems to be of little importance for the activity. This is evident if we compare luteolin (
61) with quercetin (
62) (
Table 6) and apigenin (
60) with kaempferol (
65) (
Table 7). Therefore, the keto-enol system apparently does not take part in the inhibition mechanism. The better inhibitory activity of fisetin (
66) and rhamnetin (
67) should be attributed to structural differences in ring A. Interestingly, rutin (
69) compared to flavonol aglycones such as quercetin (
62) or rhamnetin (
67) is more active suggesting that sugars may play an important role, as discussed previously.
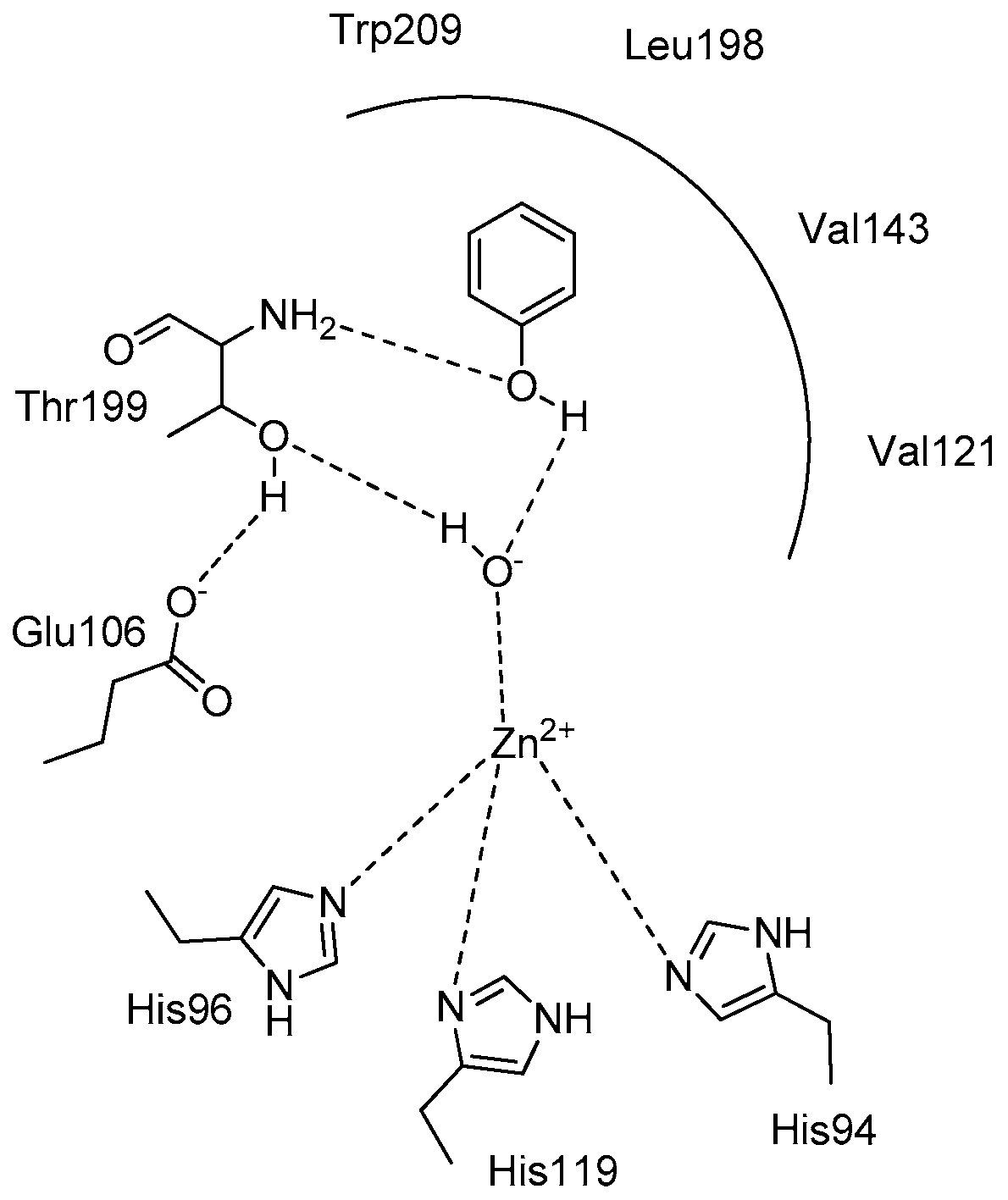
A possible explanation of the way that flavonoids interact with the CA active site was given in a very interesting docking study by Gidaro et al., [
69]. In this work a structure-based virtual screening was performed against five carbonic anhydrase isoforms using, as a ligand library, natural components of
Citrus bergamia (Bergamot) and
Allium cepa var.
tropea (red onion). Both plants are edible and part of the Calabrian (Italy) diet. By use of virtual screening, the flavone apigenin
60 and the flavanone glycoside eriocitrin (eriodictyol-7-
O-rutinoside)
73 (
Figure 16) were identified together with other eight flavonoids. In vitro tests against CA I, II, VA, IX, and XII isoforms showed that eriocitrin was the best hCA VA inhibitor with a K
I value of 0.15 μM. On the contrary, the computational prediction failed for apigenin. Docking studies revealed that eriocitrin probably binds by both the catechol and rutinoside portions. The catechol moiety is linked by additional H-bond interactions with the enzymatic residue Thr 199 located in the catalytic site, while the rutinose group connects by an extended H-bond network with several external mCA VA amino acid residues (Gln 67, Glu 69, Gln 92, Asp 171) at the entrance of the binding pocket. Apigenin instead, which was not as active as predicted, seems to interact only by its ring B which is involved in the zinc coordination into the mCA VA active site.
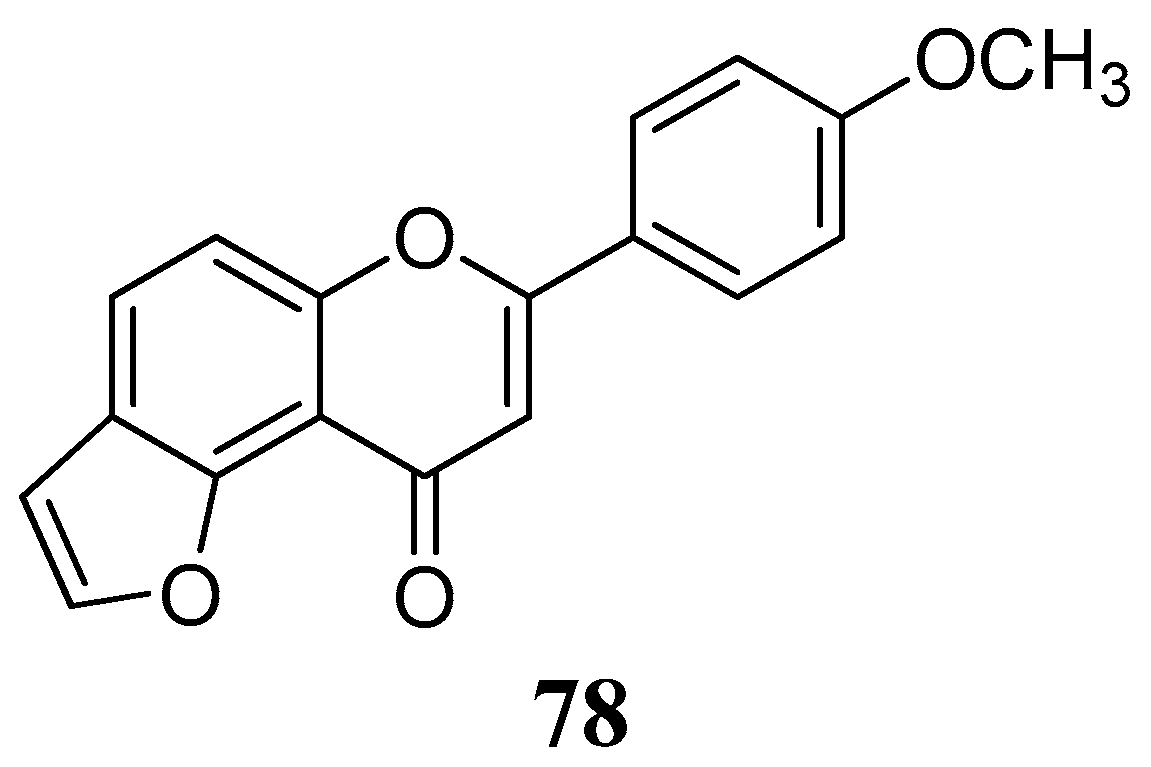
The lipophilic furanoflavonoid
78 from the bark of
Millettia ovalifolia (Fabaceae) was considered for inhibition of the cytosolic form of the bovine carbonic anhydrase-II (
Figure 17) [
70]. Despite the lack of free hydroxyl groups in its structure it displayed significant inhibitory activity with IC
50 value of 17.86 ± 0.07 μM (vs. zonisamide, 1.86 ± 0.03).
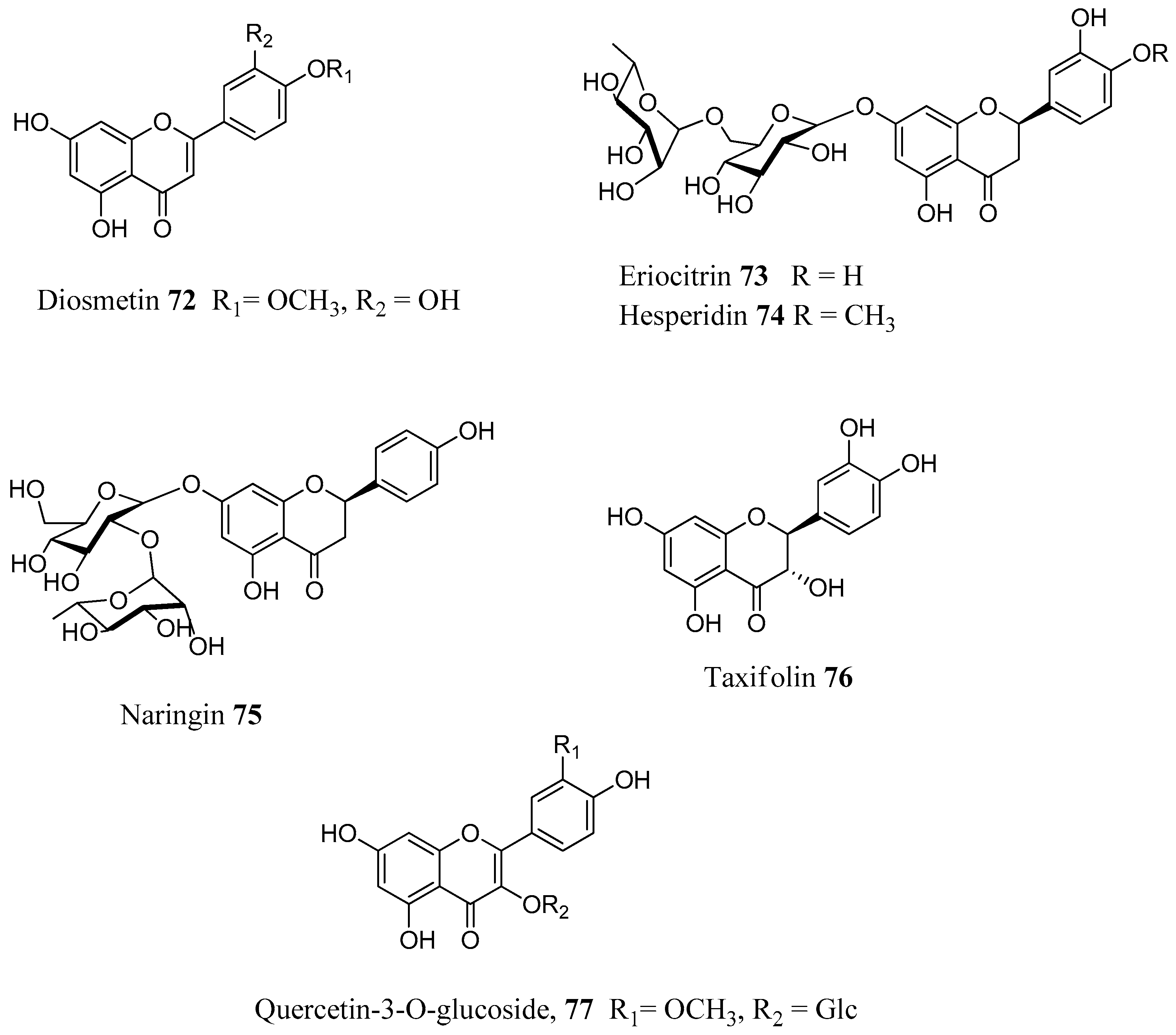
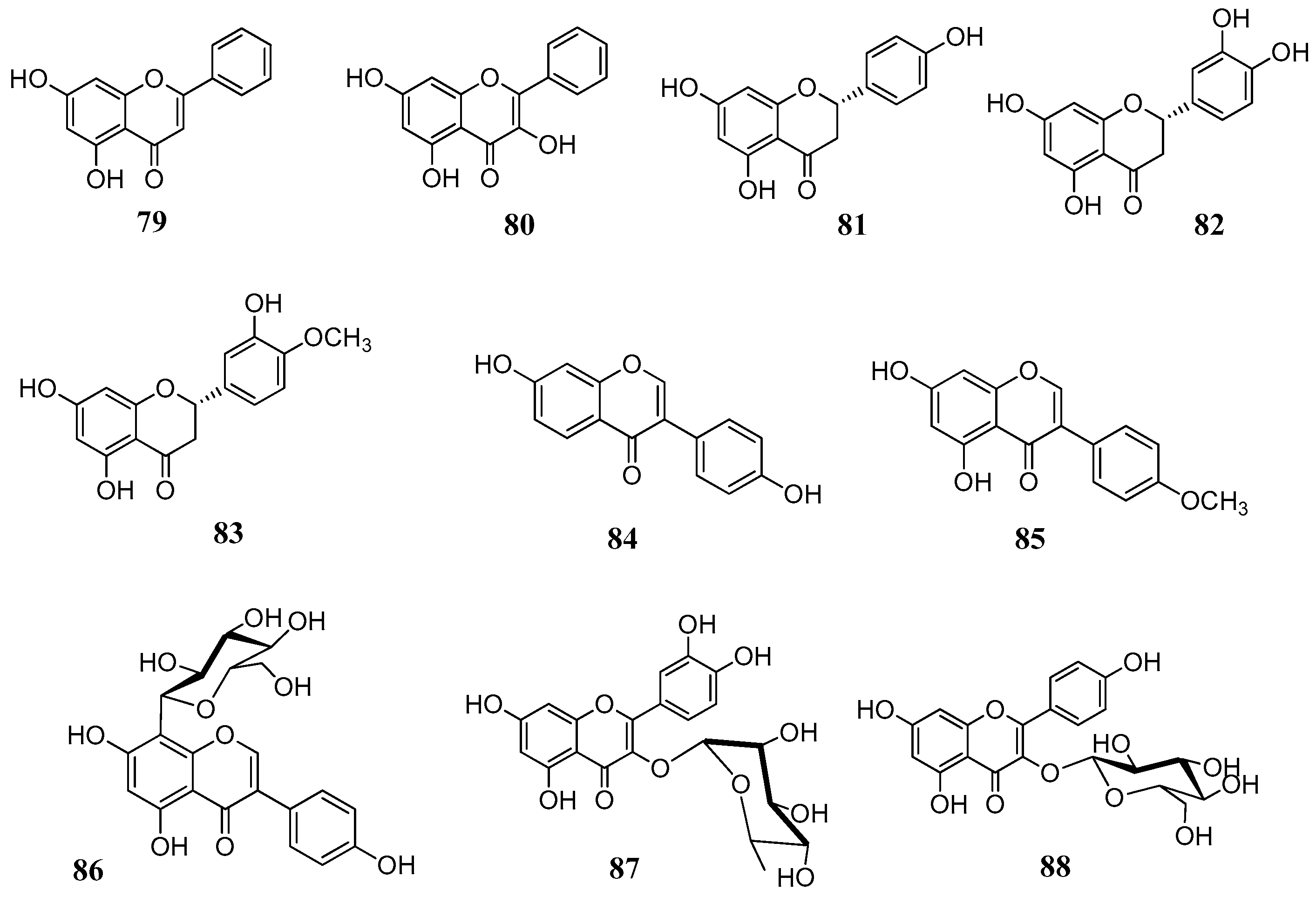
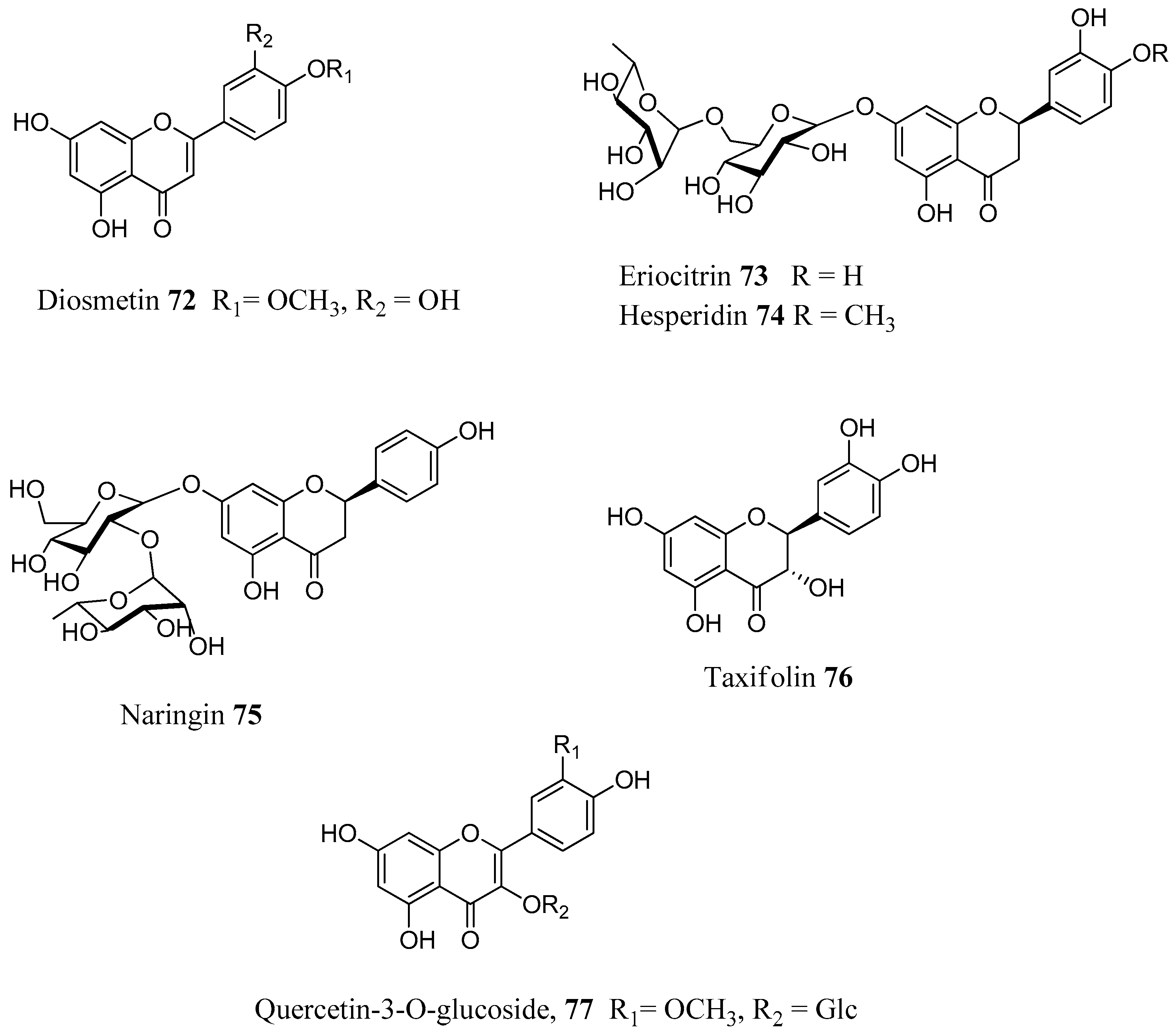
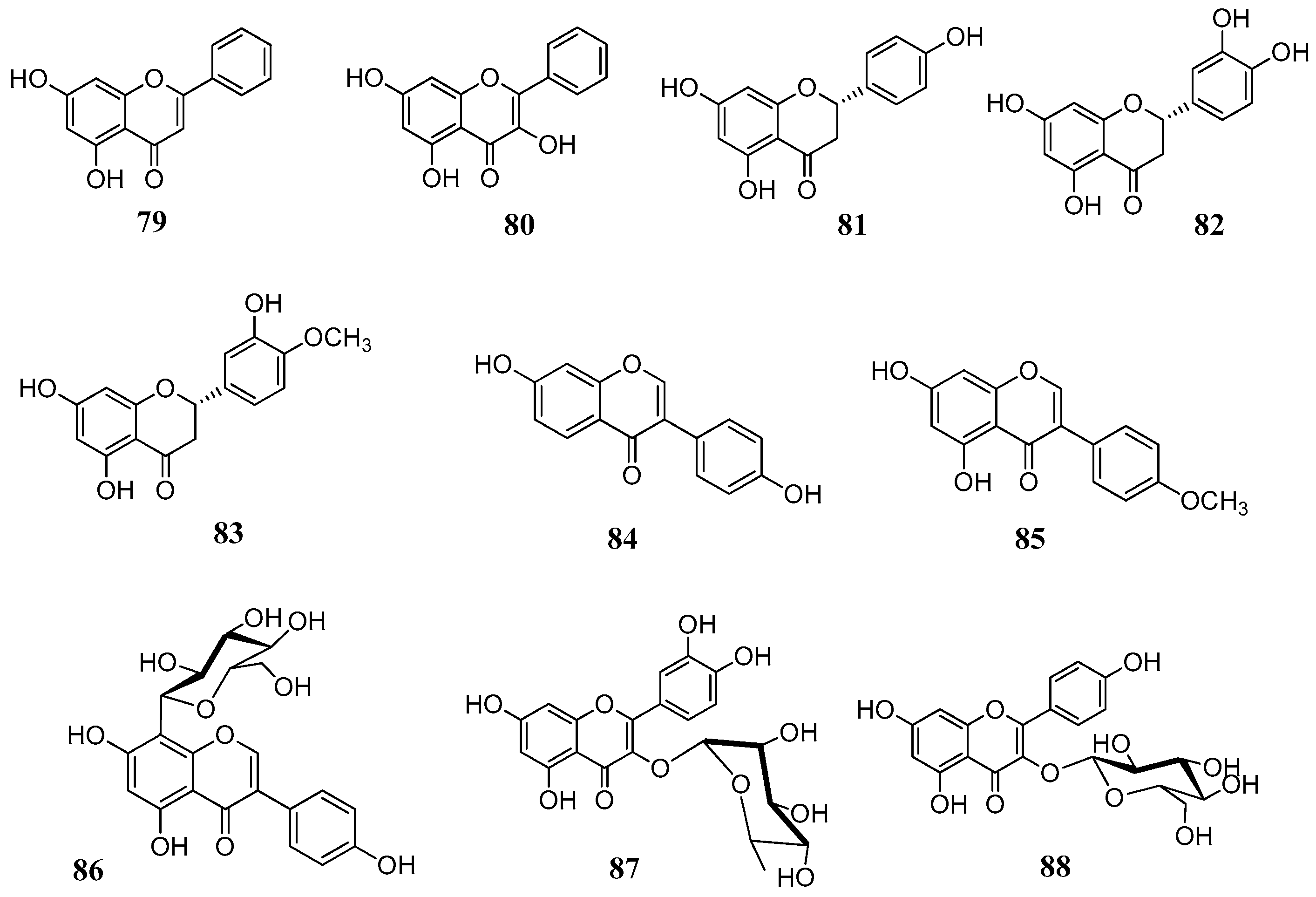
The above reported results spurred us to investigate a larger number of flavonoids against a line of human CAs. In total, 18 flavonoids (
Figure 18) were selected both from commercial sources and lab isolates [
71]. The selection of the compounds was done on the basis of the flavonoid subgroup and the substitution pattern. Flavones, flavonols, flavanones, isoflavones and some glycosylated derivatives were considered for assays. Chrysin (
79) is one of the main flavonoids of
Passiflora incarnata, a medicinal plant used phytotherapy as a mild sedative and anxiolytic [
72]. It is a flavone without any substitution of ring B. The flavone luteolin (
61), the two flavonols kaempferol (
65) and isorhamnetin (
68), as well as the flavonol glycosides quercetin-3-
O-glucoside (
77), quercetin-3-
O-rhamnoside (
87) and kaempferol-3-
O-glucoside (
88) are ubipresent flavonoids in most vegetables and fruits daily consumed, while galangin (
80) is present in honey and propolis [
73,
74]. Among the flavonoids considered were the flavanones naringenin (
81), eriodictyol (
82) and hesperitin (
83), which are abundant in edible
Citrus spp. (e.g. orange, mandarin) and possess antioxidant, anti-inflammatory and antitumoral properties [
75]. The isoflavones
84–
85 are abundant in legumes, soy and show potent estrogenic activity by interacting with the estrogenic receptors [
76,
77]. Puerarin (
86) is the main active principle of
Pueraria lobata root (Fabaceae) and it used in clinical practice in China to treat cardiovascular diseases, cerebrovascular disorders, Parkinson’s disease, Alzheimer’s disease, diabetes and diabetic complications [
78]. The flavonols
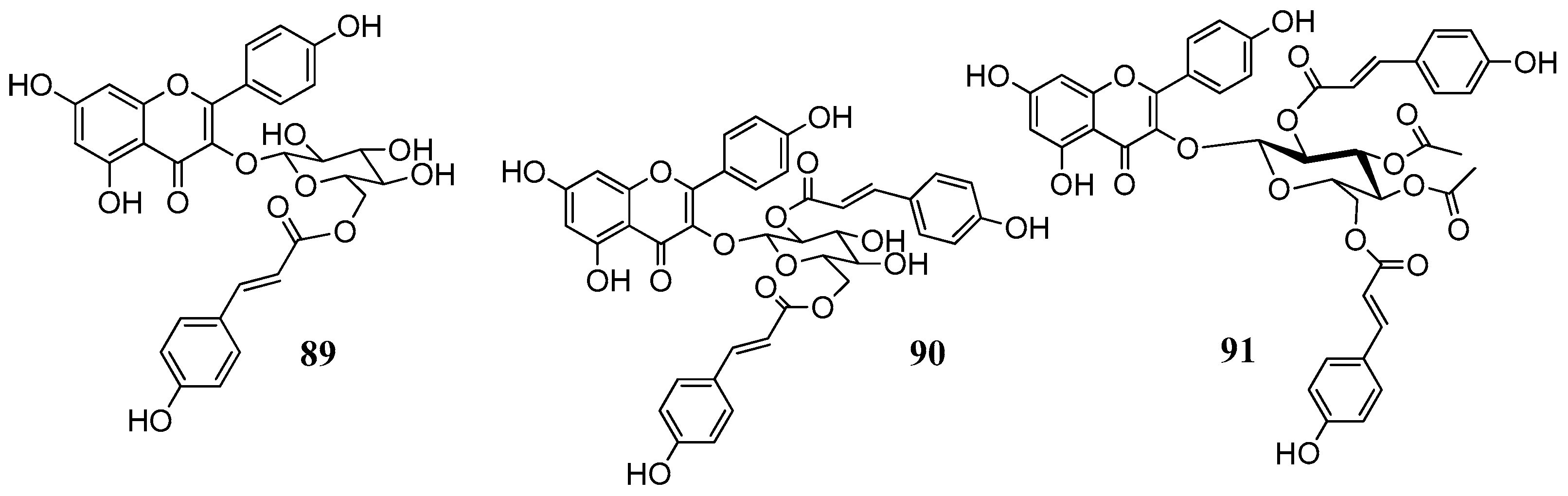
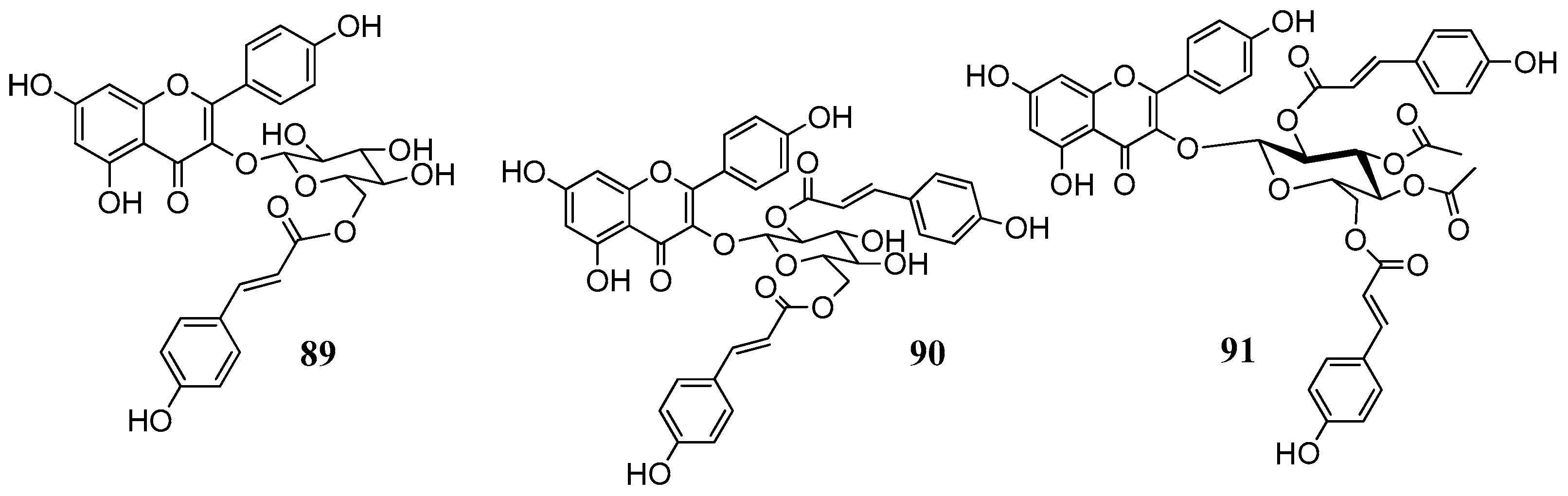
89–
91 were isolated from leaves of
Quercus ilex (Fagaceae) and belong to the group of acylated flavonoids (
Figure 19). Since they combine the flavonol nucleus and the coumaric acid moieties, they were selected with the aim to see whether we would have an additional inhibitory effect on the CAs. For the in vitro inhibitory activities five physiological relevant hCA isoforms were considered: the cytosolic I, II, VII and the membrane bound CA IV and XII. Results are displayed in
Table 10.
Although a SAR is difficult, some general remarks can be made. As can be observed from
Table 10, none of the compounds considered here show a significant inhibitory activity against the cytosolic hCA I, but rather showed selectivity against isoforms hCA IV and hCAV II. Concerning the inhibition of hCA II, the presence of ortho-hydroxyl groups on ring B seems to be important for the activity as long as the double bond in ring C remains intact. This is shown comparing luteolin with eriodictyol and taxifolin. Instead, glycosylation of the C-3 hydroxyl enhances the activity, as demonstrated by Kaempferol
-3-
O-glucoside which was the most active of all tested compounds with a K
I of 168 nM.
Results were quite surprising regarding the inhibition hCA IV. The flavanones naringenin, eriodictyol and hesperitin, lacking the C2-3 double bond as well as the glycosides 77, 87, 91 were quite effective against the with KI values close to the standard CAI acetazolamide (74.0 nM). Especially comparing the activity of compounds 65, 89–91 it could be suggested that the optimum activity of compound 91 is not due to the flavonoid scaffold but to the acetyl/coumaric functionalities which may hydrolyze and released in situ. This hypothesis requires further investigation.
The kinetic data on the cytosolic hCA VII showed that several of the investigated compounds were low nanomolar inhibitors of the hCA VII with K
I very close to the standard acetazolamide. Among the investigated constituents are the flavone luteolin (with
ortho-hydroxyl groups) the flavonol isorhamnetin, the flavanones naringenin, eriodictyol and hesperitin, the isoflavone daidzein, the quercetin flavonols
77 and
87 and the acylated glycosides
89 and
90. With the exception of catechin, this is the first time that flavonoids are tested against this particular hCA isoform [
68]. The heterogeneity of constituents does not permit any SAR conclusion or it is possible that some other common structural feature may be responsible for the inhibition, such as ring A or combination of rings A and B. Similarly, the inhibition data on the tumor associated hCA XII isoform (
Table 10) showed a complex and different SAR. Generally most of the substances were high nanomolar inhibitors with K
I values spanning between 515.0 and 134.3 nM. The best inhibitor evidenced was Kaempferol-3-
O-glucoside with a K
I value of 4.9 nM. The results in both hCAV II and XII show that flavonoids merit further attention and warrant future investigation to comprehend the mechanism of action in order to design new, selective inhibitors.
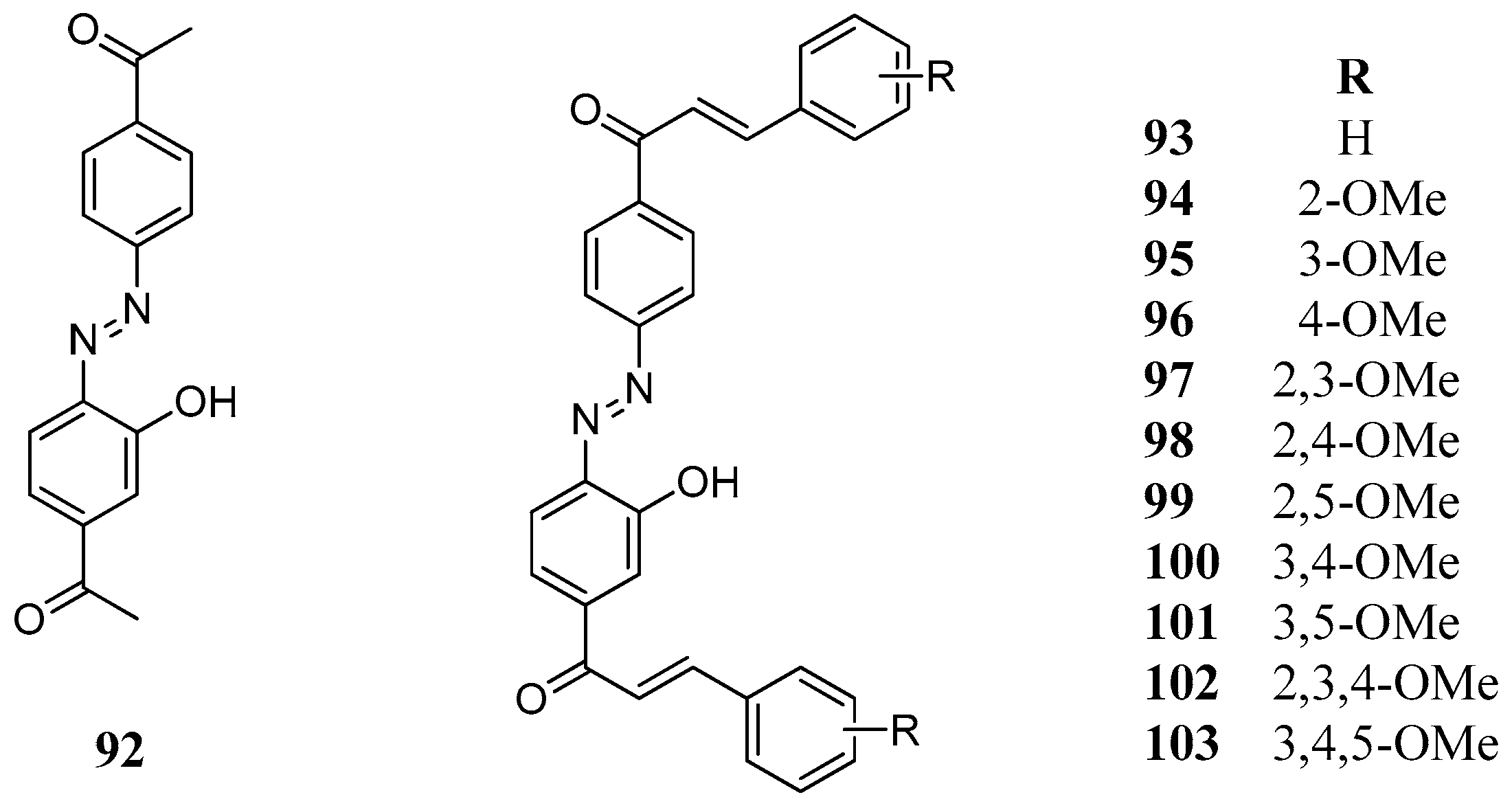
A series of synthetic bischalcones
92–
103 bearing a C–N=N–C linkage has been prepared and tested against human hCA I and II (
Figure 20) [
79]. Most of the compounds had one to three methoxy groups as substituents on their phenolic rings. Compounds
93,
97,
101, and
103 were among the best inhibitors (
Table 11). Furthermore, similarly to AZA, some of the investigated bischalcone compounds act as competitive inhibitors with 4-NPA as substrate, that is, they bind to the same regions of the active site cavity as the substrate. However, the binding site of 4-NPA itself is unknown, but it is presumed to be in the same region as that of CO
2, the physiological substrate of this enzyme.
2.5. Phenols and Polyphenols as Effective Mycobacterial CA Inhibitors
The discovery of the α-, β-, and η-families of carbonic anhydrase in parasitic bacteria, fungi protozoa and helminths with different structure than human CAs has created new opportunities in the search for new drugs against pathogens [
29]. Since many pathogen carbonic anhydrases belong to different enzyme families, which are either absent in humans (like β- and η-CAs) or have marked structural differences with the human isoforms they consist attractive targets for the development of new anti-infective agents in terms of selectivity and efficacy. Given the continuous development of resistance of several pathogens to the existing therapeutic schemes, CA inhibition represents a new, complementary mechanism of action worth of investigation. Among the most investigated CAs, are the β-CAs from
Helicobacter pylori,
Brucella suis and
Mycobacterium tuberculosis, which have been cloned and characterized [
80,
81]. Other β-CAs which have been intensely studied are the CAs from the fungal strains
Candida albicans,
Candida glabrata and
Cryptococcus neoformans: CaNce103 and CgNce103 (from
C. albida and
C. glabrata, respectively) and Can1, Can2 from
C. neoformans. In these pathogens CAs regulate the physiological concentrations of CO
2/HCO
3. Both fungal strains are adapted to extremely low concentrations of CO
2. However, once the infection occurs, elevated concentrations of CO
2 promote distinct structural and developmental procedures which influence the survival, virulence and pathogenesis of these fungi [
82].
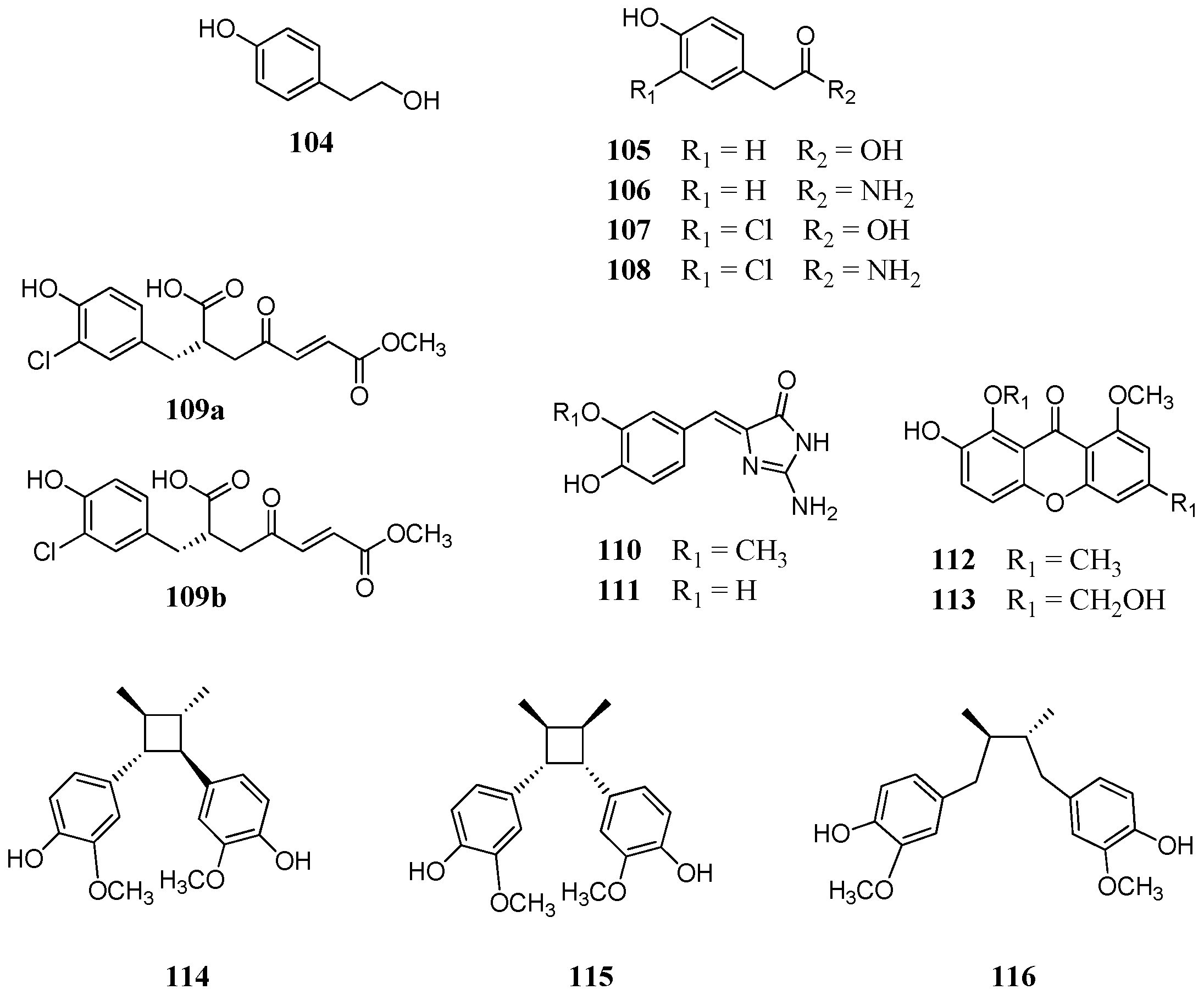
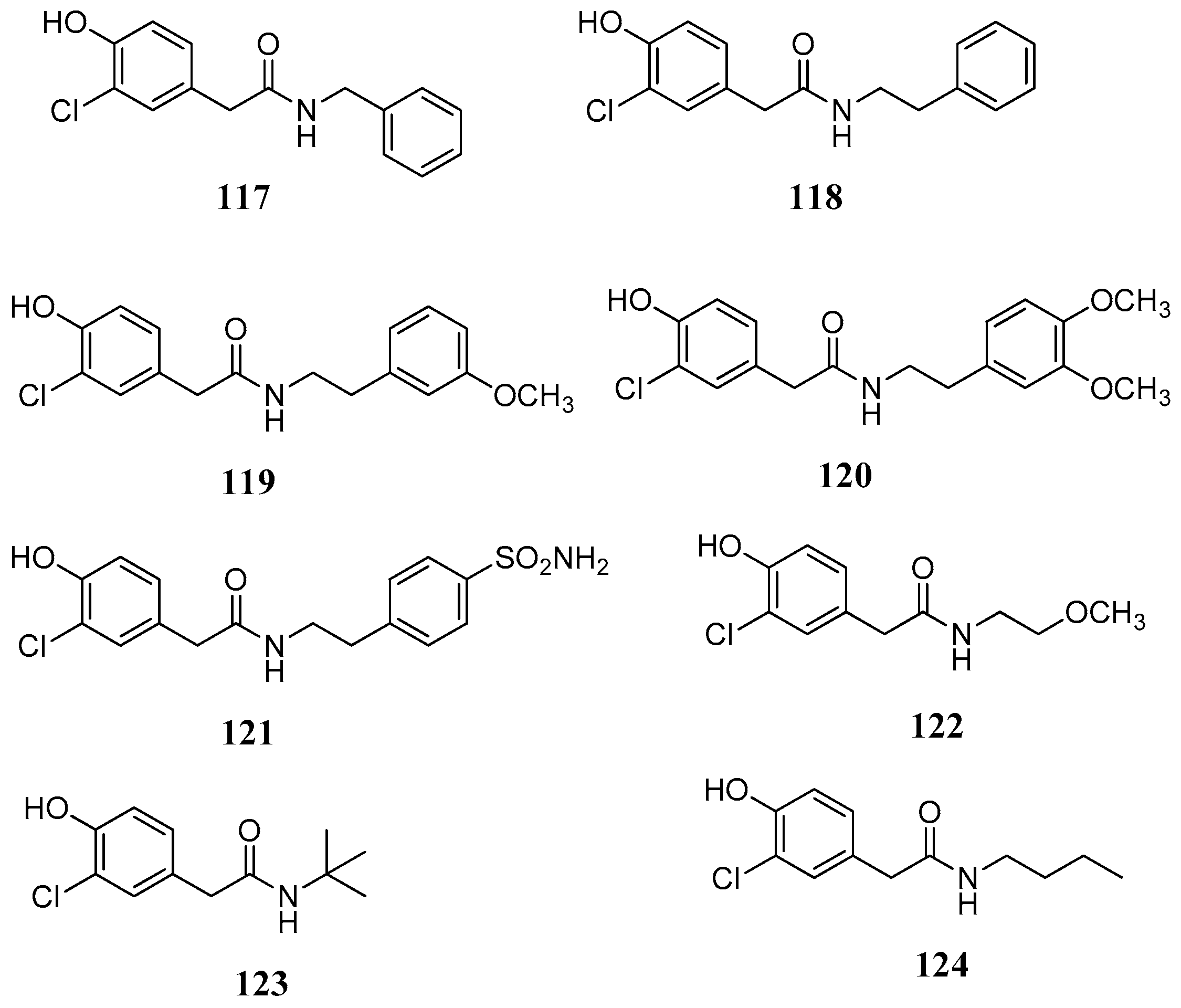
The first evidence that phenols can inhibit pathogenic CAs came from the screening of a natural product library [
83] against β-CAs from
M. tuberculosis,
C. albicans, and
C. neoformans. Screening of libraries of plant extracts and isolated constituents, although time consuming, is a valuable tool in the exploration of new scaffolds in order to discover new chemotypes to combat diseases. The Queensland Compound Library reported by Davis et al., [
83] comprised a wide range of natural and synthetic products incorporating the phenol group. Overall 21 constituents
104–
124 were selected and tested (
Figure 21 and
Figure 22) including eight fungal (compounds
104–
109a,
112,
113), two ascidian (compounds
107,
108), and three plant (compounds
111–
113) secondary metabolites. The tested structures were diverse comprising a series of simple mono- or disubstituted phenols
104–
108, (−)-xylariamide A (
109a) and its synthetic enantiomer (+)-xylariamide A (
109b), polyandrocarpamine A (
110), polyandrocarpamine B (
111), xanthones
112 and
113, endiandrin A (
114), endiandrin B (
115) and (−)-dihydroguaiaretic acid
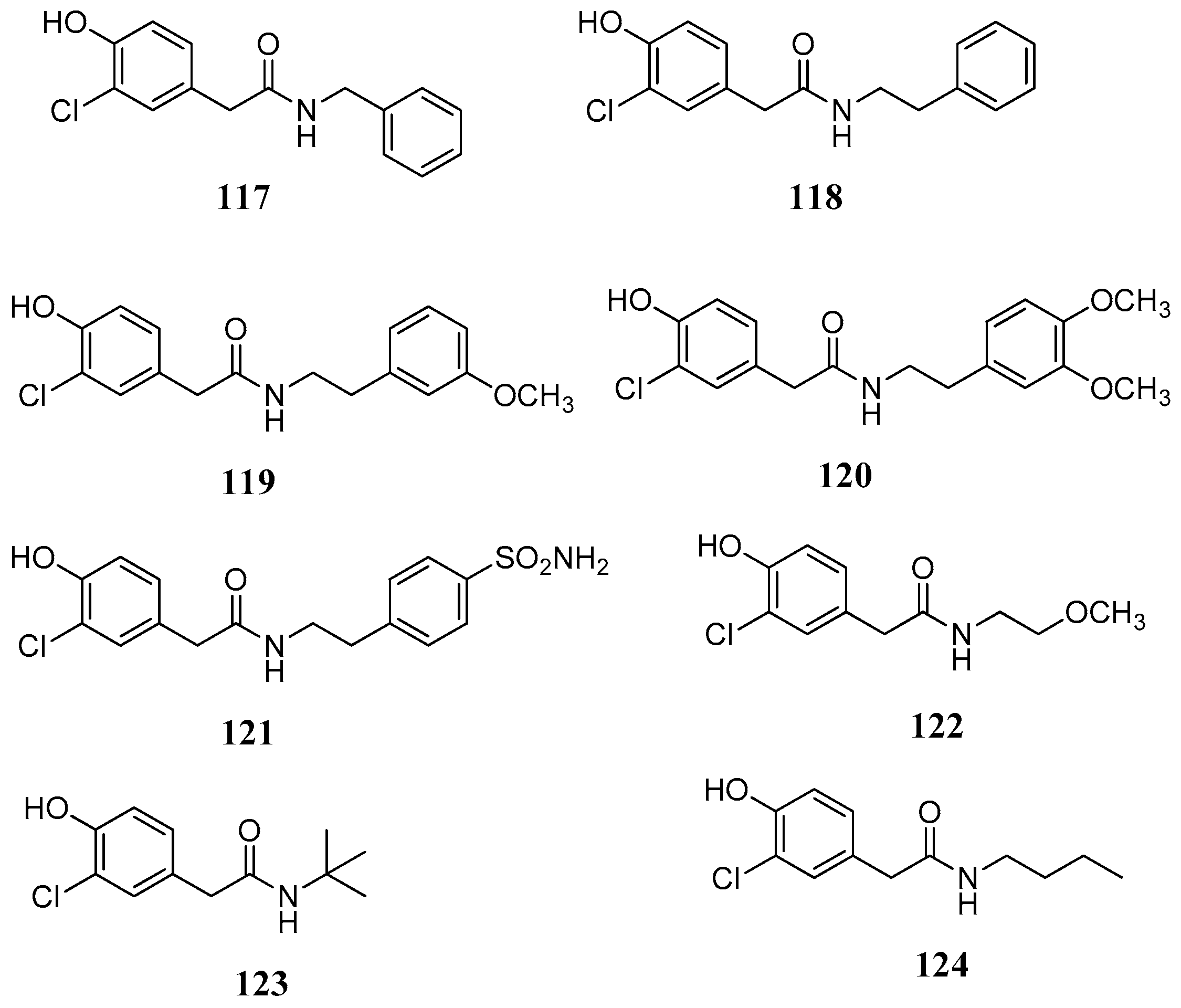
116. The synthetic phenolic compounds
117–
124 (
Figure 22) were eight secondary amides inspired by the fungal metabolite
108.
Inhibition studies over human CAs were carried out in parallel in order to assess the selectivity of the tested compounds. As it can be observed from
Table 12, several phenolic natural products were selective inhibitors of mycobacterial and fungal β-CAs.
Among the natural products tested the best inhibitors was (−)-dihydroguaiaretic acid (
116) which inhibited β-CA in the submicromolar range with up to 495-fold selectivity over hCA I and 371-fold selectivity over hCA II. From the library of synthetic derivatives, compound
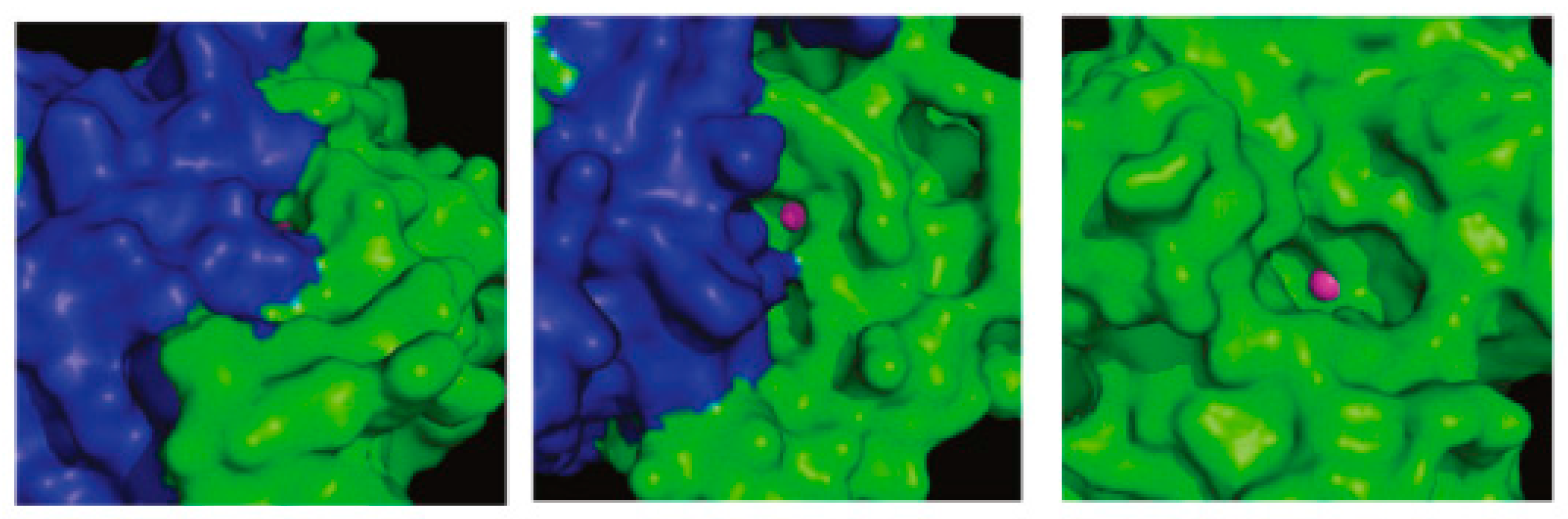
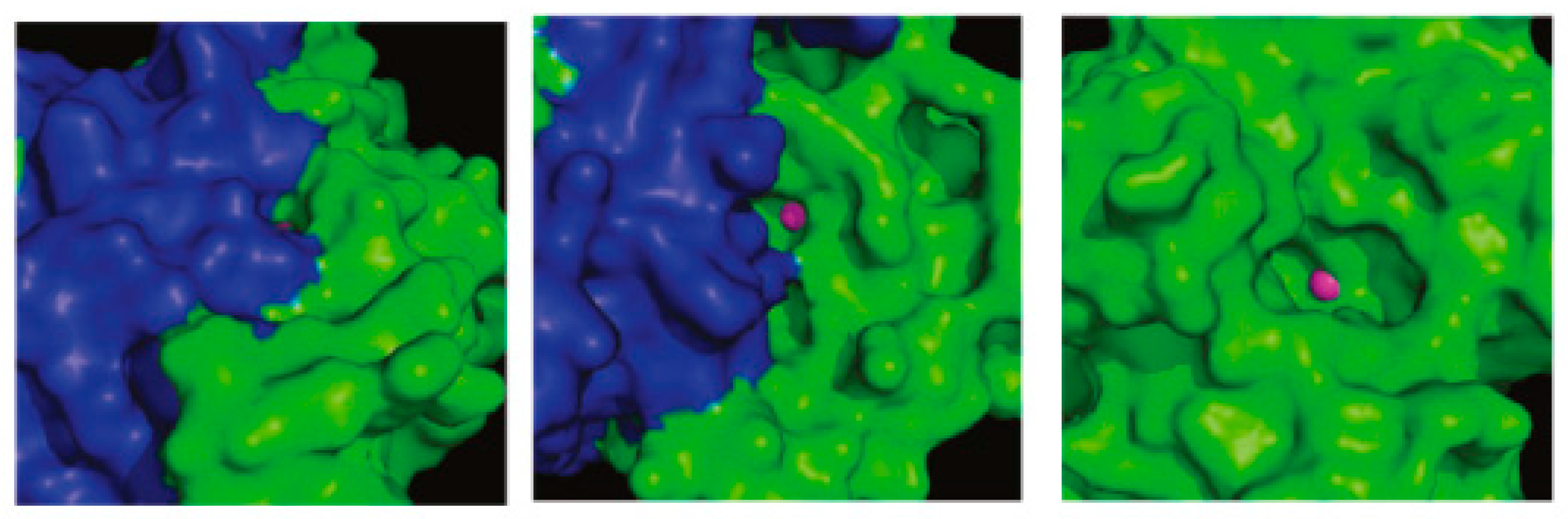
108 was a low micromolar inhibitor of the fungal CAs and displayed 130- to 280-fold selectivity over the two human CAs. These compounds were the first non-sulfonamide inhibitors that displayed β- over α-CA enzyme selectivity. This selectivity could be explained if the different structure of α- and β-isozymes is taken into consideration. Although both enzymes contain a zinc ion in the active site, they have several differences. Unlike α-CAs, the β-class of CAs exist as dimers (
Figure 23). Whilst the catalytically important residues in the active site of β-CAs are provided exclusively from one monomer, access to the catalytic center may be constituted by residues from both monomers leaving a cleft of limited space for the inhibitor to enter. Therefore, it seems unlikely that the phenolic hydroxyl groups of the tested constituents directly interact with the active site zinc of the pathogen CAs, especially for the spacious one
116. It was suggested that the compounds might cause either monomerization or considerable conformational changes in the access area to the active site. To prove this concept crystallization of the adduct, and X-ray analysis is necessary. Unfortunately none of the tested compounds gave crystals with Can2, Nce103, Rv3273, and Rv1284 enzymes.
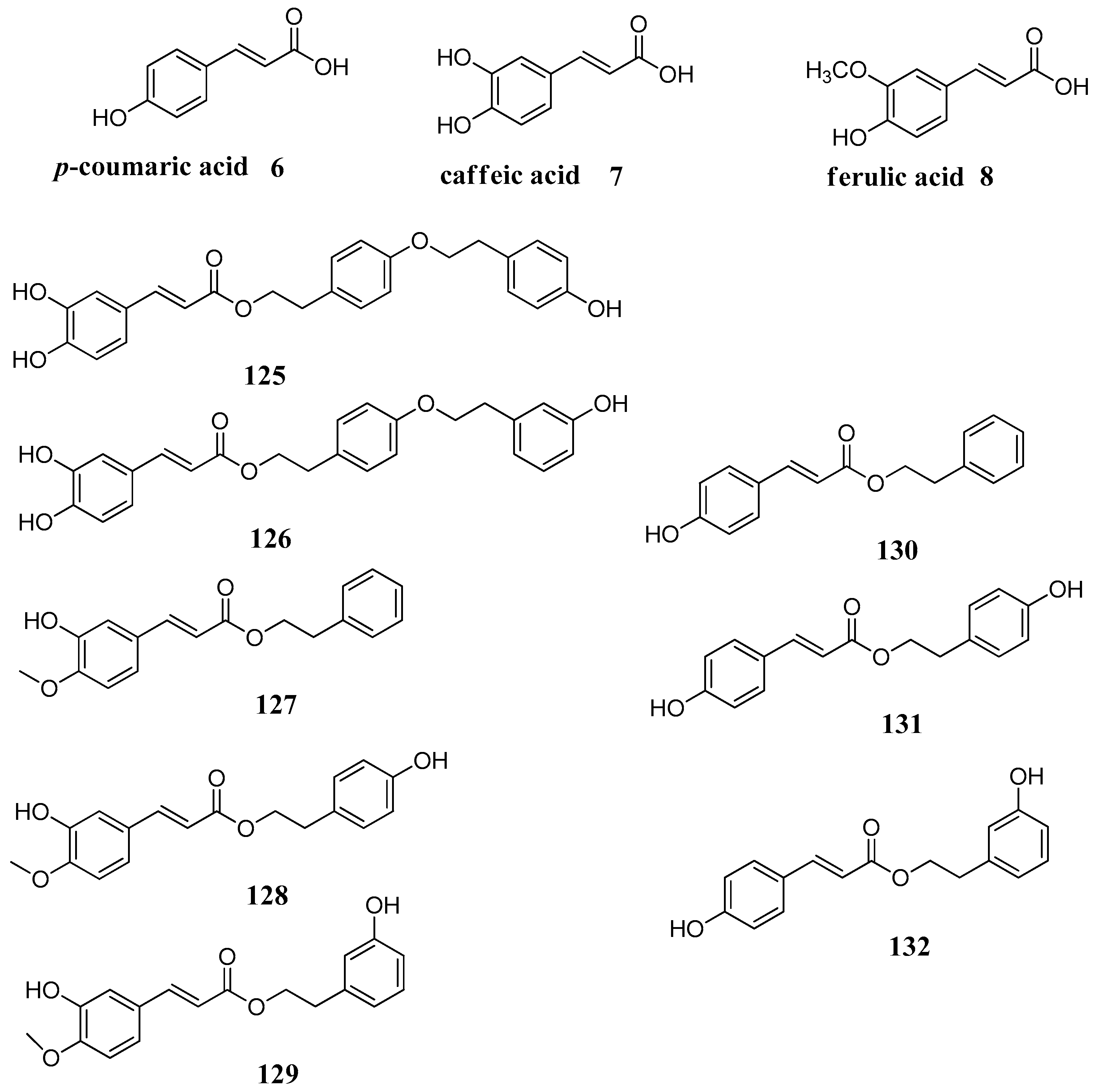
A series of phenolic acids
6–
8 and some of their esters
125–
132, derivatives of
p-coumaric (
6), caffeic (
7) and ferulic acid (
8), was investigated for the inhibition of three β-carbonic anhydrases (CAs, EC 4.2.1.1) from the pathogenic bacterium
Mycobacterium tuberculosis, Rv1248, Rv3588 and Rv3273 β-CAs (
Figure 24) [
85]. Some of these compounds were low micromolar inhibitors of the pathogenic enzymes and they did not show inhibitory activity against the human widespread cytosolic isoforms CA I and II. As it can be observed from
Table 13, simple phenolic acids
6–
8 are active against Mtb β-CAs but suffer of low selectivity. The presence of additional phenolic rings in the structure makes the compounds more selective as it increases the ratio of β- over α-CA inhibitory activity. This is possibly due to additional interactions with the dimeric structure of the β-CA.
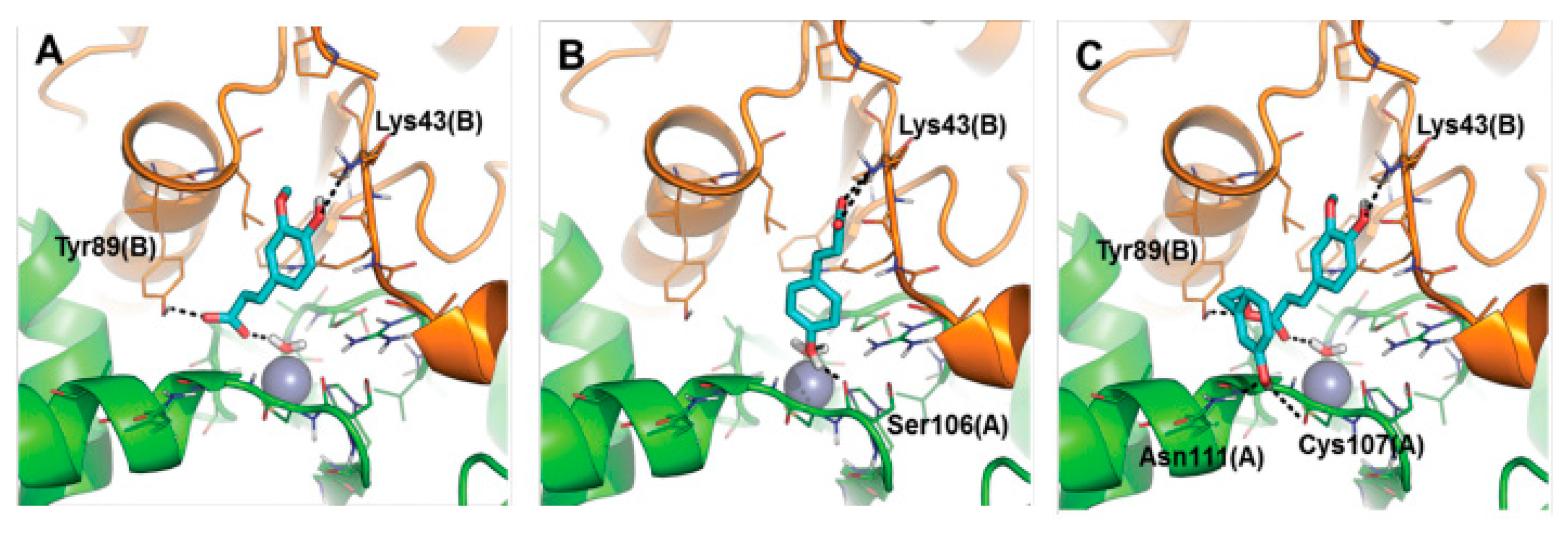
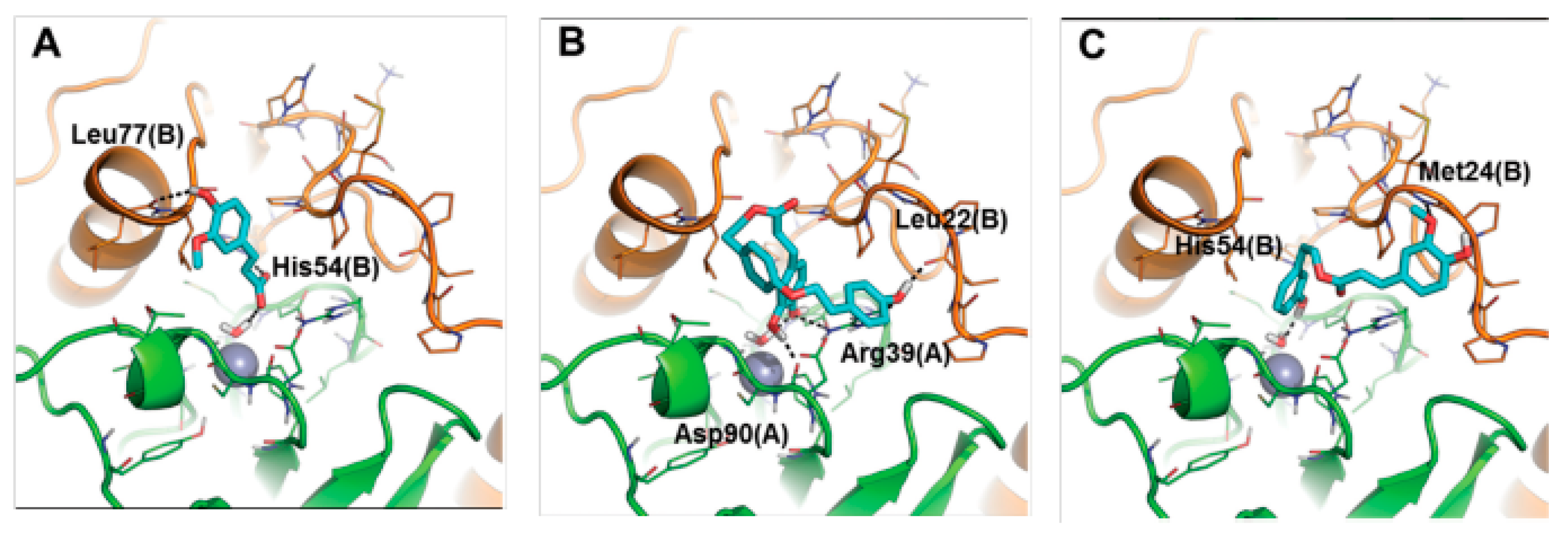
Unfortunately it was not possible to obtain good quality crystals which would permit the study of the adduct by X-ray crystallography. To overcome this problem, the binding mode of these inhibitors to the bacterial enzymes Rv3588 and Rv1248 was investigated by computational approaches (
Figure 25 and
Figure 26). A protocol consisting of classic MD and SMD simulations with molecular docking and rescoring was applied. It was proposed that the inhibitors anchor to the zinc-coordinated water molecule from the CA active site interfering with the nucleophilic attack of the zinc hydroxide on the substrate CO
2. These compounds may be considered as interesting anti-mycobacterial lead compounds.








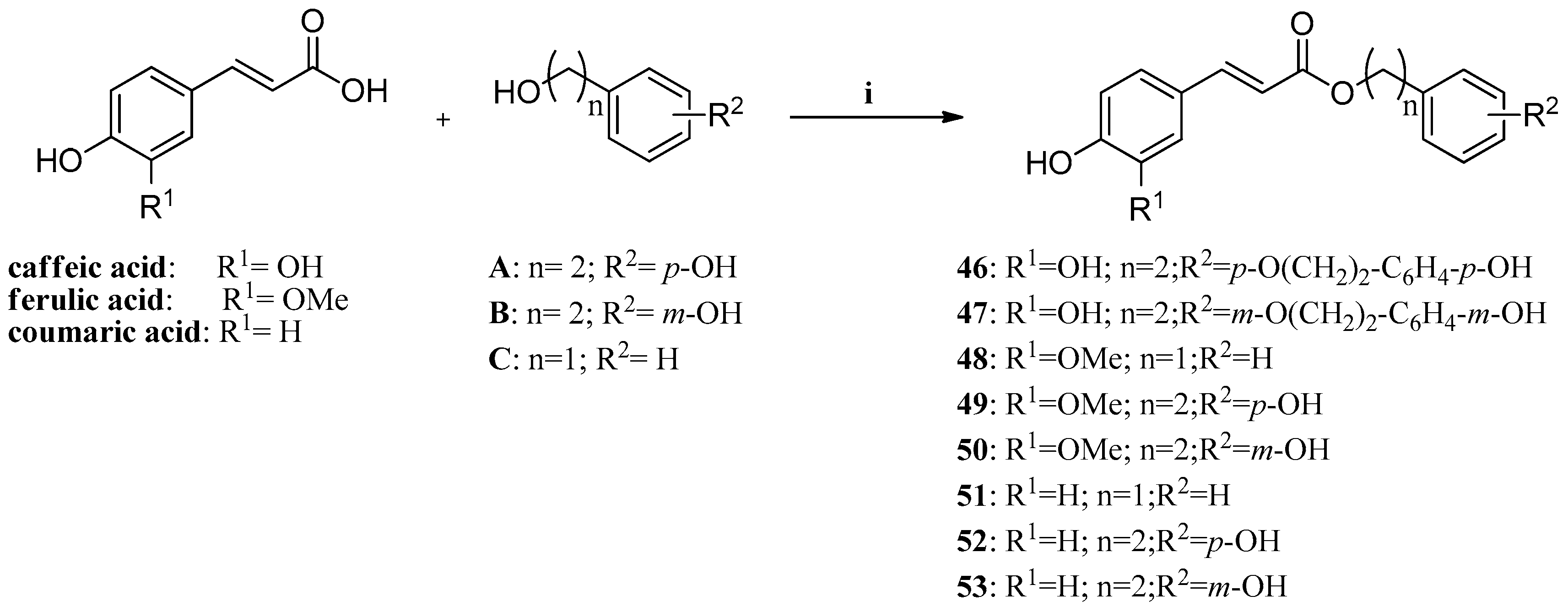







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}