
Selective and Efficient Generation of ortho-Brominated para-Substituted Phenols in ACS-Grade Methanol

Abstract
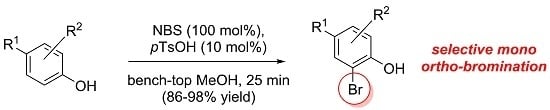
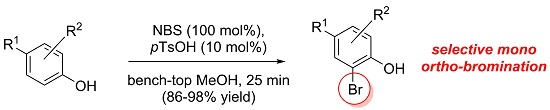
:
1. Introduction
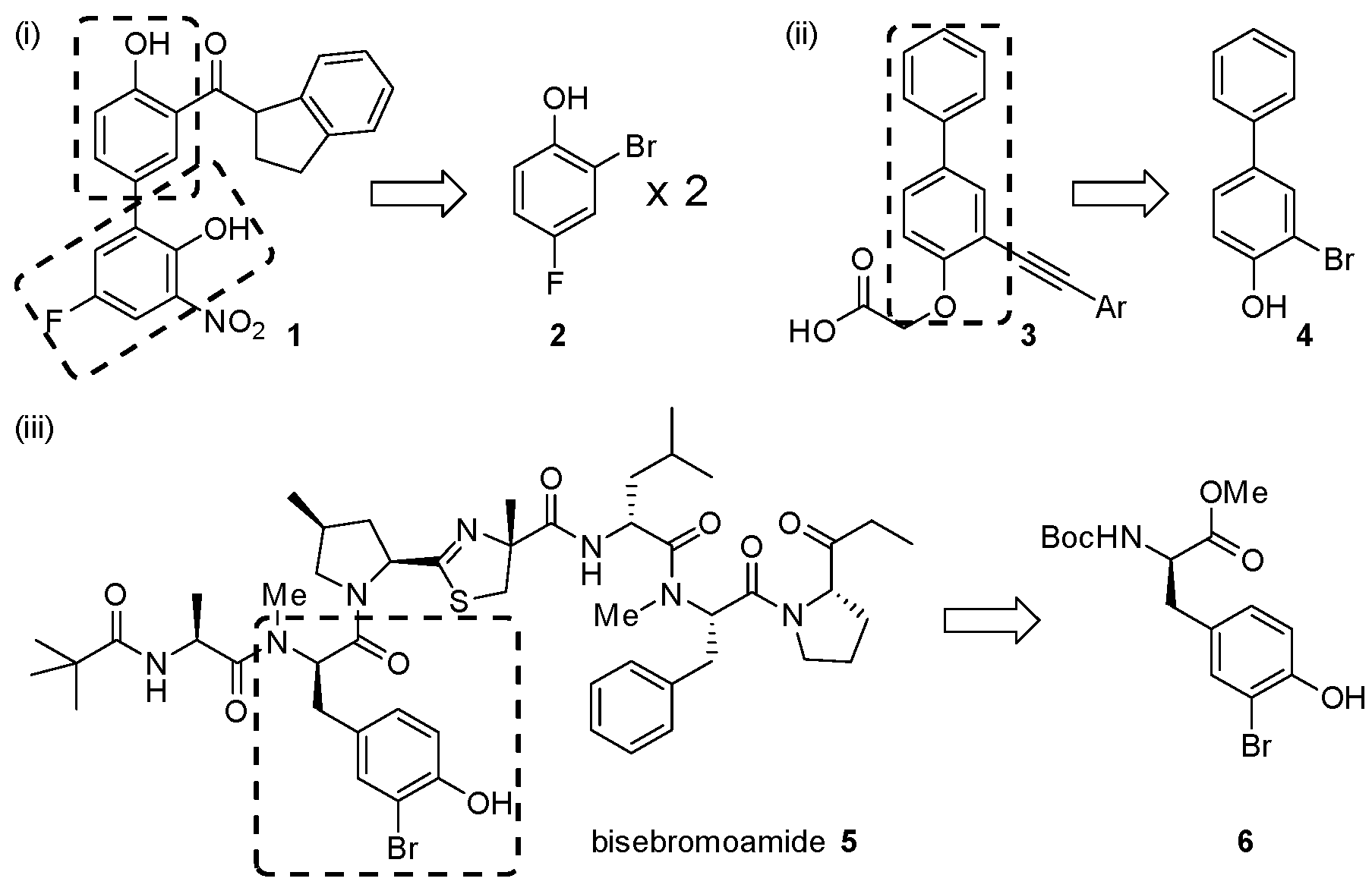

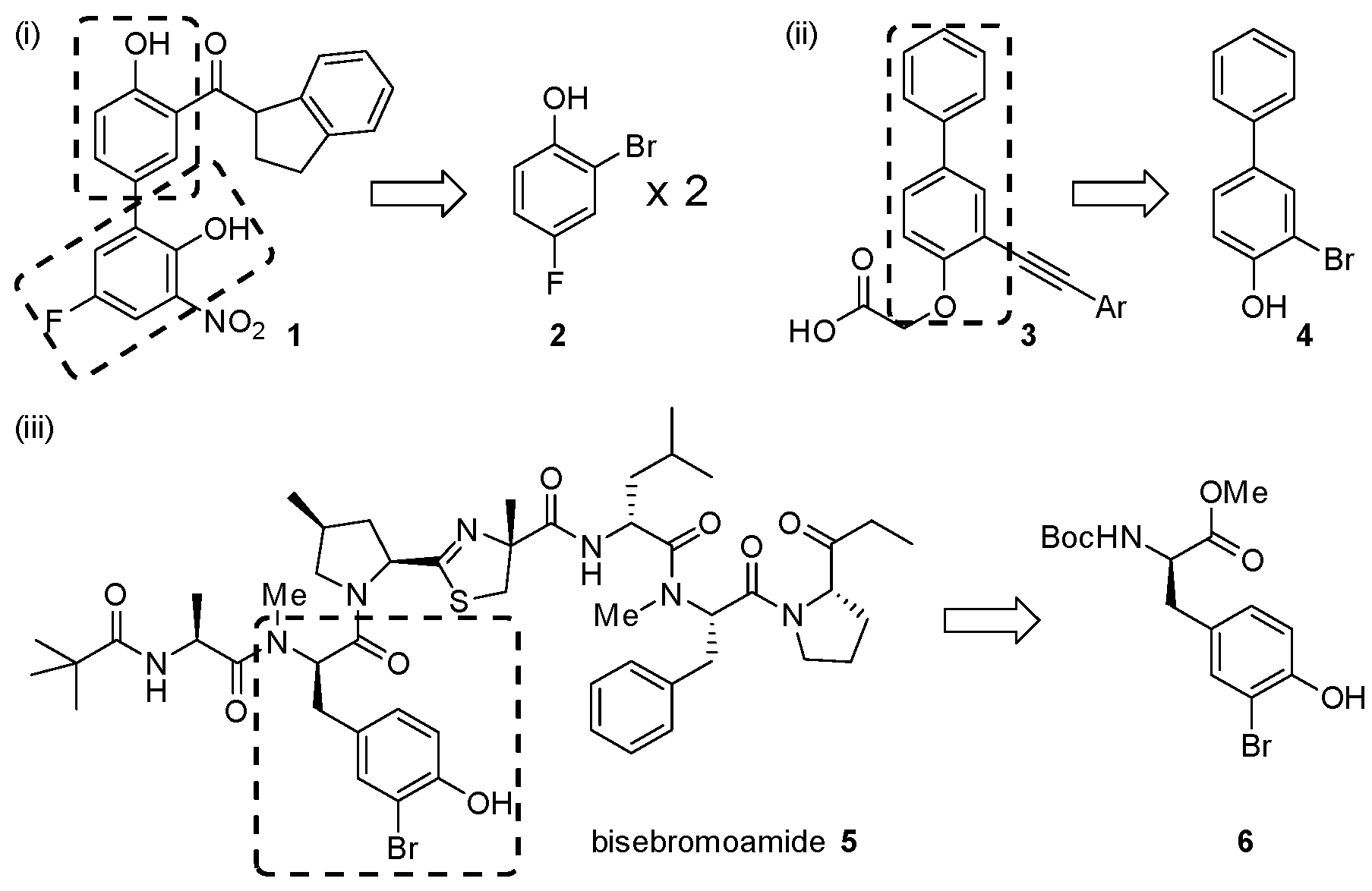
2. Results and Discussion
2.1. Investigation of the Mono ortho-Bromination Flow Reaction
2.2. Investigation of Batch Reaction Conditions Using ACS-Grade Methanol


{kind=link}
{kind=link}
{kind=link}
| Entry | NBS (mol %) | pTsOH (mol %) | Ratio (Relative % at 285 nm) | Isolated Yield 10 (%) | ||
|---|---|---|---|---|---|---|
| 9 | 10 | 11 | ||||
| 1 a,b | 100 | 10 | 6 | 87 | 7 | -- |
| 2 b | 100 | 10 | 5 | 93 | 2 | -- |
| 3 | 100 | 10 | 3 | 94 | 3 | 92% |
| 4 | 110 | 10 | 0 | 88 | 12 | 86% |
| 5 | 100 | 0 | 15 | 77 | 8 | 74% |
| 6 | 100 | 20 | 3 | 93 | 4 | 90% |
| Entry | Starting Material a | Major Product | Ratio (SM:mono:di) b | Isolated Yield c (%) |
|---|---|---|---|---|
| 1 | p-Cresol 9 |  | 1:95:4 | 92 |
| 2 | 4-Fluorophenol |  | 4:89:7 | 86 |
| 3 | Vanillin |  | 5:95:-- | 90 |
| 4 | 2-Naphthol |  | 0:99:1 | 98 |
| 5 | 4-tert-Butylphenol |  | 2:92:6 | 90 |
| 6 | [1,1′-Biphenyl]-4-ol |  | 4:90:6 | 89 |
| 7 | 3′-Fluoro-6′-methoxy-[1,1′-biphenyl]-4-ol |  | 4:92:4 | 90 |
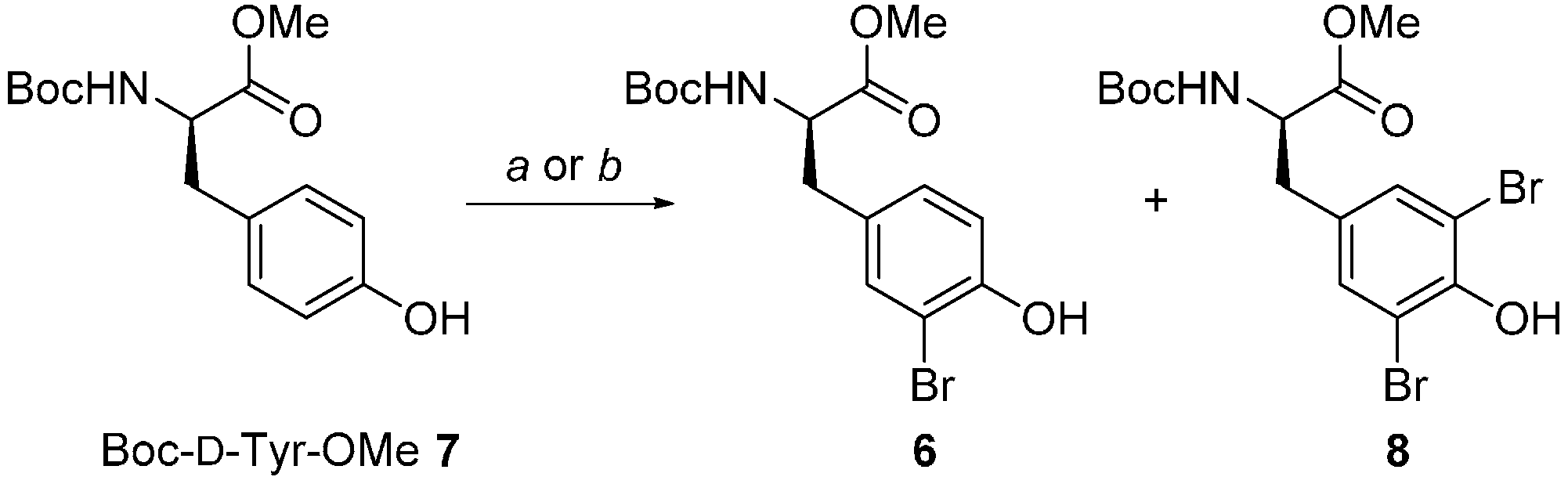
| 8 | Boc-Tyr-OMe ent-7 |  | 2:94:4 | 89 |
3. Experimental Section
3.1. General Information
3.2. General Procedure for Batch Reaction Conditions in ACS-Grade Methanol
3.3. Characterization of Products
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Daum, S.; Erdmann, F.; Fischer, G.; Féaux-de-Lacroix, B.; Hessamian-Alinejad, A.; Houben, S.; Frank, W.; Braun, M. Aryl indanyl ketones: Efficient inhibitors of the human peptidyl prolyl cis/trans isomerase Pin1. Angew. Chem. Int. Ed. 2006, 45, 7454–7458. [Google Scholar] [CrossRef] [PubMed]
- Crosignani, S.; Prêtre, A.; Jorand-Lebrun, C.; Fraboulet, G.; Seenisamy, J.; Augustine, J.K.; Missotten, M.; Humbert, Y.; Cleva, C.; Abla, N.; et al. Discovery of potent, selective, and orally bioavailable alkynylphenoxyacetic acid CRTH2 (DP2) receptor antagonists for the treatment of allergic inflammatory diseases. J. Med. Chem. 2011, 54, 7299–7317. [Google Scholar] [CrossRef] [PubMed]
- Teruya, T.; Sasaki, H.; Fukazawa, H.; Suenaga, K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp.: Isolation, stereostructure, and biological activity. Org. Lett. 2009, 11, 5062–5065. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.G.; Liu, Y.Q.; Kwong, S.Q.; Xu, Z.S.; Ye, T. Total synthesis and stereochemical reassignment of bisebromoamide. Org. Lett. 2010, 12, 3018–3021. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Yu, S.Y.; Jin, M.Z.; Xia, H.G.; Ma, D.W. Total synthesis and cytotoxicity of bisebromoamide and its analogues. Tetrahedron Lett. 2011, 52, 2124–2127. [Google Scholar] [CrossRef]
- Boys, S.K. Tyrosine Derivatives and Their Anti-Cancer Applications. Ph.D. Thesis, The University of Edinburgh, Edinburgh, UK, 2012. [Google Scholar]
- Johnston, H.J. Development of Novel Analogues of the Anti-Proliferative Marine Natural Product Bisebromoamide: Synthesis and Structure Activity Relationship Studies. Ph.D. Thesis, The University of Edinburgh, Edinburgh, UK, 2014. [Google Scholar]
- Johnston, H.J.; McWhinnie, F.S.; Landi, F.; Hulme, A.N. Flexible, phase-transfer catalyzed approaches to 4-substituted prolines. Org. Lett. 2014, 16, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.J.; Hulme, A.N. A facile, inexpensive and scalable route to thiol-protected α-methyl cysteine. Synlett 2013, 24, 591–594. [Google Scholar]
- Zhou, C.-Y.; Li, J.; Peddibhotla, S.; Romo, D. Mild arming and derivatization of natural products via an In(OTf)3-catalyzed arene iodination. Org. Lett. 2010, 12, 2104–2107. [Google Scholar] [CrossRef] [PubMed]
- Chhattise, P.K.; Ramaswamy, A.V.; Waghmode, S.B. Regioselective, photochemical bromination of aromatic compounds using N-bromosuccinimide. Tetrahedron Lett. 2008, 49, 189–194. [Google Scholar] [CrossRef]
- Bovonsombat, P.; Khanthapura, P.; Krause, M.M.; Leykajarakul, J. Facile syntheses of 3-halo and mixed 3,5-dihalo analogues of N-acetyl-l-tyrosine via sulfonic acid-catalysed regioselective monohalogenation. Tetrahedron Lett. 2008, 49, 7008–7011. [Google Scholar] [CrossRef]
- Bovonsombat, P.; Ali, R.; Khan, C.; Leykajarakul, J.; Pla-on, K.; Aphimanchindakul, S.; Pungcharoenpong, N.; Timsuea, N.; Arunrat, A.; Punpongjareorn, N. Facile p-toluenesulfonic acid-promoted para-selective monobromination and chlorination of phenol and analogues. Tetrahedron 2010, 66, 6928–6935. [Google Scholar] [CrossRef]
- Verner, E.; Katz, B.A.; Spencer, J.R.; Allen, D.; Hataye, J.; Hruzewicz, W.; Hui, H.C.; Kolesnikov, A.; Li, Y.; Luong, C.; et al. Development of serine protease inhibitors displaying a multicentered short (<2.3 Å) hydrogen bond binding mode: Inhibitors of urokinase-type plasminogen activator and factor Xa. J. Med. Chem. 2001, 44, 2753–2771. [Google Scholar]
- Hartman, R.L.; McMullen, J.P.; Jensen, K.F. Deciding whether to go with the flow: Evaluating the merits of flow reactors for synthesis. Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, F.; Seeberger, P.H. Highly efficient continuous flow reactions using singlet oxygen as a “green” reagent. Org. Lett. 2011, 13, 5008–5011. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.P.; Elliott, L.D.; Booker-Milburn, K.I. Flow photochemistry: Old light through new windows. Beilstein J. Org. Chem. 2012, 8, 2025–2052. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, F.; O’Brien, A.G.; Götze, S.; Seeberger, P.H.; Hartmann, L. Synthesis of carbohydrate-functionalised sequence-defined oligo(amidoamine)s by photochemical thiol-ene coupling in a continuous flow reactor. Chem. Eur. J. 2013, 19, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- McQuade, D.T.; Seeberger, P.H. Applying flow chemistry: Methods, materials, and multistep synthesis. J. Org. Chem. 2013, 78, 6384–6389. [Google Scholar] [CrossRef] [PubMed]
- Bovonsombat, P.; McNelis, E. Ring halogenations of polyalkylbenzenes with N-halosuccinimide and acid catalysts. Synthesis 1993, 237–241. [Google Scholar] [CrossRef]
- Mahajan, T.; Kumar, L.; Dwivedi, K.; Agarwal, D.D. Efficient and facile chlorination of industrially-important aromatic compounds using NaCl/p-TsOH/NCS in aqueous media. Ind. Eng. Chem. Res. 2012, 51, 3881–3886. [Google Scholar] [CrossRef]
- Stock, R.I.; Nandi, L.G.; Nicoleti, C.R.; Schramm, A.D.S.; Meller, S.L.; Heying, R.S.; Coimbra, D.F.; Andriani, K.F.; Caramori, G.F.; Bortoluzzi, A.J.; et al. Synthesis and solvatochromism of substituted 4-(nitrostyryl)phenolate dyes. J. Org. Chem. 2015, 80, 7971–7983. [Google Scholar] [CrossRef] [PubMed]
- Mangas-Sánchez, J.; Busto, E.; Gotor-Fernández, V.; Gotor, V. Straightforward synthesis of enantiopure 2,3-dihydrobenzofurans by a sequential stereoselective biotransformation and chemical intramolecular cyclization. Org. Lett. 2010, 12, 3498–3501. [Google Scholar] [CrossRef] [PubMed]
- Adimurthy, S.; Ramachandraiah, G.; Bedekar, A.V.; Ghosh, S.; Ranu, B.C.; Ghosh, P.K. Eco-friendly and versatile brominating reagent prepared from a liquid bromine precursor. Green Chem. 2006, 8, 916–922. [Google Scholar] [CrossRef]
- Huang, G.-J.; Bhaskar-Reddy, M.V.; Kuo, P.-C.; Huang, C.-H.; Shih, H.-C.; Lee, E.J.; Yang, M.-L.; Leu, Y.-L.; Wu, T.-S. A Concise synthesis of viscolin, and its anti-inflammatory effects through the suppression of iNOS, COX-2, ERK phosphorylation and proinflammatory cytokines expressions. Eur. J. Med. Chem. 2012, 48, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Environmentally benign electrophilic and radical bromination “on water”: H2O2-HBr system versus N-bromosuccinimide. Tetrahedron 2009, 65, 4429–4439. [Google Scholar] [CrossRef]
- Schmidt, B.; Holter, F. Suzuki-Miyaura cross coupling reactions with phenoldiazonium salts. Org. Biomol. Chem. 2011, 9, 4914–4920. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiev, D.; Saes, B.W.H.; Johnston, H.J.; Boys, S.K.; Healy, A.; Hulme, A.N. Selective and Efficient Generation of ortho-Brominated para-Substituted Phenols in ACS-Grade Methanol. Molecules 2016, 21, 88. https://doi.org/10.3390/molecules21010088
Georgiev D, Saes BWH, Johnston HJ, Boys SK, Healy A, Hulme AN. Selective and Efficient Generation of ortho-Brominated para-Substituted Phenols in ACS-Grade Methanol. Molecules. 2016; 21(1):88. https://doi.org/10.3390/molecules21010088
Chicago/Turabian StyleGeorgiev, David, Bartholomeus W. H. Saes, Heather J. Johnston, Sarah K. Boys, Alan Healy, and Alison N. Hulme. 2016. "Selective and Efficient Generation of ortho-Brominated para-Substituted Phenols in ACS-Grade Methanol" Molecules 21, no. 1: 88. https://doi.org/10.3390/molecules21010088