
Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs

Abstract
:1. Introduction
1.1. Drug-Like Properties
1.2. Chemical Structure Properties and Drug-Like Properties
2. Results and Discussion
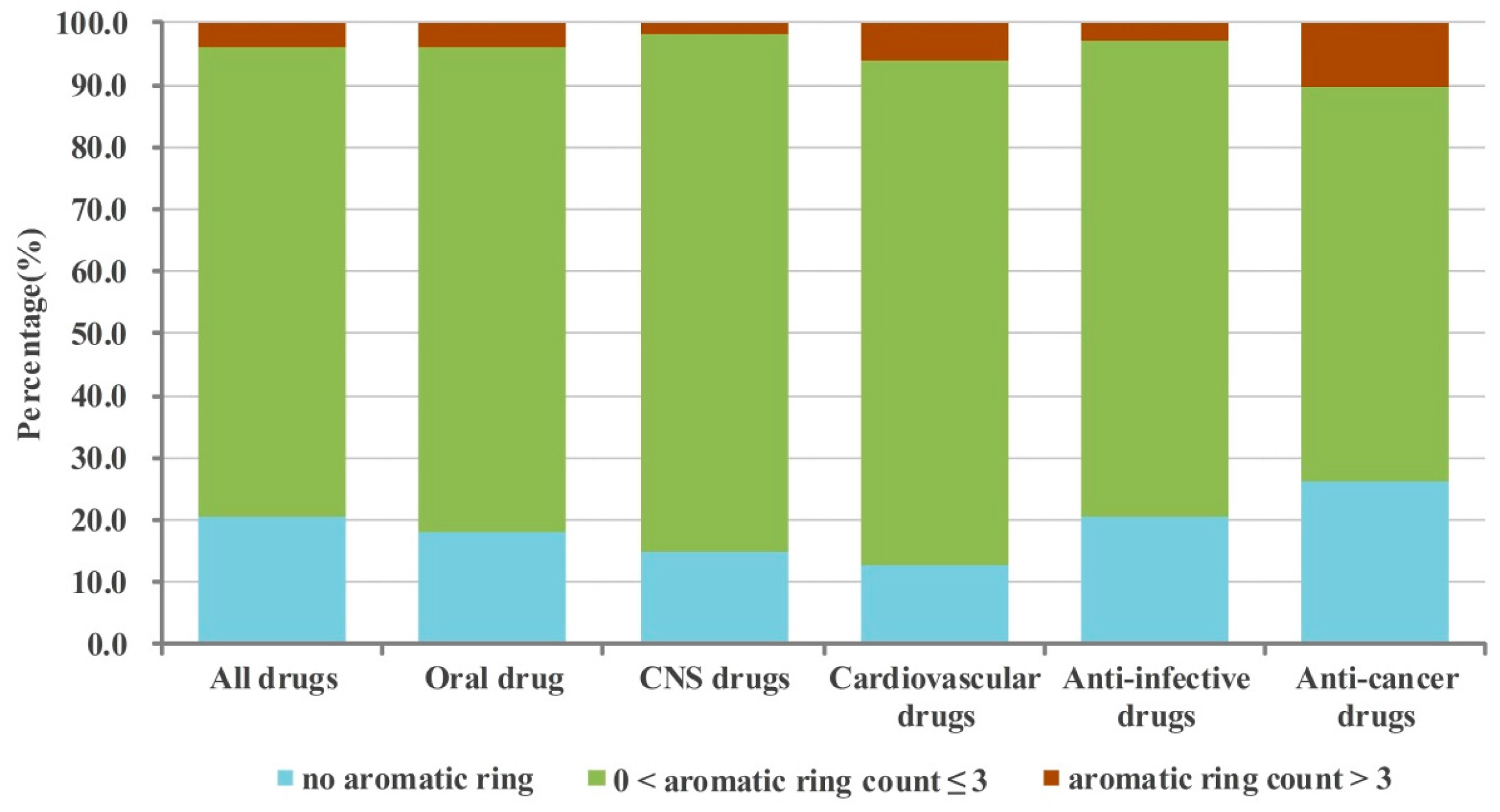
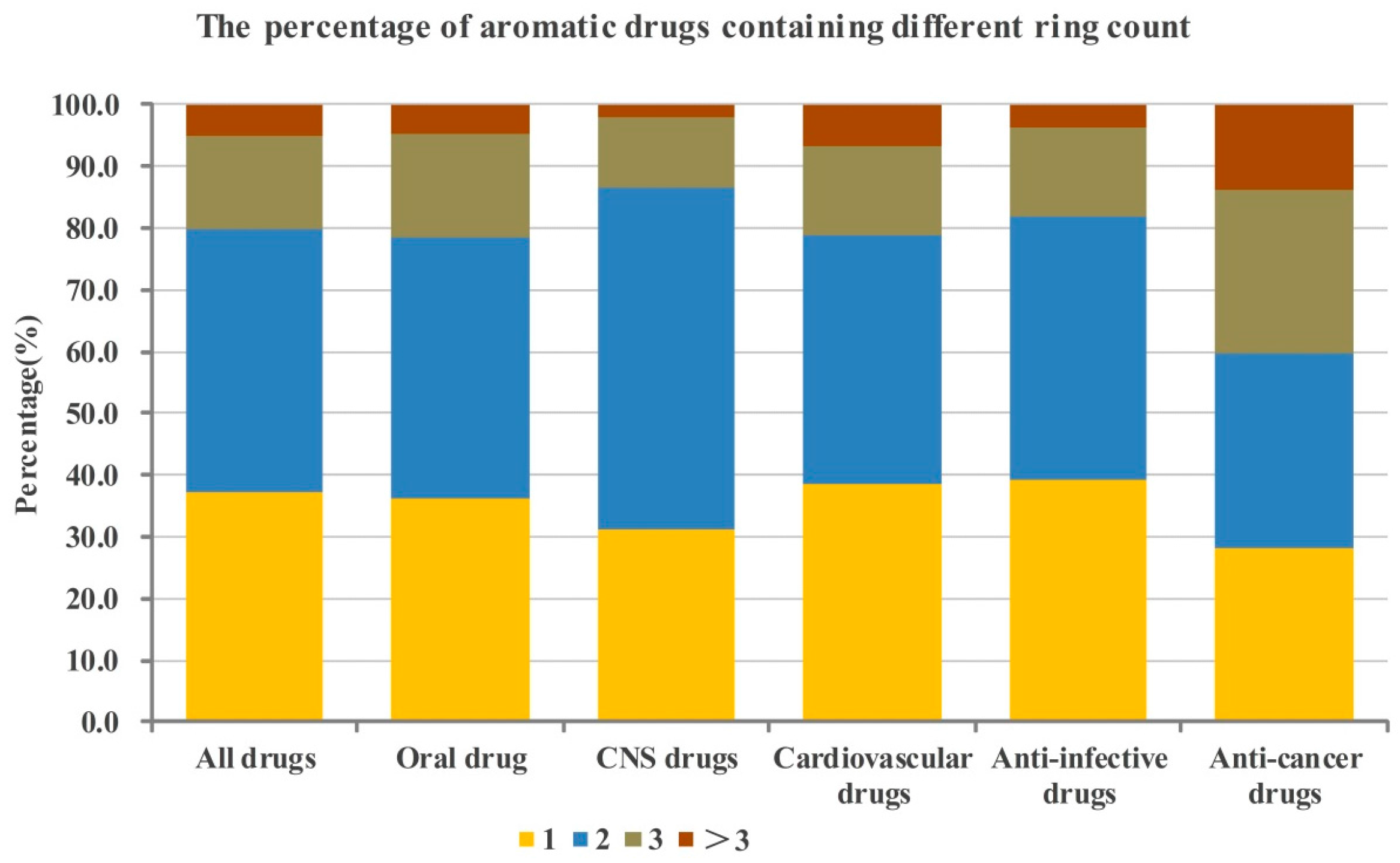
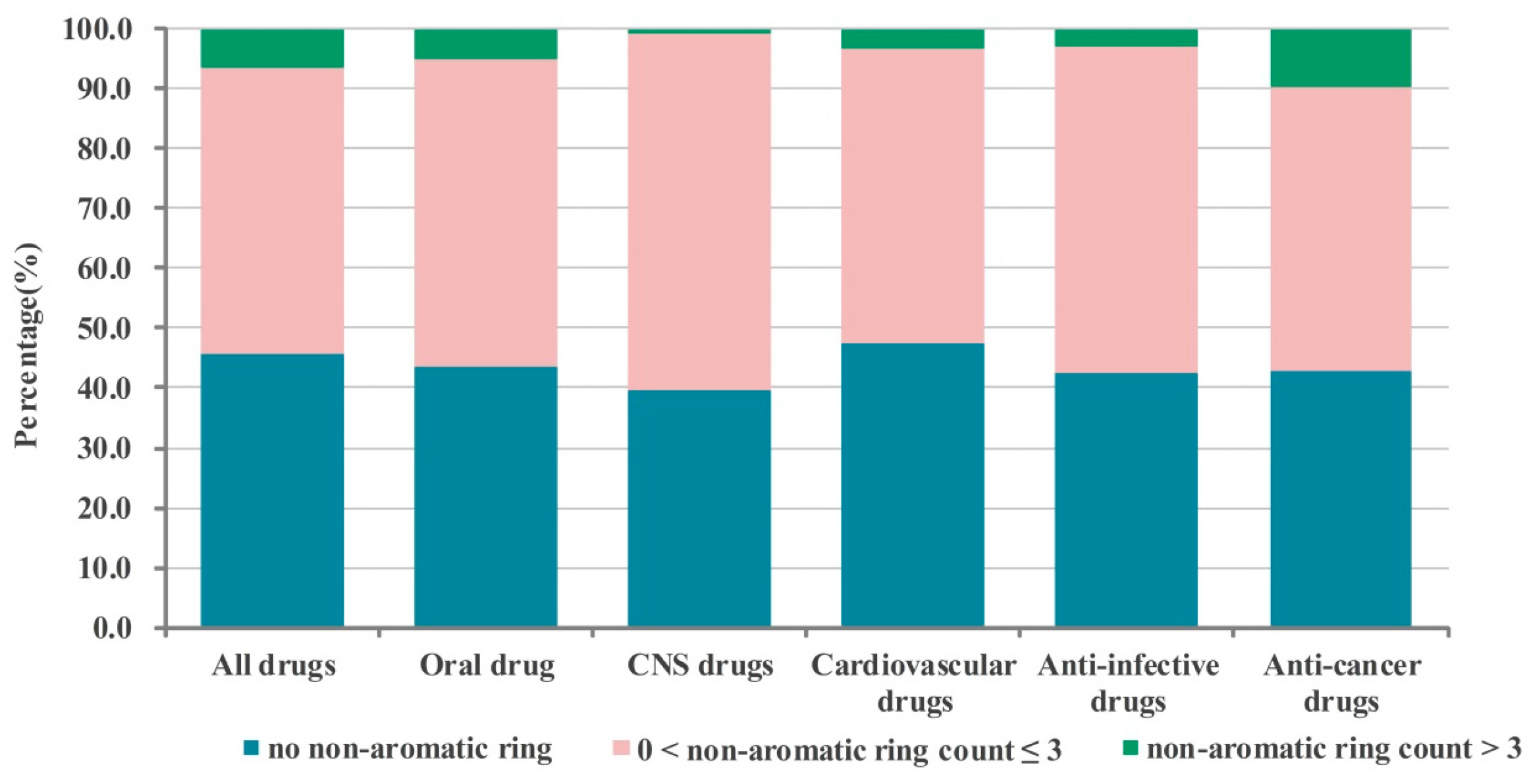
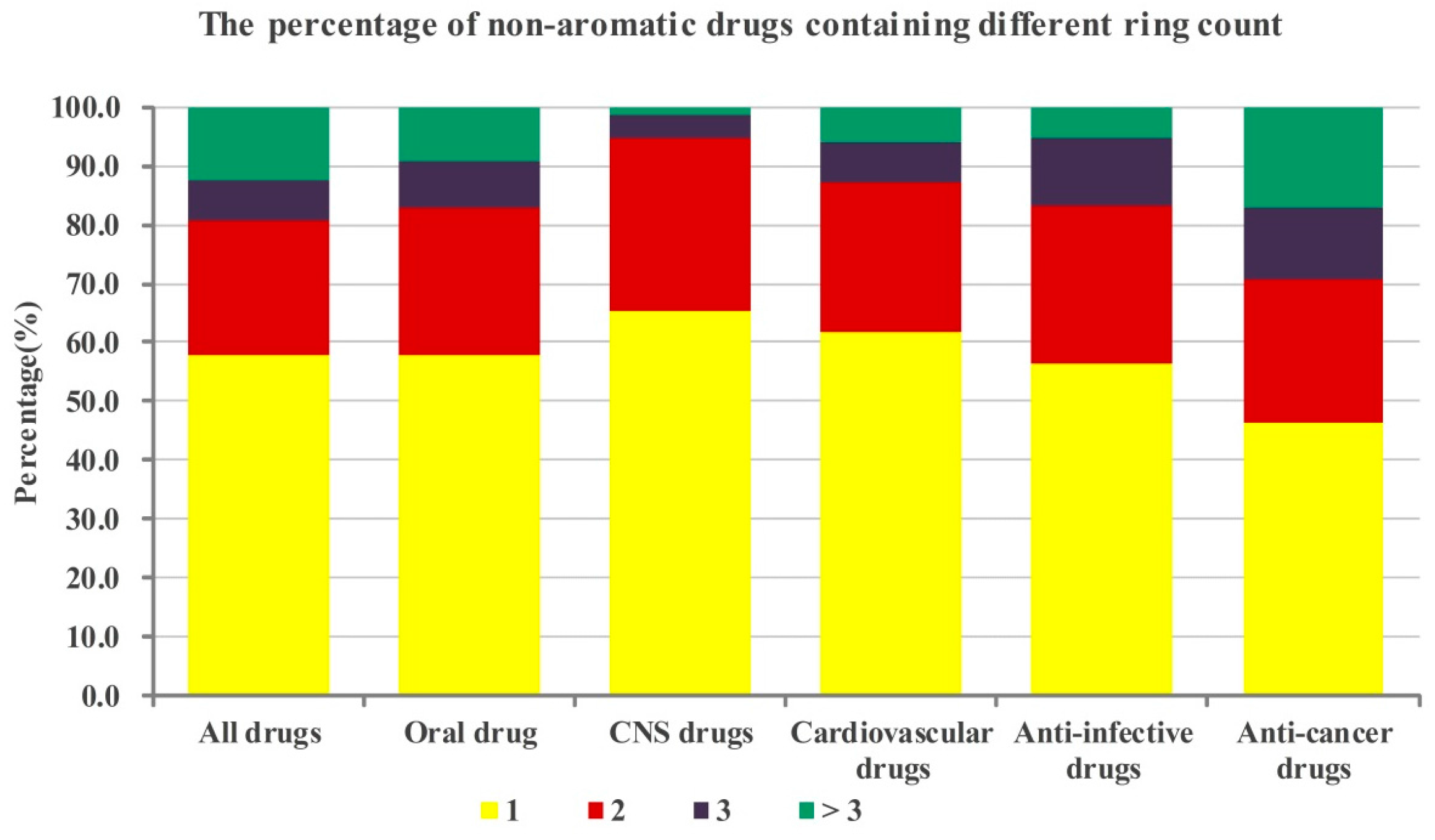
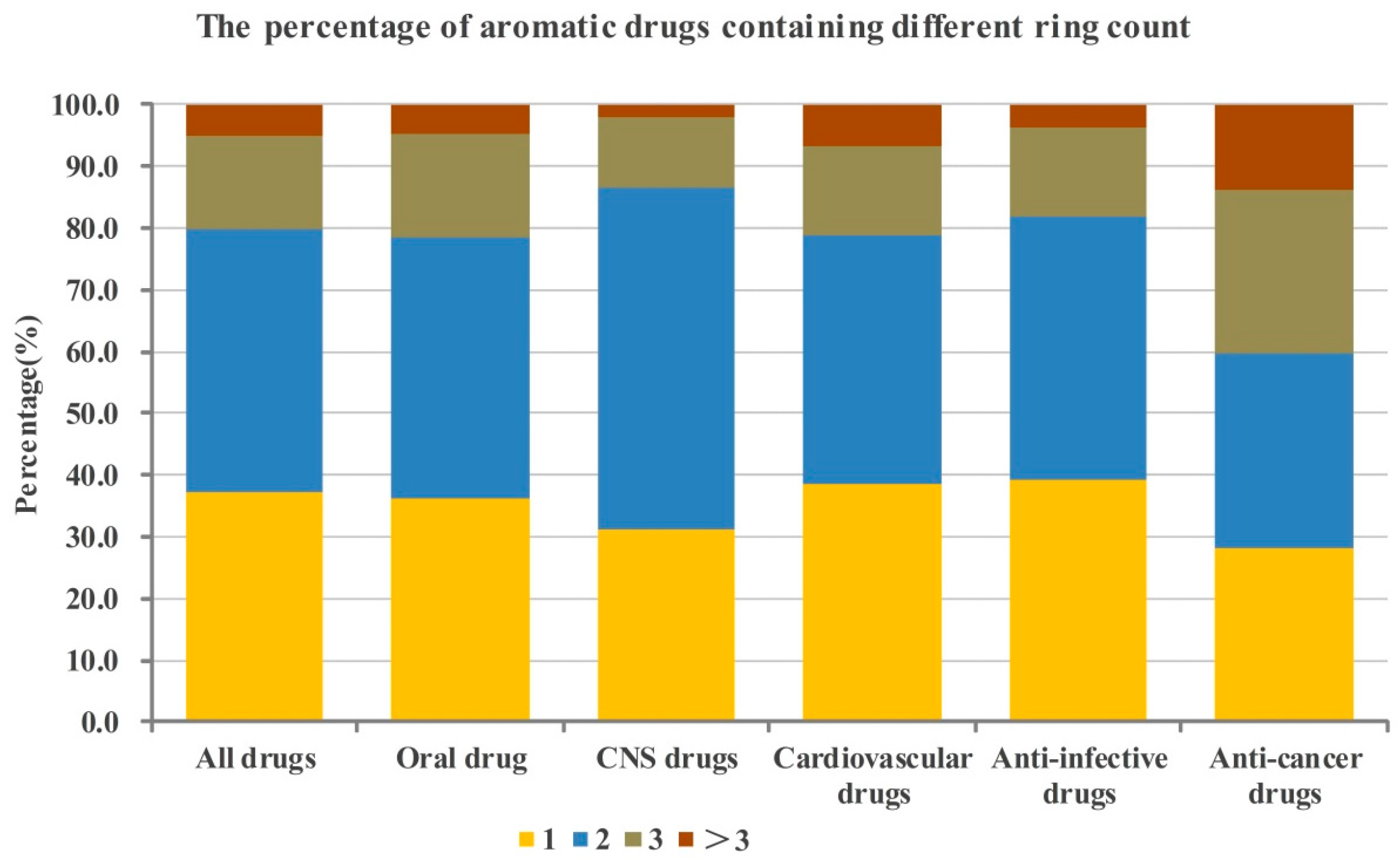
2.1. Numbers of Aromatic and Non-Aromatic Rings Analysis of Our Approved Drugs Database


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Aromatic Rings | All Drugs | Oral Drug | CNS Drugs | Cardiovascular Drugs | Anti-Infective Drugs | Anti-Cancer Drugs |
|---|---|---|---|---|---|---|
| 0 | 1245 | 164 | 138 | 112 | 174 | 108 |
| 1 | 2041 | 313 | 298 | 343 | 281 | 92 |
| 2 | 2341 | 365 | 529 | 360 | 307 | 103 |
| 3 | 826 | 145 | 110 | 129 | 104 | 86 |
| >3 | 272 | 40 | 20 | 60 | 26 | 45 |
| Number of Non-Aromatic Rings | All Drugs | Oral Drug | CNS Drugs | Cardiovascular Drugs | Anti-Infective Drugs | Anti-Cancer Drugs |
|---|---|---|---|---|---|---|
| 0 | 2985 | 435 | 416 | 466 | 372 | 182 |
| 1 | 2164 | 343 | 443 | 332 | 293 | 117 |
| 2 | 854 | 147 | 199 | 138 | 140 | 61 |
| 3 | 258 | 47 | 29 | 35 | 60 | 31 |
| >3 | 464 | 55 | 8 | 33 | 27 | 43 |
| bridge rings | 166 | 24 | 27 | 17 | 13 | 8 |



2.1.1. Numbers of Aromatic Rings Analysis
2.1.2. Numbers of Non-aromatic Rings Analysis
2.1.3. Crossed Analysis of the Numbers of Aromatic and Non-aromatic Rings
| Classes | Number of Rings a | 0 | 1 | 2 | 3 | >3 |
|---|---|---|---|---|---|---|
| All drugs | 0 | 5.8 | 12.1 | 16.5 | 6.6 | 2.3 |
| 1 | 3.9 | 11.1 | 11.8 | 3.6 | 1.1 | |
| 2 | 2.4 | 4.2 | 4.2 | 1.3 | 0.4 | |
| 3 | 0.9 | 1.4 | 1.2 | 0.3 | 0.1 | |
| >3 | 5.2 | 0.9 | 0.3 | 0.3 | 0 | |
| Oral drugs | 0 | 4.5 | 11.1 | 15.9 | 7.5 | 2.4 |
| 1 | 3.6 | 11.6 | 12.4 | 4.1 | 1.0 | |
| 2 | 2.1 | 4.5 | 5.1 | 1.9 | 0.4 | |
| 3 | 1.0 | 2.1 | 1.1 | 0.1 | 0.1 | |
| >3 | 4.4 | 0.5 | 0.2 | 0.2 | 0 | |
| CNS drugs | 0 | 5.3 | 8.8 | 18.3 | 3.8 | 0.8 |
| 1 | 5.2 | 12.0 | 17.4 | 4.2 | 0.7 | |
| 2 | 1.4 | 4.9 | 9.7 | 1.5 | 0.2 | |
| 3 | 0.1 | 0.6 | 1.7 | 0.2 | 0 | |
| >3 | 0.3 | 0.2 | 0.1 | 0.1 | 0.1 | |
| Cardiovascular drugs | 0 | 3.6 | 14.1 | 17.3 | 7.1 | 3.4 |
| 1 | 2.0 | 12.8 | 12.3 | 3.1 | 2.3 | |
| 2 | 2.4 | 5.3 | 4.2 | 1.5 | 0.2 | |
| 3 | 0.5 | 0.9 | 1.2 | 0.9 | 0 | |
| >3 | 2.6 | 0.5 | 0.2 | 0 | 0 | |
| Anti-infective drugs | 0 | 3.3 | 10.5 | 17.3 | 7.8 | 2.1 |
| 1 | 4.4 | 13.0 | 11.9 | 2.7 | 0.3 | |
| 2 | 6.3 | 4.4 | 3.8 | 0.9 | 0.1 | |
| 3 | 3.3 | 2.2 | 0.7 | 0.1 | 0.3 | |
| >3 | 1.9 | 0.9 | 0.2 | 0 | 0 | |
| Anti-cancer drugs | 0 | 11.1 | 9.5 | 7.7 | 7.9 | 5.0 |
| 1 | 6.6 | 6.6 | 6.8 | 5.0 | 1.6 | |
| 2 | 3.2 | 2.3 | 2.5 | 2.9 | 3.0 | |
| 3 | 0.7 | 1.4 | 4.1 | 0.7 | 0.2 | |
| >3 | 2.9 | 1.1 | 2.2 | 3.0 | 0.4 |
| Classes a | Only Aromatic Rings | Only Non-Aromatic Rings | Both Aromatic and Non-Aromatic Rings | No Rings | Bridge Rings |
|---|---|---|---|---|---|
| All drugs | 37.5 (12.1, 16.5) b | 12.4 | 42.0 (11.1, 11.8) c | 5.8 | 2.4 |
| Oral drug | 36.9 (11.1, 15.9) b | 11.1 | 45.2 (11.6, 12.4) c | 4.1 | 2.3 |
| CNS drugs | 31.7 (8.8, 18.3) b | 7.0 | 53.6 (12.0, 17.4) c | 5.3 | 2.4 |
| Cardiovascular drugs | 42.0 (14.1, 17.3) b | 7.3 | 45.3 (12.8, 12.3) c | 3.6 | 1.7 |
| Anti-infective drugs | 37.8 (10.5, 17.3) b | 15.9 | 41.5 (13.0, 11.9) c | 3.3 | 1.4 |
| Anti-cancer drugs | 30.1 (9.5, 7.7, 7.9) d | 13.3 | 43.7 (6.6, 6.8, 5.0) e | 11.1 | 1.8 |
2.2. Numbers of Special Functional Groups Analysis of Our Approved Drugs Database
| Functional Groups | All Drugs | Oral Drug | CNS Drugs | Cardiovascular Drugs | Anti-Infective Drugs | Anti-Cancer Drugs | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 a | 2 a | >2 a | 1 a | 2 a | >2 a | 1 a | 2 a | >2 a | 1 a | 2 a | >2 a | 1 a | 2 a | >2 a | 1 a | 2 a | >2 a | |
| -F | 5.4 | 1.6 | 0.4 | 6.8 | 2.1 | 0.4 | 9.2 | 2.0 | 0 | 2.9 | 1.2 | 0.2 | 6.4 | 2.0 | 1.2 | 6.6 | 1.6 | 0.2 |
| -CF3 | 2.5 | 0.2 | 0.1 | 2.8 | 0.4 | 0 | 3.9 | 0.4 | 0.1 | 2.3 | 0.1 | 0.2 | 0.9 | 0.1 | 0 | 1.8 | 0 | 0 |
| -CN | 2.4 | 0.2 | 0 | 2.1 | 0.5 | 0 | 2.0 | 0 | 0 | 3.8 | 0.3 | 0 | 2.0 | 0.3 | 0 | 1.1 | 0.5 | 0 |
| -NO2 | 2.0 | 0.1 | 0 | 3.3 | 0.1 | 0 | 1.5 | 0 | 0 | 2.5 | 0 | 0 | 5.1 | 0.8 | 0 | 2.0 | 0.5 | 0 |
| -NH2 | 9.3 | 1.1 | 0.5 | 11.1 | 1.2 | 0 | 6.8 | 0.5 | 0 | 7.1 | 0.3 | 0 | 23.2 | 2.7 | 2.5 | 17.6 | 3.2 | 0.5 |
| -OH | 20.0 | 7.2 | 6.1 | 19.1 | 6.2 | 6.5 | 14.0 | 2.9 | 1.2 | 24.1 | 6.2 | 4.8 | 19.0 | 7.2 | 13.3 | 14.3 | 11.3 | 15.8 |
| -SH | 0.3 | 0 | 0 | 0.5 | 0.1 | 0 | 0.1 | 0 | 0 | 0.5 | 0 | 0 | 0 | 0 | 0 | 0.2 | 0 | 0 |
| -CHO | 0.7 | 0 | 0 | 0.7 | 0 | 0 | 0.2 | 0 | 0 | 0.4 | 0 | 0.1 | 1.9 | 0 | 0 | 0.7 | 0 | 0 |
| -COOH | 12.9 | 1.7 | 0.3 | 16.8 | 1.0 | 0.1 | 3.6 | 0.6 | 0.2 | 12.3 | 2.2 | 0 | 26.9 | 2.5 | 0.1 | 7.7 | 2.7 | 0.9 |
| -CONHOH | 0.2 | 0 | 0 | 0.4 | 0 | 0 | 0.1 | 0 | 0 | 0.1 | 0 | 0 | 0 | 0.1 | 0 | 1.1 | 0 | 0 |
| -COOR | 16.1 | 3.6 | 1.0 | 14.1 | 4.5 | 0.8 | 9.4 | 1.3 | 0.2 | 14.7 | 7.6 | 2.0 | 13.6 | 3.5 | 1.3 | 13.1 | 3.8 | 4.8 |
| -CONH2 | 3.5 | 0.1 | 0.1 | 4.5 | 0.1 | 0 | 5.5 | 0.4 | 0 | 3.0 | 0.2 | 0 | 5.2 | 0 | 0 | 3.4 | 0 | 0 |
| -SO3H | 0.7 | 0.2 | 0 | 0.3 | 0.2 | 0 | 0.2 | 0.3 | 0 | 0.3 | 0 | 0 | 1.2 | 0.3 | 0.1 | 0.2 | 0 | 0 |
| -SO2NH2 | 1.3 | 0.1 | 0 | 2.6 | 0.1 | 0 | 0.7 | 0 | 0 | 0.7 | 0 | 0 | 0.9 | 0.1 | 0 | 0.7 | 0 | 0 |
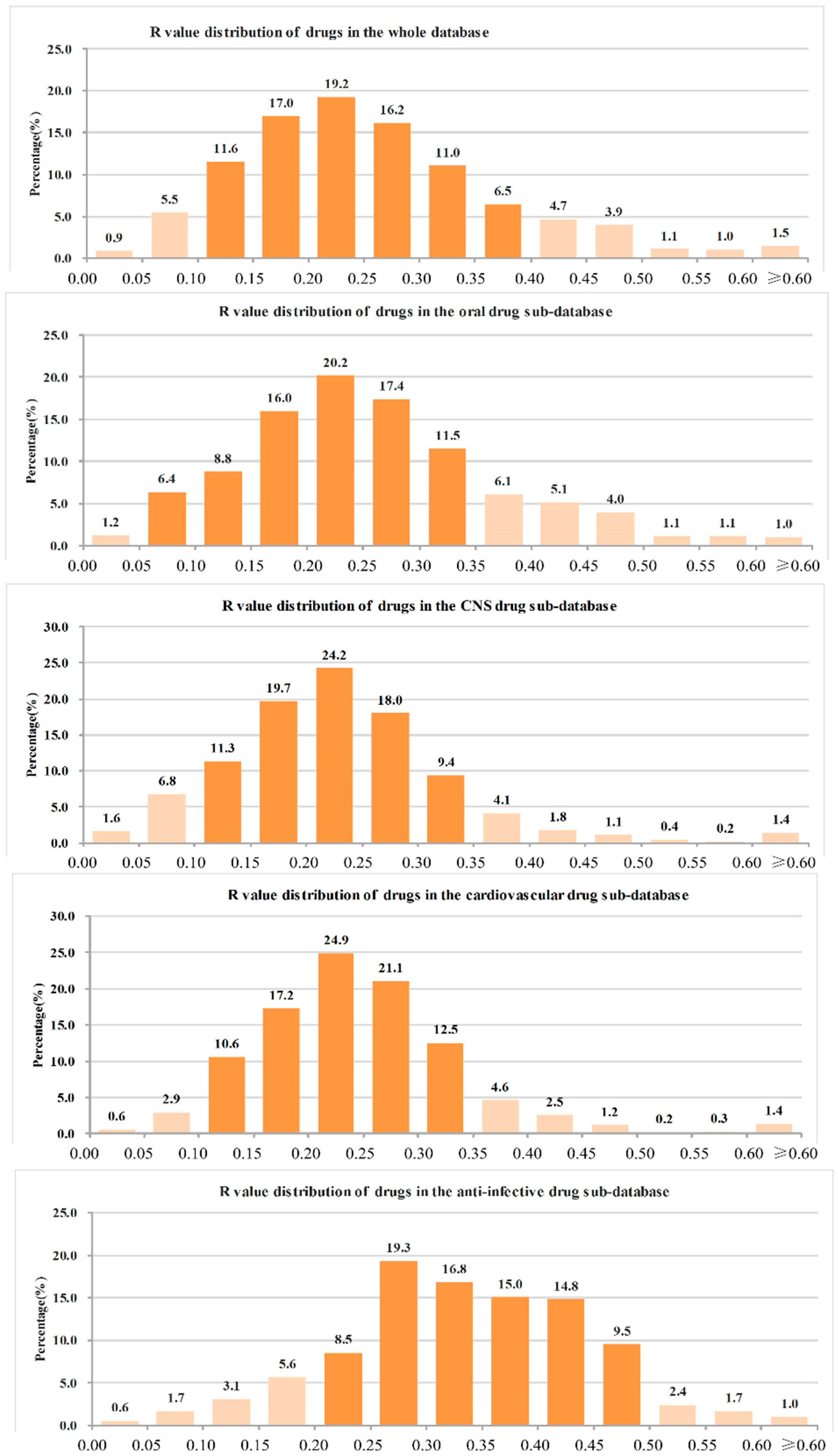
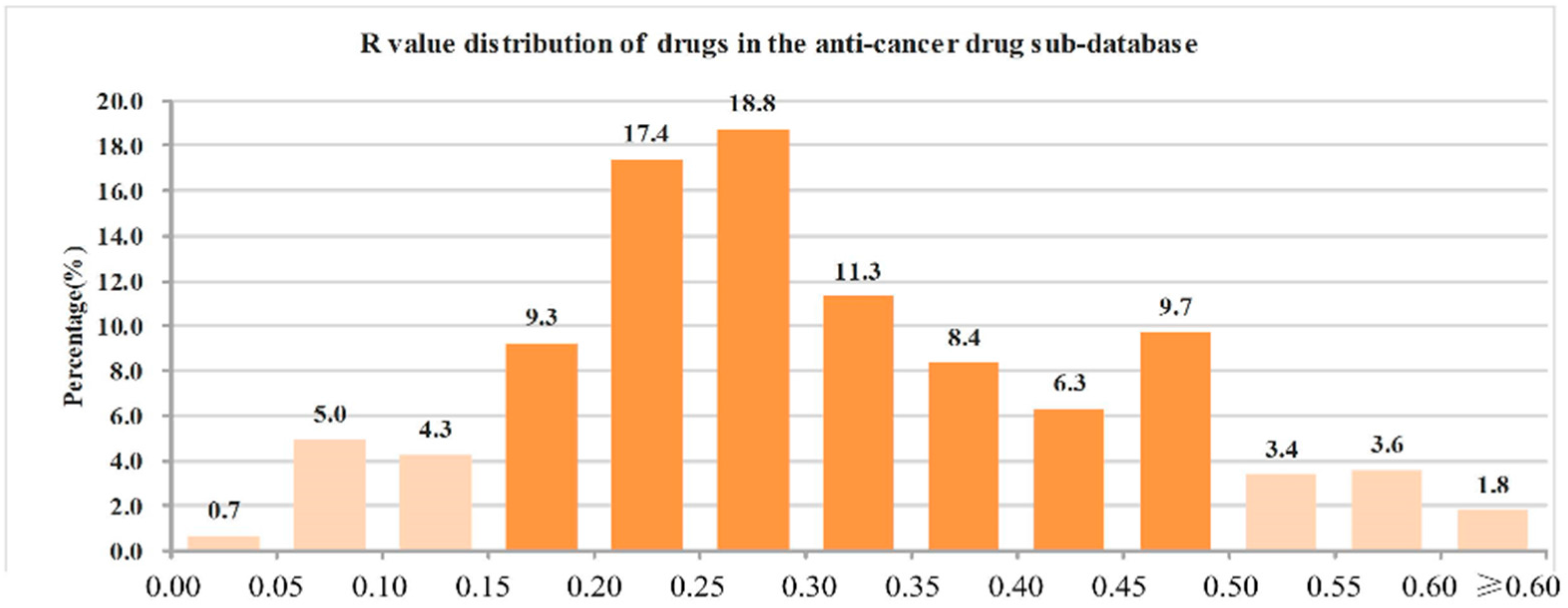
2.3. Heavy Atoms Proportion Analysis of Our Approved Drugs Database
| Classes | Number of Drugs | Mean | Minimum | Maximum |
|---|---|---|---|---|
| All drugs | 6891 | 0.26 | 0 | 1 |
| Oral drugs | 1051 | 0.26 | 0.04 | 0.82 |
| CNS drugs | 1122 | 0.24 | 0.04 | 0.80 |
| Cardiovascular drugs | 1021 | 0.25 | 0.04 | 0.80 |
| Anti-infective drugs | 905 | 0.34 | 0.02 | 0.86 |
| Anti-cancer drugs | 442 | 0.32 | 0 | 0.80 |
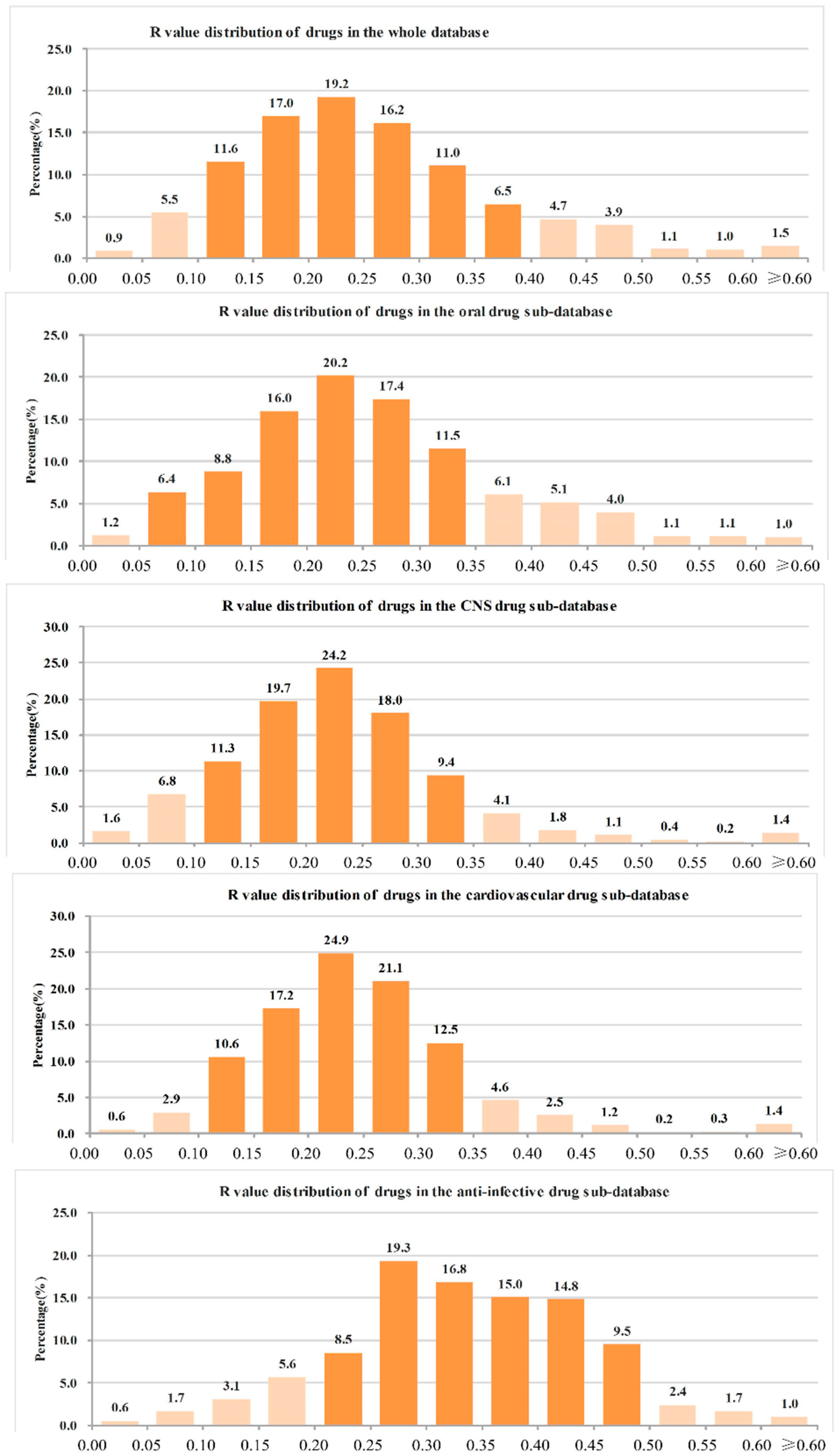

- (1)
- The ranges of R values covering more than 90% of drugs were as follows: all drugs: 0.05–0.45 (91.6%); oral drugs: 0.05–0.45 (91.4%); CNS drugs: 0.05–0.40 (93.5%); cardiovascular drugs: 0.10–0.40 (90.9%); anti-infective drugs: 0.10–0.50 (92.7%); anti-cancer drugs: 0.05–0.50 (90.9%).
- (2)
- The range of R values covering more than 70% of drugs were as follows: all drugs: 0.10–0.35 (75.0%); oral drugs: 0.10–0.35 (73.8%); CNS drugs: 0.10–0.30 (73.3%); cardiovascular drugs: 0.15–0.35 (75.7%); anti-infective drugs: 0.20–0.45 (74.5%); anti-cancer drugs: 0.15–0.45 (71.5%).
- (3)
- The R values of more than 90% of drugs were not more than 0.50, and the details were as follows: all drugs: 96.4%; oral drugs: 96.7%; CNS drugs: 98.0%; cardiovascular drugs: 98.1%; anti-infective drugs: 94.9%; anti-cancer drugs: 91.2%.
- (4)
- The R value of more than 70% of drugs was not more 0.40, and the details were as follows: all drugs: 87.8%; oral drugs: 87.5%; CNS drugs: 95.1%; cardiovascular drugs: 94.4%; anti-infective drugs: 70.6%; anti-cancer drugs: 75.1%.
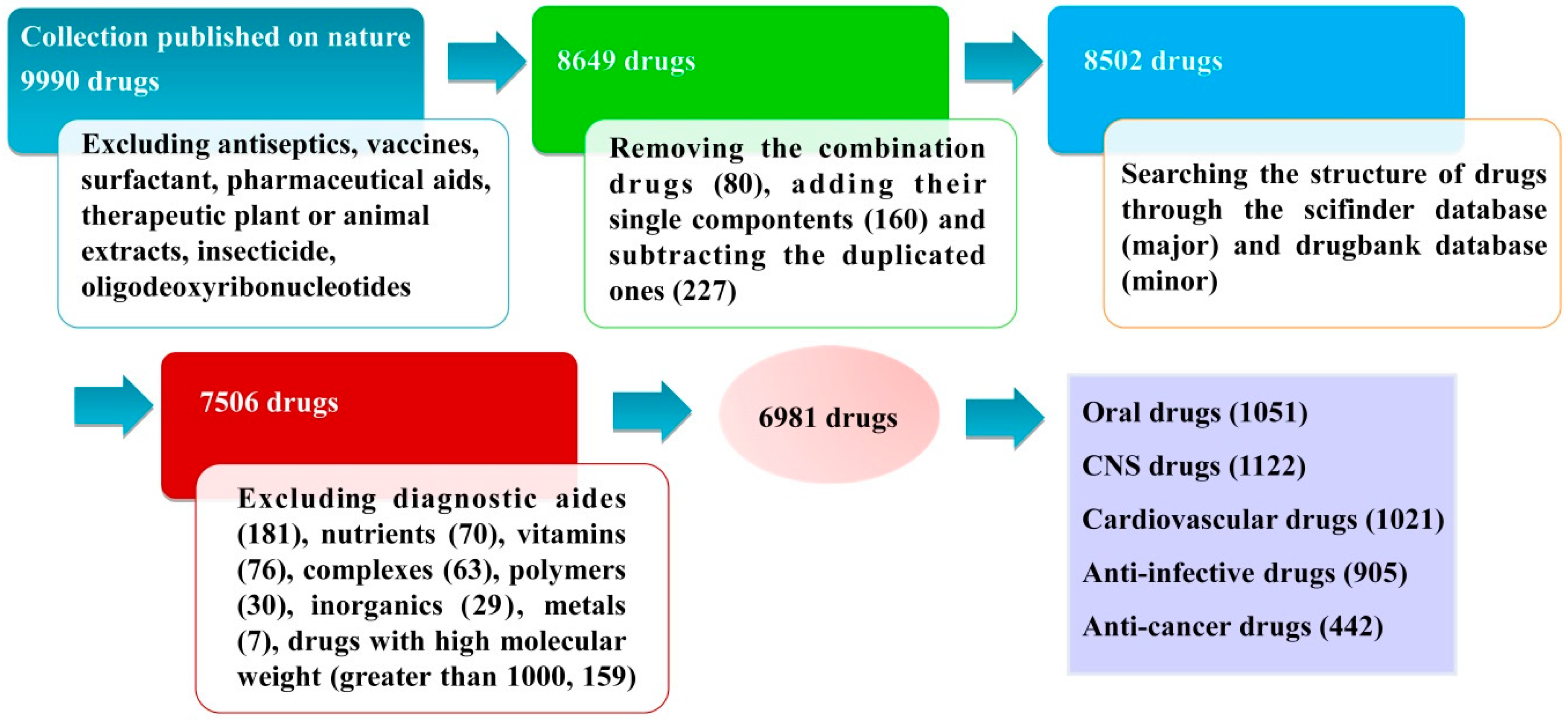
3. Database Construction
3.1. Database Source and Data Collection
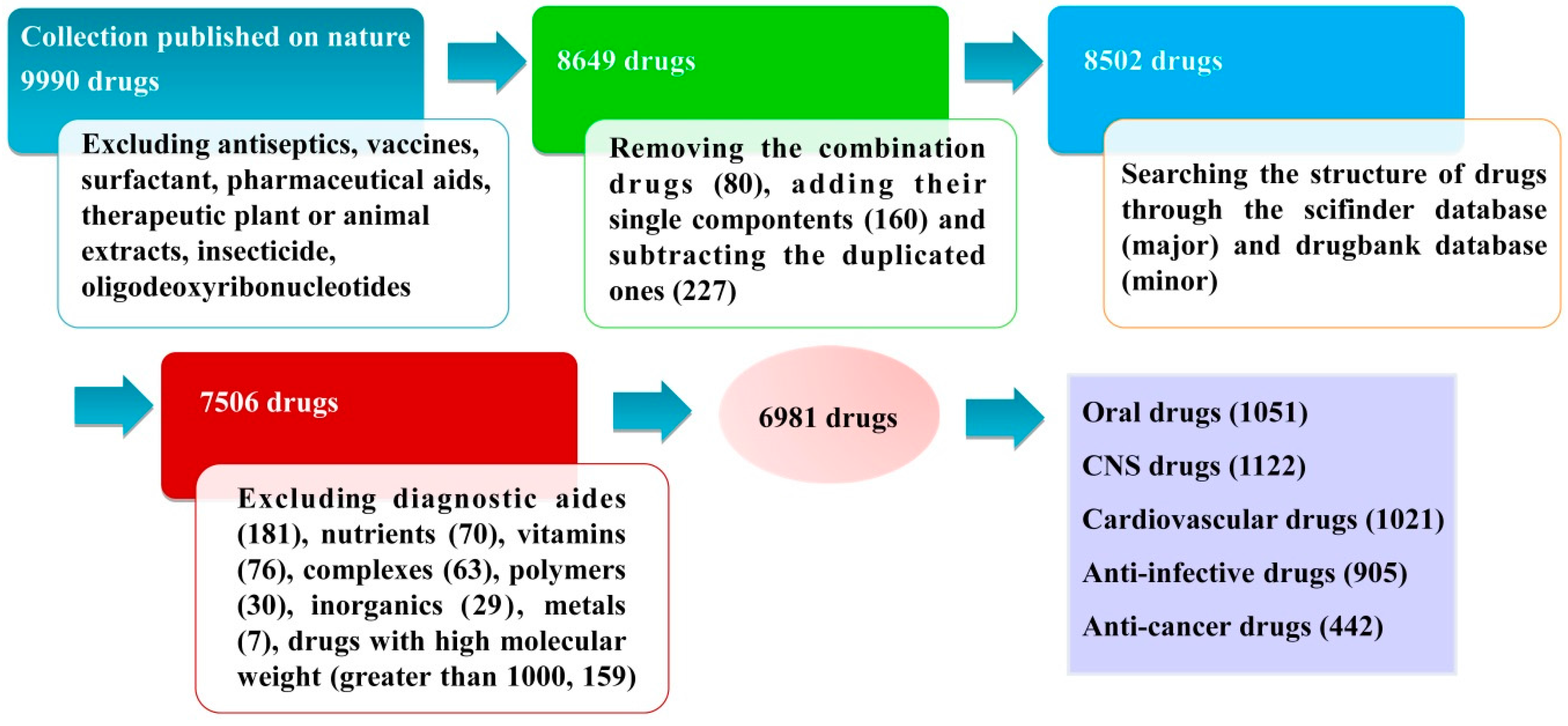
3.2. Database Classification
3.2.1. Oral Drugs
3.2.2. CNS Drugs
3.2.3. Cardiovascular Drugs
3.2.4. Anti-Infective Drugs
3.2.5. Anti-Cancer Drugs
4. Conclusions
| The best numbers of aromatic and non-aromatic rings of candidate drugs are 2 and 1, respectively. |
| The best functional groups of candidate drugs are usually -OH, -COOR and –COOH. |
| The -F functional group is beneficial to CNS drugs. |
| The -NH2 functional group is beneficial to anti-infective drugs and anti-cancer drugs. |
| The best R value interval of candidate drugs is in the range of 0.05–0.50 (preferably 0.10–0.35). |
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Price Waterhouse Coopers (PWC). From Vision to Decision. Pharma 2020 Report; PWC: London, UK, 2012; Available online: http://www.pwc.com/gx/en/industries/pharmaceuticals-life-sciences/pharma-2020/vision-to-decision.html (accessed on 16 November 2015).
- Sugiyama, Y. Druggability: Selecting optimized drug candidates. Drug Discov. Today 2005, 10, 1577–1579. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Borchardt, R.T. Pharmaceutical Profiling in Drug Discovery for Lead Selection; American Association of Pharmaceutical Scientists: Arlington, VA, USA, 2004. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Davis, A.M. Time-related differences in the physical property profiles of oral drugs. J. Med. Chem. 2004, 47, 6338–6348. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A rule of three for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Wenlock, M.C.; Austin, R.P.; Barton, P.; Davis, A.M.; Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 2003, 46, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Ellens, H.; Eddy, E.P.; Lee, C.-P.; Dougherty, P.; Lago, A.; Xiang, J.-N.; Elliott, J.D.; Cheng, H.-Y.; Ohlstein, E.; Smith, P.L. In vitro permeability screening for identification of orally bioavailable endothelin receptor antagonists. Adv. Drug Deliver. Rev. 1997, 23, 99–109. [Google Scholar] [CrossRef]
- Pardridge, W.M. Transport of small molecules through the blood-brain barrier: Biology and methodology. Adv. Drug Deliv. Rev. 1995, 15, 5–36. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Smith, D.A.; Beaumont, K.; Walker, D.K. Property-based design: Optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Campbell, M.; Borthwick, A.D.; Brown, D.; Burns-Kurtis, C.L.; Chan, C.; Convery, M.A.; Crowe, M.C.; Dayal, S.; Diallo, H.; et al. Structure- and property-based design of factor Xa inhibitors: Pyrrolidin-2-ones with acyclic alanyl amides as P4 motifs. Bioorg. Med. Chem. Lett. 2006, 16, 5953–5957. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Borthwick, A.D.; Brown, D.; Burns-Kurtis, C.L.; Campbell, M.; Chan, C.; Charbaut, M.; Chung, C.W.; Convery, M.A.; Kelly, H.A.; et al. Structure and property based design of factor Xa inhibitors: Pyrrolidin-2-ones with biaryl P4 motifs. Bioorg. Med. Chem. Lett. 2008, 18, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Eljack, F.T.; Eden, M.R.; Kazantzi, V.; Qin, X.Y.; El-Halwagi, M.A. Simultaneous process and molecular design—A property based approach. Aiche J. 2007, 53, 1232–1239. [Google Scholar]
- Jiang, H.; Zuo, Y.; Zhang, L.; Li, J.D.; Zhang, A.M.; Li, Y.B.; Yang, X.C. Property-based design: Optimization and characterization of polyvinyl alcohol (PVA) hydrogel and PVA-matrix composite for artificial cornea. J. Mater. Sci. Mater. Med. 2014, 25, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.E.; Alimzhanov, M.; Bao, L.; Block, M.H.; Chuaqui, C.; Cooke, E.L.; Denz, C.R.; Hird, A.; Huang, S.; Larsen, N.A.; et al. Structure and property based design of pyrazolo 1,5-a pyrimidine Inhibitors of CK2 kinase with activity in vivo. ACS Med. Chem. Lett. 2013, 4, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.D.; Wilson, M.W.; Abe, A.; Shu, L.M.; George, C.H.; Kirchhoff, P.; Showalter, H.D.H.; Xiang, J.M.; Keep, R.F.; Shayman, J.A. Property-based design of a glucosylceramide synthase inhibitor that reduces glucosylceramide in the brain. J. Lipid Res. 2012, 53, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Fraley, M.E.; Hoffman, W.F.; Arrington, K.L.; Hungate, R.W.; Hartman, G.D.; McFall, R.C.; Coll, K.E.; Rickert, K.; Thomas, K.A.; McGaughey, G.B. Property-based design of KDR kinase inhibitors. Curr. Med. Chem. 2004, 11, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Balakin, K.V.; Tkachenko, S.E.; Lang, S.A.; Okun, I.; Ivashchenko, A.A.; Savchuk, N.P. Property-based design of GPCR-targeted library. J. Chem. Inf. Comp. Sci. 2002, 42, 1332–1342. [Google Scholar] [CrossRef]
- Abraham, M.H.; Ibrahim, A.; Zissimos, A.M.; Zhao, Y.H.; Comer, J.; Reynolds, D.P. Application of hydrogen bonding calculations in property based drug design. Drug Discov. Today 2002, 7, 1056–1063. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L. Drug-Like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization; Elsevier Inc.: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Bemis, G.W.; Murcko, M.A. Properties of known drugs. 2. Side chains. J. Med. Chem. 1999, 42, 5095–5099. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.X.; Luo, X.M.; Chen, G.; Zhu, W.L.; Shen, J.H.; Chen, K.X.; Jiang, H.L. A new rapid and effective chemistry space filter in recognizing a druglike database. J. Chem. Inf. Model. 2005, 45, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; Macdonald, S.J.F. The impact of aromatic ring count on compound developability—Are too many aromatic rings a liability in drug design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; Macdonald, S.J.; Young, R.J.; Pickett, S.D. The impact of aromatic ring count on compound developability: further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discov. Today 2011, 16, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; Macdonald, S.J.F.; Peace, S.; Pickett, S.D.; Luscombe, C.N. The developability of heteroaromatic and heteroaliphatic rings—Do some have a better pedigree as potential drug molecules than others? MedChemComm 2012, 3, 1062. [Google Scholar] [CrossRef]
- Ward, S.E.; Beswick, P. What does the aromatic ring number mean for drug design? Expert Opin. Drug Dis. 2014, 9, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.D.; MacCoss, M.; Lawson, A.D. Rings in drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; Desai, R.; Reynisson, J. Known drug space as a metric in exploring the boundaries of drug-like chemical space. Eur. J. Med. Chem. 2009, 44, 5006–5011. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Begue, J.P.; Bonnet-Delpon, D. Why fluorine is an invaluable tool in medicinal chemistry. Chim. Oggi. 2005, 23, 3–5. [Google Scholar]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.L. Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J. Fluorine Chem. 2006, 127, 1013–1029. [Google Scholar] [CrossRef]
- Smith, B.R.; Eastman, C.M.; Njardarson, J.T. Beyond C, H, O, and N! analysis of the elemental composition of U.S. FDA approved drug architectures. J. Med. Chem. 2014, 57, 9764–9773. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-mining for sulfur and fluorine: An evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Sullivan, D.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef] [PubMed]
- SciFinder. Available online: https://scifinder.cas.org (accessed on 17 November 2015).
- DrugBank Database. Available online: http://Www.Drugbank.Ca (accessed on 17 November 2015).
- Thomson Reuters Integrity. Available online: https://integrity.thomson-pharma.com (accessed on 17 November 2015).
- Thomson Reuters Cortellis. Available online: https://cortellis.thomsonreuterslifesciences.com (accessed on 17 November 2015).
- Reichel, A. The role of blood-brain barrier studies in the pharmaceutical industry. Curr. Drug Metab. 2006, 7, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, F.; Ni, W.; Xu, X.; Wang, H.; Wang, J.; Ji, M.; Li, J. Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs. Molecules 2016, 21, 75. https://doi.org/10.3390/molecules21010075
Mao F, Ni W, Xu X, Wang H, Wang J, Ji M, Li J. Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs. Molecules. 2016; 21(1):75. https://doi.org/10.3390/molecules21010075
Chicago/Turabian StyleMao, Fei, Wei Ni, Xiang Xu, Hui Wang, Jing Wang, Min Ji, and Jian Li. 2016. "Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs" Molecules 21, no. 1: 75. https://doi.org/10.3390/molecules21010075