
Cross-Species, Amplifiable EST-SSR Markers for Amentotaxus Species Obtained by Next-Generation Sequencing


Abstract
:1. Introduction
2. Results and Discussion
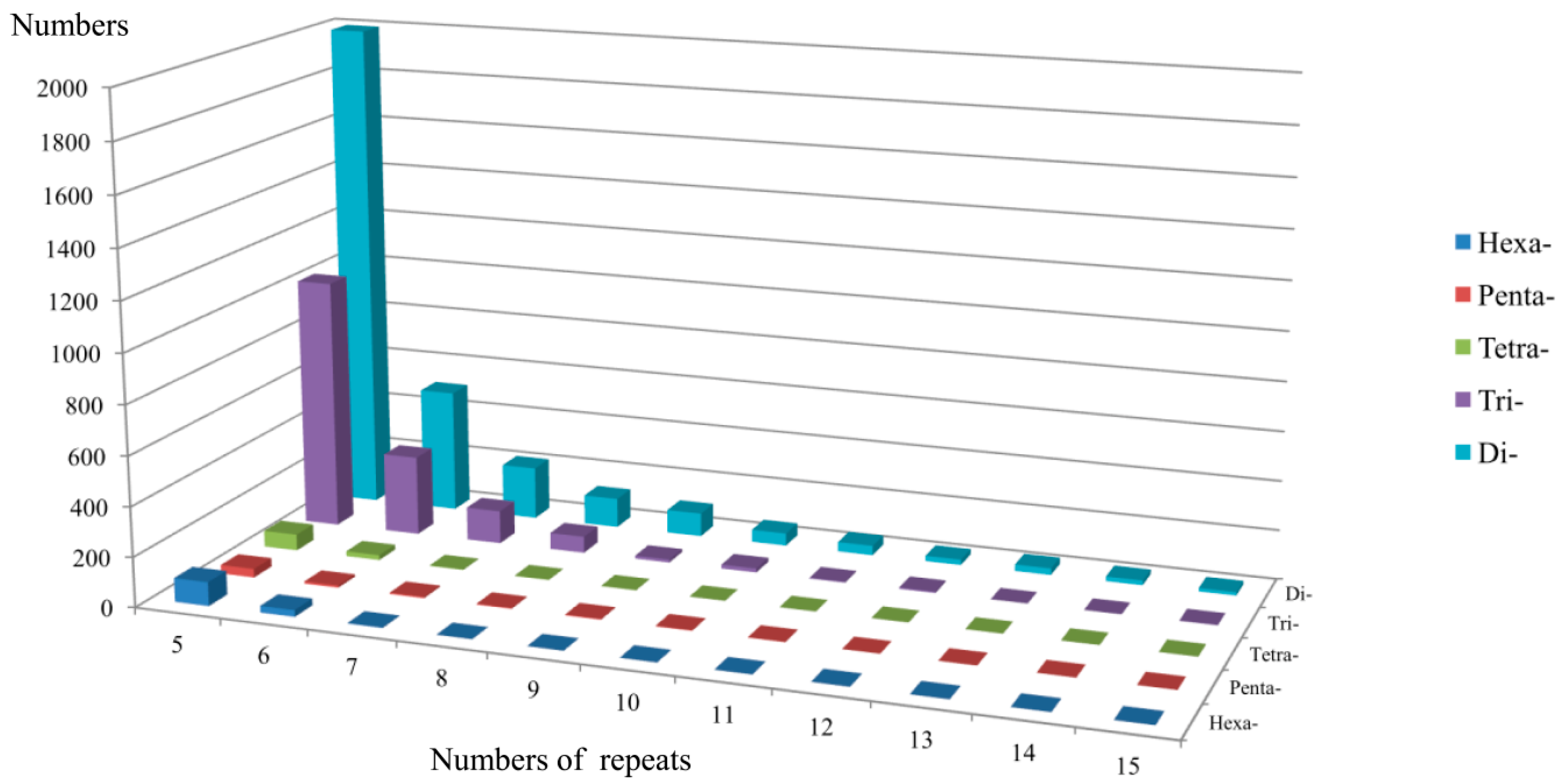
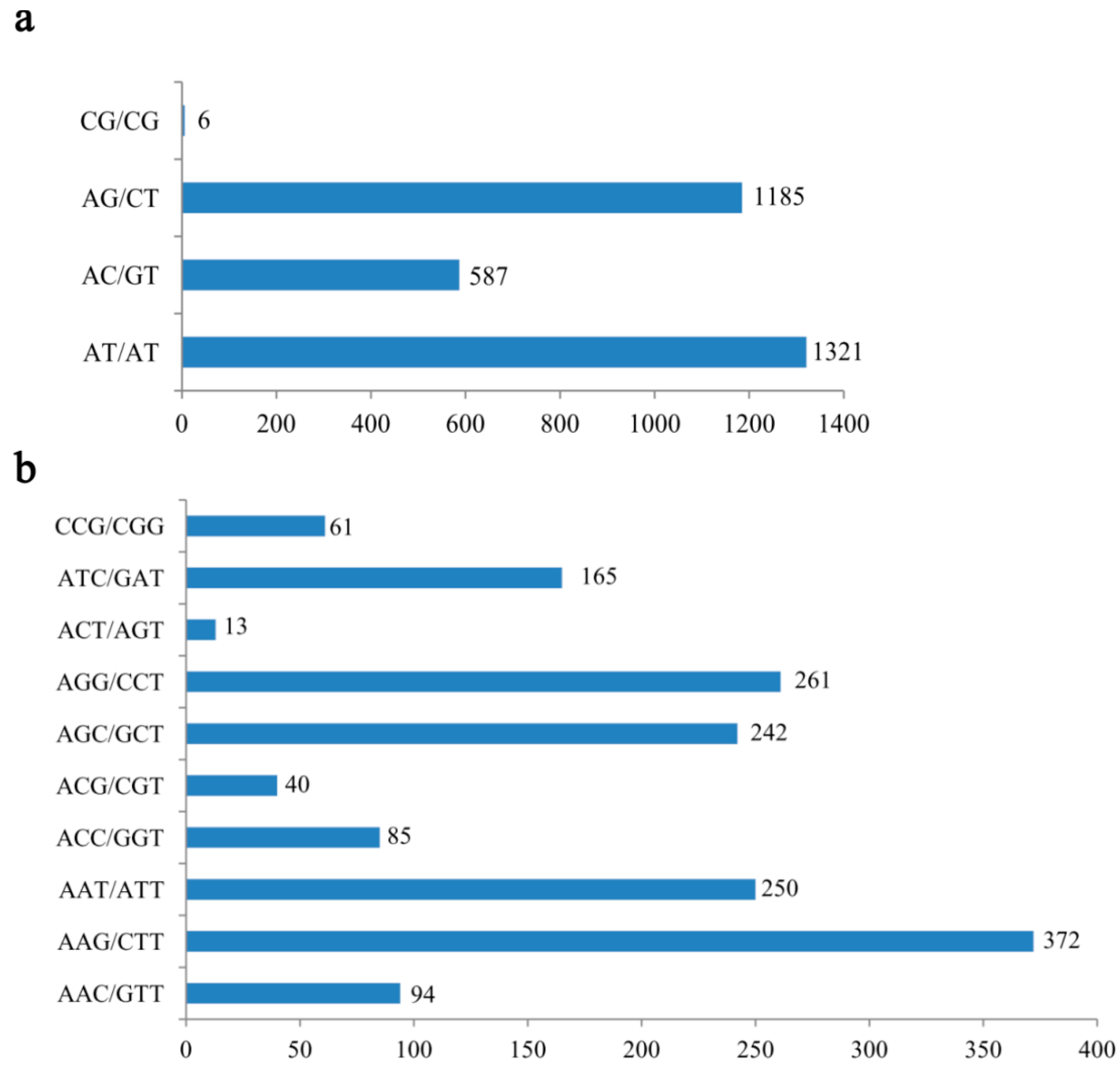
2.1. Frequency Distribution of Various SSRs in the Transcriptome
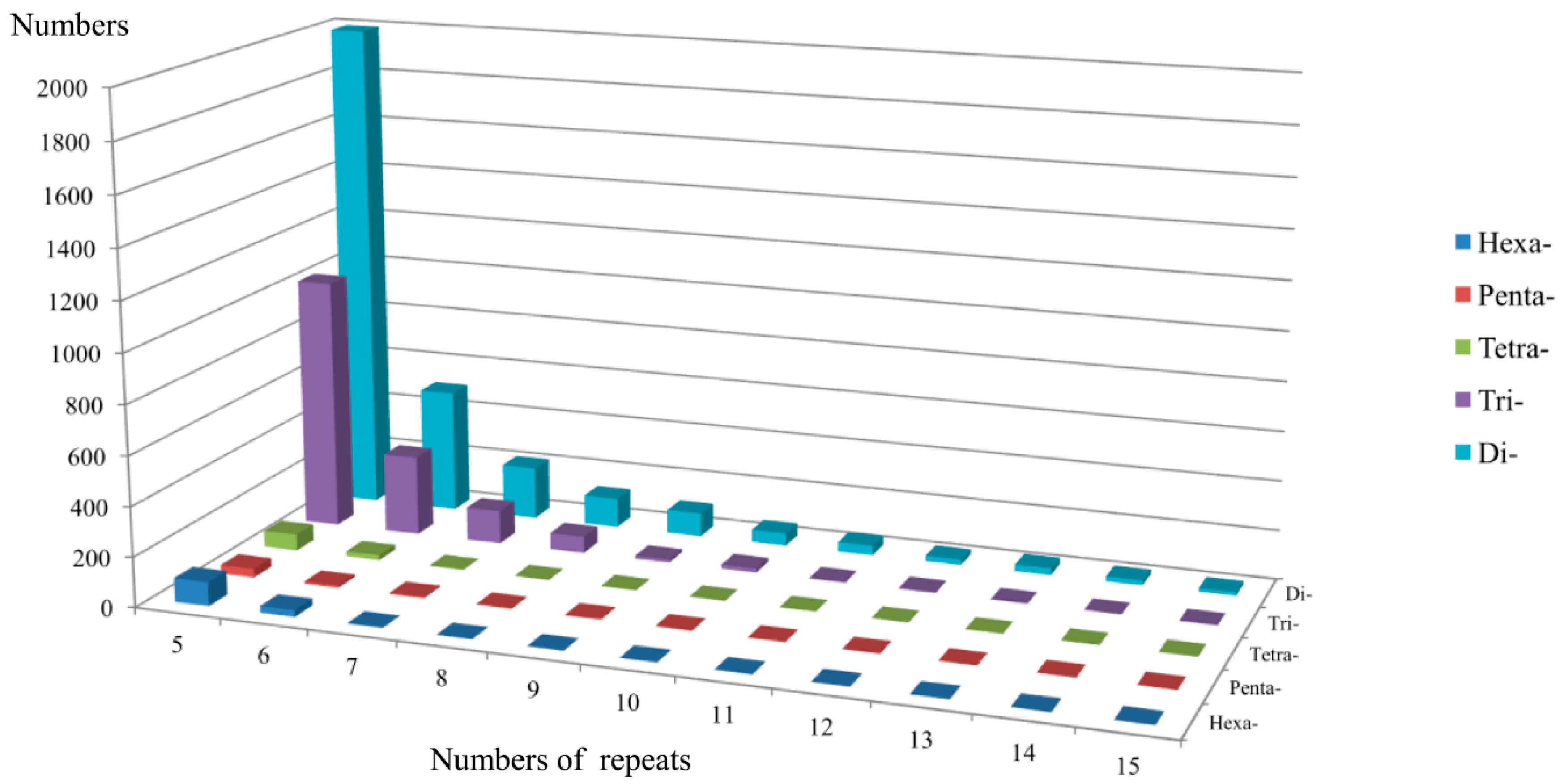
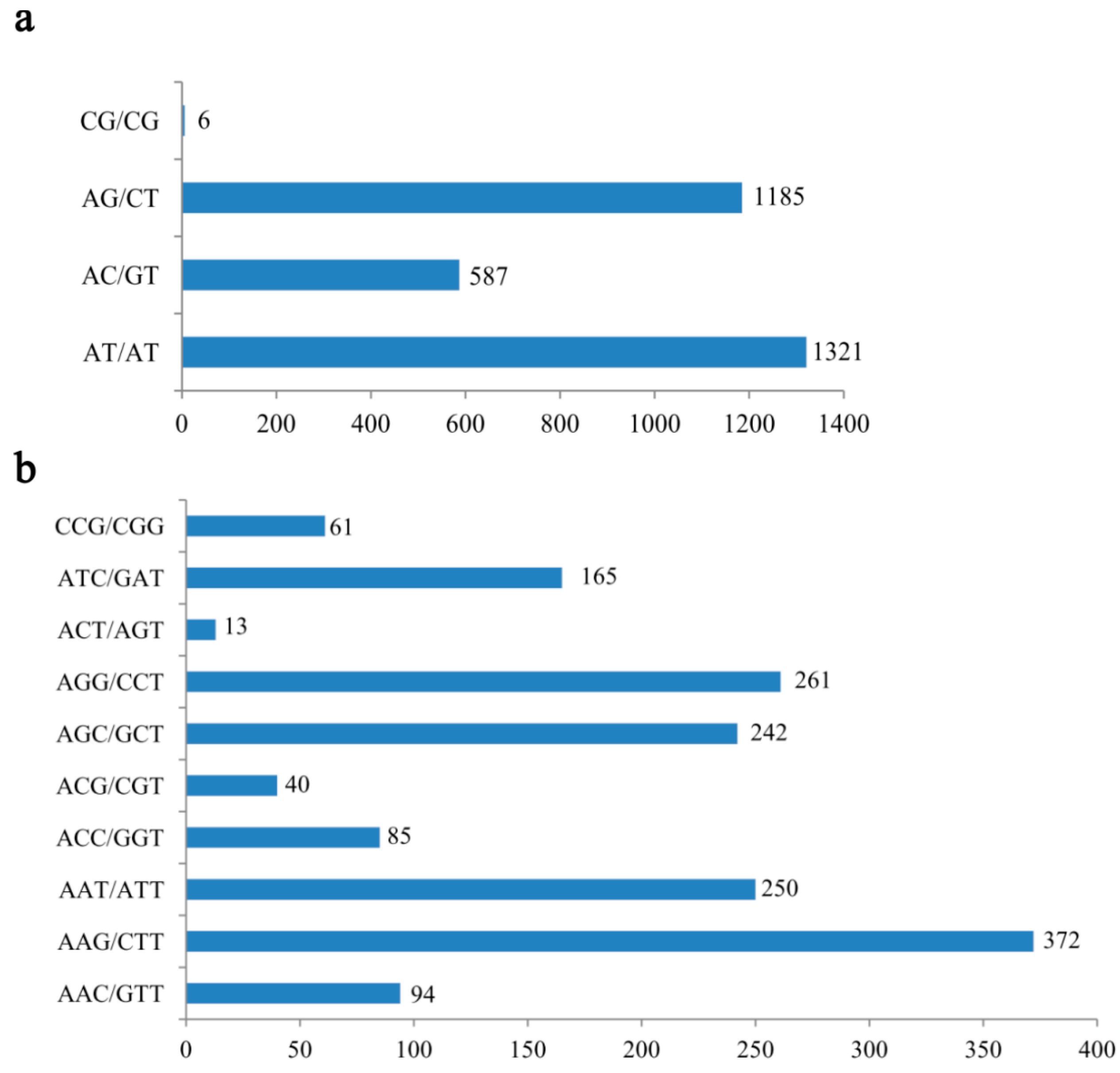
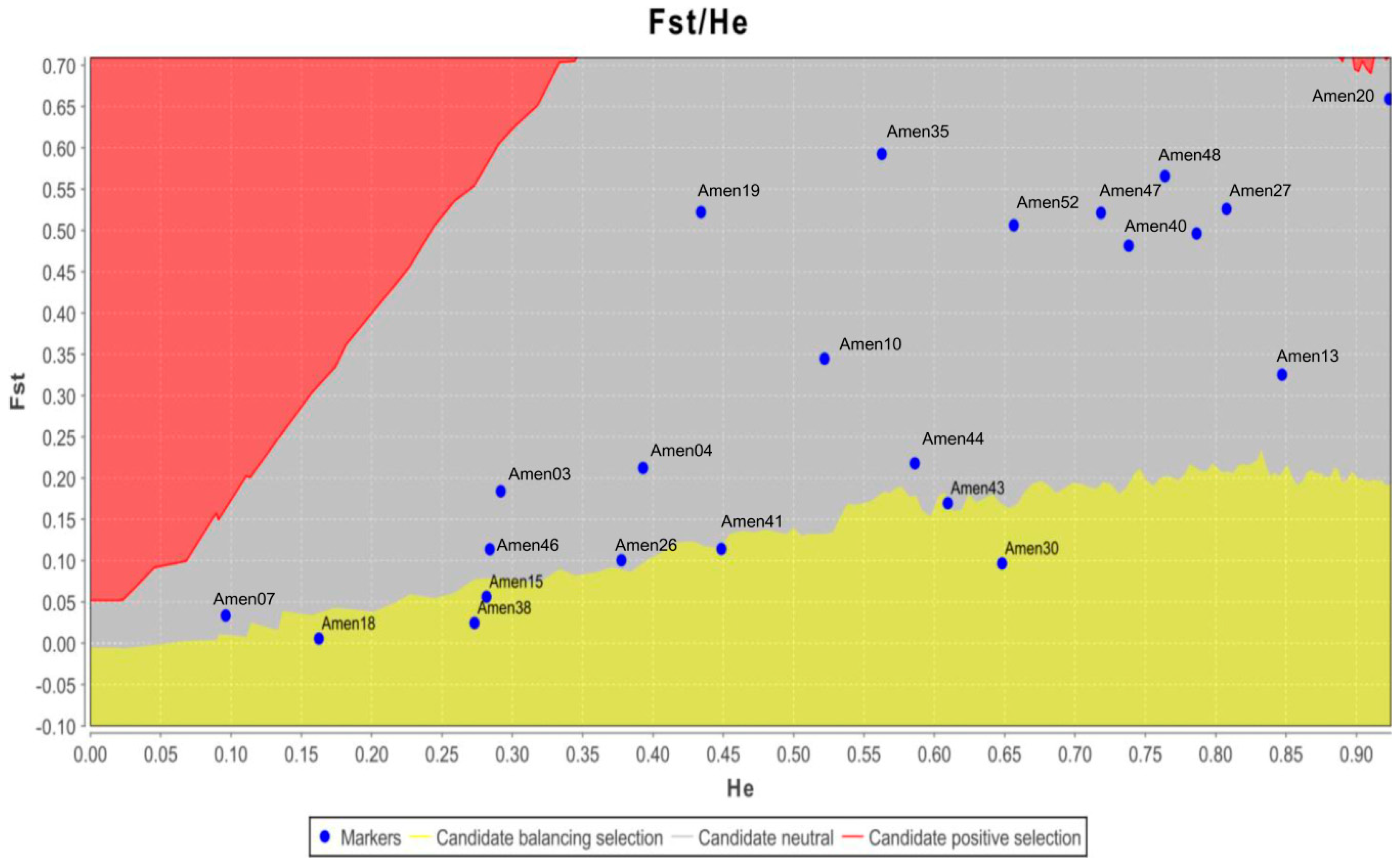
2.2. EST-SSR Marker Polymorphism and Cross-Species Transferability
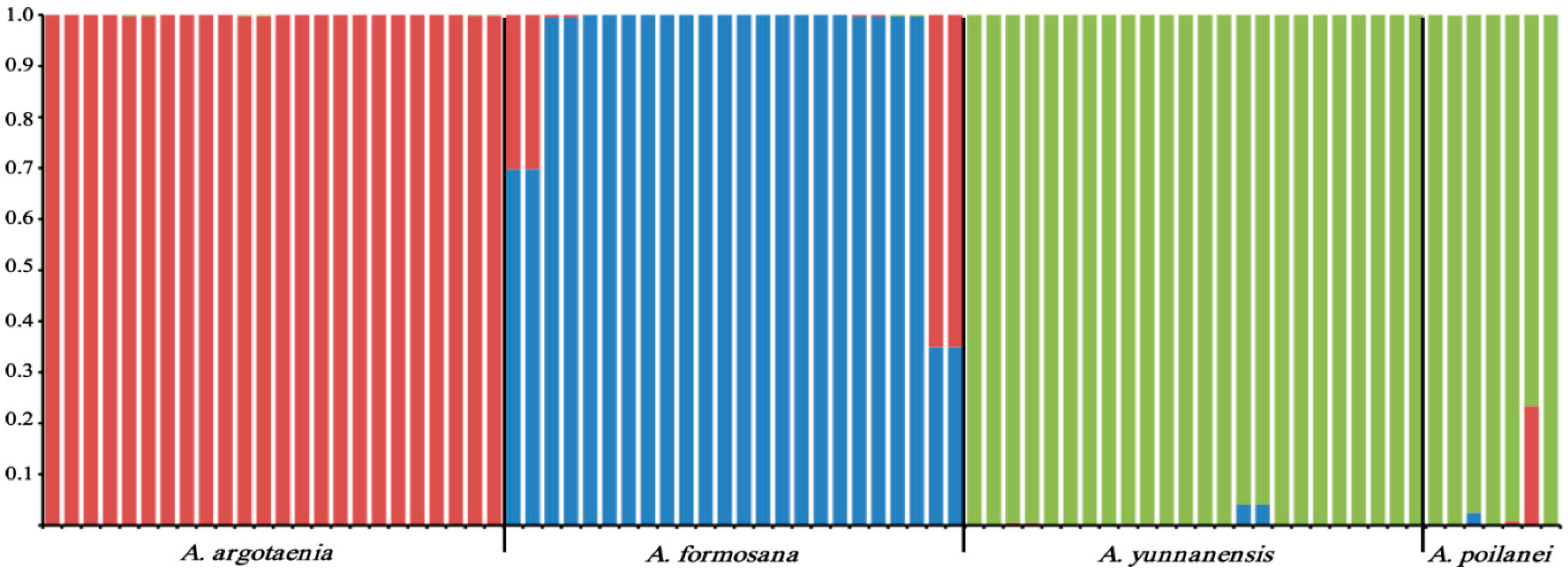
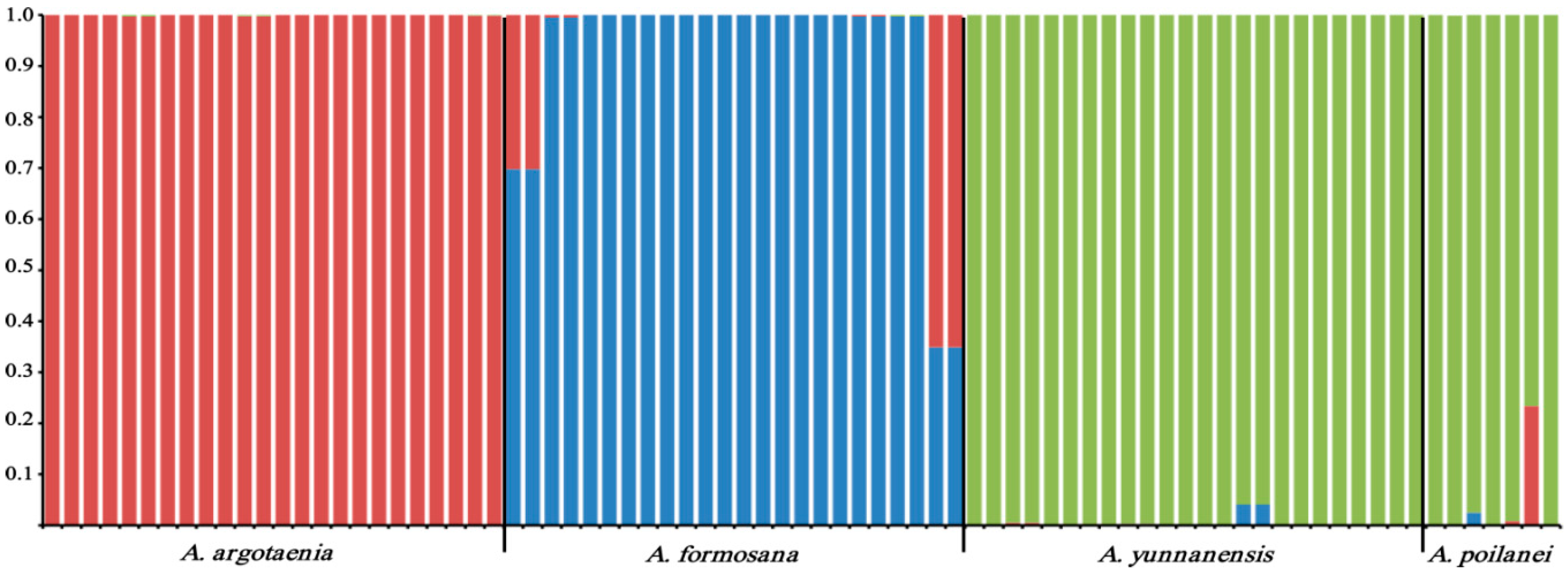
2.3. Genetic Diversity and Population Genetic Structure Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. argotaenia | A. formosana | A. yunnanensis | A. poilanei | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | A | Ho | He | PIC | HWE p-value | A | Ho | He | PIC | HWE p-value | A | Ho | He | PIC | HWE p-value | A | Ho | He | PIC | HWE p-value |
| Amen03 | 2.000 | 0.000 | 0.153 | 0.141 | 0.001 | 1.000 | 0.000 | 0.000 | 0.000 | - | 3.000 | 0.182 | 0.517 | 0.422 | 0.057 | 2.000 | 0.286 | 0.245 | 0.215 | 0.659 |
| Amen04 | 2.000 | 0.167 | 0.500 | 0.375 | 0.021 | 1.000 | 0.000 | 0.000 | 0.000 | - | 3.000 | 0.417 | 0.344 | 0.307 | 0.842 | 2.000 | 0.429 | 0.337 | 0.280 | 0.471 |
| Amen07 | 2.000 | 0.250 | 0.219 | 0.195 | 0.621 | 1.000 | 0.000 | 0.000 | 0.000 | - | 2.000 | 0.167 | 0.153 | 0.141 | 0.753 | 1.000 | 0.000 | 0.000 | 0.000 | - |
| Amen10 | 2.000 | 0.167 | 0.375 | 0.305 | 0.054 | 1.000 | 0.000 | 0.000 | 0.000 | - | 2.000 | 0.100 | 0.375 | 0.305 | 0.020 | 2.000 | 0.833 | 0.486 | 0.368 | 0.080 |
| Amen13 | 2.000 | 0.909 | 0.496 | 0.373 | 0.006 | 4.000 | 0.667 | 0.543 | 0.480 | 0.003 | 4.000 | 0.917 | 0.740 | 0.692 | 0.000 | 2.000 | 1.000 | 0.500 | 0.375 | 0.008 |
| Amen15 | 3.000 | 0.500 | 0.406 | 0.371 | 0.721 | 1.000 | 0.000 | 0.000 | 0.000 | - | 2.000 | 0.500 | 0.375 | 0.305 | 0.248 | 3.000 | 0.286 | 0.255 | 0.240 | 0.978 |
| Amen18 | 2.000 | 0.083 | 0.080 | 0.077 | 0.880 | 2.000 | 0.167 | 0.375 | 0.305 | 0.174 | 1.000 | 0.000 | 0.000 | 0.000 | - | 2.000 | 0.143 | 0.133 | 0.124 | 0.839 |
| Amen19 | 3.000 | 0.333 | 0.500 | 0.449 | 0.000 | 2.000 | 0.000 | 0.219 | 0.195 | 0.005 | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - |
| Amen20 | 2.000 | 0.417 | 0.413 | 0.328 | 0.977 | 2.000 | 0.000 | 0.153 | 0.141 | 0.001 | 1.000 | 0.000 | 0.000 | 0.000 | - | 3.000 | 0.000 | 0.571 | 0.501 | 0.003 |
| Amen24 | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - |
| Amen25 | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - |
| Amen26 | 2.000 | 0.333 | 0.278 | 0.239 | 0.488 | 2.000 | 0.083 | 0.080 | 0.077 | 0.880 | 3.000 | 0.583 | 0.434 | 0.369 | 0.565 | 3.000 | 0.857 | 0.541 | 0.453 | 0.268 |
| Amen27 | 2.000 | 0.000 | 0.500 | 0.375 | 0.001 | 2.000 | 0.000 | 0.278 | 0.239 | 0.001 | 1.000 | 0.000 | 0.000 | 0.000 | - | 3.000 | 0.286 | 0.653 | 0.580 | 0.066 |
| Amen30 | 4.000 | 0.750 | 0.670 | 0.606 | 0.562 | 2.000 | 0.000 | 0.494 | 0.372 | 0.003 | 4.000 | 0.500 | 0.618 | 0.562 | 0.174 | 3.000 | 0.429 | 0.449 | 0.406 | 0.497 |
| Amen35 | 2.000 | 0.000 | 0.444 | 0.346 | 0.001 | 2.000 | 0.000 | 0.165 | 0.152 | 0.001 | 3.000 | 0.250 | 0.226 | 0.212 | 0.970 | 1.000 | 0.000 | 0.000 | 0.000 | - |
| Amen38 | 2.000 | 0.091 | 0.351 | 0.290 | 0.014 | 2.000 | 0.000 | 0.298 | 0.253 | 0.001 | 1.000 | 0.000 | 0.000 | 0.000 | - | 3.000 | 0.429 | 0.357 | 0.325 | 0.914 |
| Amen40 | 2.000 | 0.636 | 0.434 | 0.340 | 0.122 | 1.000 | 0.000 | 0.000 | 0.000 | - | 3.000 | 0.250 | 0.538 | 0.432 | 0.133 | 3.000 | 0.429 | 0.500 | 0.427 | 0.808 |
| Amen41 | 3.000 | 0.667 | 0.486 | 0.424 | 0.392 | 1.000 | 0.000 | 0.000 | 0.000 | - | 4.000 | 0.714 | 0.541 | 0.502 | 0.904 | 2.000 | 0.857 | 0.490 | 0.370 | 0.047 |
| Amen42 | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - | 1.000 | 0.000 | 0.000 | 0.000 | - |
| Amen43 | 4.000 | 0.500 | 0.535 | 0.498 | 0.075 | 2.000 | 0.100 | 0.255 | 0.222 | 0.055 | 3.000 | 0.545 | 0.533 | 0.432 | 0.838 | 3.000 | 0.714 | 0.602 | 0.523 | 0.214 |
| Amen44 | 2.000 | 0.167 | 0.278 | 0.239 | 0.166 | 2.000 | 0.000 | 0.486 | 0.368 | 0.001 | 4.000 | 0.667 | 0.517 | 0.482 | 0.809 | 2.000 | 0.571 | 0.408 | 0.325 | 0.290 |
| Amen46 | 2.000 | 0.083 | 0.469 | 0.359 | 0.004 | 1.000 | 0.000 | 0.000 | 0.000 | - | 2.000 | 0.182 | 0.165 | 0.152 | 0.740 | 2.000 | 0.429 | 0.337 | 0.280 | 0.471 |
| Amen47 | 2.000 | 0.364 | 0.397 | 0.318 | 0.782 | 1.000 | 0.000 | 0.000 | 0.000 | - | 4.000 | 0.200 | 0.575 | 0.526 | 0.034 | 2.000 | 0.286 | 0.245 | 0.215 | 0.659 |
| Amen48 | 2.000 | 0.000 | 0.500 | 0.375 | 0.001 | 2.000 | 0.200 | 0.180 | 0.164 | 0.725 | 1.000 | 0.000 | 0.000 | 0.000 | - | 2.000 | 0.000 | 0.490 | 0.370 | 0.008 |
| Amen50 | 3.000 | 0.000 | 0.406 | 0.371 | 0.001 | 2.000 | 0.000 | 0.375 | 0.305 | 0.046 | 3.000 | 0.000 | 0.594 | 0.511 | 0.001 | 2.000 | 0.000 | 0.375 | 0.305 | 0.046 |
| Amen52 | 2.000 | 0.000 | 0.165 | 0.152 | 0.001 | 4.000 | 0.000 | 0.678 | 0.623 | 0.000 | 2.000 | 0.000 | 0.444 | 0.346 | 0.001 | 1.000 | 0.000 | 0.000 | 0.000 | - |
| Mean a | 2.348 ± 0.198 | 0.279 ± 0.057 | 0.394 ± 0.029 | 0.328 ± 0.087 | - | 1.783 ± 0.177 | 0.053 ± 0.030 | 0.1993 ± 0.044 | 0.1693 ± 0.145 | - | 2.478 ± 0.234 | 0.2683 ± 0.058 | 0.3343 ± 0.051 | 0.291 ± 0.184 | - | 2.174 ± 0.149 | 0.359 ± 0.067 | 0.347 ± 0.043 | 0.291 ± 0.135 | - |
| Mean b | 2.167 ± 0.090 | 0.250 ± 0.064 | 0.370 ± 0.028 | 0.317 ± 0.070 | - | 1.778 ± 0.222 | 0.053 ± 0.038 | 0.175 ± 0.051 | 0.152 ± 0.145 | - | 2.556 ± 0.258 | 0.258 ± 0.067 | 0.342 ± 0.057 | 0.300 ± 0.177 | - | 2.000 ± 0.162 | 0.348 ± 0.083 | 0.343 ± 0.051 | 0.281 ± 0.140 | - |
3. Experimental Section
3.1. Plant Material and DNA Extraction
3.2. cDNA Preparation, Illumina Sequencing, and de Novo Assembly
3.3. Detection of EST-SSR Markers and Primer Design
3.4. DNA Amplification and Genotyping
3.5. Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Farjon, A. A Handbook of the World’s Conifers; Brill: Leiden, The Netherlands, 2010. [Google Scholar]
- Royer, D.L.; Hickey, L.J.; Wing, S.L. Ecological conservatism in the “living fossil” Ginkgo. Paleobiology 2003, 29, 84–104. [Google Scholar] [CrossRef]
- Zhou, Q.X. Studies on Systematics of the Taxaceae. Ph.D. Thesis, Kunming Institute of Botany: Kunming, China, 2001. [Google Scholar]
- Zhou, Q.X.; Gu, Z.J.; Yue, Z.S. Karyomorphology and relationships of Amentotaxus. Pilg. Acta Phytotaxon. Sin. 2000, 38, 522–527. [Google Scholar]
- Zonneveld, B.J.M. Genome sizes of 172 species, covering 64 out of the 67 genera, range from 8 to 72 picogram. Nord. J. Bot. 2012, 30, 490–502. [Google Scholar] [CrossRef]
- The IUCN Red List of Threatened Species, Version 2013.2; Available online: http://www.iucnredlist.org (accessed on 2 November 2015).
- Ge, X.J.; Zhou, X.L.; Li, Z.C.; Hsu, T.W.; Schaal, B.A.; Chiang, T.Y. Genetic structure of the relict Amentotaxus argotaenia complex (Taxaceae) based on ISSR fingerprinting. J. Plant Res. 2005, 118, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.J.; Hung, K.H.; Ko, Y.Z.; Hsu, T.W.; Gong, X.; Chiang, T.Y.; Chiang, Y.C. Genetic Divergence and Biogeographical Patterns in Amentotaxus argotaenia Species Complex. Plant. Mol. Biol. Rep. 2015, 33, 264–280. [Google Scholar] [CrossRef]
- Rao, V.R.; Hodgkin, T. Genetic diversity and conservation and utilization of plant genetic resources. Plant. Cell. Tissue Organ Cult. 2002, 68, 1–19. [Google Scholar]
- Mian, M.A.R.; Saha, M.C.; Hopkins, A.A.; Wang, Z. Use of tall fescue EST-SSR markers in phylogenetic analysis of cool-season forage grasses. Genome 2005, 48, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, K.; Fujii, S.; Ishizuka, W.; Goto, S.; Tsumura, Y. Development of 32 EST-SSR markers for Abies firma (Pinaceae) and their transferability to related species. Appl. Plant. Sci. 2013, 1, 1200464. [Google Scholar]
- Chung, J.W.; Kim, T.S.; Suresh, S.; Lee, S.Y.; Cho, G.T. Development of 65 Novel Polymorphic cDNA-SSR Markers in Common Vetch (Vicia sativa subsp. sativa) Using Next Generation Sequencing. Molecules 2013, 18, 8376–8392. [Google Scholar] [PubMed]
- Sato, M.; Hasegawa, Y.; Mishima, K.; Takata, K. Isolation and characterization of 22 EST-SSR markers for the genus Thujopsis. (Cupressaceae). Appl. Plant. Sci. 2015, 3, 1400101. [Google Scholar]
- Fageria, M.S.; Rajora, O.P. Effects of harvesting of increasing intensities on genetic diversity and population structure of white spruce. Evol. Appl. 2013, 6, 778–794. [Google Scholar] [CrossRef]
- Castoe, T.A.; Poole, A.W.; Gu, W.; de Koning, A.P.J.; Daza, J.M.; Smith, E.N.; Pollock, D.D. Rapid identification of thousands of copperhead snake (Agkistrodon contortrix) microsatellite loci from modest amounts of 454 shotgun genome sequence. Mol. Ecol. Resour. 2010, 10, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; Poole, A.W.; de Koning, A.P.J.; Jones, K.L.; Tomback, D.F.; Oyler-McCance, S.J.; Fike, J.A.; Lance, S.L.; Streicher, J.W.; Smith, E.N.; et al. Rapid Microsatellite Identification from Illumina Paired-End Genomic Sequencing in Two Birds and a Snake. PLoS ONE 2015, 10, 0030953. [Google Scholar]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Ranade, S.S.; Lin, Y.C.; Van de Peer, Y.; García-Gil, M. Comparative in silico analysis of EST-SSRs in angiosperm and gymnosperm tree genera. BMC Plant. Biol. 2014, 14, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Ji, K. Designing Pinus massoniana SSR Primers from Pinus. EST Sequences. Mol. Plant. Breed. 2009, 7, 833–838. [Google Scholar]
- Xiang, X.; Zhang, Z.; Wang, Z.; Zhang, X.; Wu, G. Transcriptome sequencing and development of EST-SSR markers in Pinus dabeshanensis, an endangered conifer endemic to China. Mol. Breed. 2015, 35, 158. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.; Lian, C. De Novo Transcriptome Sequencing Analysis of cDNA Library and Large-Scale Unigene Assembly in Japanese Red Pine (Pinus densiflora). Int. J. Mole Sci. 2015, 16, 29047–29059. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Moriguchi, Y.; Uchiyama, K.; Ujino-Ihara, T.; Futamura, N.; Sakurai, T.; Shinohara, K.; Tsumura, Y. A second generation framework for the analysis of microsatellites in expressed sequence tags and the development of EST-SSR markers for a conifer, Cryptomeria japonica. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.S.; Chen, C.T.; Ko, Y.Z.; Ge, X.J.; Hung, K.H.; Hsu, T.W.; Chiang, Y.C. Isolation and characterization of 15 microsatellite loci in four endangered Amentotaxus species (Taxaceae). Am. J. Bot. 2012, 99, e157–e159. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.Z. The Study of Populations Genetics and Conservation in Amentotaxus argotaenia Complex. Master’s Thesis, National Sun Yat-sen University, Kaohsiung, Taiwan, 2012. [Google Scholar]
- Yang, J.B.; Li, H.T.; Li, D.Z.; Liu, J.; Gao, L.M. Isolation and characterization of microsatellite markers in the endangered species Taxus wallichiana using the FIASCO method. HortScience 2009, 44, 2043–2045. [Google Scholar]
- Guan, L.; Suharyanto; Shiraishi, S. Isolation and characterization of tetranucleotide microsatellite loci in Pinus massoniana (Pinaceae). Am. J. Bot. 2011, 98, e216–e217. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.D.; Eggler, P.; Seaton, G.; Rossetto, M.; Ablett, E.M.; Lee, L.S.; Henry, R.J. Analysis of SSRs derived from grape ESTs. Theor. Appl. Genet. 2000, 100, 723–726. [Google Scholar] [CrossRef]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within genes: Structure; function; and evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Ganopoulos, I.; Aravanopoulos, F.; Argiriou, A.; Tsaftaris, A. Genome and population dynamics under selection and neutrality: An example of S-allele diversity in wild cherry (Prunus avium L.). Tree Genet. Genom. 2012, 8, 1181–1190. [Google Scholar] [CrossRef]
- Shen, D.F.; Bo, W.H.; Xu, F.; Wu, R.L. Genetic diversity and population structure of the Tibetan poplar (Populus szechuanica var. tibetica) along an altitude gradient. BMC Genet. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, Y.; Kang, K.S.; Lee, K.Y.; Lee, S.W.; Kim, Y.Y. Genetic variation of Picea jezoensis populations in South Korea revealed by chloroplast, mitochondrial and nuclear DNA markers. J. Plant. Res. 2009, 122, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Benkman, C.W.; Jenkins, B.; Buerkle, C.A. Low levels of population genetic structure in Pinus. contorta (Pinaceae) across a geographic mosaic of co-evolution. Am. J. Bot. 2011, 98, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.H.; Chen, J.M.; Gituru, R.W.; Wang, Q.F. Gene flow in populations of the endangered aquatic fern Ceratopteris pteridoides in China as revealed by ISSR markers. Aquat. Bot. 2007, 87, 69–74. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. Ser. B. 1996, 351, 1291–1298. [Google Scholar] [CrossRef]
- Loveless, M.D.; Hamrick, J.L. Ecological determinants of genetic structure of plant populations. Annu. Rev. Ecol. Syst. 1984, 15, 65–95. [Google Scholar] [CrossRef]
- Bauert, M.R.; Kalin, M.; Baltisberger, M.; Edwards, P.J. No genetic variation detected within isolated relic populations of Saxifraga. cernua in the Alps using RAPD markers. Mol. Ecol. 1998, 7, 1519–1527. [Google Scholar] [CrossRef]
- Ci, X.Q.; Chen, J.Q.; Li, Q.M.; Li, J. AFLP and ISSR analysis reveals high genetic variation and inter-population differentiation in fragmented populations of the endangered Litsea szemaois (Lauraceae) from Southwest China. Plant. Syst. Evol. 2008, 273, 237–246. [Google Scholar] [CrossRef]
- Gordon, S.P.; Sloop, C.M.; Davis, H.G.; Cushman, J.H. Population genetic diversity and structure of two rare vernal pool grasses in central California. Conserv. Genet. 2012, 13, 117–130. [Google Scholar] [CrossRef]
- Zhao, X.F.; Ma, Y.P.; Sun, W.B.; Wen, X.; Milne, R. High genetic diversity and low differentiation of Michelia coriacea (Magnoliaceae), a critically endangered endemic in southeast Yunnan, China. Int. Mol. Sci. 2012, 13, 4396–4411. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Li, R.; Fan, W.; Tian, G.; Zhu, H.; He, L.; Cai, J.; Huang, Q.; Cai, Q.; Li, B.; Bai, Y.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Da Maia, L.C.; Palmieri, D.A.; de Souza, V.Q.; Kopp, M.M.; de Carvalho, F.I.; Costa de Oliveira, A. SSR locator: Tool for simple sequence repeat discovery integrated with primer design and pcr simulation. Int. J. Plant. Genom. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Untergrasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. Genepop (version-1.2)—Population-Genetics Software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar]
- Antao, T.; Lopes, A.; Lopes, R.; Beja-Pereira, A.; Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform. 2008, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A. Adaptation and speciation: What can FST tell us? Trends Ecol. Evol. 2005, 20, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Sample Availability: Samples are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.-Y.; Chiang, T.-Y.; Chiang, Y.-C.; Hsu, H.-M.; Ge, X.-J.; Huang, C.-C.; Chen, C.-T.; Hung, K.-H. Cross-Species, Amplifiable EST-SSR Markers for Amentotaxus Species Obtained by Next-Generation Sequencing. Molecules 2016, 21, 67. https://doi.org/10.3390/molecules21010067
Li C-Y, Chiang T-Y, Chiang Y-C, Hsu H-M, Ge X-J, Huang C-C, Chen C-T, Hung K-H. Cross-Species, Amplifiable EST-SSR Markers for Amentotaxus Species Obtained by Next-Generation Sequencing. Molecules. 2016; 21(1):67. https://doi.org/10.3390/molecules21010067
Chicago/Turabian StyleLi, Chiuan-Yu, Tzen-Yuh Chiang, Yu-Chung Chiang, Hsin-Mei Hsu, Xue-Jun Ge, Chi-Chun Huang, Chaur-Tzuhn Chen, and Kuo-Hsiang Hung. 2016. "Cross-Species, Amplifiable EST-SSR Markers for Amentotaxus Species Obtained by Next-Generation Sequencing" Molecules 21, no. 1: 67. https://doi.org/10.3390/molecules21010067