
New Phragmalin-Type Limonoids from Chukrasia tabularis and Their α-Glucosidase Inhibitory Activity

Abstract
:1. Introduction
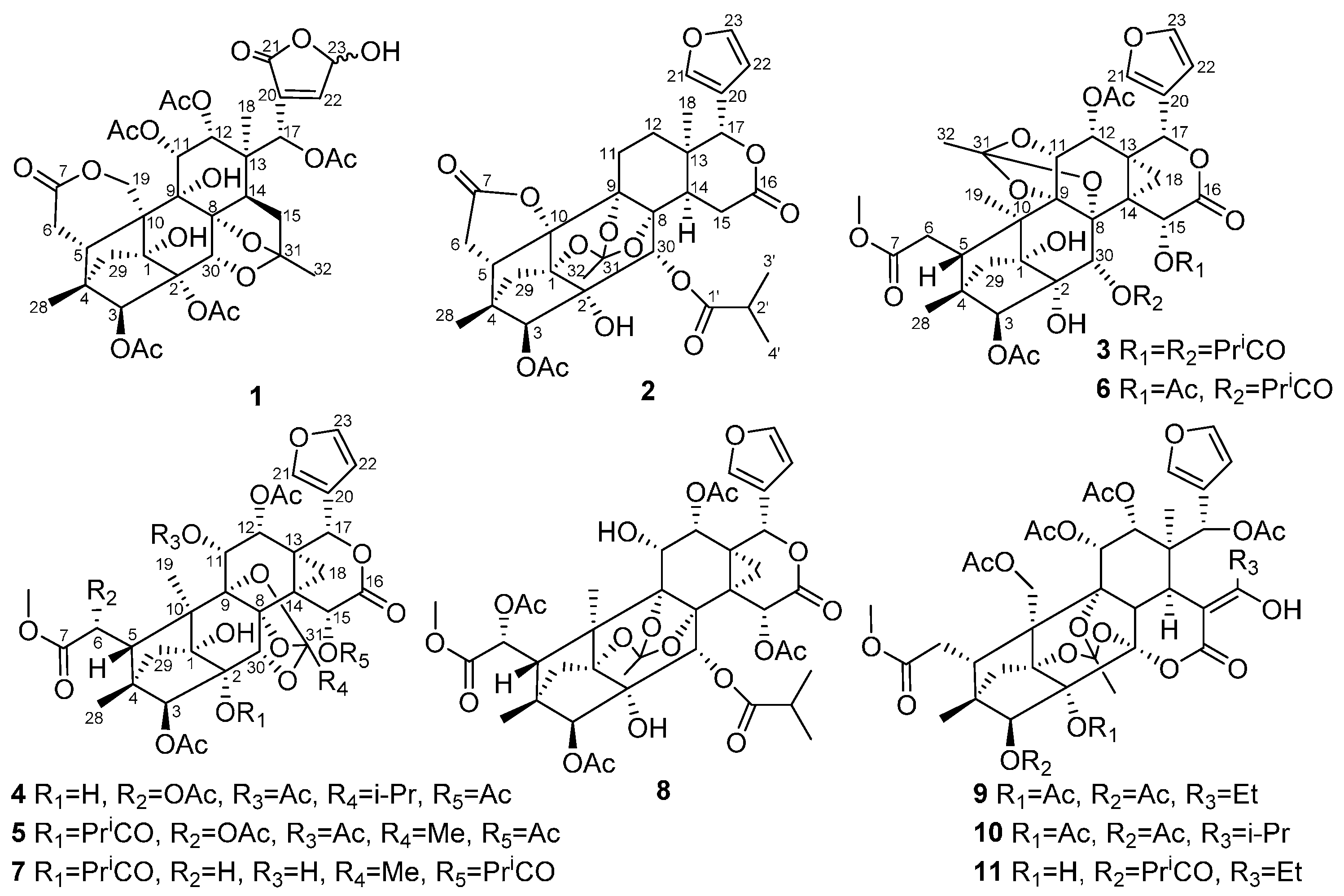
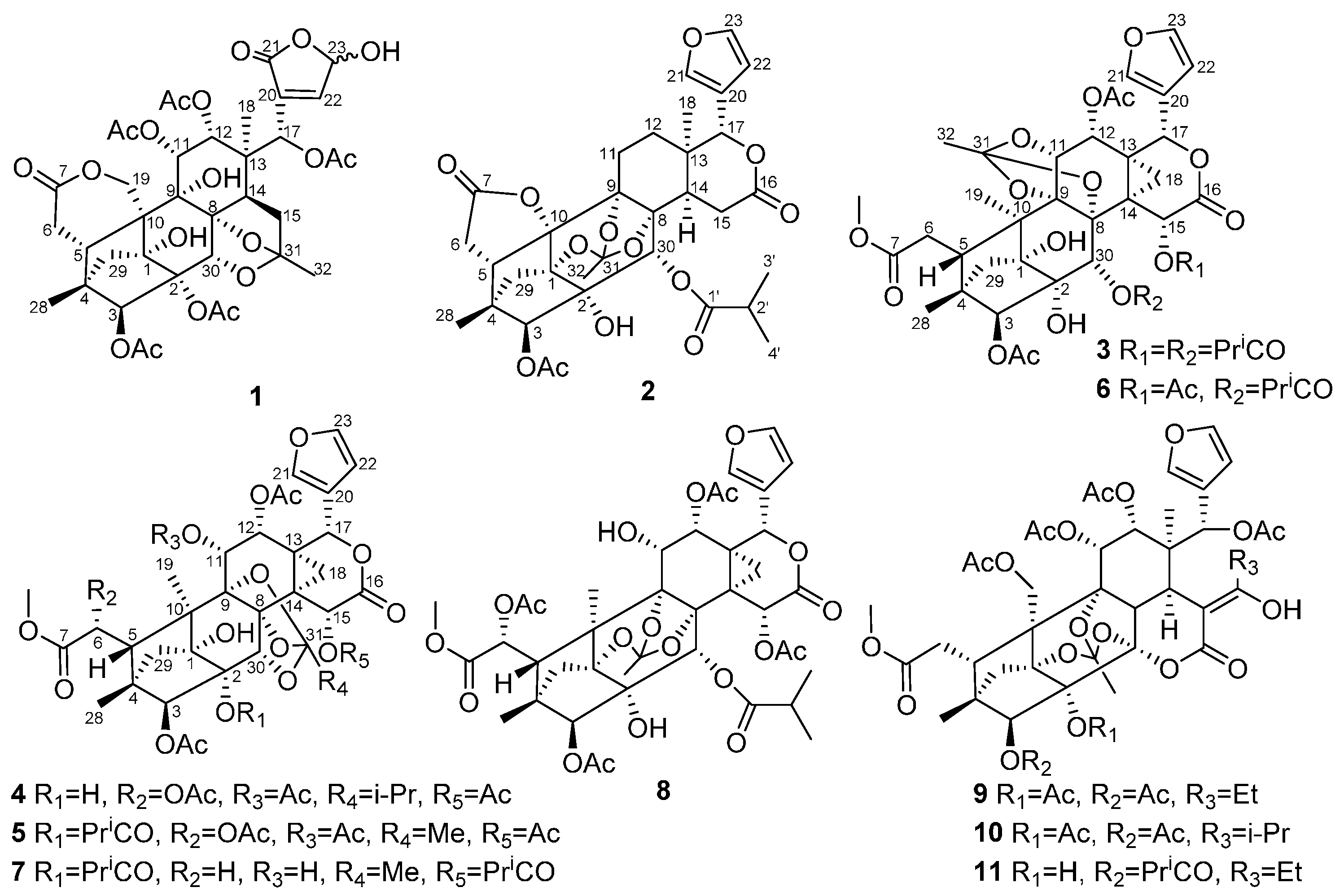
2. Results and Discussion
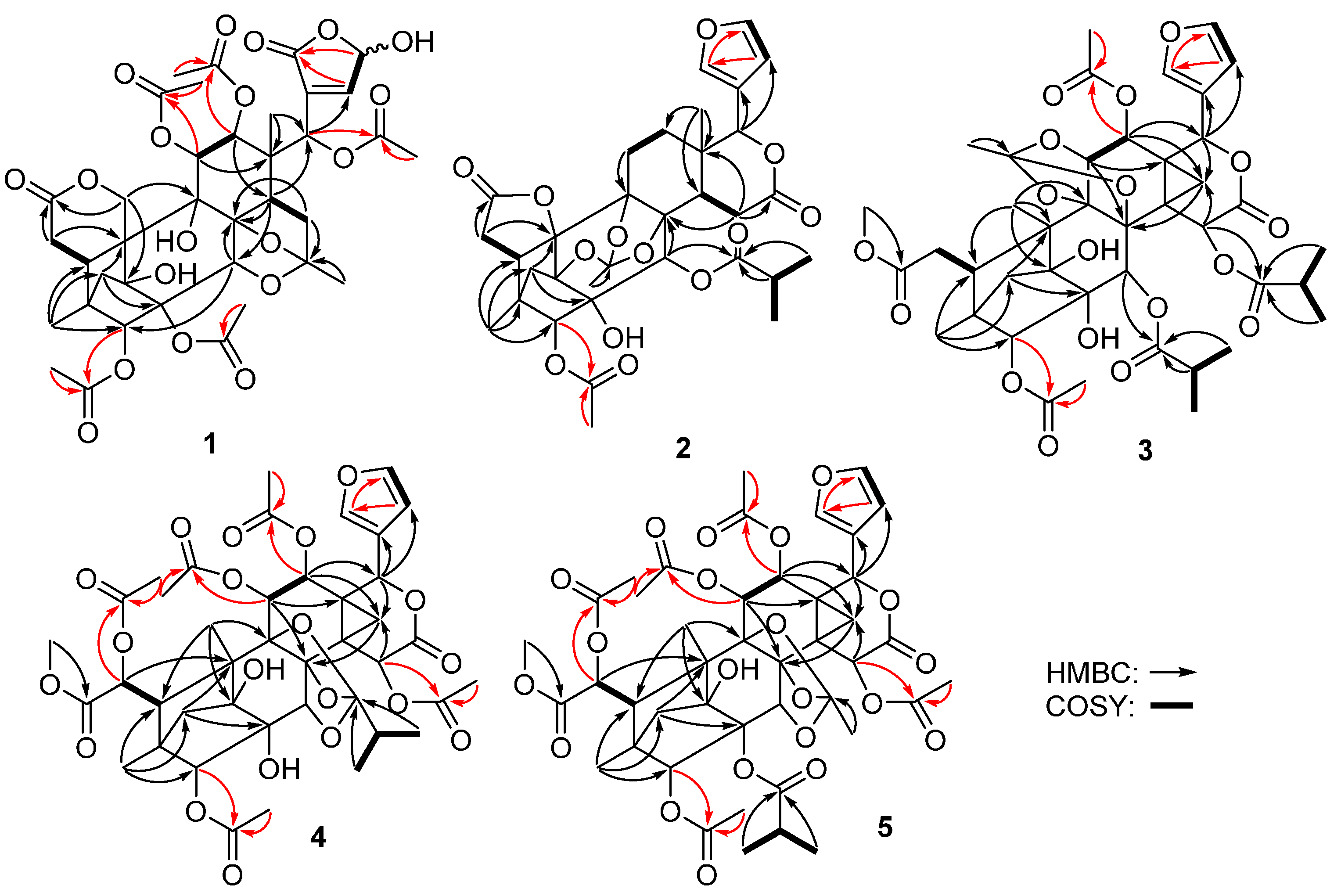
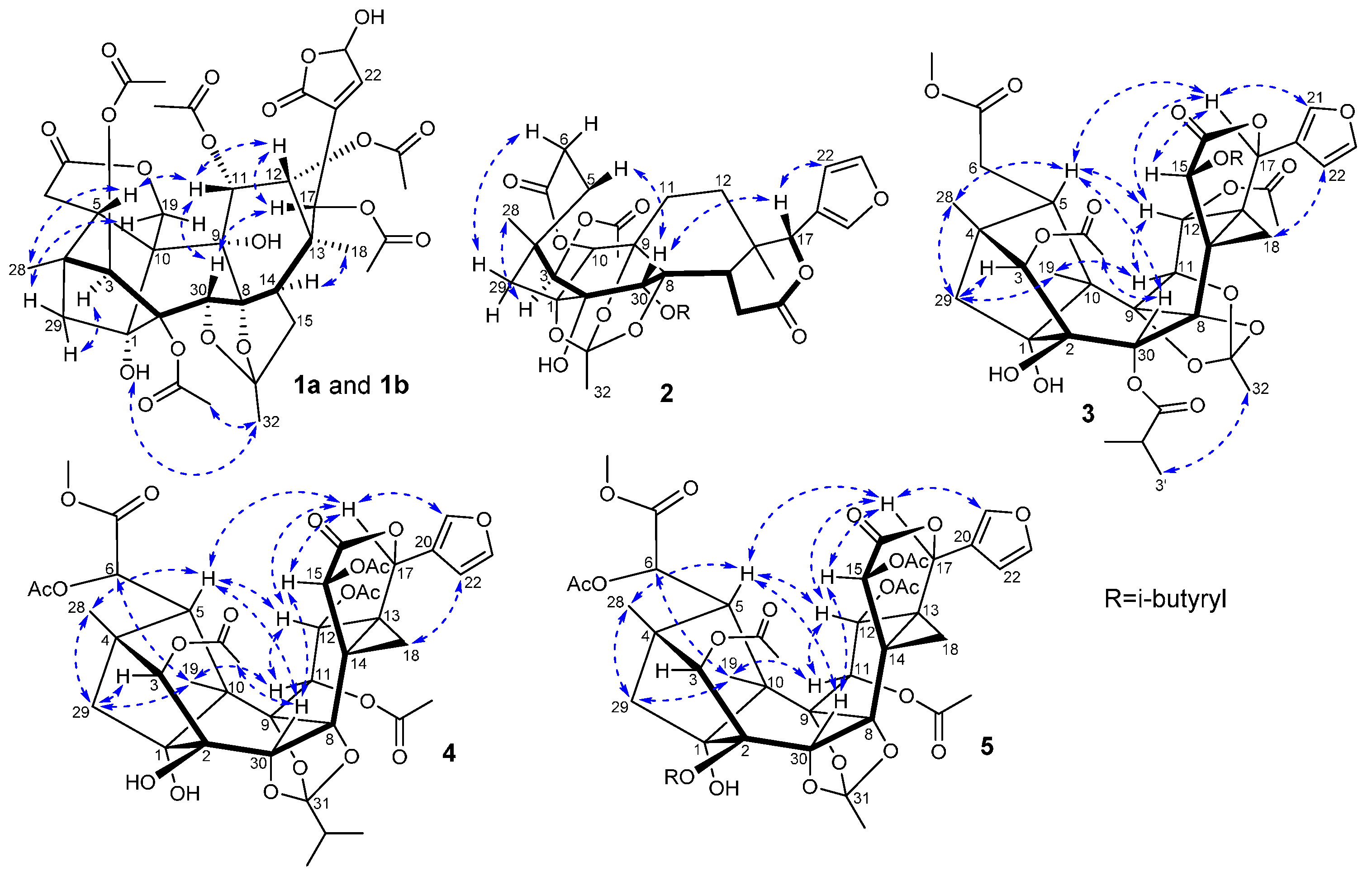

{kind=link}
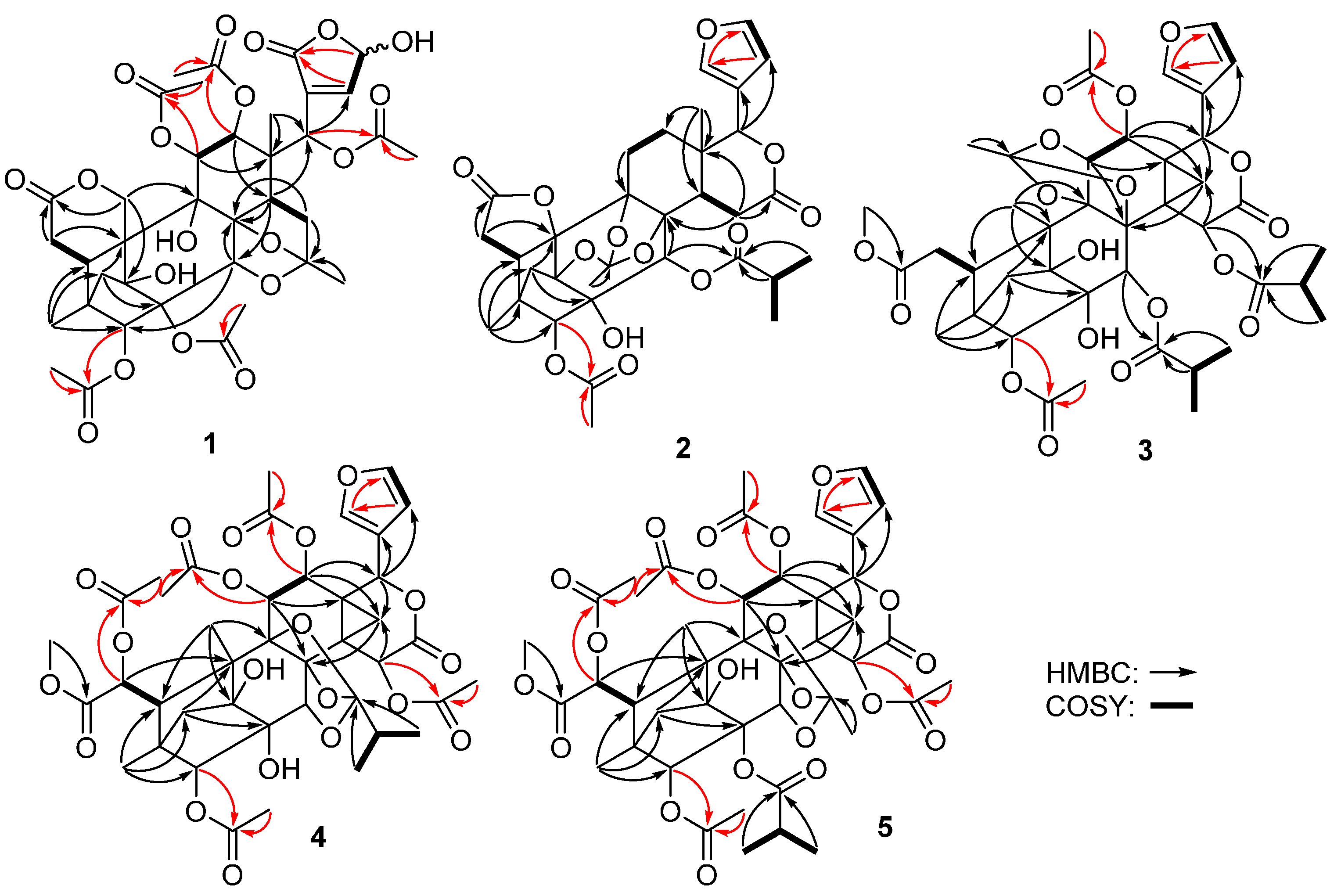{kind=link}
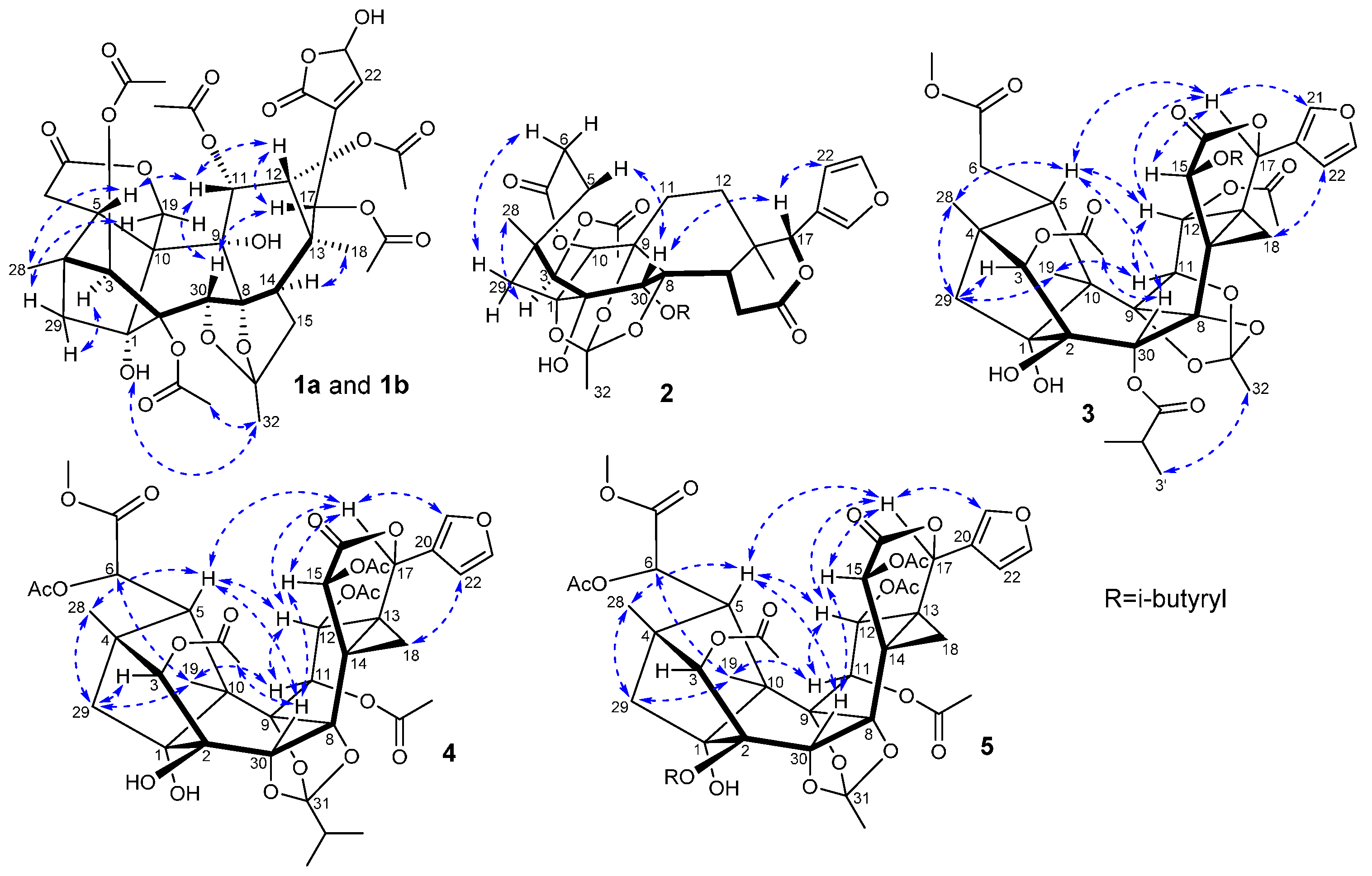{kind=link}
| No. | 1a | 1b | ||||
|---|---|---|---|---|---|---|
| δC a | δH b | ROESY c | δC a | δH b | ROESY c | |
| 1 | 85.7 s | 85.7 s | ||||
| 2 | 81.1 s | 81.1 s | ||||
| 3 | 83.4 d | 5.31 (s) | 29b | 83.3 d | 5.31 (s) | 29b |
| 4 | 45.7 s | 45.8 s | ||||
| 5 | 40.8 d | 2.00 (m) | 11, 28 | 40.7 d | 2.00 (m) | 11, 28 |
| 6a | 31.5 t | 2.31 (m, 2H) | 31.5 t | 2.31 (m, 2H) | ||
| 6b | ||||||
| 7 | 172.7 s | 172.8 s | ||||
| 8 | 89.3 s | 89.4 s | ||||
| 9 | 75.0 s | 75.2 s | ||||
| 10 | 52.6 s | 52.6 s | ||||
| 11 | 70.6 d | 5.47 (d, 3.8) | 5, 12, 30 | 70.6 d | 5.54 (d, 3.8) | 5, 12, 30 |
| 12 | 71.2 d | 5.38 (d, 3.8) | 11, 17 | 71.4 d | 5.38 (d, 3.8) | 11, 17 |
| 13 | 41.8 s | 41.8 s | ||||
| 14 | 44.6 d | 3.21 (dd, 12.2, 7.1) | 18 | 44.5 d | 3.21 (dd, 12.2, 7.1) | 18 |
| 15a | 35.4 t | 2.62 (dd, 11.9, 7.1) | 35.4 t | 2.63 (dd, 11.9, 7.1) | ||
| 15b | 1.94 (dd, 12.2, 11.9) | 1.94 (dd, 12.2, 11.9) | ||||
| 17 | 72.0 d | 5.72 (s) | 12, 30, 3-OAc | 72.1 d | 5.73 (s) | 12, 30, 3-OAc |
| 18 | 18.8 q | 1.02 (s) | 14, 9-OH | 18.8 q | 1.02 (s) | 14, 9-OH |
| 19a | 69.5 t | 5.00 (d, 12.5) | 1-OH | 69.5 t | 4.97 (d, 12.5) | 1-OH |
| 19b | 4.18 (dd, 12.5, 4.7) | 29a | 4.18 (dd, 12.5, 4.7) | 29a | ||
| 20 | 133.2 s | 133.5 s | ||||
| 21 | 167.7 s | 167.7 s | ||||
| 22 | 147.5 d | 7.38 (br s) | 148.3 d | 7.34 (br s) | ||
| 23 | 95.7 d | 6.09 (t, 10.9) | 96.2 d | 6.09 (t, 10.9) | ||
| 28 | 15.2 q | 0.90 (s) | 5 | 15.2 q | 0.90 (s) | 5 |
| 29a | 38.9 t | 2.09 (d, 11.8) | 19b | 38.9 t | 2.09 (d, 11.8) | 19b |
| 29b | 2.02 (d, 11.8) | 3 | 2.02 (d, 11.8) | 3 | ||
| 30 | 70.9 d | 4.45 (s) | 11, 17 | 70.9 d | 4.42 (s) | 11, 17 |
| 31 | 111.3 s | 111.7 s | ||||
| 32 | 18.8 q | 1.65 (s) | 1-OH, 2-OAc | 18.8 q | 1.65 (s) | 1-OH, 2-OAc |
| 2-OAc | 170.2 s | 2.09 (s) | 32 | 170.1 s | 2.09 (s) | 32 |
| 20.9 q | 20.9 q | |||||
| 3-OAc | 168.7 s | 2.45 (s) | 17 | 168.9 s | 2.45 (s) | 17 |
| 21.0 q | 21.0 q | |||||
| 11-OAc | 171.5 s | 2.13 (s) | 171.2 s | 2.12 (s) | ||
| 20.8 q | 20.8 q | |||||
| 12-OAc | 170.5 s | 2.08 (s) | 170.5 s | 2.08 (s) | ||
| 20.3 q | 20.3 q | |||||
| 17-OAc | 170.7 s | 2.11 (s) | 170.7 s | 2.11 (s) | ||
| 20.2 q | 20.2 q | |||||
| 1-OH | 4.86 (s) | 32, 19a | 4.85 (s) | 32, 19a | ||
| 9-OH | 3.32 (s) | 18 | 3.30 (s) | 18 | ||
| No. | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| δC a | δH b | ROESY c | δC a | δH b | ROESY c | |
| 1 | 84.8 s | 83.0 s | ||||
| 2 | 79.8 s | 76.7 s | ||||
| 3 | 82.8 d | 4.83 (s) | 28 | 85.9 d | 5.49 (s) | 29b |
| 4 | 44.0 s | 44.1 s | ||||
| 5 | 39.1 d | 2.94 (d, 8.4) | 30 | 38.1 d | 2.58 (d, 11.9) | 12, 17, 28 |
| 6a | 30.2 t | 2.77 (d, 12.6) | 29a | 33.1 t | 2.66 (d, 12.3) | |
| 6b | 2.59 (dd, 12.6, 8.4) | 2.45 (d, 12.3 | ||||
| 7 | 174.3 s | 173.9 s | ||||
| 8 | 86.5 s | 78.5 s | ||||
| 9 | 84.5 s | 90.6 s | ||||
| 10 | 86.4 s | 45.1 s | ||||
| 11a | 22.9 t | 1.64 (overlapped) | 75.0 d | 4.17 (d, 3.6) | 12, 19 | |
| 11b | 2.03 (m) | |||||
| 12a | 29.0 t | 1.53 (m) | 66.7 d | 5.14 (br d, 3.6) | 5, 11, 17 | |
| 12b | 1.41 (overlapped) | |||||
| 13 | 34.9 s | 31.3 s | ||||
| 14 | 42.6 d | 2.10 (dd, 10.6, 2.1) | 31.1 s | |||
| 15a 15b | 29.9 t | 3.15 (dd, 19.6, 2.1) | 69.4 d | 7.16 (br d, 2.8) | 17, 30 | |
| 2.72 (dd, 19.6, 10.6) | ||||||
| 16 | 169.8 s | 167.1 s | ||||
| 17 | 78.6 d | 5.32 (s) | 22, 30 | 70.2 d | 6.42 (s) | 5, 12, 15, 21 |
| 18a | 20.3 q | 1.15 (s) | 18.8 t | 2.64 (dd, 7.0, 3.1) | ||
| 18b | 1.44 (d, 7.0) | |||||
| 19 | 14.4 q | 1.31 (s) | 11, 29a | |||
| 20 | 121.0 s | 122.3 s | ||||
| 21 | 141.0 d | 7.47 (br s) | 142.2 d | 7.47 (br s) | 17 | |
| 22 | 109.7 d | 6.40 (br s) | 17 | 109.9 d | 6.50 (br d, 1.6) | |
| 23 | 143.6 d | 7.43 (br s) | 143.4 d | 7.39 (br t, 1.6) | ||
| 28 | 14.3 q | 1.01 (s) | 3 | 14.8 q | 0.83 (s) | 5, 29b |
| 29a | 39.5 t | 1.87 (s, 2H) | 6b | 39.0 t | 1.92 (s, 2H) | 19 |
| 29b | 3, 28 | |||||
| 30 | 69.8 d | 5.64 (s) | 5, 17 | 69.4 d | 5.39 (s) | 15, 3-OAc |
| 31 | 119.8 s | 119.9 s | 1.66 (s) | 3′ | ||
| 32 | 21.0 q | 1.75 (s) | 16.4 q | |||
| 3-OAc | 170.1 s | 2.19 (s) | 169.3 s | 2.22 s | 30 | |
| 21.6 q | 21.2 q | |||||
| 12-OAc | 170.9 s | 1.66 (s) | ||||
| 20.0 q | ||||||
| 7-OCH3 | 52.6 q | 3.75 (s) | ||||
| 15-OCOCHMe2 | ||||||
| 1’ | 177.9 s | |||||
| 2’ | 34.2 d | 2.92 (m) | ||||
| 3’ | 19.9 q | 1.32 (d, 7.0) | ||||
| 4’ | 18.0 q | 1.25 (d, 7.0) | ||||
| 30-OCOCHMe2 | ||||||
| 1’ | 175.4 s | 173.9 s | ||||
| 2’ | 34.6 d | 2.56-2.61 (m) | 34.0 d | 2.51 (m) | ||
| 3’ | 18.2 q | 1.11 (d, 7.0) | 19.5 q | 1.19 (d, 7.0) | 32 | |
| 4’ | 19.3 q | 1.19 (d, 7.0) | 18.9 q | 1.17 (d, 7.0) | ||
| 1-OH | 2.85 (s) | |||||
| 2-OH | 2.85 (s) | 3.38 (s) | ||||
| No. | 4 | 5 | ||||
|---|---|---|---|---|---|---|
| δC a | δH b | ROESY c | δC a | δH b | ROESY c | |
| 1 | 84.6 s | 83.9 s | ||||
| 2 | 76.0 s | 83.1 s | ||||
| 3 | 85.5 d | 5.36 (s) | 29b | 85.8 d | 5.27 (s) | |
| 4 | 44.4 s | 44.6 s | ||||
| 5 | 44.7 d | 2.81 (br s) | 12, 17, 28, 30 | 43.9 d | 2.80 (s) | 12, 17, 28, 30 |
| 6 | 71.3 d | 6.26 (br s) | 19 | 71.2 d | 6.22 (s) | 19 |
| 7 | 172.1 s | 172.1 s | ||||
| 8 | 86.6 s | 86.5 s | ||||
| 9 | 84.2 s | 84.7 s | ||||
| 10 | 49.4 s | 49.5 s | ||||
| 11 | 67.1 d | 5.66 (d, 4.9) | 12, 15, 19 | 67.0 d | 5.61 (d, 4.9) | 12, 15, 19 |
| 12 | 66.7 d | 5.42 (d, 4.9) | 5, 11, 17 | 66.5 d | 5.47 (d, 4.9) | 5, 11, 17 |
| 13 | 29.6 s | 29.8 s | ||||
| 14 | 25.1 s | 24.9 s | ||||
| 15 | 69.7 d | 6.94 (br s) | 11, 17, 30 | 70.5 d | 6.99 (d, 2.6) | 11, 17, 30 |
| 16 | 166.0 s | 165.7 s | ||||
| 17 | 72.1 d | 6.50 (s) | 5, 12, 15, 21 | 71.8 d | 6.44 (s) | 5, 12, 15, 21 |
| 18a | 16.2 t | 2.70 (dd, 7.2, 2.5) | 22 | 17.8 t | 2.71 (dd, 7.2, 2.6) | |
| 18b | 1.51 (d, 7.2) | 1.43 (br d, 7.2) | ||||
| 19 | 17.8 q | 1.37 (s) | 6, 11, 29a | 17.6 q | 1.36 (s) | 6, 11, 29a |
| 20 | 122.2 s | 122.2 s | ||||
| 21 | 142.1 d | 7.49 (br s) | 17 | 142.1 d | 7.52 (br s) | 17 |
| 22 | 109.7 d | 6.49 (br d, 1.4) | 18b | 109.9 d | 6.51 (br d, 1.3) | |
| 23 | 143.4 d | 7.38 (br t, 1.6) | 143.4 d | 7.38 (br t, 1.7) | ||
| 28 | 15.6 q | 0.96 (s) | 5, 29b | 15.6 q | 0.92 (s) | 5, 29b |
| 29a | 40.2 t | 2.16 (d, 11.0) | 19 | 40.8 t | 1.71 (br d, 11.4) | 19 |
| 29b | 1.83 (d, 11.0) | 3, 28 | 2.28 (br d, 11.4) | 28 | ||
| 30 | 79.4 d | 4.09 (s) | 5, 15, 3-OAc | 76.0 d | 5.05 (s) | 5, 15 |
| 31 | 119.4 s | 116.2 s | ||||
| 32 | 29.2 d | 2.15 (m) | 15.8 q | 1.66 (s) | ||
| 33 | 17.1 q | 1.07 (d, 7.0) | ||||
| 34 | 17.0 q | 1.05 (d, 7.0) | ||||
| 3-OAc | 169.0 s | 2.21 (s) | 30 | 168.6 s | 2.33 (s) | |
| 21.1 q | 20.6 q | |||||
| 6-OAc | 169.3 s | 2.21 (s) | 168.9 s | 2.20 (s) | ||
| 21.3 q | 21.1 q | |||||
| 11-OAc | 169.2 s | 2.05 (s) | 169.0 s | 2.07 (s) | ||
| 20.9 q | 21.2 q | |||||
| 12-OAc | 170.1 s | 1.53 (s) | 170.1 s | 1.54 (s) | ||
| 19.3 q | 19.3 q | |||||
| 15-OAc | 169.0 s | 2.23 (s) | 169.2 s | 2.22 (s) | ||
| 21.1 q | 20.9 q | |||||
| 7-OCH3 | 53.7 q | 3.79 (s) | 53.7 q | 3.79 (s) | ||
| 2-OCOCHMe2 | ||||||
| 1’ | 175.9 s | |||||
| 2’ | 34.6 d | 2.50-2.55 (m) | ||||
| 3’ | 18.9 q | 1.17 (d, 7.0) | ||||
| 4’ | 18.9 q | 1.20 (d, 7.0) | ||||
| 1-OH | 3.28 (s) | 3.50 (s) | ||||
| 2-OH | 3.47 (s) | |||||
| Compound | IC50 Value (mM) a | Compound | IC50 Value (mM) a |
|---|---|---|---|
| 1 | – | 7 | – |
| 2 | 0.06 ± 0.008 | 8 | 0.20 ± 0.057 |
| 3 | 0.04 ± 0.002 | 9 | – |
| 4 | 0.52 ± 0.039 | 10 | – |
| 5 | 1.09 ± 0.040 | 11 | – |
| 6 | – | Acarbose b | 0.95 ± 0.092 |
3. Experimental Section
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. α-Glucosidase Inhibitory Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, S.K.; Chen, B.Y.; Li, H. Flora Republicae Popularis Sinicae. In Chinese Flora (Zhongguo Zhiwu Zhi); Science Press: Beijing, China, 1997; Volume 43, pp. 47–49. [Google Scholar]
- Editorial Committee of the Administration Bureau of Traditional Chinese Medicine. Chemical composition and structure. In Chinese Material Medica (Zhonghua Bencao), 1st ed.; Shanghai Science and Technology Press: Shanghai, China, 1999; Volume 5, pp. 31–32. [Google Scholar]
- Tan, Q.G.; Luo, X.D. Meliaceous limonoids: Chemistry and Biological Activities. Chem. Rev. 2011, 111, 7437–7522. [Google Scholar] [CrossRef] [PubMed]
- Ragettli, T.; Tamm, C. Die Chukrasine A, B, C, D und E, fünf neue Tetranortriterpene aus Chukrasia tabularis A. Juss. Helv. Chim. Acta 1978, 61, 1814–1831. [Google Scholar] [CrossRef]
- Connolly, J.D.; Labbe, C.; Rycroft, D.S. Tetranortriterpenoids and related substances. Part 20. New tetranortriterpenoids from the seeds of Chukrasia tabularis (Meliaceae); simple esters of phragmalin and 12α-acetoxy phragmalin. J. Chem. Soc. Perkin Trans. 1978, 1, 285–288. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.S.; Cao, W.J.; Kong, L.Y. A new phragmalin-type limonoid from Chukrasia tabularis var. velutina. Chin. J. Nat. Med. 2011, 9, 98–100. [Google Scholar]
- Fan, C.Q.; Wang, X.N.; Yin, S.; Zhang, C.R.; Wang, F.D.; Yue, J.M. Tabularisins A–D, phragmalin ortho esters with new skeleton isolated from the seeds of Chukrasia tabularis. Tetrahedron 2007, 63, 6741–6747. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.S.; Luo, J.G.; Wang, X.B.; Kong, L.Y. Velutabularins A–J, phragmalin-type limonoids with novel cyclic moiety from Chukrasia tabularis var. velutina. Tetrahedron 2011, 67, 2942–2948. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.S.; Wang, X.B.; Huang, X.F.; Luo, J.G.; Kong, L.Y. Chukvelutilides A–F, phragmalin limonoids from the stem barks of Chukrasia tabularis var. velutina. Tetrahedron 2009, 65, 3425–3431. [Google Scholar] [CrossRef]
- Zhang, C.R.; Yang, S.P.; Liao, S.G.; Fan, C.Q.; Wu, Y.; Yue, J.M. Chuktabularins A–D, four new limonoids with unprecedented carbon skeletons from the stem bark of Chukrasia tabularis. Org. Lett. 2007, 9, 3383–3386. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.S.; Luo, J.G.; Wang, X.B.; Kong, L.Y. Chukvelutins A–C, 16-Norphragmalin Limonoids with Unprecedented Skeletons from Chukrasia tabularis var. velutina. Org. Lett. 2009, 11, 2281–2284. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.S.; Wang, X.B.; Luo, J.G.; Kong, L.Y. Chuktabularins E–T, 16-Norphragmalin Limonoids from Chukrasia tabularis var. velutina. J. Nat. Prod. 2010, 73, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.R.; Fan, C.Q.; Zhang, L.; Yang, S.P.; Wu, Y.; Lu, Y.; Yue, J.M. Chuktabrins A and B, two novel limonoids fom the twigs and leaves of Chukrasia tabularis. Org. Lett. 2008, 10, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.L.; Di, Y.T.; Fang, X.; Liu, E.D.; Liu, H.Y.; He, H.P.; Li, S.L.; Li, S.F.; Hao, X.J. Tabulvelutin A, the first 19-nor limonoid with unprecedented ring system from Chukrasia tabularis var. velutina. Tetrahedron Lett. 2011, 52, 3083–3085. [Google Scholar] [CrossRef]
- Liu, H.B.; Zhang, H.; Li, P.; Gao, Z.B.; Yue, J.M. Chukrasones A and B: Potential Kv1.2 Potassium Channel Blockers with New Skeletons from Chukrasia tabularis. Org. Lett. 2012, 14, 4438–4441. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, M.; Abdelgaleil, S.A.M.; Saad, M.M.G.; Huang, R.C.; Doe, M.; Iwagawa, T. Phragmalin limonoids from Chukrasia tabularis. Phytochemistry 2004, 65, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.S.; Wang, X.B.; Luo, J.G.; Kong, L.Y. Phragmalin-type limonoid orthoesters from Chukrasia tabularis var. velutina. Chem. Pharm. Bull. 2011, 59, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.; Wang, J.S.; Lu, J.; Wang, X.B.; Luo, J.G.; Kong, L.Y. Twelve Novel and Diverse 16-Norphragmalin-Type Limonoids from Chukrasia tabularis var. velutina. Chem. Pharm. Bull. 2012, 60, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.; Wang, J.S.; Lu, J.; Kong, L.Y. Three new C-15-isobutyryl 16-norphragmalin-type limonoids from Chukrasia tabularis var. velutina. Phytochem. Lett. 2012, 5, 249–252. [Google Scholar] [CrossRef]
- Zhang, C.R.; Yang, S.P.; Chen, X.Q.; Wu, Y.; Zhen, X.C.; Yue, J.M. Limonoids from the twigs and leaves of Chukrasia tabularis. Helv. Chim. Acta 2008, 91, 2338–2350. [Google Scholar] [CrossRef]
- Liu, H.B.; Zhang, H.; Li, P.; Wu, Y.; Gao, Z.B.; Yue, J.M. Kv1.2 potassium channel inhibitors from Chukrasia tabularis. Org. Biomol. Chem. 2012, 10, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.D.; Wu, S.H.; Wu, D.G.; Ma, Y.B.; Qi, S.H. Novel antifeeding limonoids from Dysoxylum hainanense. Tetrahedron 2002, 58, 7797–7804. [Google Scholar] [CrossRef]
- Mohamad, K.; Hirasawa, Y.; Lim, C.S.; Awang, K.; Hadi, A.H.A.; Takeya, K.; Morita, H. Ceramicine A and walsogyne A, novel limonoids from two species of Meliaceae. Tetrahedron Lett. 2008, 49, 4276–4278. [Google Scholar] [CrossRef]
- Sakamoto, A.; Tanaka, Y.; Inoue, T.; Kikuchi, T.; Kajimoto, T.; Muraoka, O.; Yamada, T.; Tanaka, R. Andirolides Q–V from the flower of andiroba (Carapa guianensis, Meliaceae). Fitoterapia 2013, 90, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, J.; Wang, Q.; Kong, L.Y. Two new limonoids from the stem barks of Chukrasia tabularis var. velutina. J. Asian Nat. Prod. Res. 2011, 13, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.R.; Yang, S.P.; Zhu, Q.; Liao, S.G.; Wu, Y.; Yue, J.M. Nortriterpenoids from Chukrasia tabularis var. velutina. J. Nat. Prod. 2007, 70, 1616–1619. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, X.D.; Song, Y.W.; Liu, J.W. A microplate-based screening method for alpha-glucosidase inhibitors. Chin. J. Clin. Pharmacol. Ther. 2005, 10, 1128–1134. [Google Scholar]
- Sample Availability: Samples of the compounds 1–11 are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, J.-L.; Wang, J.; Kong, F.-D.; Liu, Z.-Q.; Wang, P.; Gai, C.-J.; Jiang, B.; Mei, W.-L.; Dai, H.-F. New Phragmalin-Type Limonoids from Chukrasia tabularis and Their α-Glucosidase Inhibitory Activity. Molecules 2016, 21, 58. https://doi.org/10.3390/molecules21010058
Peng J-L, Wang J, Kong F-D, Liu Z-Q, Wang P, Gai C-J, Jiang B, Mei W-L, Dai H-F. New Phragmalin-Type Limonoids from Chukrasia tabularis and Their α-Glucosidase Inhibitory Activity. Molecules. 2016; 21(1):58. https://doi.org/10.3390/molecules21010058
Chicago/Turabian StylePeng, Jun-Lin, Jun Wang, Fan-Dong Kong, Zi-Qi Liu, Pei Wang, Cui-Juan Gai, Bei Jiang, Wen-Li Mei, and Hao-Fu Dai. 2016. "New Phragmalin-Type Limonoids from Chukrasia tabularis and Their α-Glucosidase Inhibitory Activity" Molecules 21, no. 1: 58. https://doi.org/10.3390/molecules21010058