
Design, Synthesis and Biological Evaluation of Novel Substituted N,N′-Diaryl ureas as Potent p38 Inhibitors

Abstract
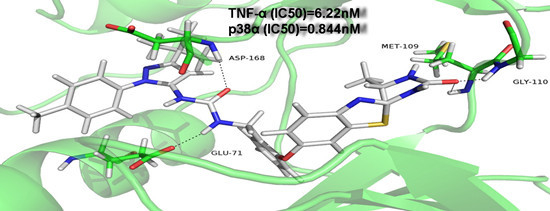
:
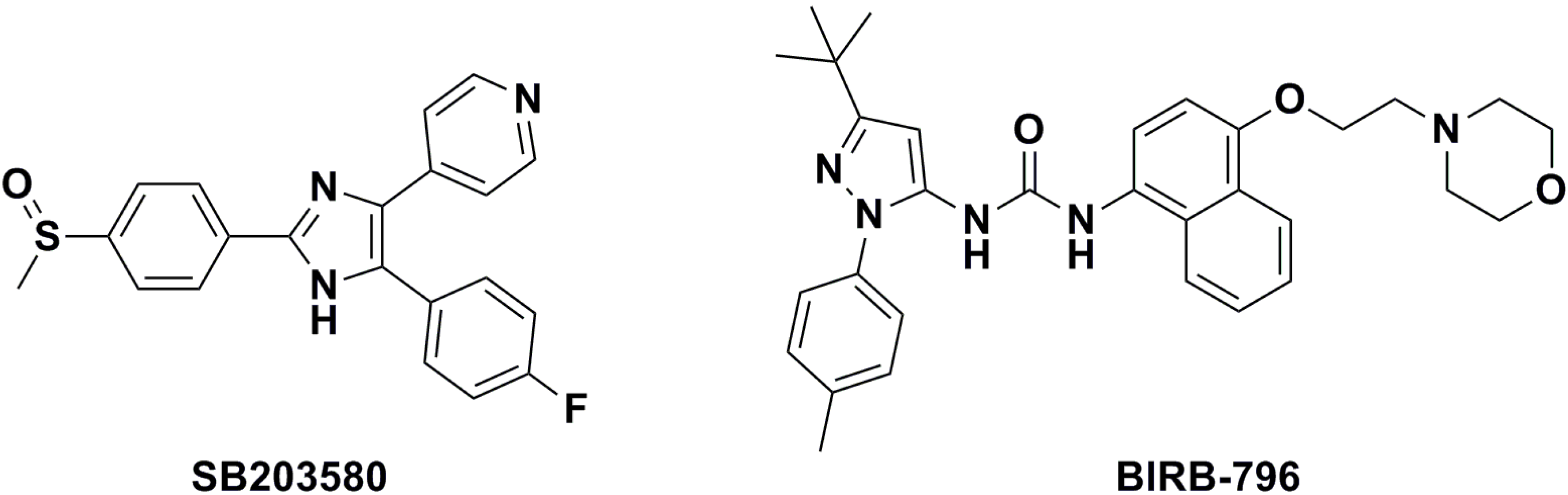
1. Introduction
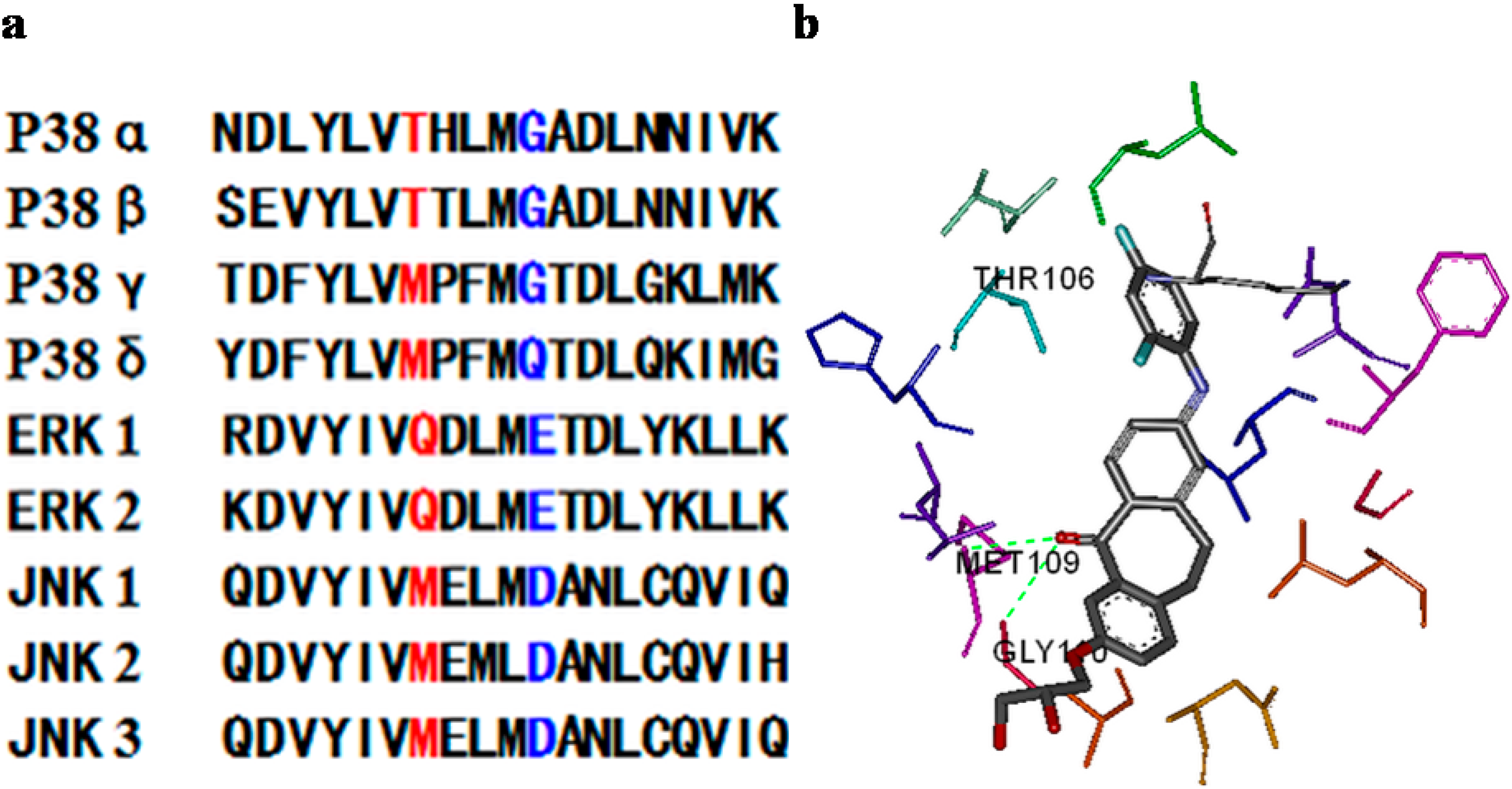
2. Results and Discussion


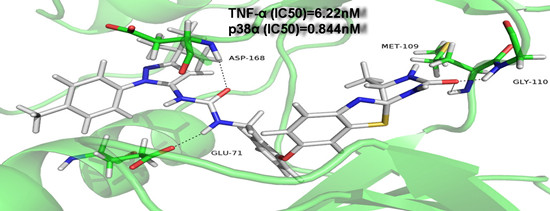{kind=link}
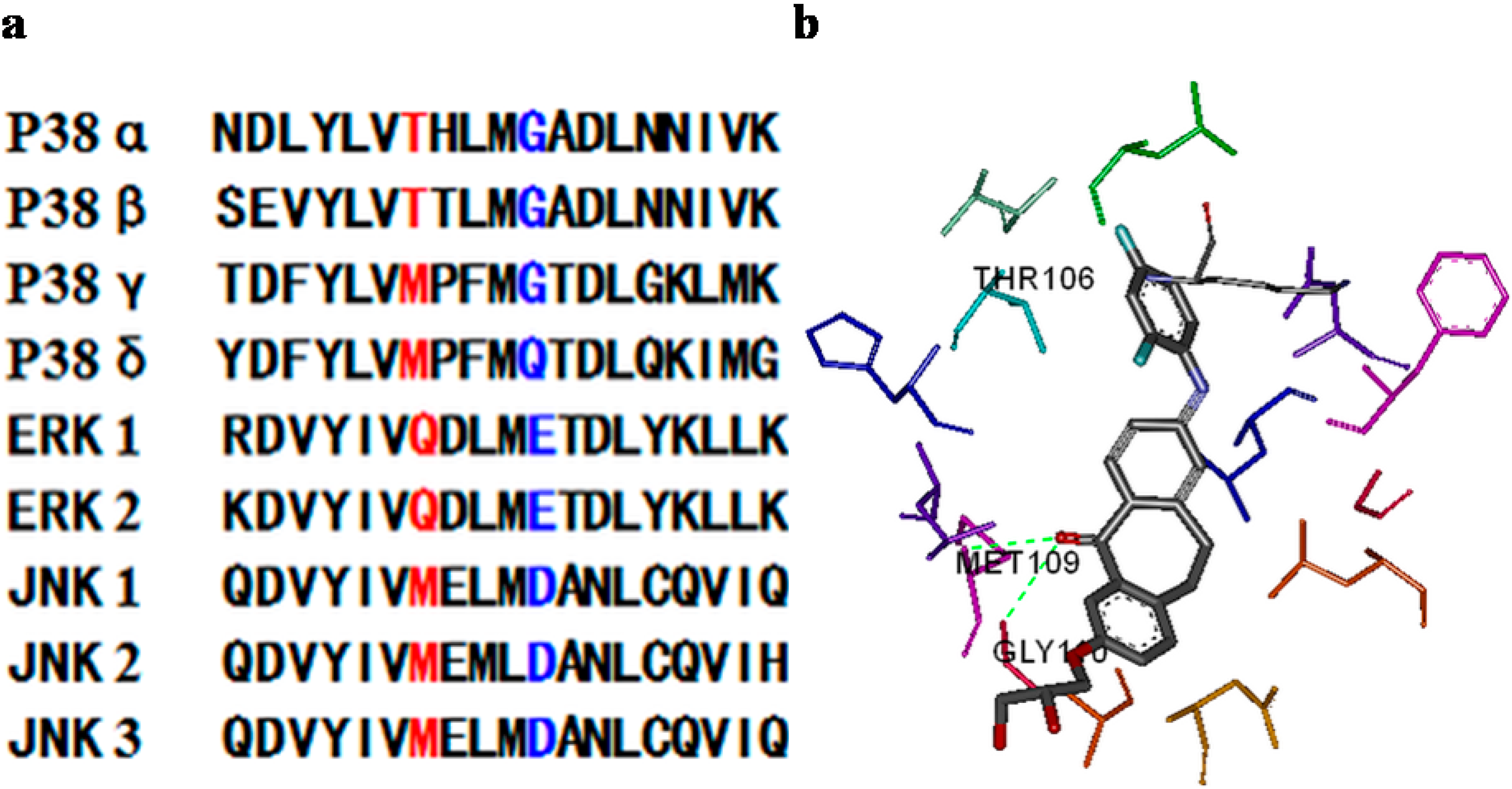{kind=link}
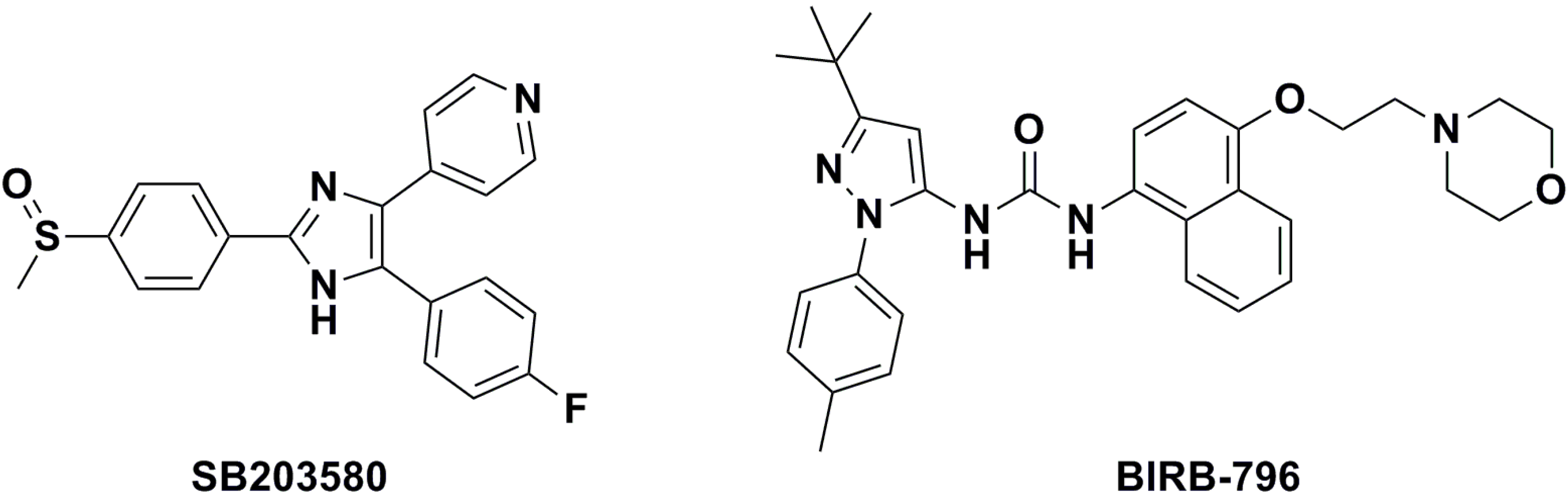{kind=link}
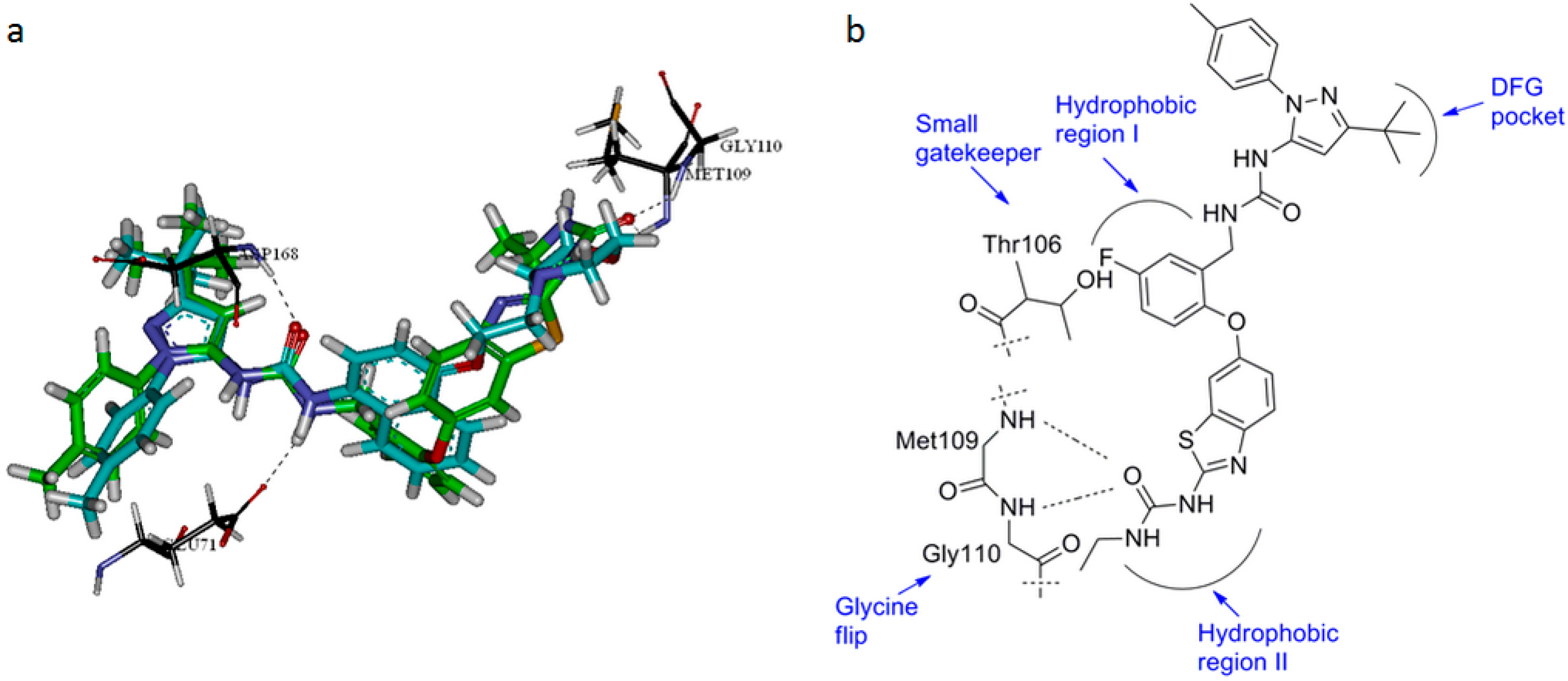{kind=link}
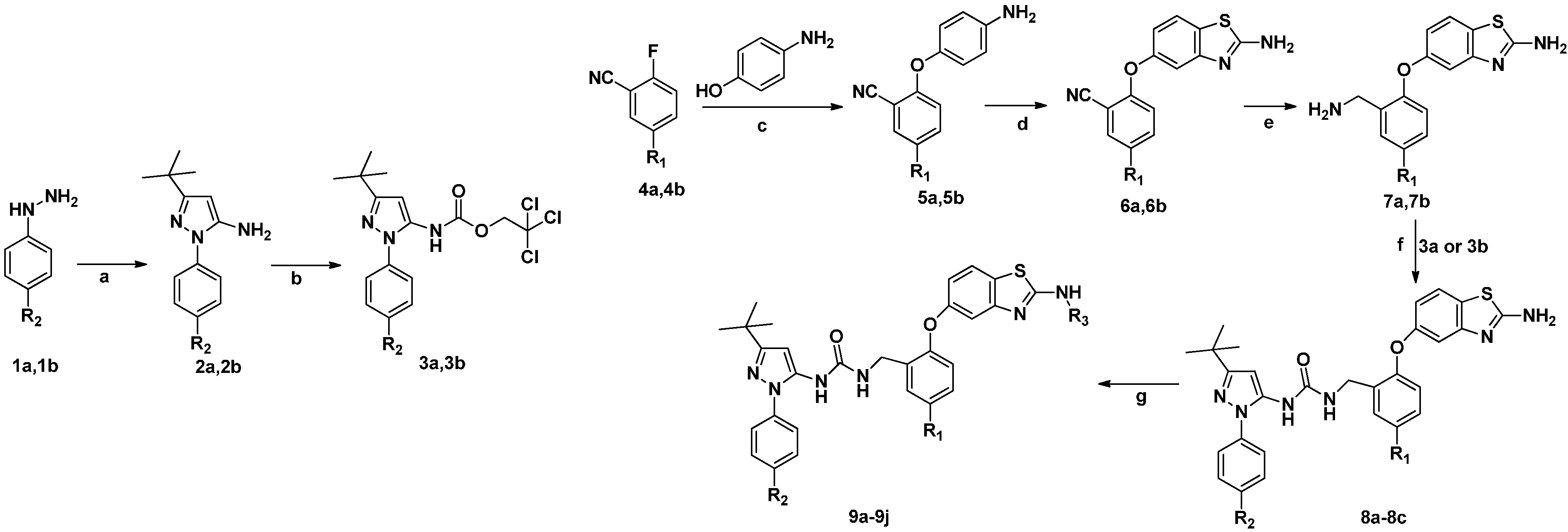{kind=link}
| Compound No. | R1 | R2 | R3 | p38α IC50 (nM) a | TNF-α IC50 (nM) b |
|---|---|---|---|---|---|
| BIRB-796 | — | — | — | 13.74 ± 2.01 | n.d. c |
| SB203580 | — | — | — | 13.89 ± 1.17 | 800 ± 13.15 |
| 8a | H | NO2 | H | 2.878 ± 0.01 | 31.60 ± 5.27 |
| 8b | F | Methyl | H | 1.108 ± 0.20 | 8.94 ± 1.02 |
| 9a | H | NO2 |  | 1.309 ± 0.09 | 33.16 ± 1.17 |
| 9b | H | Methyl |  | 6.165 ± 0.07 | 62.11 ± 5.01 |
| 9c | H | Methyl |  | 1.023 ± 0.09 | 18.32 ± 4.20 |
| 9d | H | Methyl |  | 1.501 ± 0.24 | 8.86 ± 3.32 |
| 9e | H | Methyl |  | 3.288 ± 0.25 | 90.21 ± 6.12 |
| 9f | H | Methyl |  | 4.499 ± 0.04 | 56.44 ± 4.09 |
| 9g | F | Methyl |  | 0.844 ± 1.04 | 6.22 ± 2.07 |
| 9h | F | Methyl |  | 12.38 ± 0.18 | 995.97 ± 9.11 |
| 9i | F | Methyl |  | 7.538 ± 1.99 | 106.62 ± 4.10 |
| 9j | F | Methyl |  | 4.277 ± 1.14 | 52.82 ± 8.03 |
| Compound No. | IC50 (nM) | |||
|---|---|---|---|---|
| p38α | p38β | p38γ | p38δ | |
| 9d | 1.501 ± 0.20 | 17.54 ± 1.25 | 191 ± 7.20 | 230.9 ± 7.15 |
| 9g | 2.844 ± 0.65 | 20.07 ± 1.40 | 424.3 ± 5.75 | 948.4 ± 8.35 |
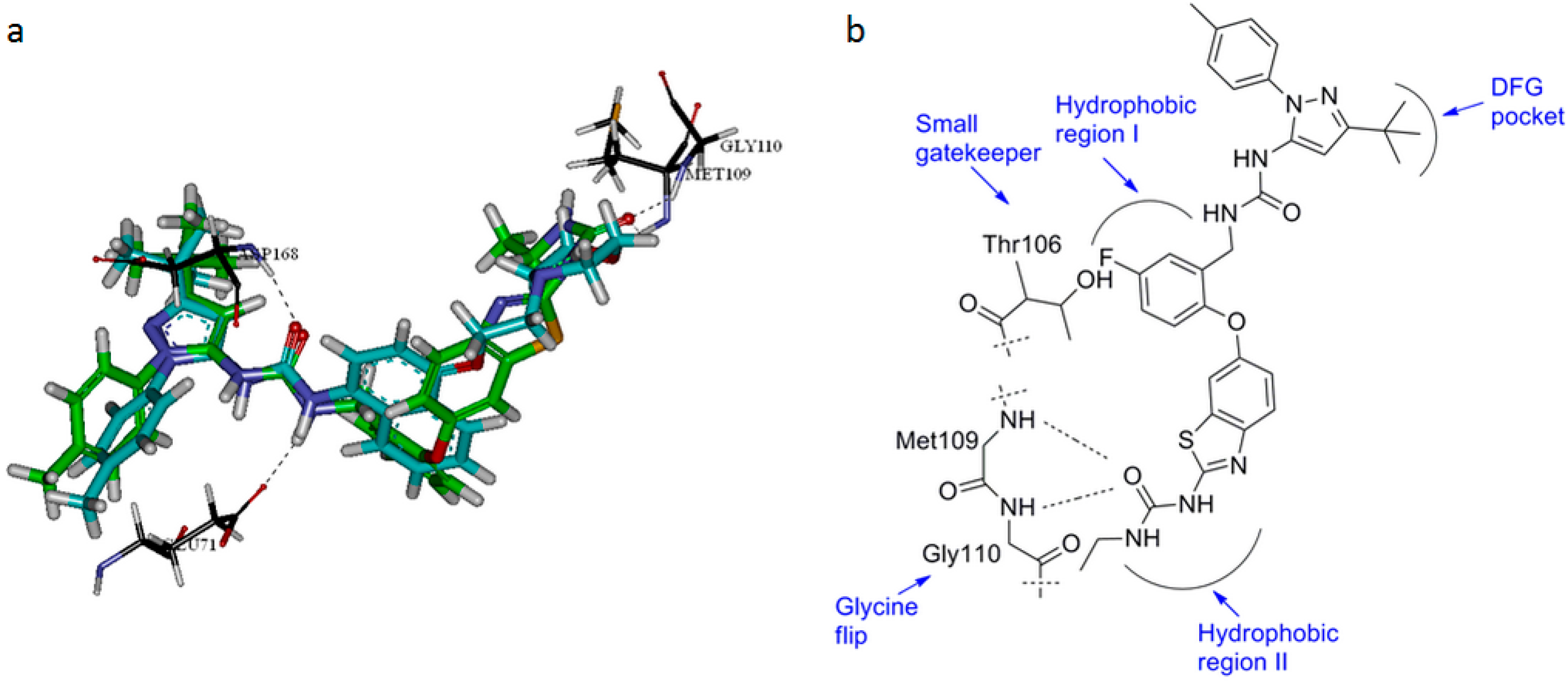
3. Experimental Section
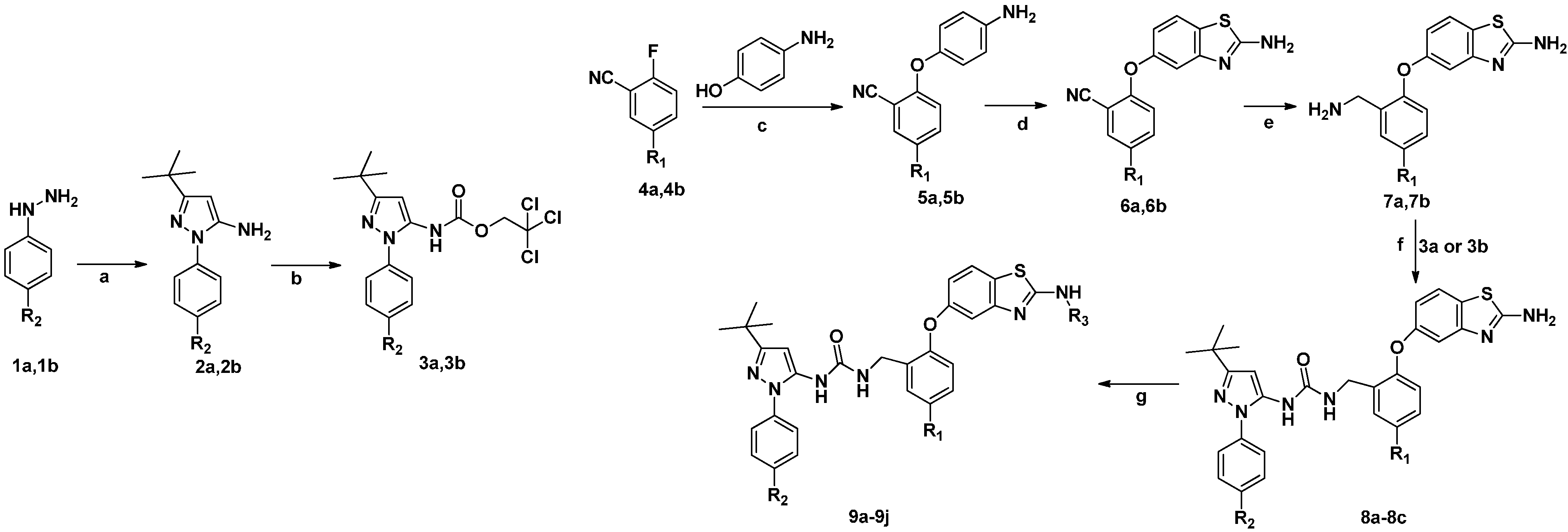
3.1. General Synthetic Information and Synthesis Procedures
3.2. Chemistry
3.3. Pharmacology
3.3.1. In Vitro Pharmacological Activity. IC50 Determi Nation of Inhibition of TNF-α Release from Isolated Human Peripheral Blood Mononuclear Cells (PBMCs) after LPS Stimulation
3.3.2. p38α MAP Kinase Activity was Assessed Based on the Rate of Phosphorylation of ATF-2 (Activation Transcription Factor 2) in an in Vitro Assay
3.3.3. Kinase Selectivity Assay of Selected Compounds (9d, 9g)
3.3.4. Docking (Discovery Studio)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Rutgeerts, P.; D’Haens, G.; Targan, S.; Vasiliauskas, E.; Hanauer, S.B.; Present, D.H.; Mayer, L.; Van Hogezand, R.A.; Braakman, T.; DeWoody, K.L.; et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999, 117, 761–769. [Google Scholar] [CrossRef]
- Foster, M.L.; Halley, F.; Souness, J.E. Potential of p38 inhibitors in the treatment of rheumatoid arthritis. Drug News Perspect. 2000, 13, 488–497. [Google Scholar] [PubMed]
- Dominguez, C.; Powers, D.A.; Tamayo, N. p38 map kinase inhibitors: Many are made, but few are chosen. Curr. Opin. Drug Discov. Dev. 2005, 8, 421–430. [Google Scholar]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Laufer, S.A.; Margutti, S. Isoxazolone based inhibitors of p38 map kinases. J. Med. Chem. 2008, 51, 2580–2584. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.L.; Boehm, J.C.; Kassis, S.; Gorycki, P.D.; Webb, E.F.; Hall, R.; Sorenson, M.; Lee, J.C.; Ayrton, A.; Griswold, D.E.; et al. Pyrimidinylimidazole inhibitors of csbp/p38 kinase demonstrating decreased inhibition of hepatic cytochrome p450 enzymes. Bioorg. Med. Chem. Lett. 1998, 8, 3111–3116. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Dominguez, C. Map kinase p38 inhibitors: Clinical results and an intimate look at their interactions with p38alpha protein. Curr. Med. Chem. 2005, 12, 2979–2994. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.P.; McCaffrey, P.G.; Hsiao, K.; Pazhanisamy, S.; Galullo, V.; Bemis, G.W.; Fitzgibbon, M.J.; Caron, P.R.; Murcko, M.A.; Su, M.S. The structural basis for the specificity of pyridinylimidazole inhibitors of p38 map kinase. Chem. Biol. 1997, 4, 423–431. [Google Scholar] [CrossRef]
- Lisnock, J.; Tebben, A.; Frantz, B.; O’Neill, E.A.; Croft, G.; O’Keefe, S.J.; Li, B.; Hacker, C.; de Laszlo, S.; Smith, A.; et al. Molecular basis for p38 protein kinase inhibitor specificity. Biochemistry 1998, 37, 16573–16581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Canagarajah, B.J.; Boehm, J.C.; Kassisa, S.; Cobb, M.H.; Young, P.R.; Abdel-Meguid, S.; Adams, J.L.; Goldsmith, E.J. Structural basis of inhibitor selectivity in map kinases. Structure 1998, 6, 1117–1128. [Google Scholar] [CrossRef]
- Tong, L.; Pav, S.; White, D.M.; Rogers, S.; Crane, K.M.; Cywin, C.L.; Brown, M.L.; Pargellis, C.A. A highly specific inhibitor of human p38 map kinase binds in the ATP pocket. Nat. Struct. Biol. 1997, 4, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, C.E.; Patel, S.B.; Becker, J.W.; Cameron, P.M.; Zaller, D.; Pikounis, V.B.; O’Keefe, S.J.; Scapin, G. Structural basis for p38alpha map kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat. Struct. Biol. 2003, 10, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Fischer, S.; Schollmeyer, D.; Schattel, V.; Grutter, C.; Rauh, D.; Laufer, S.A. Design, synthesis, and biological evaluation of novel disubstituted dibenzosuberones as highly potent and selective inhibitors of p38 mitogen activated protein kinase. J. Med. Chem. 2012, 55, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, P.F.; Pargellis, C.; Regan, J. The non-diaryl heterocycle classes of p38 map kinase inhibitors. Curr. Top. Med. Chem. 2002, 2, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.; Breitfelder, S.; Cirillo, P.; Gilmore, T.; Graham, A.G.; Hickey, E.; Klaus, B.; Madwed, J.; Moriak, M.; Moss, N.; et al. Pyrazole urea-based inhibitors of p38 map kinase: From lead compound to clinical candidate. J. Med. Chem. 2002, 45, 2994–3008. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.; Capolino, A.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Hickey, E.; Kroe, R.R.; Madwed, J.; Moriak, M.; Nelson, R.; et al. Structure-activity relationships of the p38alpha map kinase inhibitor 1-(5-tert-butyl-2-p-tolyl-2H-pyrazol-3-yl)-3-[4-(2-morpholin-4-yl-ethoxy)naph- thalen-1-yl]urea (birb-796). J. Med. Chem. 2003, 46, 4676–4686. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.; Pargellis, C.A.; Cirillo, P.F.; Gilmore, T.; Hickey, E.R.; Peet, G.W.; Proto, A.; Swinamer, A.; Moss, N. The kinetics of binding to p38 map kinase by analogues of birb-796. Bioorg. Med. Chem. Lett. 2003, 13, 3101–3104. [Google Scholar] [CrossRef]
- Dietrich, J.; Hulme, C.; Hurley, L.H. The design, synthesis, and evaluation of 8 hybrid dfg-out allosteric kinase inhibitors: A structural analysis of the binding interactions of gleevec, nexavar, and birb-796. Bioorg. Med. Chem. 2010, 18, 5738–5748. [Google Scholar] [CrossRef] [PubMed]
- Traxler, P.; Furet, P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- Patel, R.V.; Patel, P.K.; Kumari, P.; Rajani, D.P.; Chikhalia, K.H. Synthesis of benzimidazolyl-1,3,4-oxadiazol-2ylthio-n-phenyl (benzothiazolyl) acetamides as antibacterial, antifungal and antituberculosis agents. Eur. J Med. Chem. 2012, 53, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Tae, J.; Lee, K.; Rhim, H.; Choo, I.H.; Cho, H.; Park, W.K.; Keum, G.; Choo, H. Novel n-biphenyl-2-ylmethyl 2-methoxyphenylpiperazinylalkanamides as 5-ht7r antagonists for the treatment of depression. Bioorg. Med. Chem. 2014, 22, 4587–4596. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Widdowson, C.S.; Kipling, D. Microwave-assisted synthesis of n-pyrazole ureas and the p38alpha inhibitor birb-796 for study into accelerated cell ageing. Org. Biomol. Chem. 2006, 4, 4158–4164. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, X.; Zhang, J.; Wang, L.; Long, L.; Zheng, Z.; Li, S.; Zhong, W. Synthesis and biological evaluation of chromenylurea and chromanylurea derivatives as anti-tnf-alpha agents that target the p38 MAPK pathway. Molecules 2014, 19, 2004–2028. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Ohno, M.; Inoue, H.; Hayashi, S.; Aoki, T.; Hirokawa, H.; Meguro, H.; Koga, Y.; Oshida, K.; Kainoh, M.; et al. Design and synthesis of novel p38alpha map kinase inhibitors: Discovery of pyrazole-benzyl ureas bearing 2-molpholinopyrimidine moiety. Bioorg. Med. Chem. Lett. 2012, 22, 5118–5122. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Nishigaki, N.; Washio, Y.; Kano, K.; Harris, P.A.; Sato, H.; Mori, I.; West, R.I.; Shibahara, M.; Toyoda, H.; et al. Discovery of novel benzimidazoles as potent inhibitors of tie-2 and vegfr-2 tyrosine kinase receptors. J. Med. Chem. 2007, 50, 4453–4470. [Google Scholar] [CrossRef] [PubMed]
- Millan, D.S.; Bunnage, M.E.; Burrows, J.L.; Butcher, K.J.; Dodd, P.G.; Evans, T.J.; Fairman, D.A.; Hughes, S.J.; Kilty, I.C.; Lemaitre, A.; et al. Design and synthesis of inhaled p38 inhibitors for the treatment of chronic obstructive pulmonary disease. J. Med. Chem. 2011, 54, 7797–7814. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Wagner, G.; Laufer, S. New approaches to the treatment of inflammatory disorders small molecule inhibitors of p38 map kinase. Curr. Topics Med. Chem. 2006, 6, 113–149. [Google Scholar] [CrossRef]
- Iwano, S.; Asaoka, Y.; Akiyama, H.; Takizawa, S.; Nobumasa, H.; Hashimoto, H.; Miyamoto, Y. A possible mechanism for hepatotoxicity induced by birb-796, an orally active p38 mitogen-activated protein kinase inhibitor. J. Appl. Toxicol. 2011, 31, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 map kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, D.; Li, X.; Zhong, W.; Zhao, D. Design, Synthesis and Biological Evaluation of Novel Substituted N,N′-Diaryl ureas as Potent p38 Inhibitors. Molecules 2015, 20, 16604-16619. https://doi.org/10.3390/molecules200916604
Zhu D, Li X, Zhong W, Zhao D. Design, Synthesis and Biological Evaluation of Novel Substituted N,N′-Diaryl ureas as Potent p38 Inhibitors. Molecules. 2015; 20(9):16604-16619. https://doi.org/10.3390/molecules200916604
Chicago/Turabian StyleZhu, Dianxi, Xingzhou Li, Wu Zhong, and Dongmei Zhao. 2015. "Design, Synthesis and Biological Evaluation of Novel Substituted N,N′-Diaryl ureas as Potent p38 Inhibitors" Molecules 20, no. 9: 16604-16619. https://doi.org/10.3390/molecules200916604