
New Organocatalytic Asymmetric Synthesis of Highly Substituted Chiral 2-Oxospiro-[indole-3,4′- (1′,4′-dihydropyridine)] Derivatives



Abstract
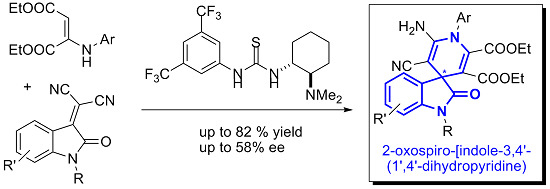
:
1. Introduction
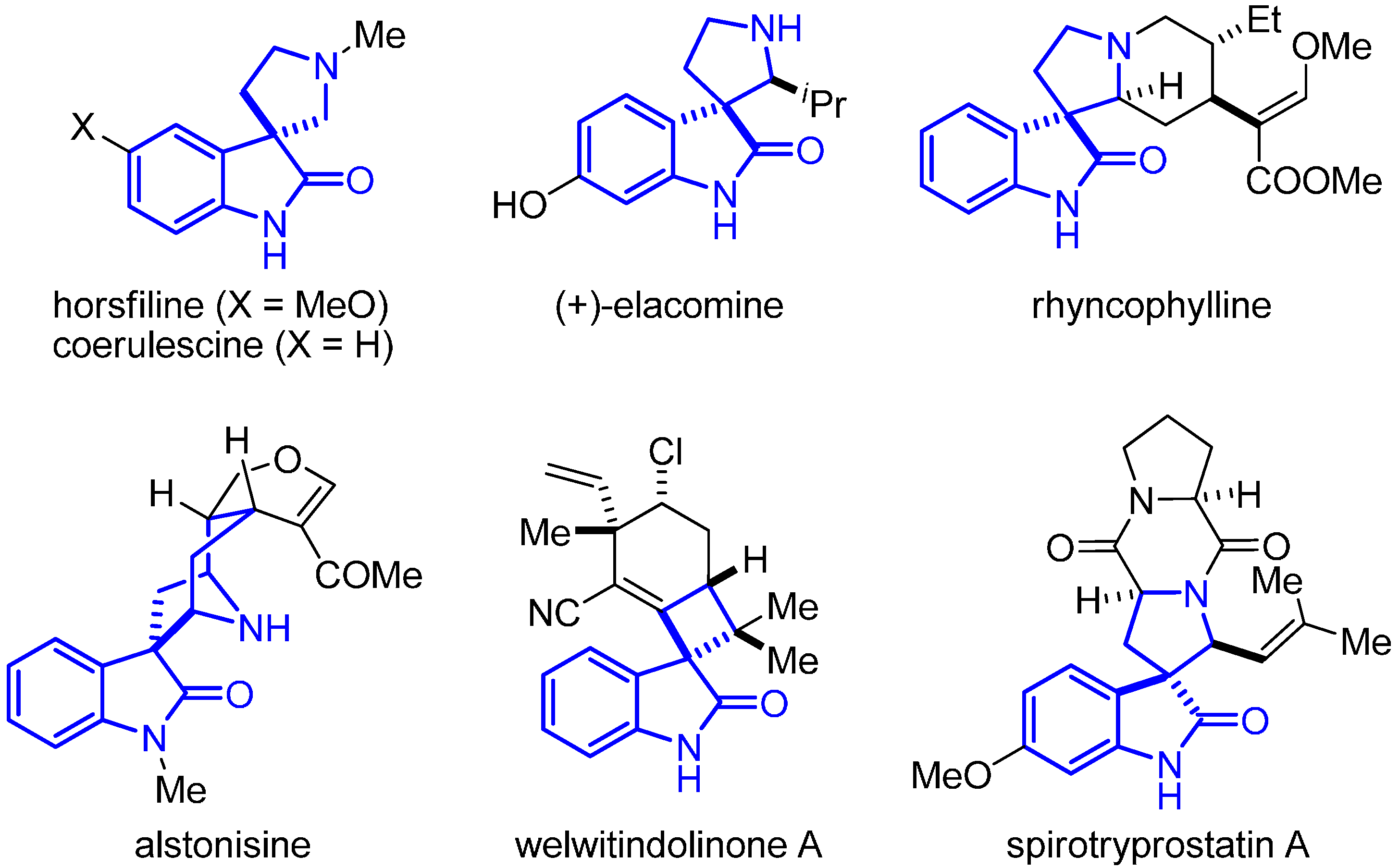
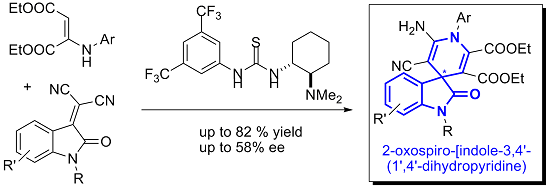
2. Results and Discussion
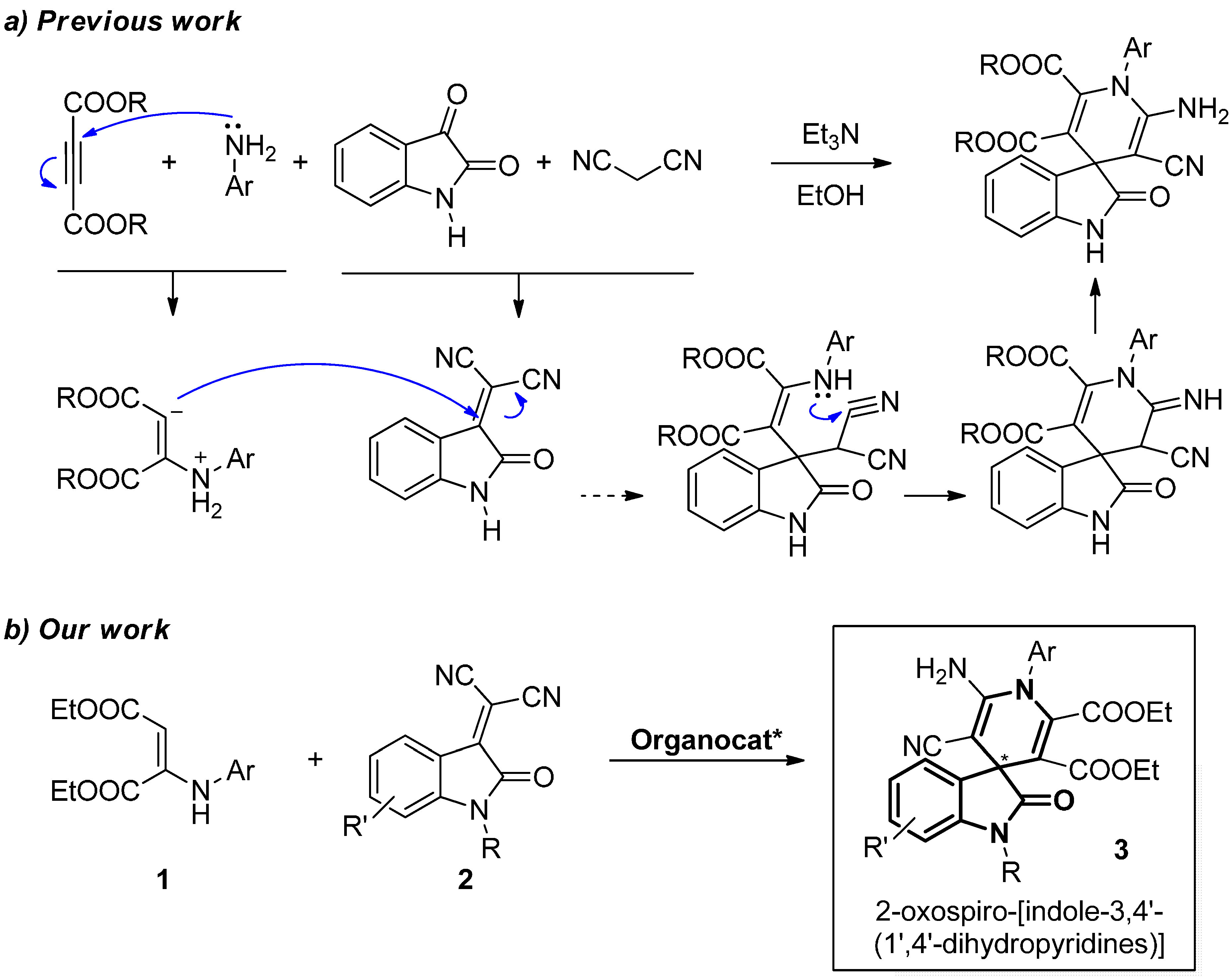
2.1. Hypothesis of Work
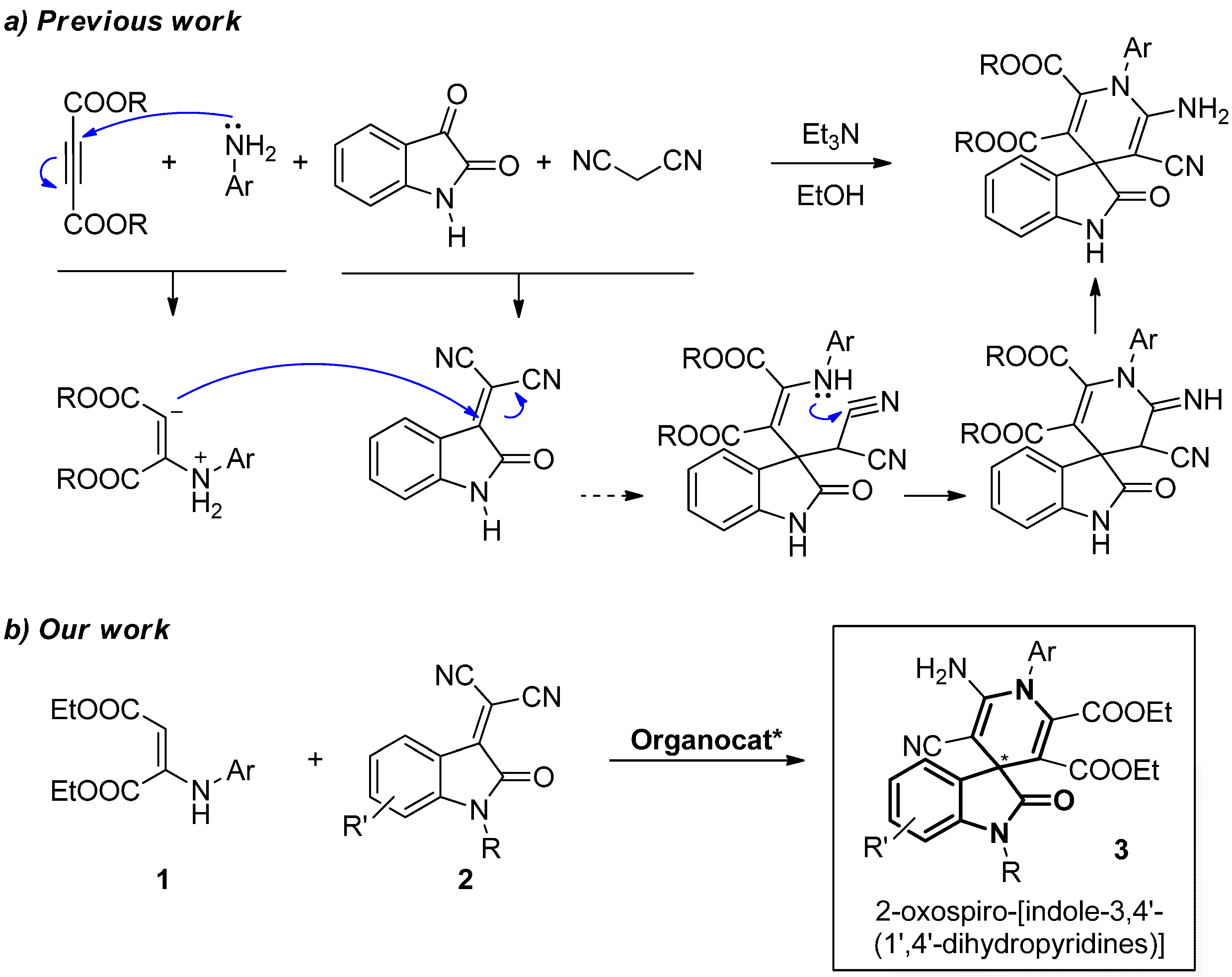
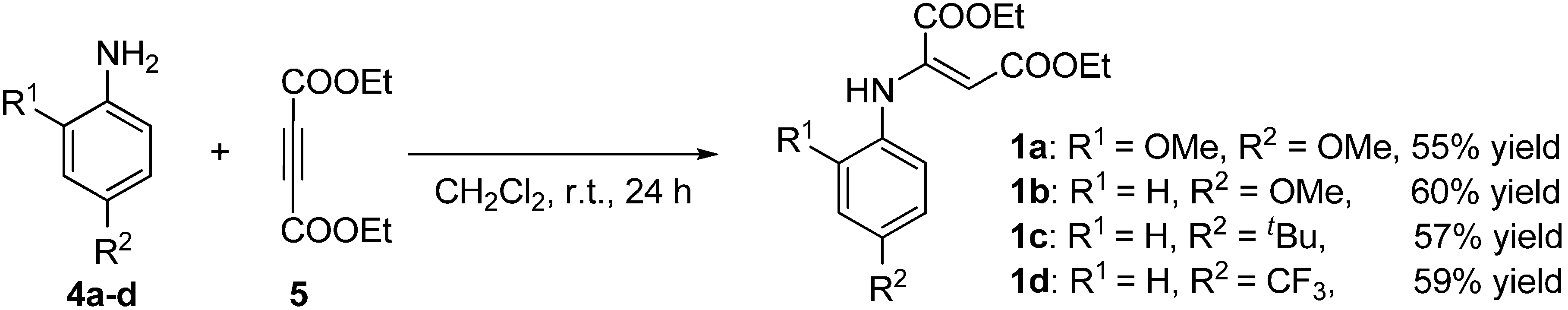
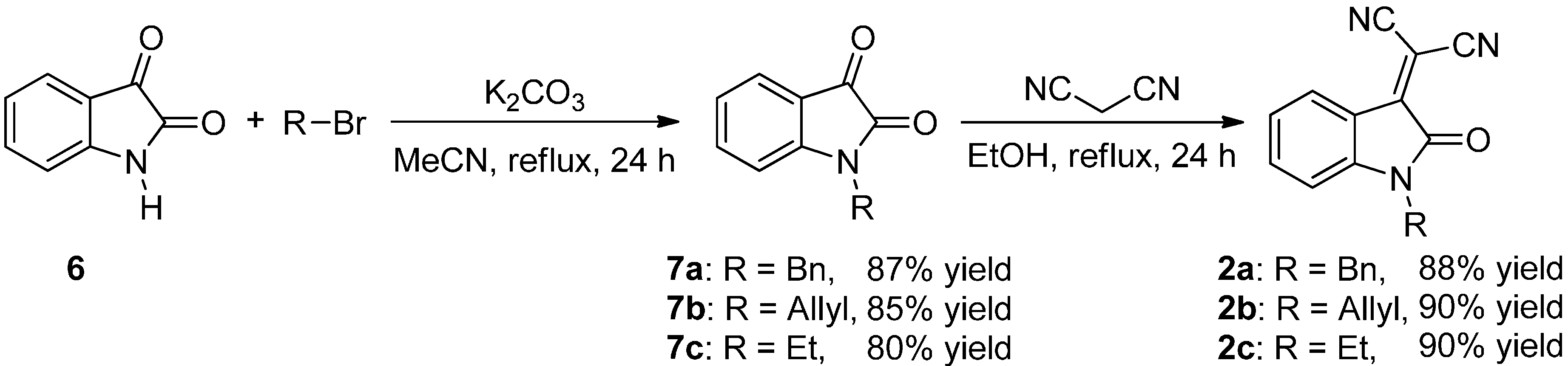
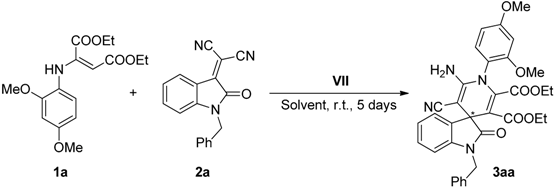
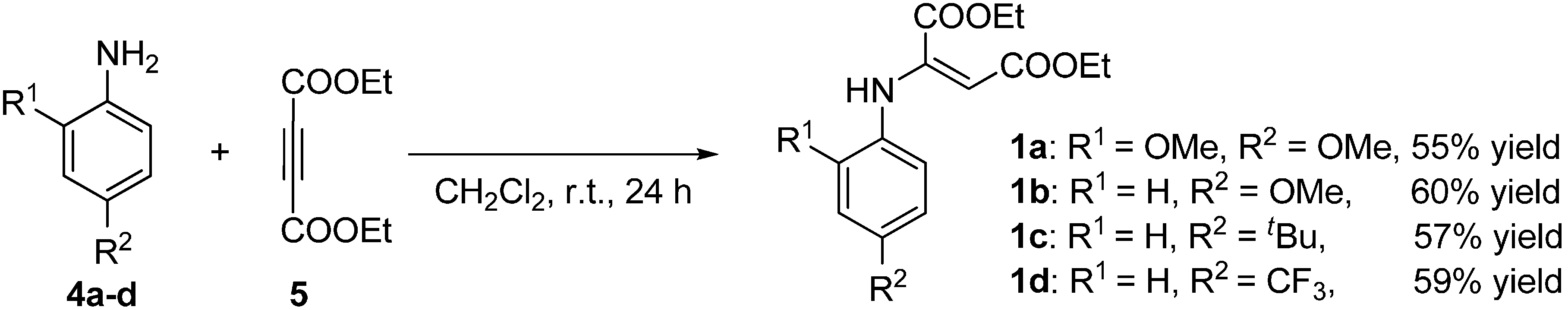
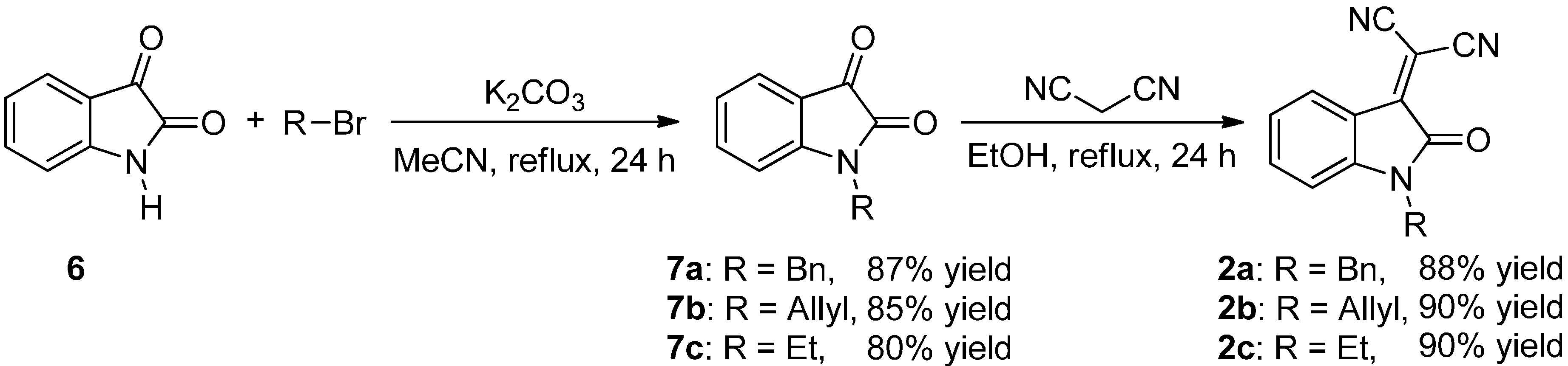
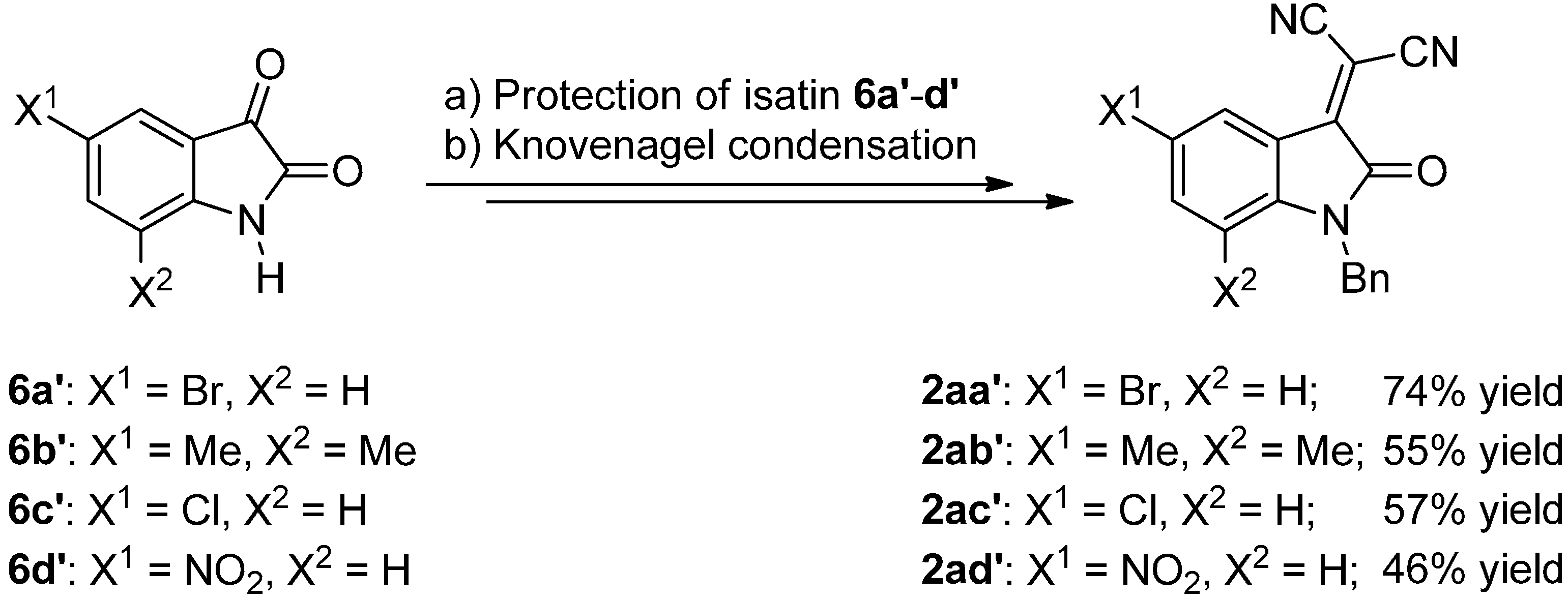
2.2. Synthesis of Starting Materials: Enamines 1 and Isatylidene Malononitriles 2
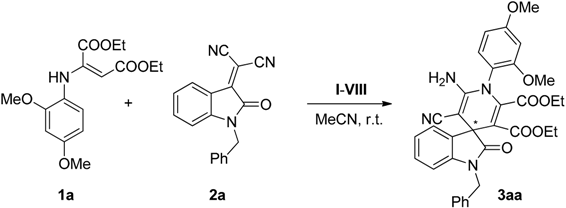
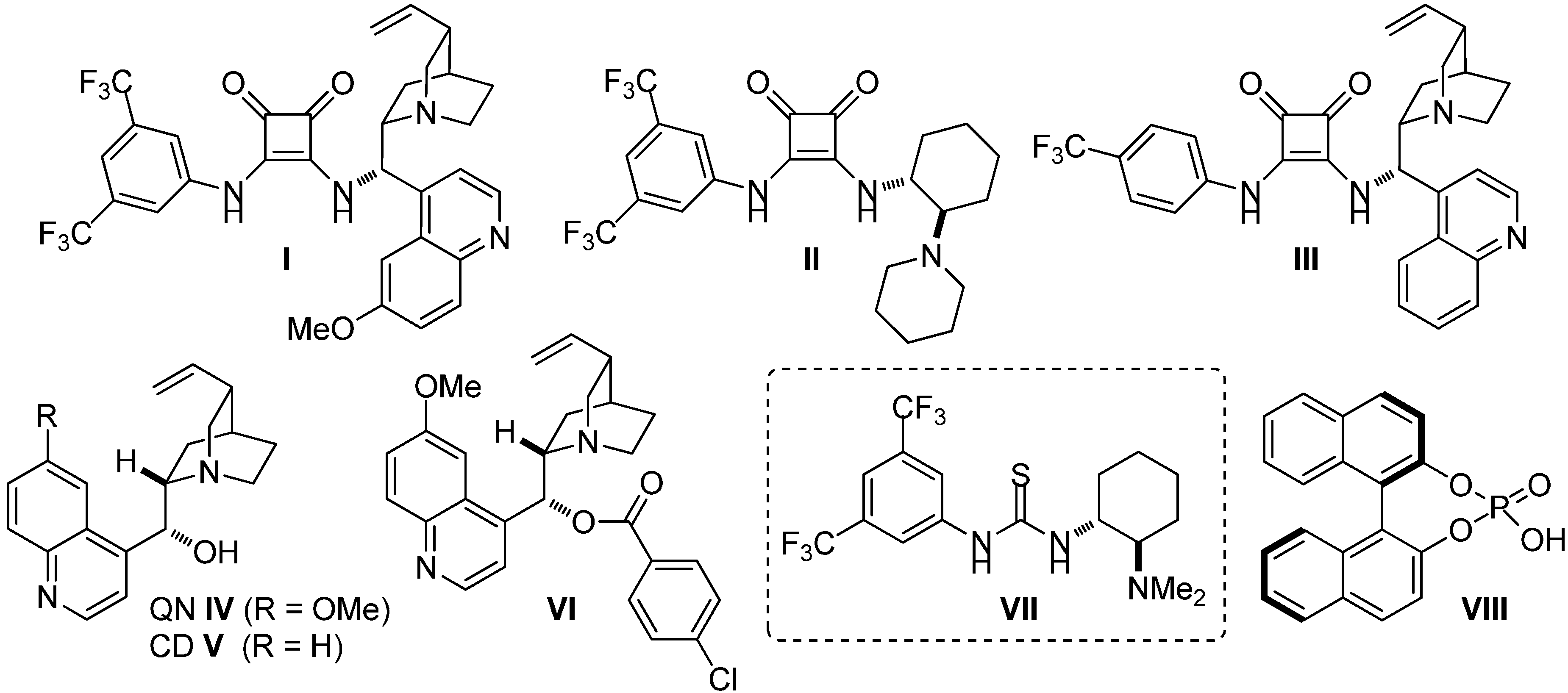
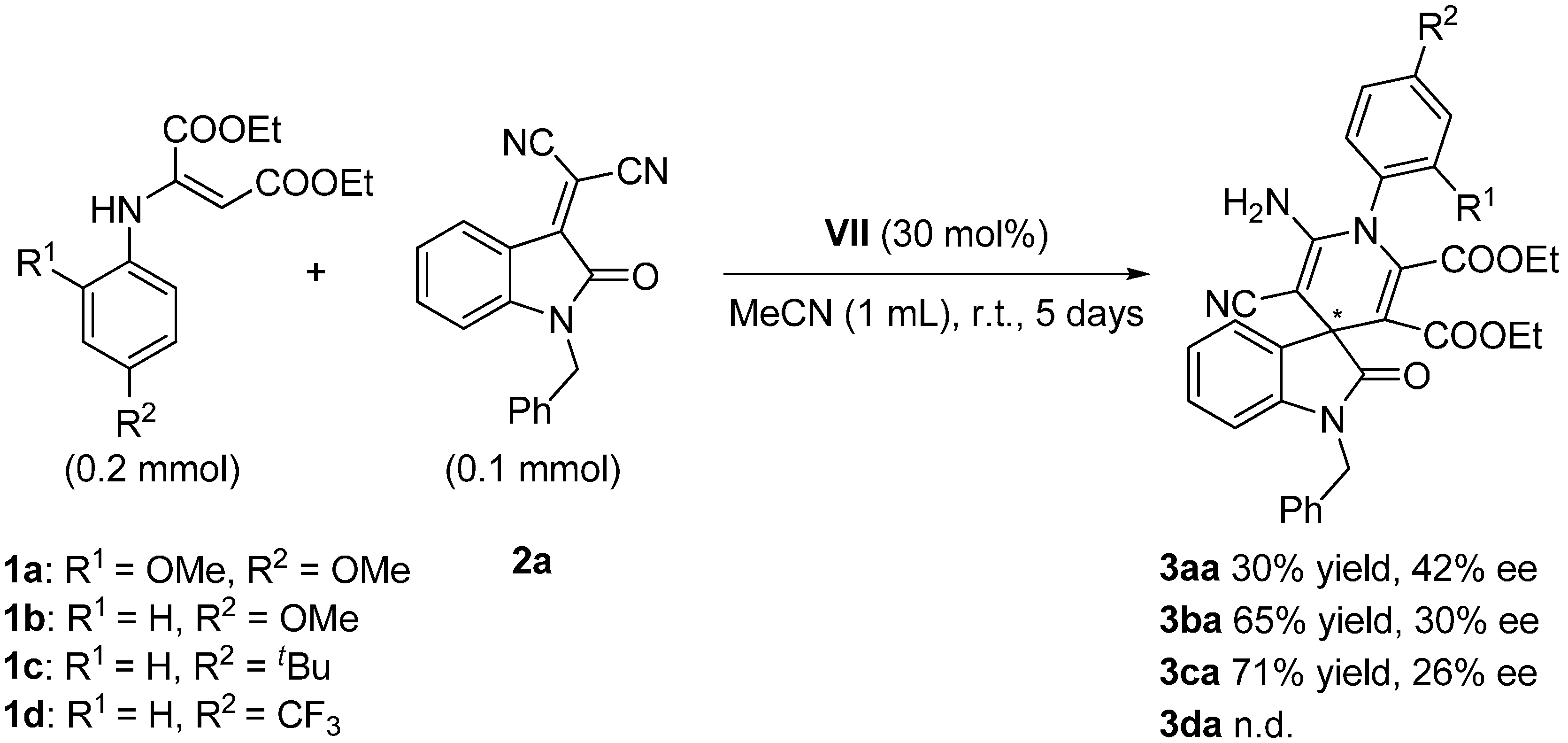
2.3. Screening


| Entry | Cat. a (mol %) | 1a (mmol) | 2a (mmol) | MeCN (mL) | t (d) | yield (%) b | ee c (%) d |
|---|---|---|---|---|---|---|---|
| 1 | I (10) | 0.2 | 0.1 | 1 | 5 | 15 | Rac. e |
| 2 | II (30) | 0.12 | 0.06 | 0.3 | 5 | 3 | 4 |
| 3 | III (30) | 0.12 | 0.06 | 0.3 | 5 | 16 | Rac. e |
| 4 | IV (30) | 0.2 | 0.1 | 1 | 5 | 77 | 5 |
| 5 | V (30) | 0.2 | 0.1 | 1 | 5 | 35 | 5 |
| 6 | VI (30) | 0.2 | 0.1 | 1 | 5 | 38 | Rac. e |
| 7 | VII (30) | 0.2 | 0.1 | 1 | 5 | 30 | 42 |
| 8 | VIII (30) | 0.2 | 0.1 | 1 | 5 | n.r. f | n.d. g |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | VII (mol %) | Solvent (mL) | yield (%) a | ee (%) b |
|---|---|---|---|---|
| 1 | 30 | MeCN (1) | 30 | 42 |
| 2 | 20 | MeCN (1) | 25 | 23 |
| 3 | 10 | MeCN (1) | 29 | 11 |
| 4 | 30 | MeCN (0.5) | 55 | 46 |
| 5 | 30 | EtOH (0.5) | 12 | 14 |
| 6 | 30 | EtOAc (0.5) | 31 | 20 |
| 7 | 30 | THF (0.5) | 13 | 16 |
| 8 | 30 | Toluene (0.5) | 29 | 14 |
| 9 | 30 | CH2Cl2 (0.5) | 49 | 26 |
| 10 | 30 | CHCl3 (0.5) | 48 | 16 |
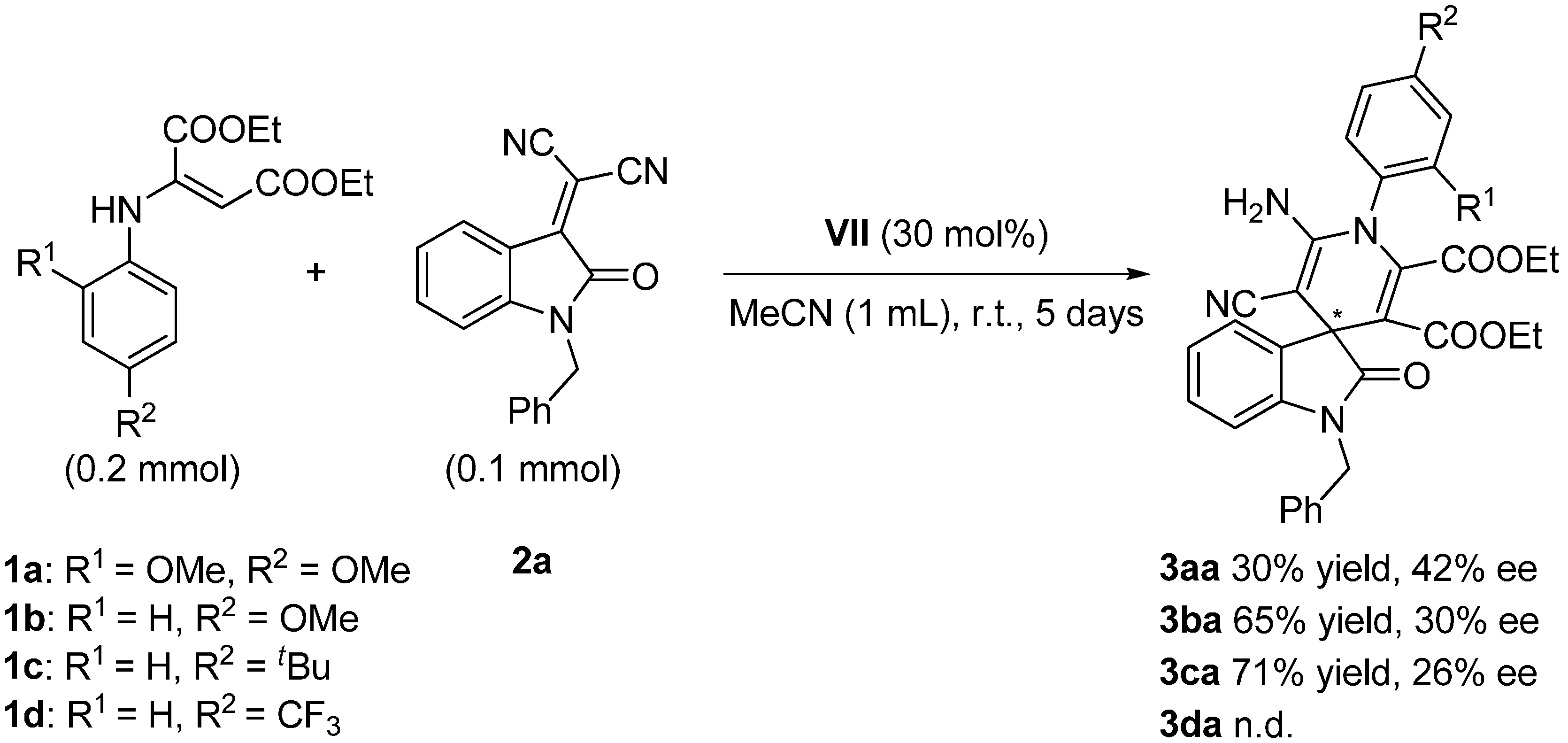
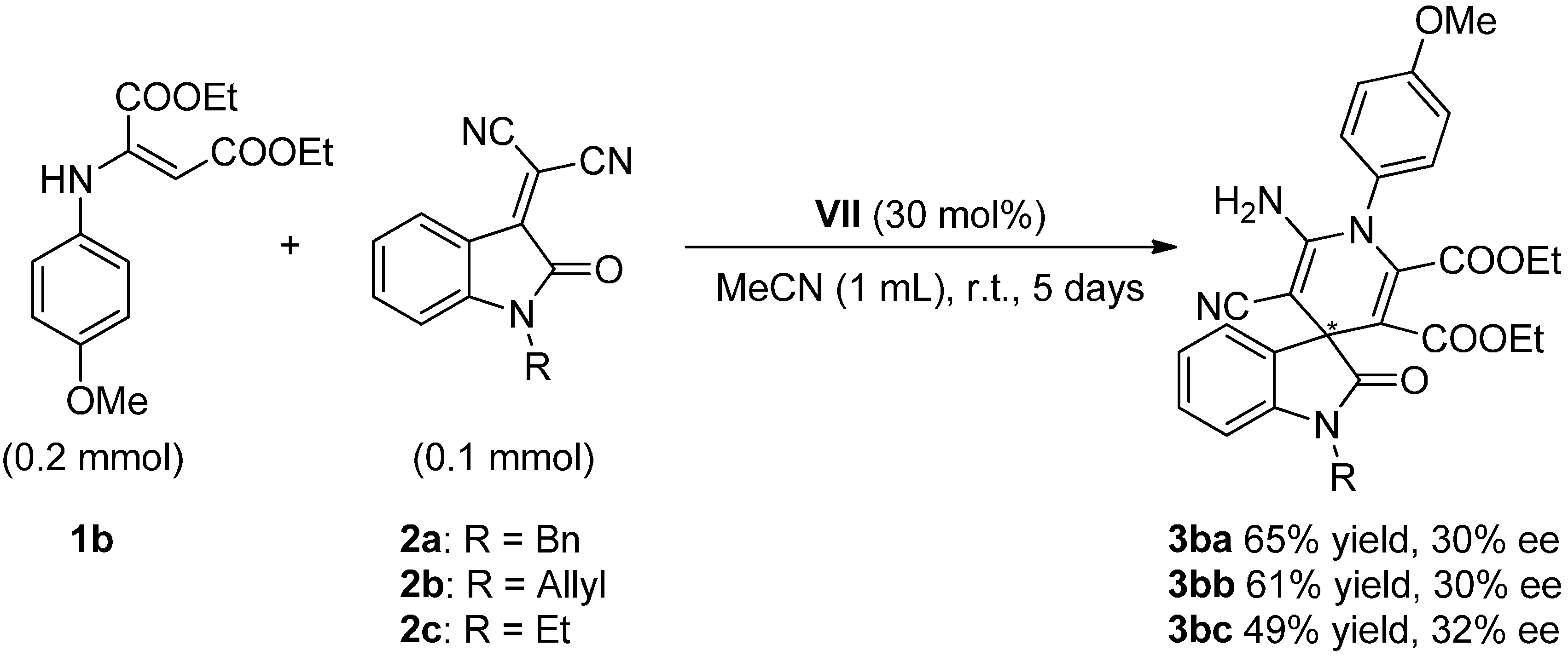
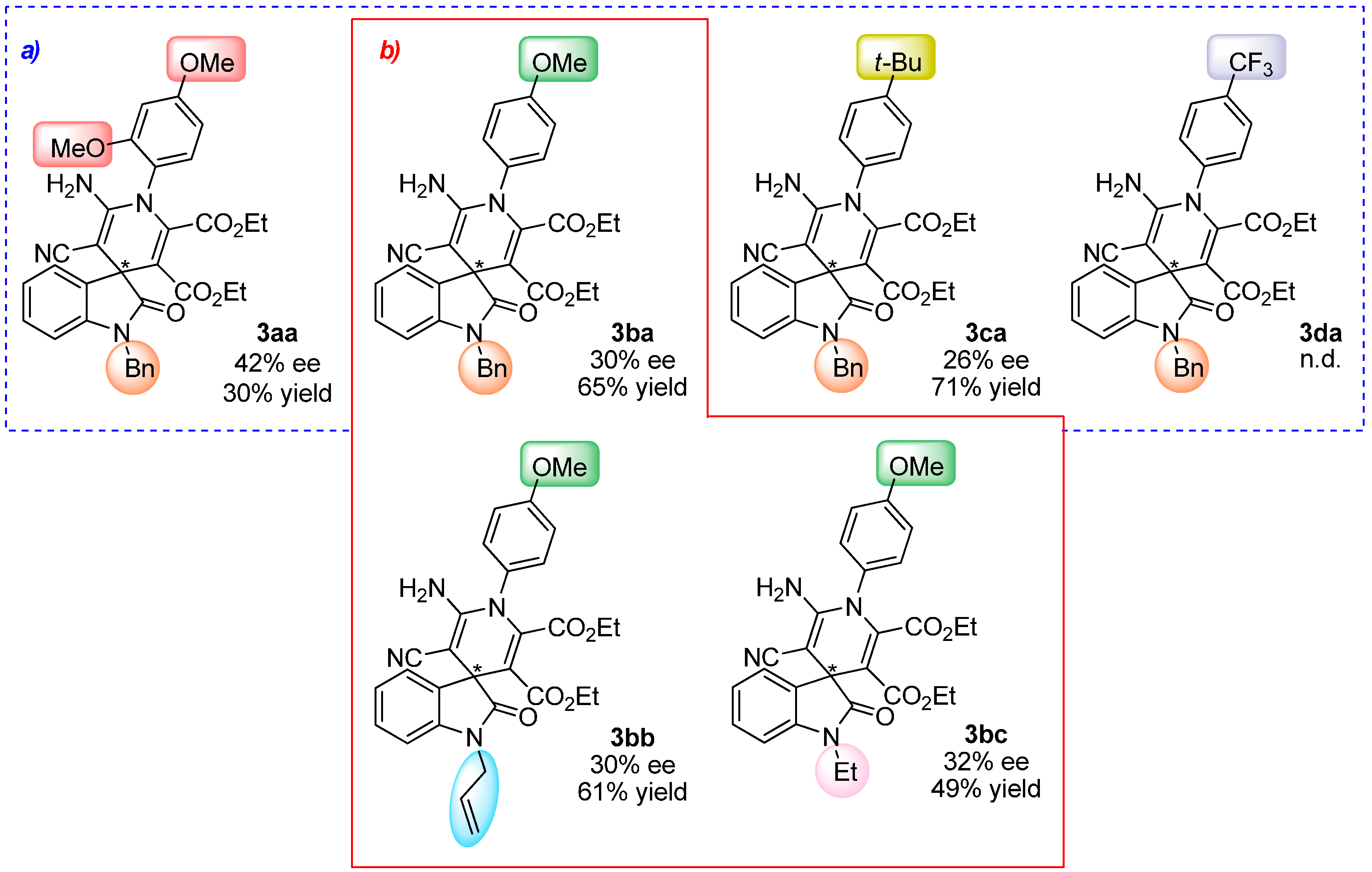
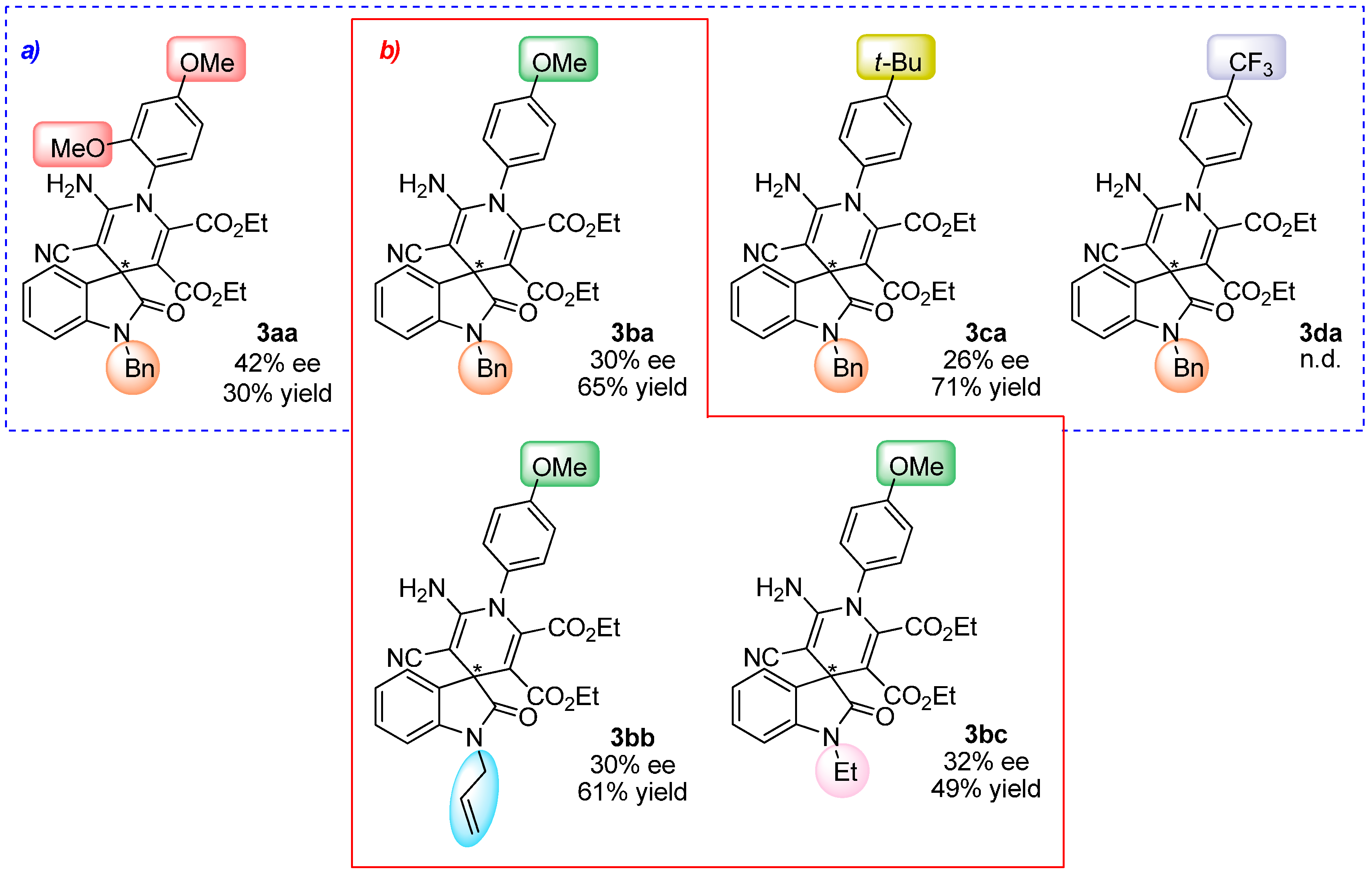
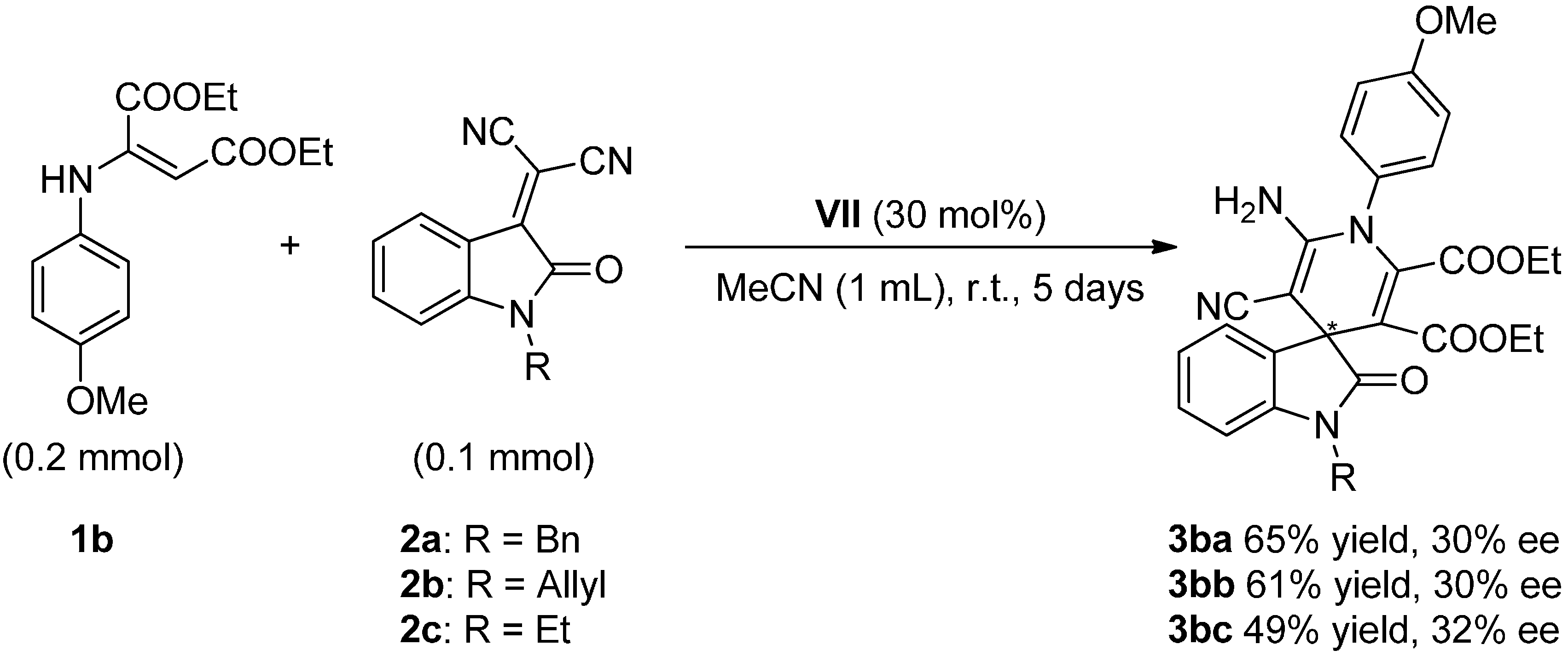
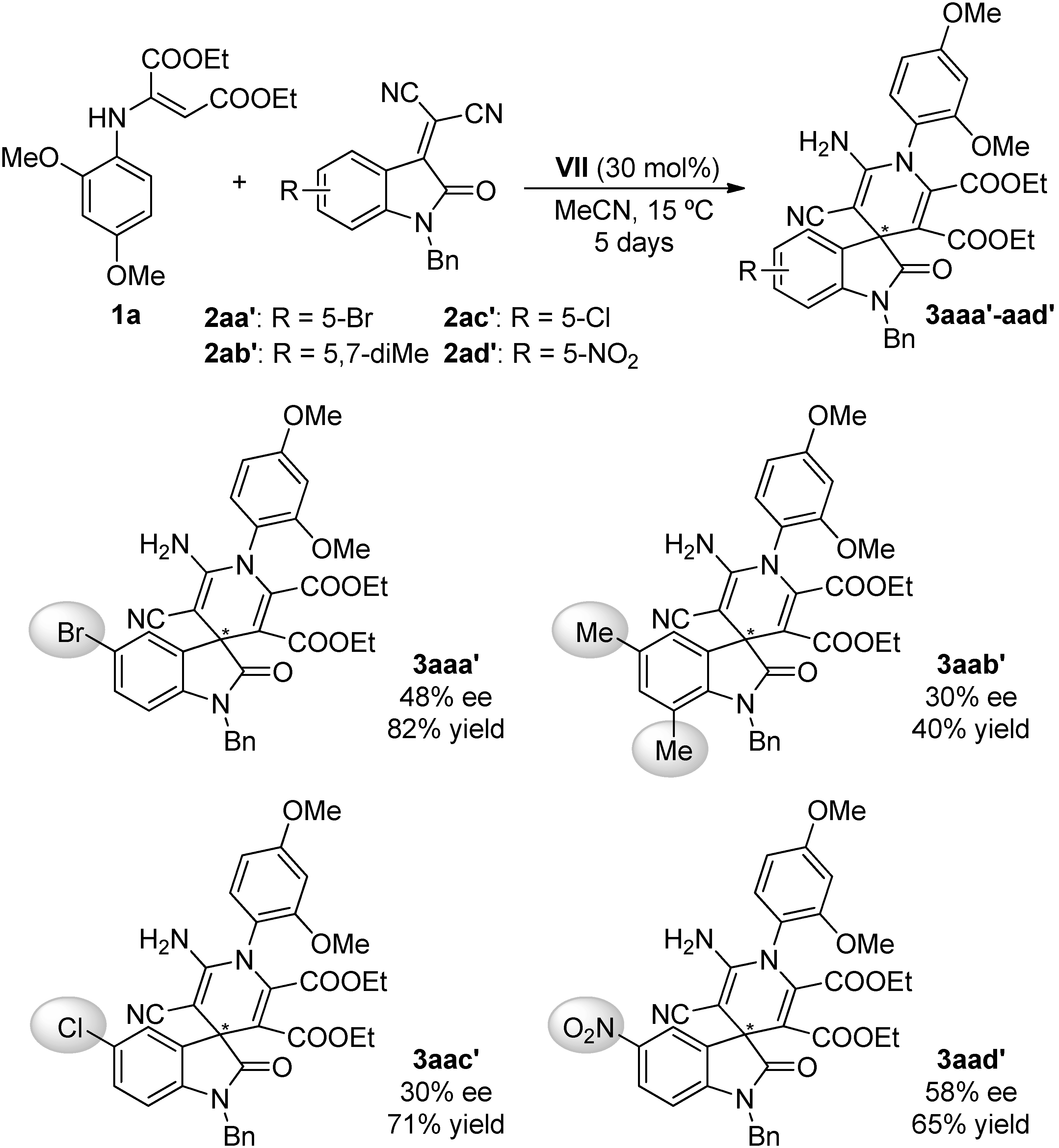
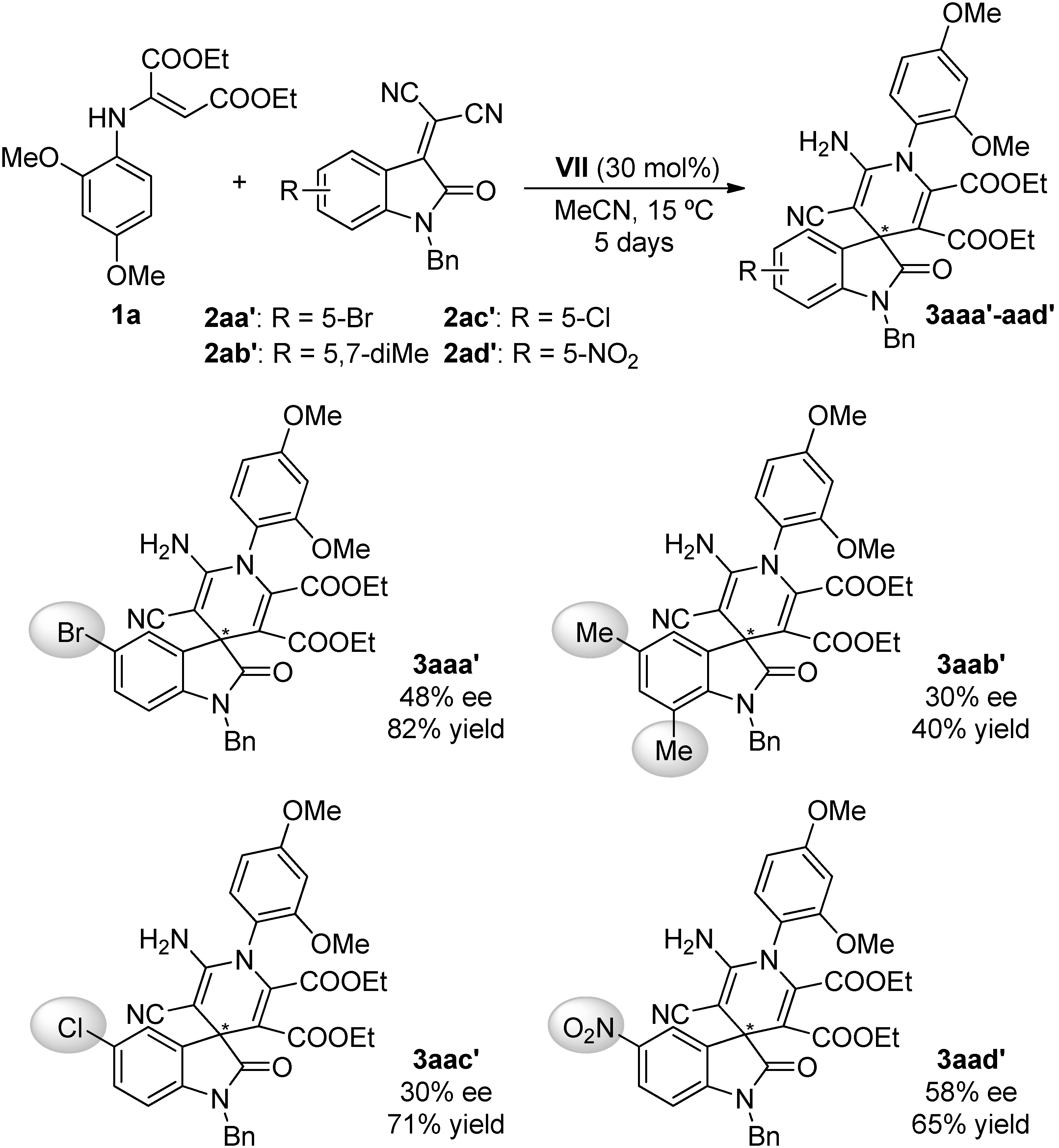
2.4. Scope of the Reaction
2.5. Mechanism of the Reaction
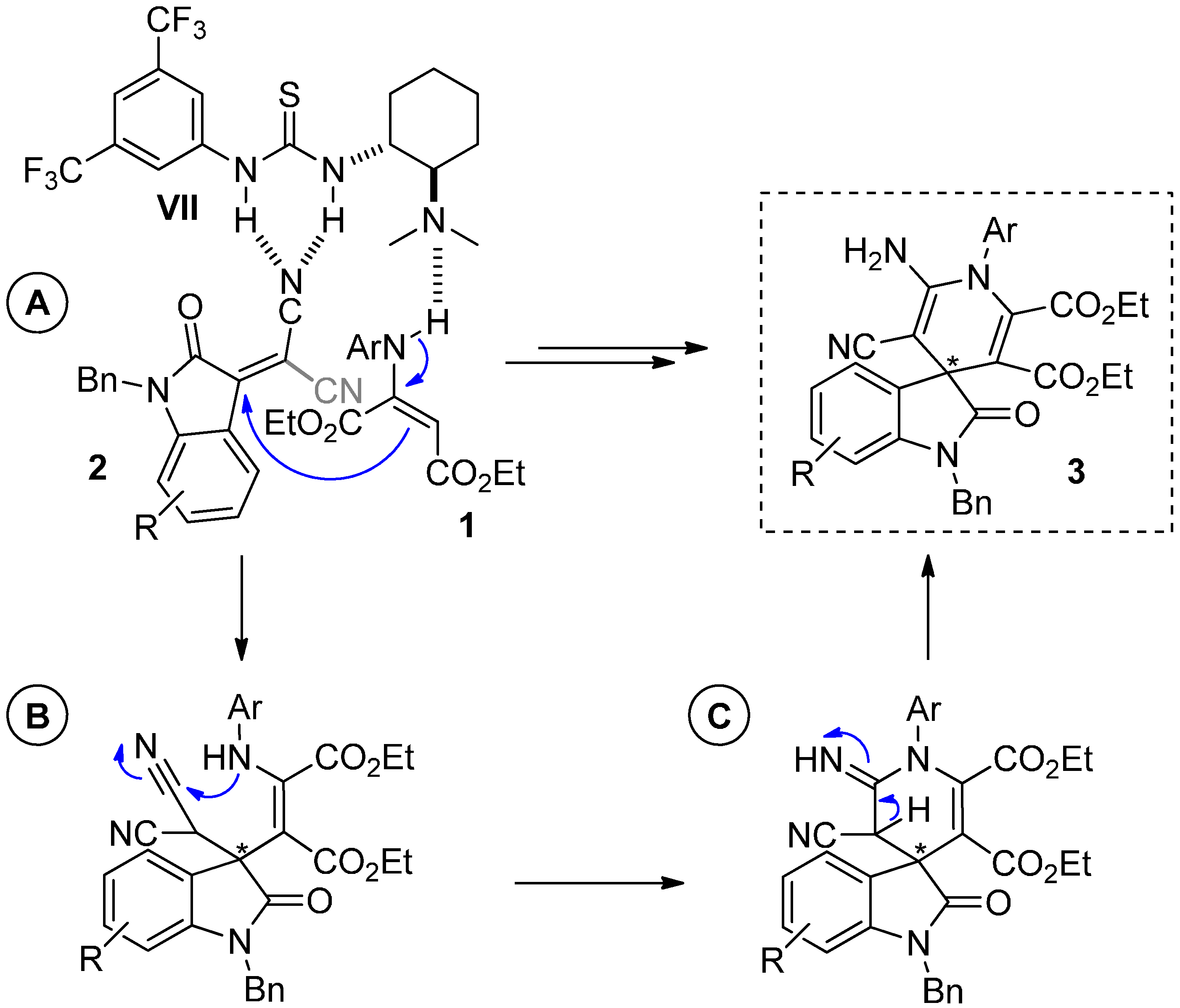
3. Experimental Section
3.1. General Experimental Methods
3.2. Materials
3.3. Synthesis and Physical, Analytical and Spectral data of Starting Materials (1 and 2) and the Final Compound (3)
3.3.1. Synthesis of E-Enamines 1a–d
3.3.2. Synthesis of Isatylidene Malononitriles 2a–c and 2aa′–ad′
Protection of Isatin (6)
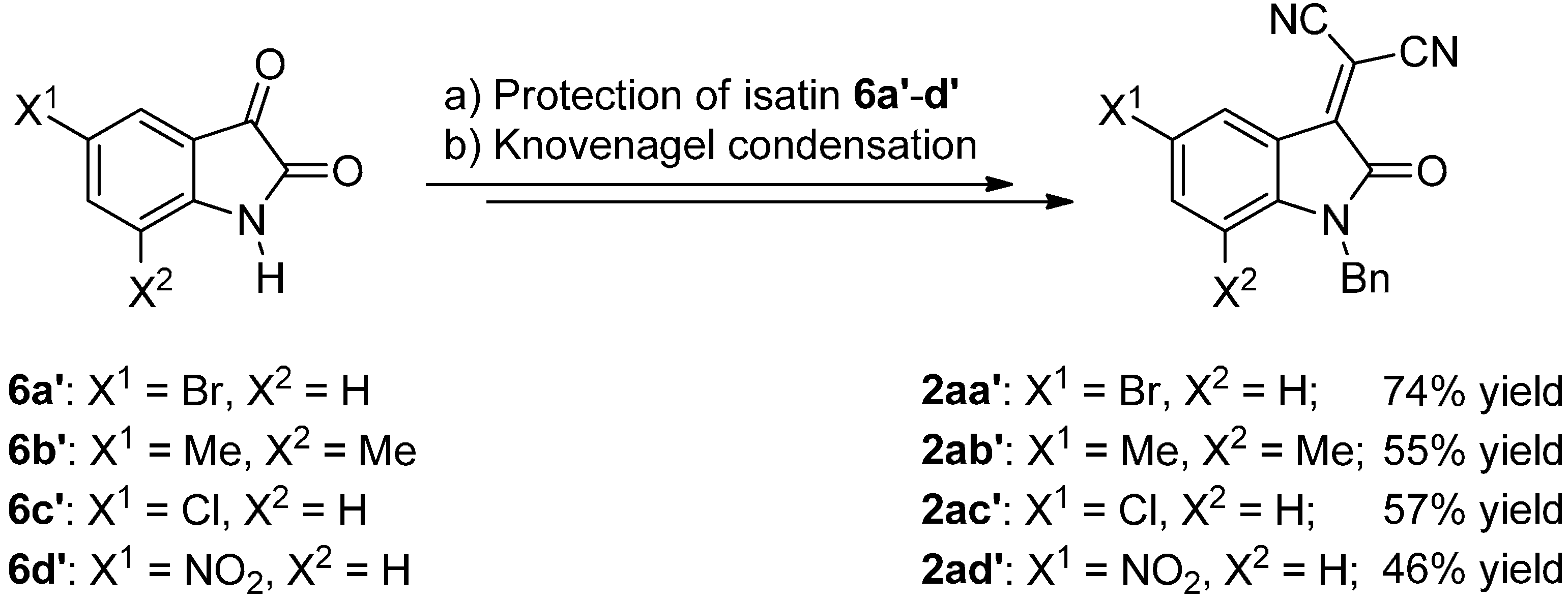
Knoevenagel Condensation
3.3.3. General Procedure for the Synthesis of Spirooxindoles 3
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Trost, B.M.; Brennan, M.K. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis 2009, 3003–3025. [Google Scholar] [CrossRef]
- Singh, G.S.; Desta, Z.Y. Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Wang, R. Recent advances in asymmetric organocatalytic construction of 3,3′-spirocyclic oxindoles. Adv. Synth. Catal. 2013, 355, 1023–1052. [Google Scholar] [CrossRef]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C.F., III. Organocatalytic asymmetric assembly reactions: Synthesis of spirooxindoles via organocascade strategies. ACS Catal. 2014, 4, 743–762. [Google Scholar] [CrossRef]
- Ball-Jones, N.R.; Badillo, J.J.; Franz, A.K. Strategies for the enantioselective synthesis of spirooxindoles. Org. Biomol. Chem. 2012, 10, 5165–5181. [Google Scholar] [CrossRef] [PubMed]
- Rottmann, M.; McNamara, C.; Yeung, B.K.S.; Lee, M.C.S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D.M.; Dharia, N.V.; Tan, J.; et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, S.; Danishefsky, S.J.; Sepp-Lorenzino, L.; Rosen, N. Total synthesis of spirotryprostatin A, leading to the discovery of some biologically promising analogues. J. Am. Chem. Soc. 1999, 121, 2147–2155. [Google Scholar] [CrossRef]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2–p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef] [PubMed]
- Antonchick, A.P.; Gerding-Reimers, C.; Catarinella, M.; Schürmann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Highly enantioselective synthesis and cellular evaluation of spirooxindoles inspired by natural products. Nat. Chem. 2010, 2, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar]
- Da Silva, J.F.M.; Garden, S.J.; Pinto, A.C. The chemistry of isatins: A review from 1975 to 1999. J. Braz. Chem. Soc. 2001, 12, 273–324. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.-L.; Zhou, J. Catalytic asymmetric synthesis of oxindoles bearing a tetrasubstituted stereocenter at the C-3 position. Adv. Synth. Catal. 2010, 352, 1381–1407. [Google Scholar] [CrossRef]
- Mohammadi, S.; Heiran, R.; Herrera, R.P.; Marqués-López, E. Isatin as a strategic motif for asymmetric catalysis. ChemCatChem 2013, 5, 2131–2148. [Google Scholar] [CrossRef]
- Eisner, U.; Kuthan, J. The chemistry of dihydropyridines. Chem. Rev. 1972, 72, 1–42. [Google Scholar] [CrossRef]
- Stout, D.M.; Meyers, A.I. Recent advances in the chemistry of dihydropyridines. Chem. Rev. 1982, 82, 223–243. [Google Scholar] [CrossRef]
- Sausins, A.; Duburs, G. Synthesis of 1,4-dihydropyridines by cyclocondensation reactions. Heterocycles 1988, 27, 269–289. [Google Scholar] [CrossRef]
- Reddy, G.M.; Shiradkar, M.; Chkravarthy, A.K. Chemical and pharmacological significance of 1,4-dihydropyridines. Curr. Org. Chem. 2007, 11, 847–852. [Google Scholar] [CrossRef]
- Wan, J.-P.; Liu, Y. Recent advances in new multicomponent synthesis of structurally diversified 1,4-dihydropyridines. RSC Adv. 2012, 2, 9763–9777. [Google Scholar] [CrossRef]
- Corey, E.J.; Guzman-Perez, A. The catalytic enantioselective construction of molecules with quaternary carbon stereocenters. Angew. Chem. Int. Ed. 1998, 37, 388–401. [Google Scholar] [CrossRef]
- Christoffers, J.; Mann, A. Enantioselective construction of quaternary stereocenters. Angew. Chem. Int. Ed. 2001, 40, 4591–4597. [Google Scholar] [CrossRef]
- Denissova, I.; Barriault, L. Stereoselective formation of quaternary carbon centers and related functions. Tetrahedron 2003, 59, 10105–10146. [Google Scholar] [CrossRef]
- Christoffers, J.; Baro, A. Stereoselective construction of quaternary stereocenters. Adv. Synth. Catal. 2005, 347, 1473–1482. [Google Scholar] [CrossRef]
- Bella, M.; Gasperi, T. Organocatalytic formation of quaternary stereocenters. Synthesis 2009, 1583–1614. [Google Scholar] [CrossRef]
- Hawner, C.; Alexakis, A. Metal-catalyzed asymmetric conjugate addition reaction: Formation of quaternary stereocenters. Chem. Commun. 2010, 46, 7295–7306. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M. Construction of asymmetric quaternary carbon centers with high enantioselectivity. Angew. Chem. Int. Ed. 2011, 50, 5998–6000. [Google Scholar] [CrossRef] [PubMed]
- Quasdorf, K.W.; Overman, L.E. Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 2014, 516, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2005. [Google Scholar]
- Dalko, P.I. (Ed.) Enantioselective Organocatalysis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2007.
- Dalko, P.I. (Ed.) Comprehensive Enantioselective Organocatalysis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2013.
- Herrera, R.P. (Ed.) Fundamentals in Organocatalysis. Past, Present and Future. Curr. Org. Chem. 2011, 15. Available online: http://www.ingentaconnect.com/content/ben/coc/2011/00000015/00000013 (accessed on 27 August 2015). [CrossRef]
- Zhang, L.-J.; Wu, Q.; Sun, J.; Yan, C.-G. Synthesis of functionalized spiro[indoline-3,4′-pyridines] and spiro[indoline-3,4′-pyridinones] via one-pot four-component reactions. Beilstein J. Org. Chem. 2013, 9, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, Y.-H.; Yan, C.-G. One-pot four-component reaction for convenient synthesis of functionalized 1-benzamidospiro[indoline-3,4′-pyridines]. Beilstein J. Org. Chem. 2014, 10, 2671–2676. [Google Scholar] [CrossRef] [PubMed]
- Kiruthika, S.E.; Lakshmi, N.V.; Banu, B.R.; Perumal, P.T. A facile strategy for the one pot multicomponent synthesis of spiro dihydropyridines from amines and activated alkynes. Tetrahedron Lett. 2011, 52, 6508–6511. [Google Scholar] [CrossRef]
- Sun, J.; Wu, Q.; Zhang, L.; Yan, C. Efficient synthesis of the functionalized spiro[indoline-3,4′-pyridine] via four-component reaction. Chin. J. Chem. 2012, 30, 1548–1554. [Google Scholar] [CrossRef]
- Tisseh, Z.N.; Ahmadi, F.; Dabiri, M.; Khavasi, H.R.; Bazgir, A. A novel organocatalytic multi-component reaction: An efficient synthesis of polysubstituted pyrano-fused spirooxindoles. Tetrahedron Lett. 2012, 53, 3603–3606. [Google Scholar] [CrossRef]
- Chen, W.-B.; Wu, Z.-J.; Pei, Q.-L.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Highly enantioselective construction of spiro[4H-pyran-3,3′-oxindoles] through a domino Knoevenagel/Michael/cyclization sequence catalyzed by cupreine. Org. Lett. 2010, 12, 3132–3135. [Google Scholar] [CrossRef] [PubMed]
- Macaev, F.; Sucman, N.; Shepeli, F.; Zveaghintseva, M.; Pogrebnoi, V. Facile and convenient one-pot process for the synthesis of spirooxindole derivatives in high optical purity using (−)-(S)-Brevicolline as an organocatalyst. Symmetry 2011, 3, 165–170. [Google Scholar] [CrossRef]
- Bernardi, L.; Fini, F.; Herrera, R.P.; Ricci, A.; Sgarzani, V. Enantioselective aza-Henry reaction using Cinchona organocatalysts. Tetrahedron 2006, 62, 375–380. [Google Scholar] [CrossRef]
- Pettersen, D.; Marcolini, M.; Bernardi, L.; Fini, F.; Herrera, R.P.; Sgarzani, V.; Ricci, A. Direct access to enantiomerically enriched α-amino phosphonic acid derivatives by organocatalytic asymmetric hydrophosphonylation of imines. J. Org. Chem. 2006, 71, 6269–6272. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R.P.; Sgarzani, V. Organocatalytic enantioselective decarboxilative addition of malonic half thioesters to imines. Adv. Synth. Catal. 2007, 349, 1037–1040. [Google Scholar] [CrossRef]
- The configuration E for the synthesized enamines was determined by H2D-NOESY.
- Thorwirth, R.; Stolle, A. Solvent-free synthesis of enamines from alkyl esters of propiolic or but-2-yne dicarboxylic acid in a ball mill. Synlett 2011, 2200–2202. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, Y.; Yabuta, T.; Takemoto, Y. Bifunctional thiourea-catalyzed enantioselective double Michael reaction of γ,δ-unsaturated β-ketoester to nitroalkene: Asymmetric synthesis of (−)-epibatidine. Tetrahedron Lett. 2004, 45, 9185–9188. [Google Scholar] [CrossRef]
- Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Enantioselective aza-Henry reaction catalyzed by a bifunctional organocatalyst. Org. Lett. 2004, 6, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. Enantio- and diastereoselective Michael reaction of 1,3-dicarbonyl compounds to nitroolefins catalyzed by a bifunctional thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, Y.; Okino, T.; Takemoto, Y. Enantioselective Michael addition to α,β-unsaturated imides catalyzed by a bifunctional organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 4032–4035. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, T.; Hoashi, Y.; Takemoto, Y. Thiourea-catalyzed asymmetric Michael addition of activated methylene compounds to α,β-unsaturated imides: Dual activation of imide by intra- and intermolecular hydrogen bonding. J. Am. Chem. Soc. 2006, 128, 9413–9419. [Google Scholar] [CrossRef] [PubMed]
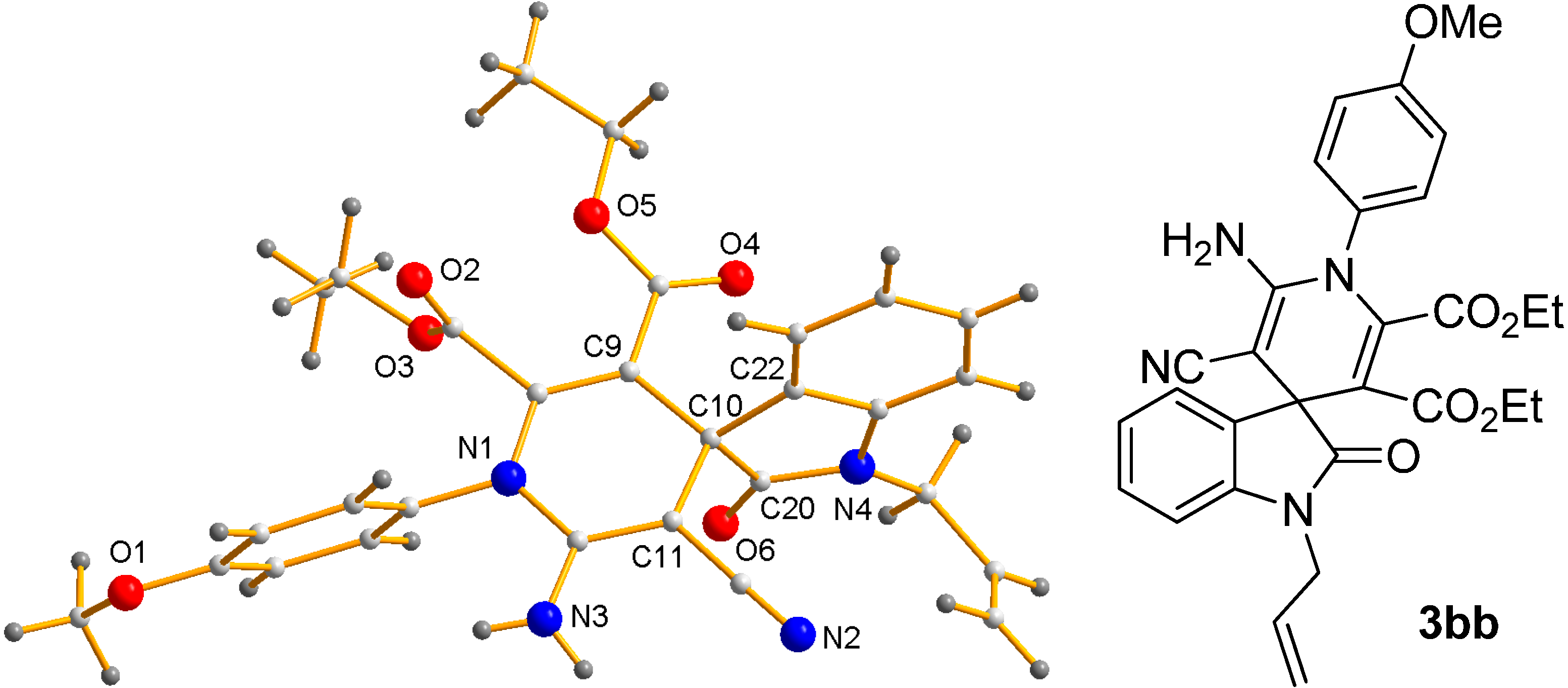
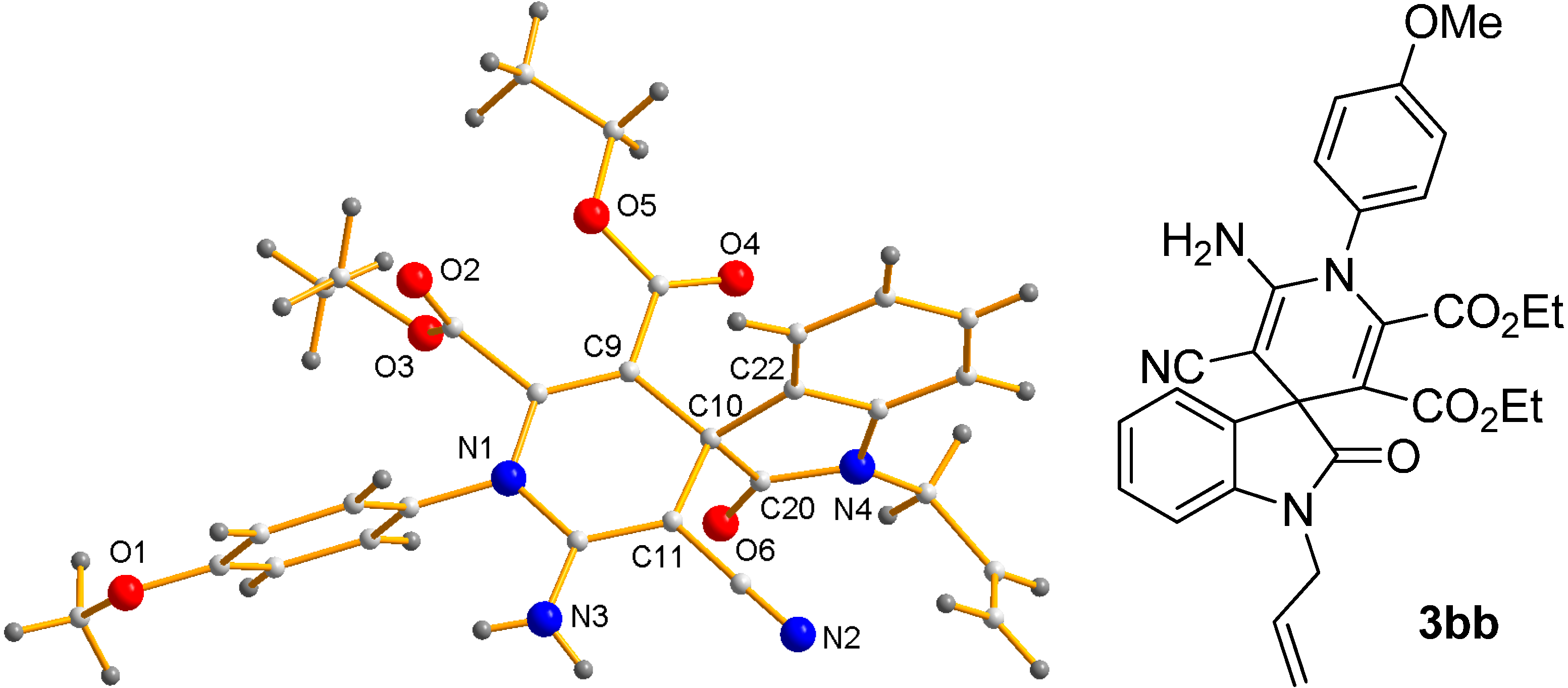
- CCDC-1411522 (3bb) Contains the Supplementary Crystallographic Data for This Paper. These Data can be Obtained Free of Charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
- Due to variations in the temperature registered at “room temperature”, it is worth to mention that the scope of the reaction was finally developed stabilizing the temperature at 15 °C in order to maintain fixed the temperature during the complete reaction time.
- Miyabe, H.; Takemoto, Y. Discovery and application of asymmetric reaction by multi-functional thioureas. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar] [CrossRef]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P. One-pot synthesis of unsymmetrical squaramides. RSC Adv. 2015, 5, 33450–33462. [Google Scholar] [CrossRef]
- Yang, W.; Du, D.-M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Lam, T.Y.; Malerich, J.P.; Rawal, V.H. Enantioselective α-amination of 1,3-dicarbonyl compounds using squaramide derivatives as hydrogen bonding catalysts. Org. Lett. 2010, 12, 2028–2031. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Peddinti, R.K. Introduction of a clean and promising protocol for the synthesis of β-amino-acrylates and 1,4-benzoheterocycles: An emerging innovation. Green Chem. 2011, 13, 3290–3299. [Google Scholar] [CrossRef]
- Redkin, R.Gr.; Shemchuk, L.A.; Chernykh, V.P.; Shishkinb, O.V.; Shishkina, S.V. Synthesis and molecular structure of spirocyclic 2-oxindole derivatives of 2-amino-4H-pyran condensed with the pyrazolic nucleus. Tetrahedron 2007, 63, 11444–11447. [Google Scholar] [CrossRef]
- Deng, H.-P.; Wei, Y.; Shi, M. Highly regio- and diastereoselective construction of spirocyclopenteneoxindoles through phosphine-catalyzed [3 + 2] annulation of Morita-Baylis-Hillman carbonates with isatylidene malononitriles. Org. Lett. 2011, 13, 3348–3351. [Google Scholar] [CrossRef] [PubMed]
- Demchuk, D.V.; Elinson, M.N.; Nikishin, G.I. “On water” Knoevenagel condensation of isatins with malononitrile. Mendeleev Commun. 2011, 21, 224–225. [Google Scholar] [CrossRef]
- Isatin 6c′ was synthetized following a previously reported protocol, see: Kathik, K.; Priyanka, K.B.; Manjula, S.; Sammaiah, G. Synthesis and evaluation of new bis-isatin derivatives for antioxidant activity. Int. J. Pharm. Pharm. Sci. 2013, 5, 224–227. [Google Scholar]
- The NMR spectra of isatin 6c′ are consistent with those reported in the literature, see: Mendonça, G.F.; Magalhães, R.R.; de Mattos, M.C.S.; Esteves, P.M. Trichloroisocyanuric acid in H2SO4: An efficient superelectrophilic reagent for chlorination of isatin and benzene derivatives. J. Braz. Chem. Soc. 2005, 16, 695–698. [Google Scholar]
- Isatin 6d′ was synthetized following a previously reported protocol, see: Sonawane, R.P.; Tripathi, R.R. The chemistry and synthesis of 1H-indole-2,3-dione (isatin) and its derivatives. Int. Lett. Chem. Phys. Astron. 2013, 7, 30–36. [Google Scholar]
- The NMR spectra of isatin 6d′ are consistent with those reported in the literature, see: Siddiqui, N.; Alam, M.S.; Stables, J.P. Synthesis and anticonvulsant properties of 1-(amino-N-arylmethanethio)-3-(1-substituted benzyl-2, 3-dioxoindolin-5-yl) urea derivatives. Eur. J. Med. Chem. 2011, 46, 2236–2242. [Google Scholar]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auria-Luna, F.; Marqués-López, E.; Mohammadi, S.; Heiran, R.; Herrera, R.P. New Organocatalytic Asymmetric Synthesis of Highly Substituted Chiral 2-Oxospiro-[indole-3,4′- (1′,4′-dihydropyridine)] Derivatives. Molecules 2015, 20, 15807-15826. https://doi.org/10.3390/molecules200915807
Auria-Luna F, Marqués-López E, Mohammadi S, Heiran R, Herrera RP. New Organocatalytic Asymmetric Synthesis of Highly Substituted Chiral 2-Oxospiro-[indole-3,4′- (1′,4′-dihydropyridine)] Derivatives. Molecules. 2015; 20(9):15807-15826. https://doi.org/10.3390/molecules200915807
Chicago/Turabian StyleAuria-Luna, Fernando, Eugenia Marqués-López, Somayeh Mohammadi, Roghayeh Heiran, and Raquel P. Herrera. 2015. "New Organocatalytic Asymmetric Synthesis of Highly Substituted Chiral 2-Oxospiro-[indole-3,4′- (1′,4′-dihydropyridine)] Derivatives" Molecules 20, no. 9: 15807-15826. https://doi.org/10.3390/molecules200915807