
A Qualitative Comparison of the Reactivities of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine and 4,5-Dichloro-1,2,3-dithiazolium Chloride


Abstract
: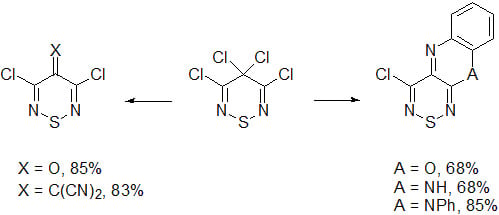
1. Introduction
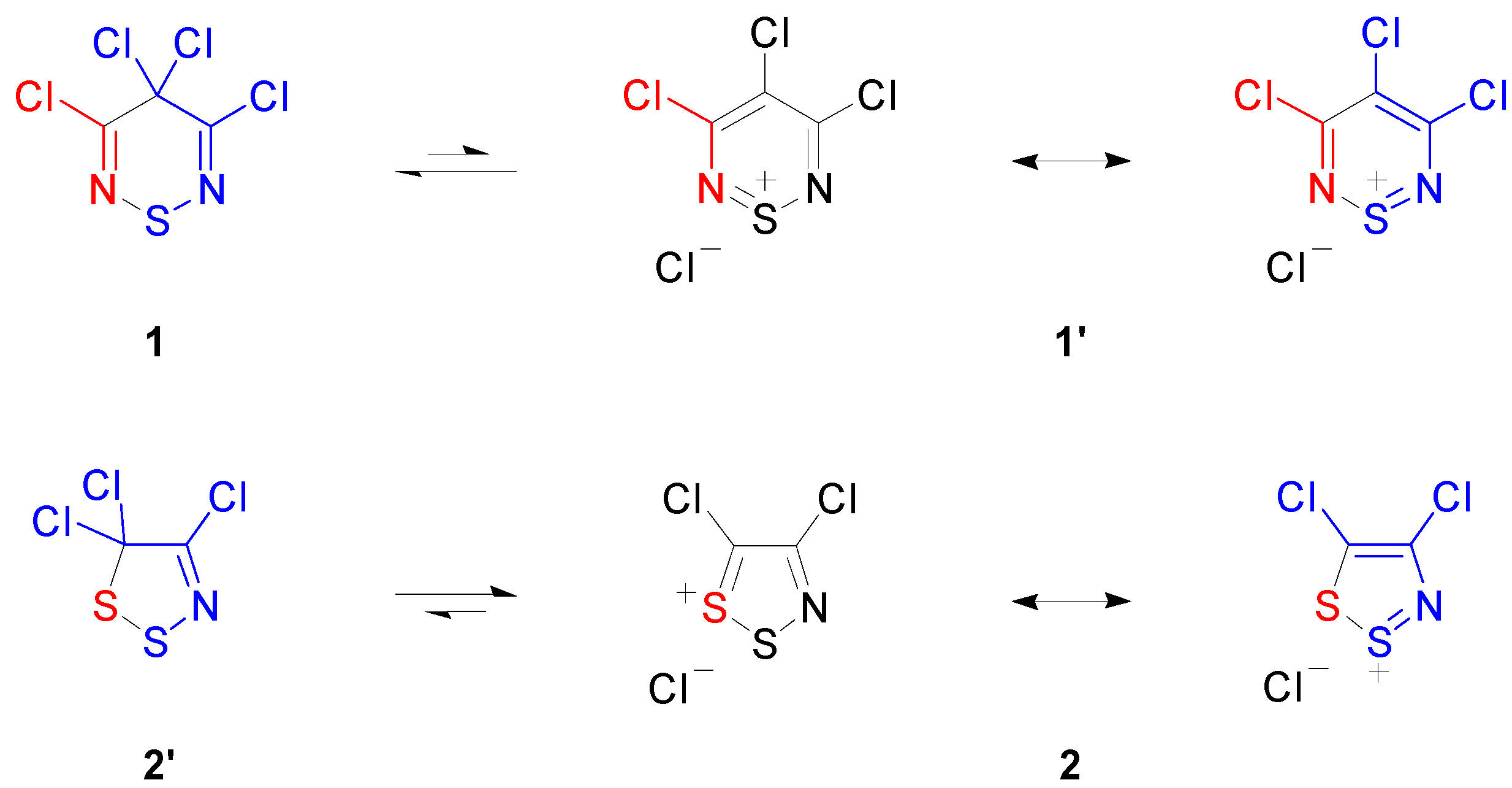

2. Results and Discussion
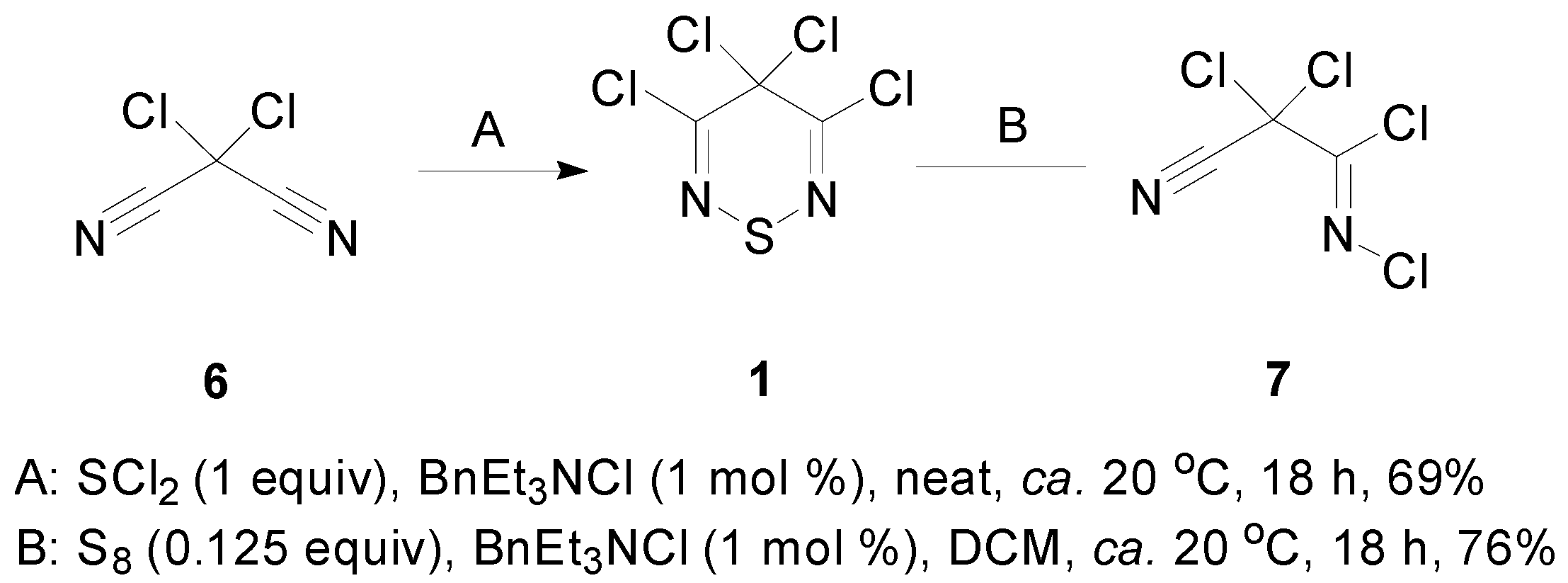
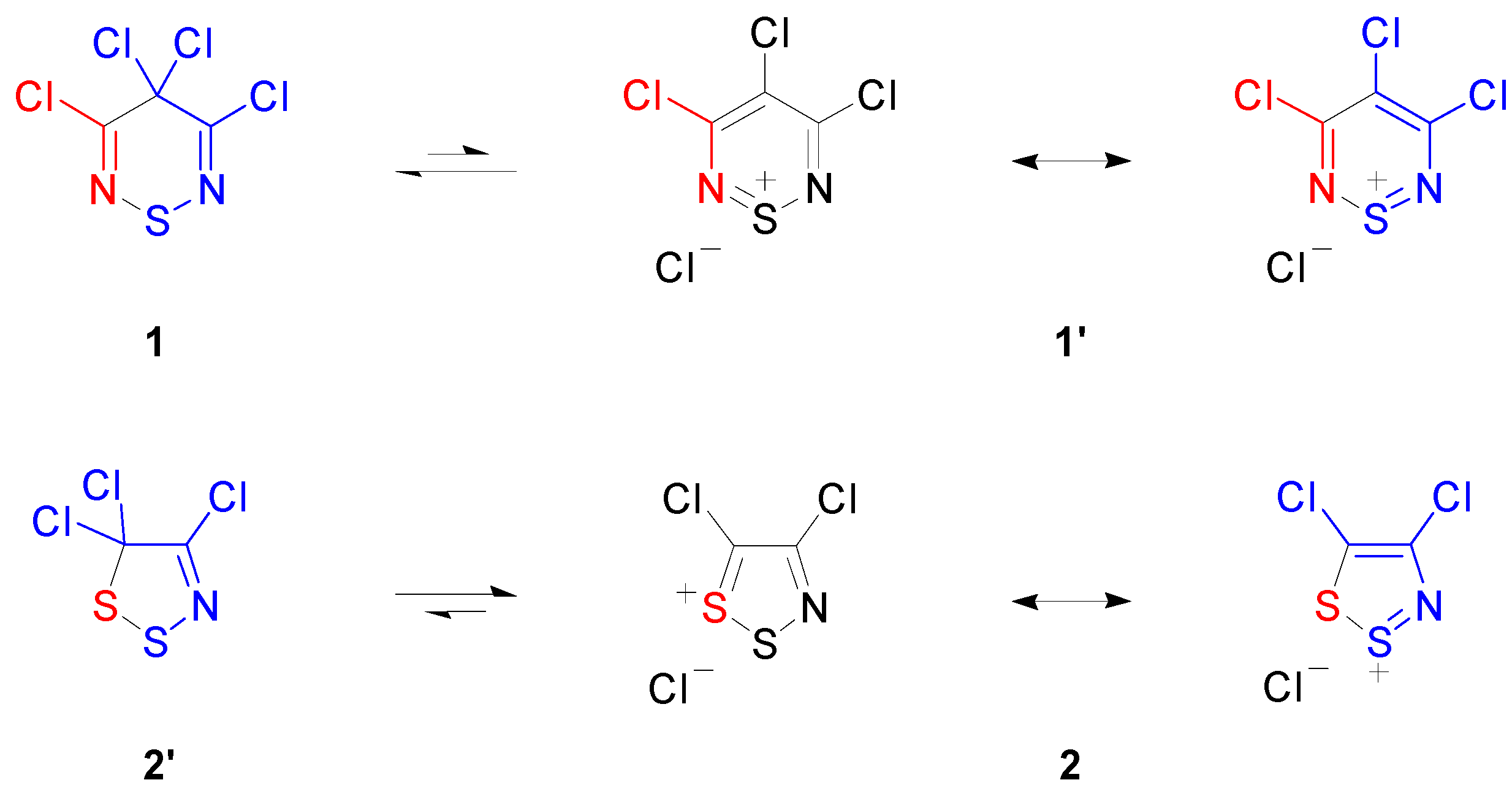
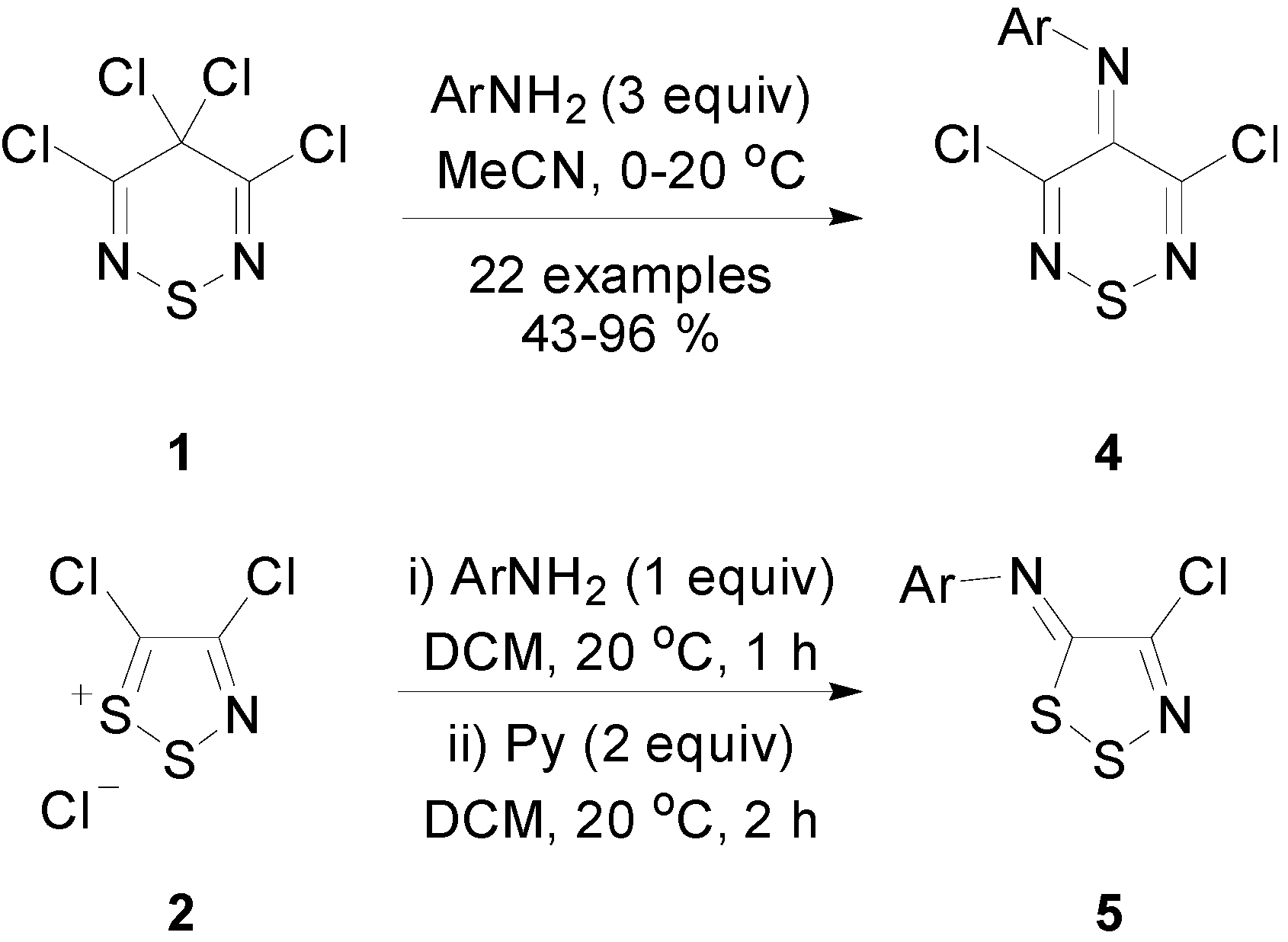
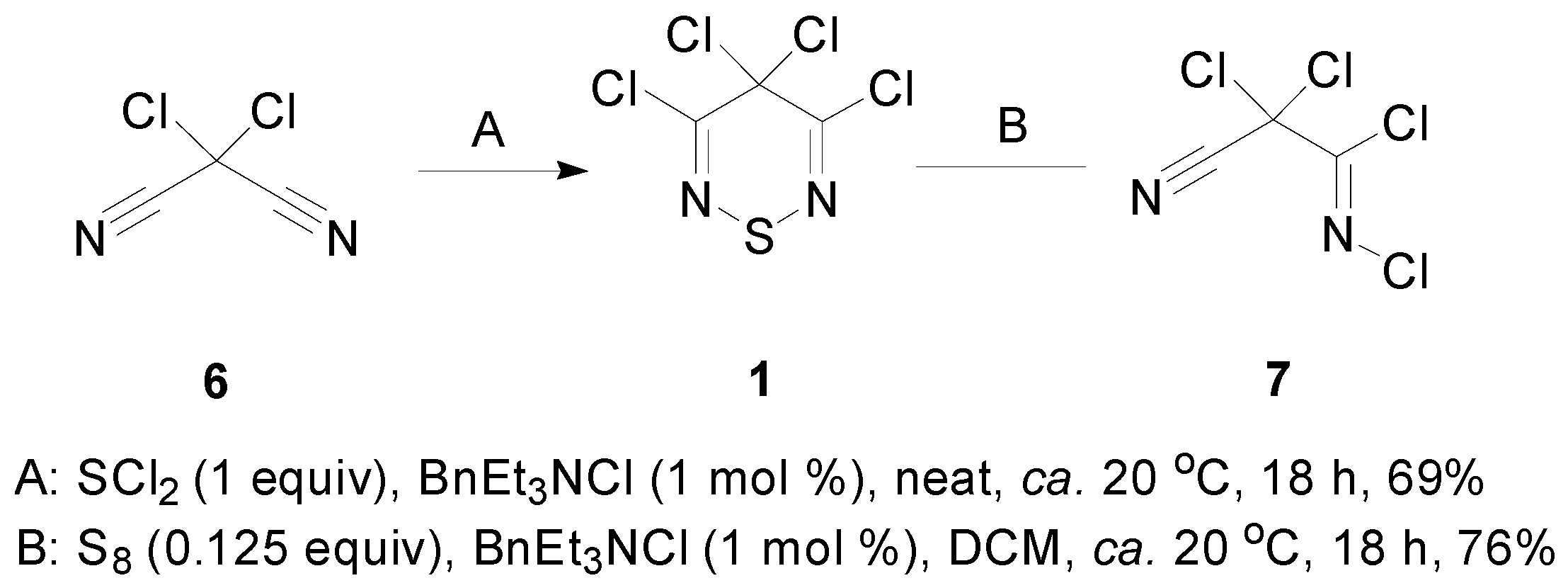
2.1. Preparation of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1)

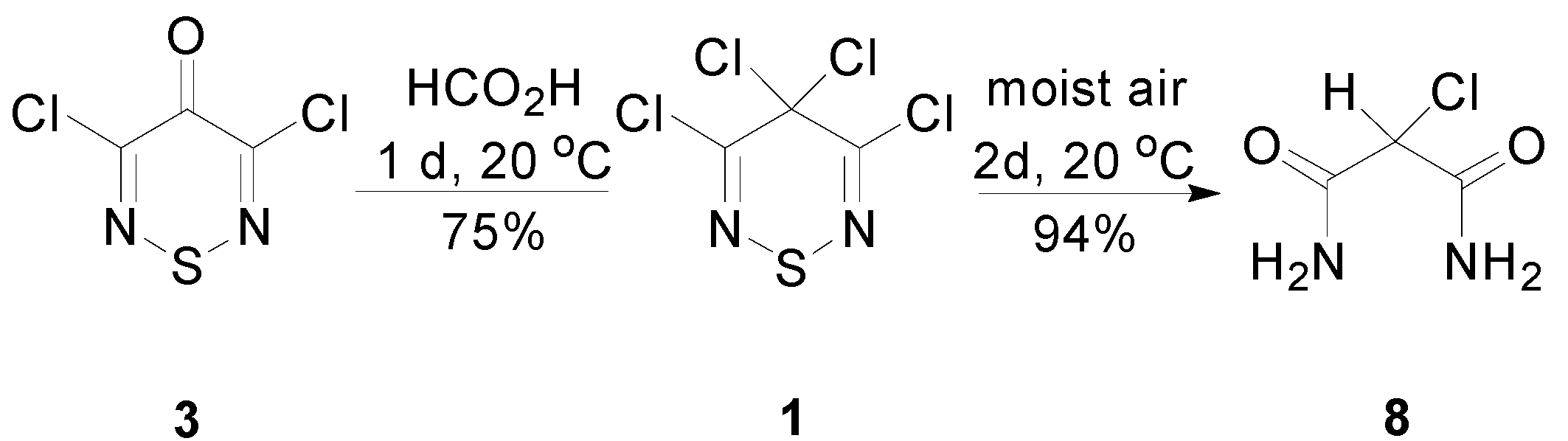
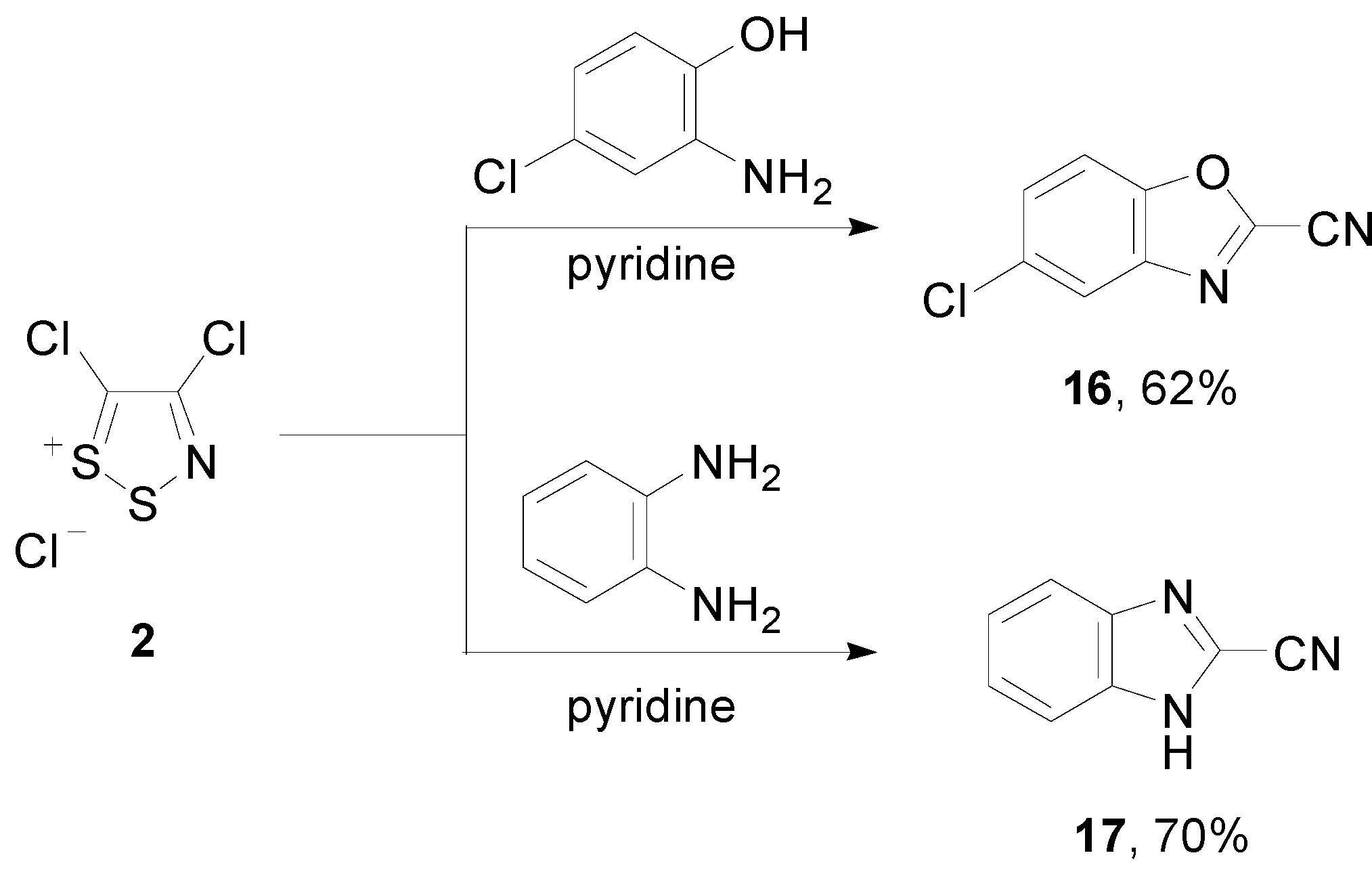
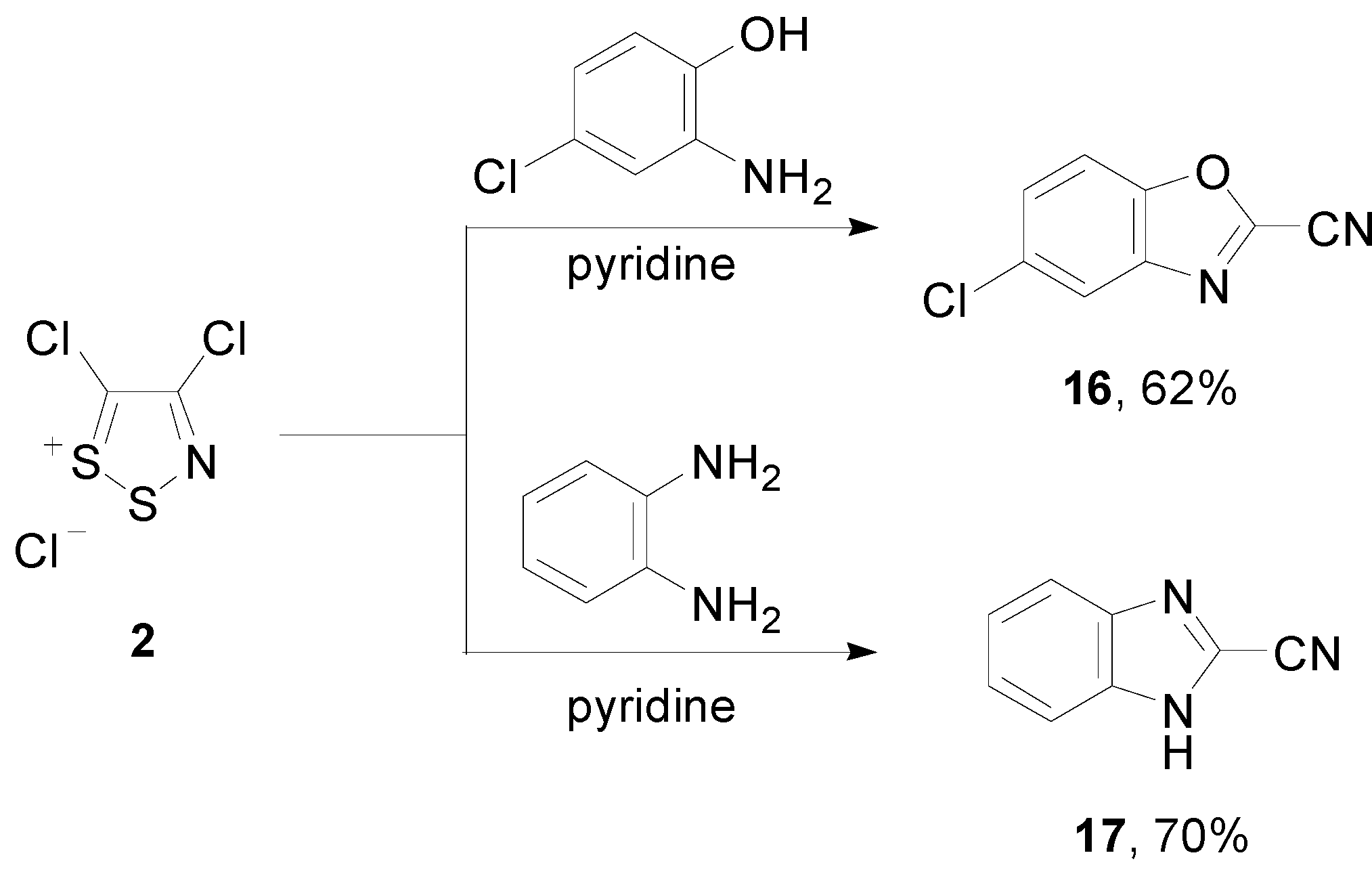
2.2. Preparation of the Thiadiazinone 3 and Dithiazolone 9


| Entry | Conditions a | Product Yields and Reaction Times | |
|---|---|---|---|
| 1 | HCO2H (98%) neat | 3 (75%), 24 h | 9 (89%), 2 h |
| 2 | HCO2H (85%) b neat | 3 (0%), c 18 h | 9 (72%), 1 h |
| 3 | AcOH neat | 3 (74%), 48 h | 9 (60%), 2 h |
| 4 | NaNO3 (1 equiv), DCM | 3 (nr), d 18 h | 9 (72%), 18 h |
| 5 | NaNO3 (1 equiv), MeCN | 3 (71%), 1.5 h | 9 (77%), 18 h |
| 6 | AgNO3 (1 equiv), MeCN | 3 (85%), 0.5 h | 9 (72%), 1 min |
| 7 | Ag2SO4 (0.5 equiv), MeCN | 3 (77%), 4 h | 9 (73%), 24 h |
| 8 | H2O (1 equiv), DMSO (1 mol %), MeCN | 3 (0%), c 20 h | 9 (86%), 1 h |
| 9 | DMSO (1 equiv), MeCN | 3 (31%), 20 h | 9 (83%), 1.5 h |
| 10 | DMSO neat | 3 (45%), 1 h | 9 (50%), 15 min |
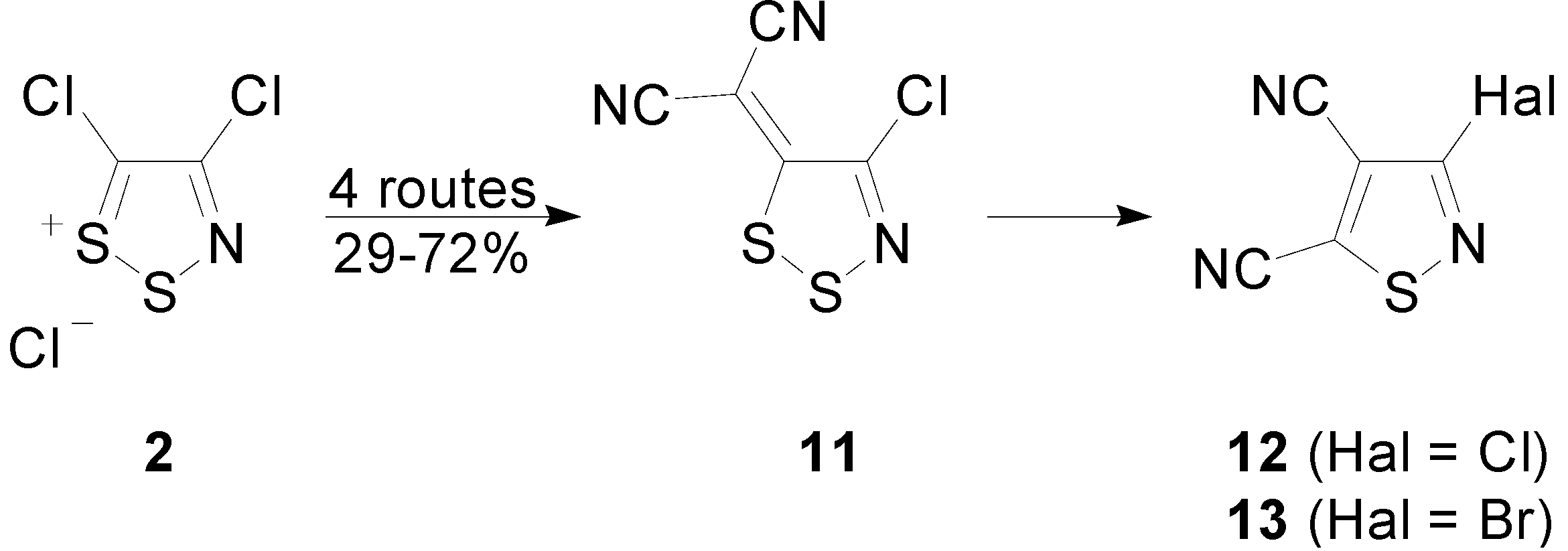
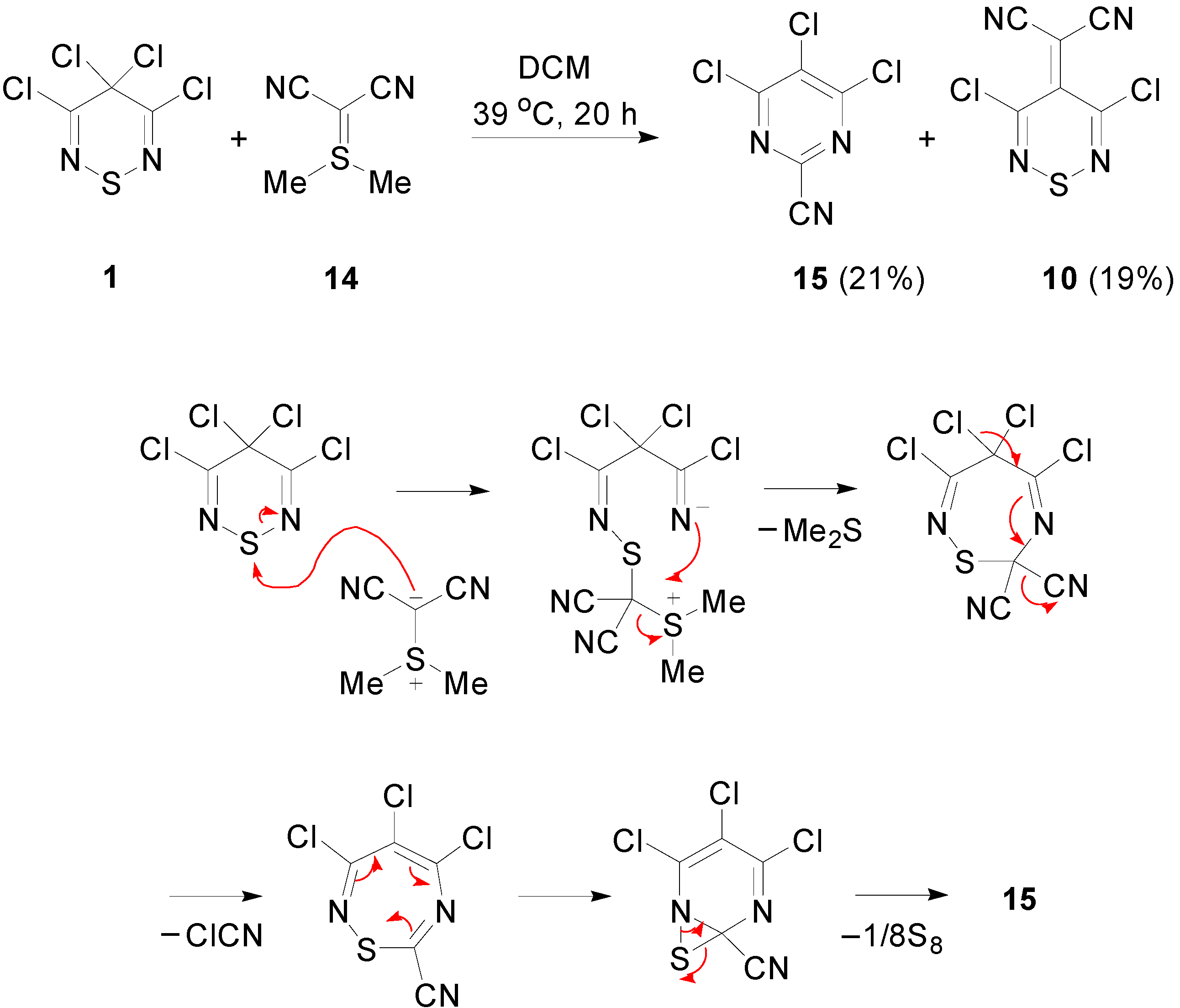
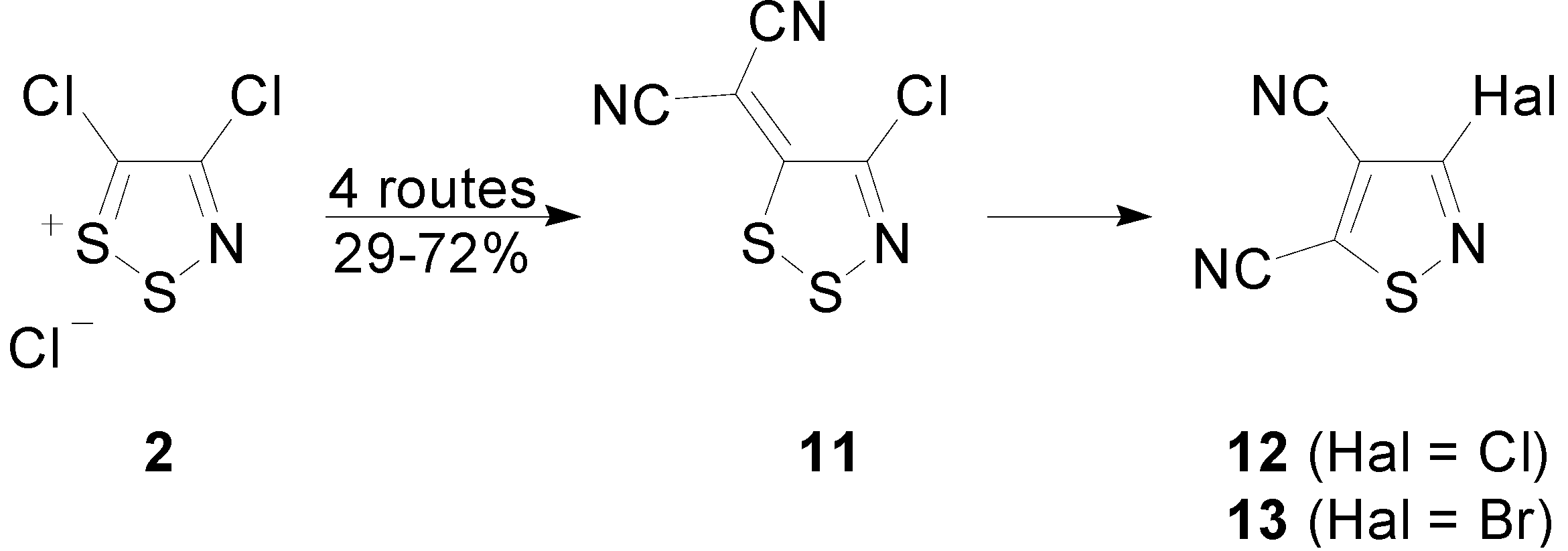
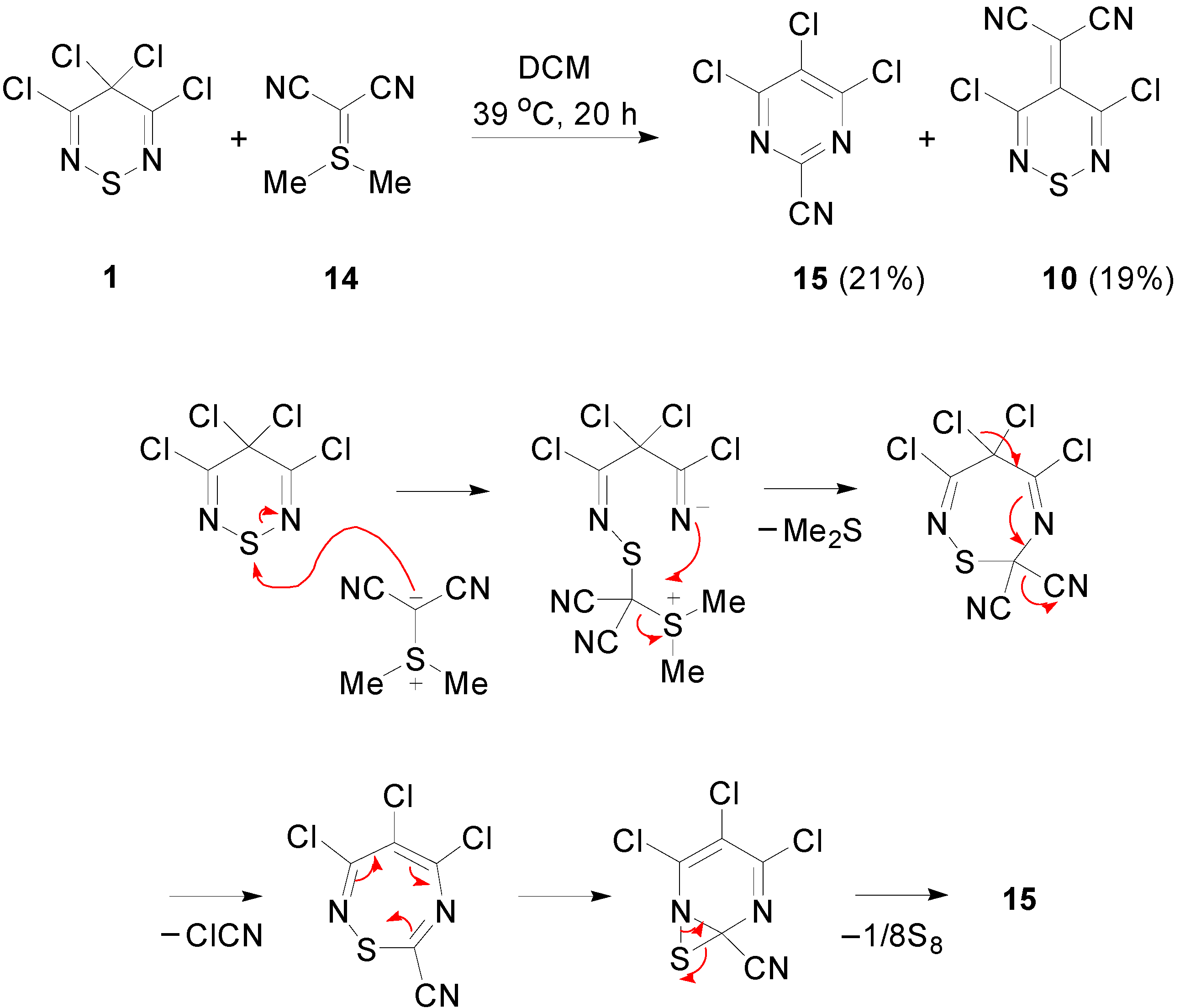
2.3. Preparation of Ylidenemalononitriles 10 and 11
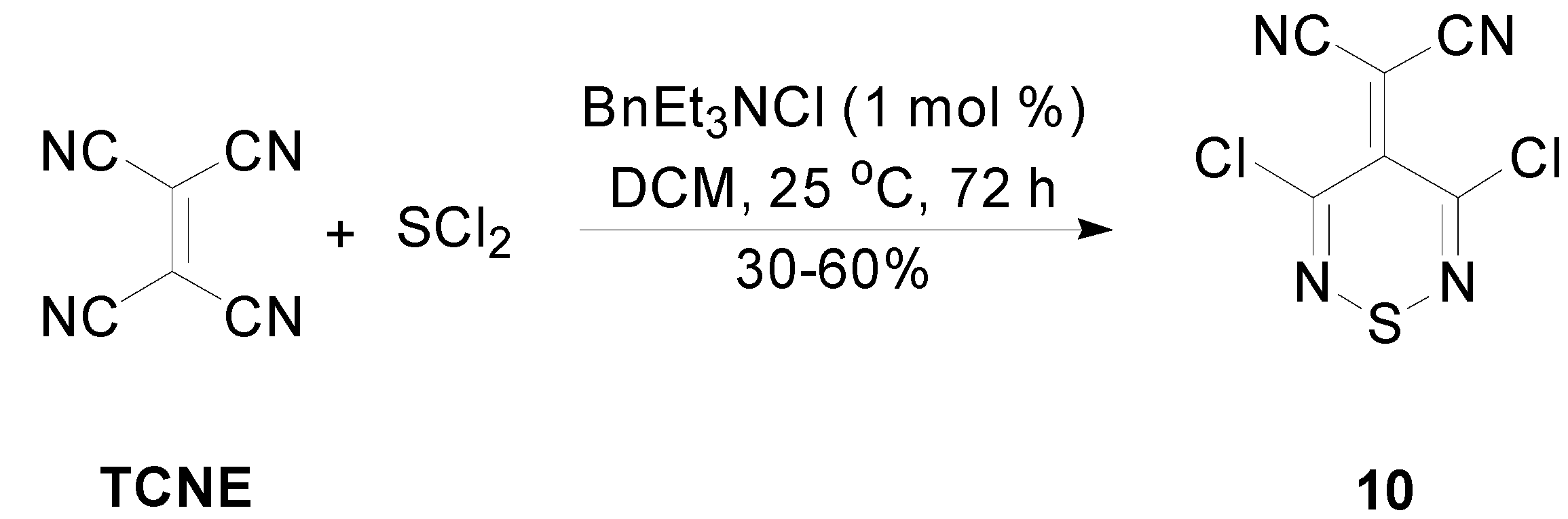

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
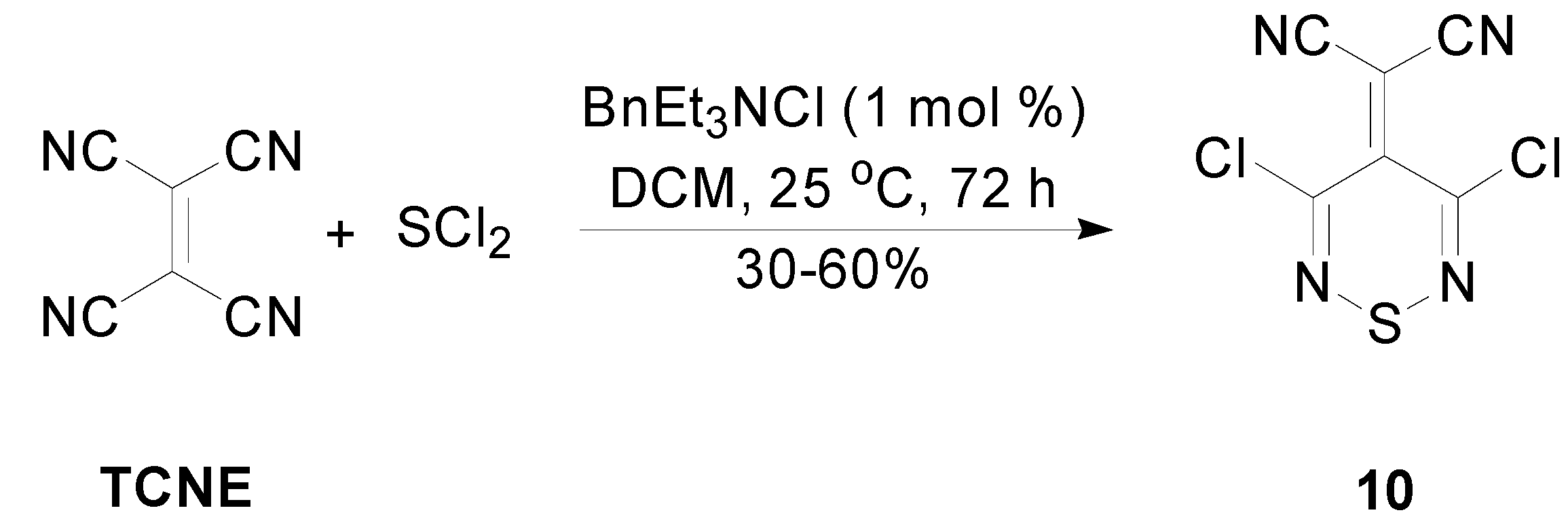{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 1 (mmol) | CH2(CN)2 (equiv) | DCM (mL) | Time (min) | Yield of 10 (%) |
|---|---|---|---|---|---|
| 1 | 0.42 | 1.1 | 1 | 10 | 78 |
| 2 | 0.42 | 1.5 | 1 | 10 | 82 |
| 3 | 0.42 | 2 | 1 | 10 | 83 |
| 4 | 1 | 1.5 | 1 | 10 | 71 |
| 5 | 2 | 1.5 | 4 | 30 | 73 |
| 6 | 4 | 1.5 | 4 | 30 | 64 a |
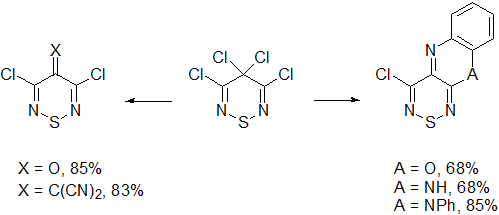
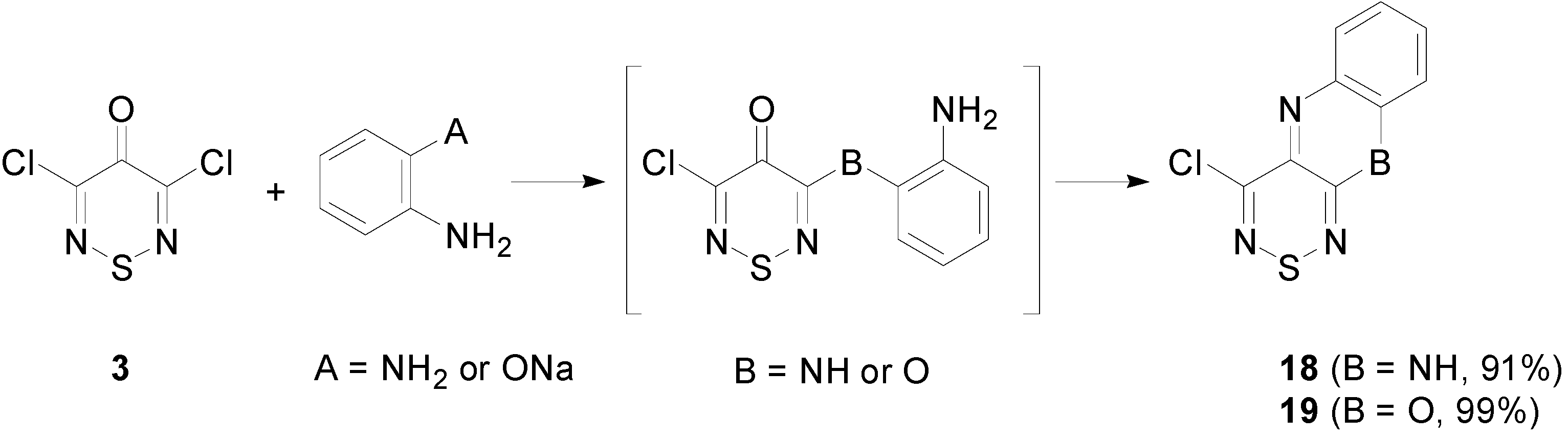
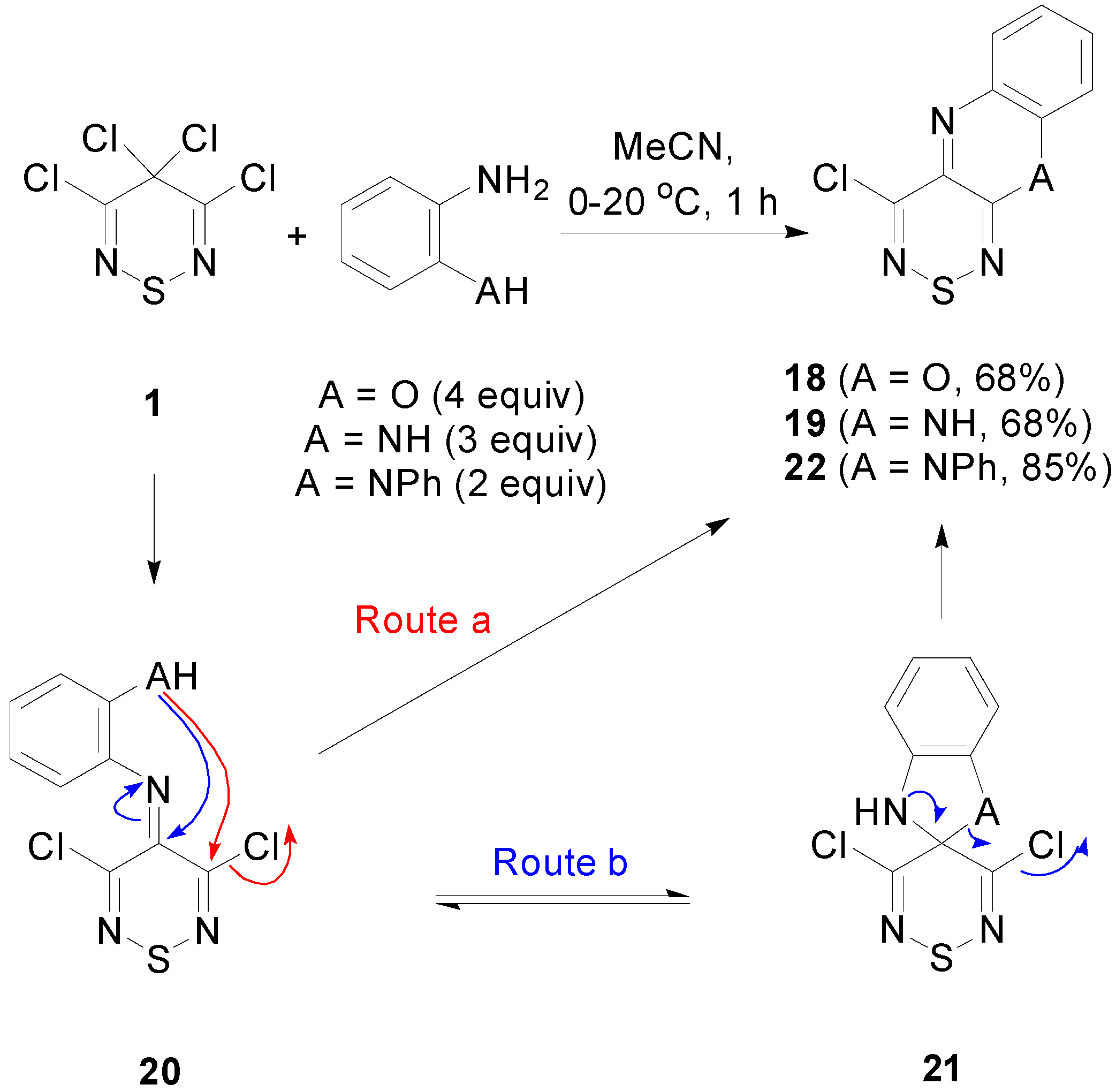
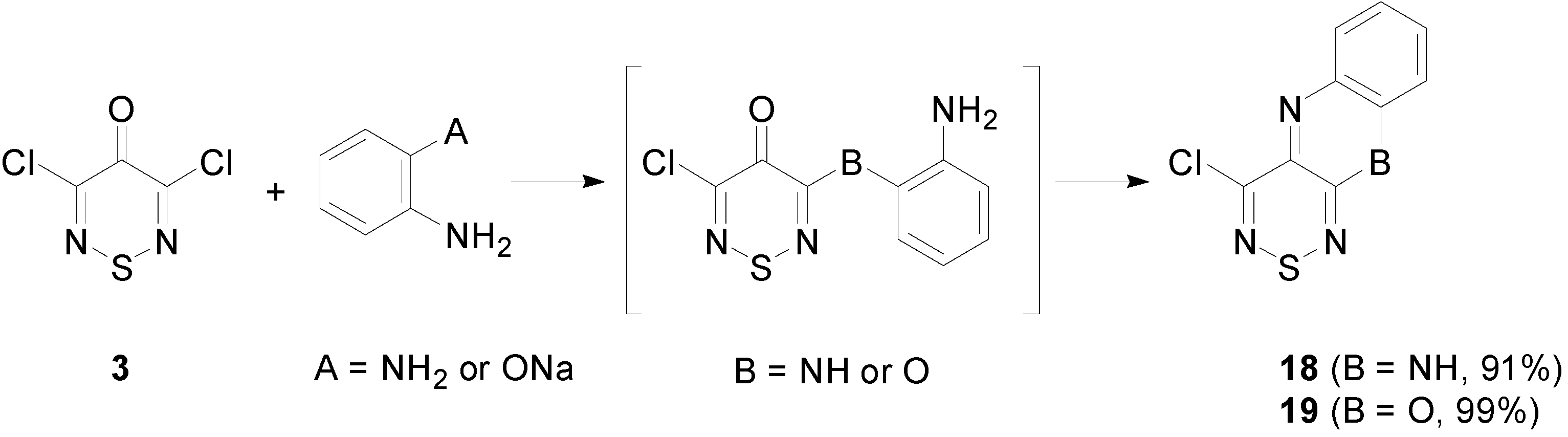
2.4. Cyclisation Reactions of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1)

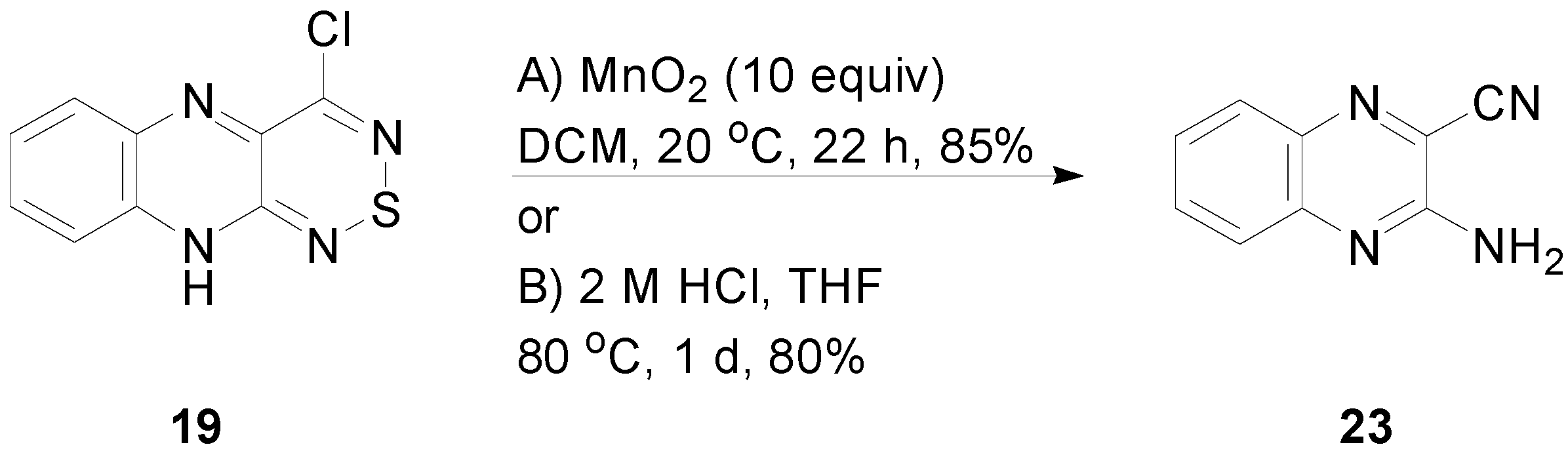
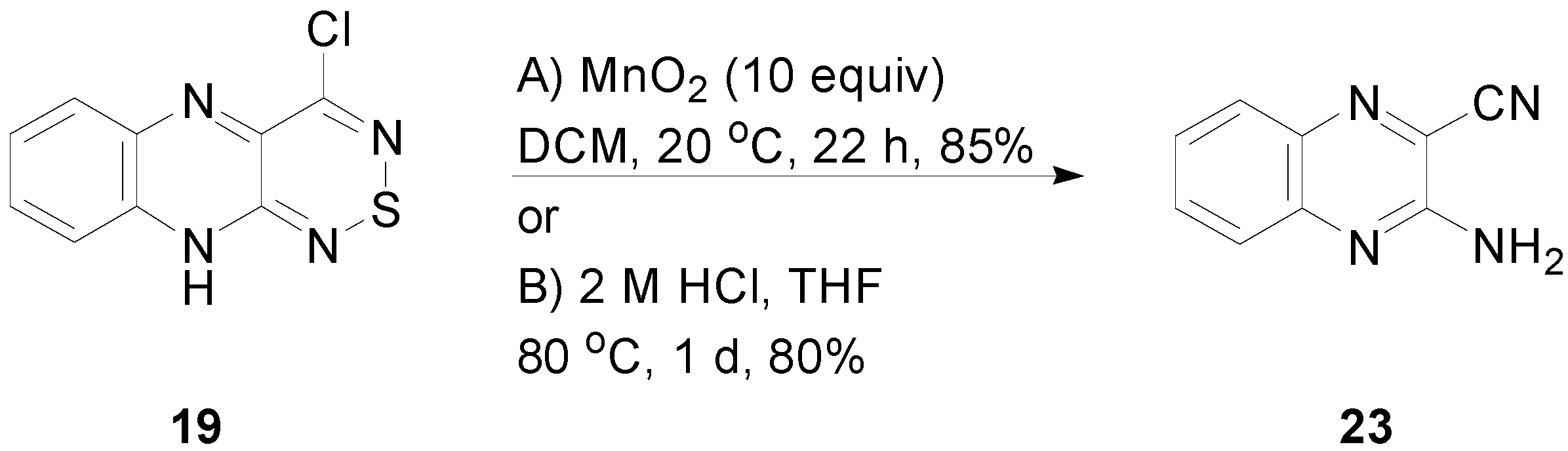
2.5. Reactivity of 4-Chloro-10H-[1,2,6]thiadiazino[3,4-b]quinoxaline (19)
3. Experimental Section
3.1. General Methods and Materials
3.2. Preparation of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1)
3.2.1. Reaction of Dichloromalononitrile with SCl2
3.2.2. Reaction of N-2,2-Trichloro-2-cyanoacetimidoyl Chloride (4) with S8
3.3. Transformations of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) to 3,5-Dichloro-4H-1,2,6-thiadiazin-4-one (3)
3.3.1. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with Formic Acid
3.3.2. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with Acetic Acid
3.3.3. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with NaNO3 in MeCN
3.3.4. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with AgNO3 in MeCN
3.3.5. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with DMSO in MeCN
3.3.6. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with Neat DMSO
3.4. Transformations of 4,5-Dichloro-1,2,3-dithiazolium Chloride (2) into 4-Chloro-5H-1,2,3-dithiazol-5-one (9)
3.4.1. Reaction of 4,5-Dichloro-1,2,3-dithiazolium Chloride (2) with Formic Acid
3.4.2. Reaction of 4,5-Dichloro-1,2,3-dithiazolium Chloride (2) with Acetic Acid
3.4.3. Reaction of 4,5-Dichloro-1,2,3-dithiazolium Chloride (2) with NaNO3 in MeCN
3.4.4. Reaction of 4,5-Dichloro-1,2,3-dithiazolium Chloride (2) with AgNO3 in MeCN
3.5. Transformations of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) to 2-(3,5-Dichloro-4H-1,2,6-thiadiazin-4-ylidene)malononitrile (10)
3.5.1. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with Malononitrile
3.5.2. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with Malononitrile, Chromatography-Free (Table 2, Entry 6)
3.5.3. Reaction of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1) with Dimethylsulfonium Dicyanomethylide (14)
3.6. Cyclisation Reactions of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine (1)
3.6.1. Synthesis of 4-Chlorobenzo[5,6][1,4]oxazino[2,3-c][1,2,6]thiadiazine (18) (General Procedure)
3.6.2. Synthesis of 4-Chloro-10H-[1,2,6]thiadiazino[3,4-b]quinoxaline (19)
3.6.3. Synthesis of 4-Chloro-10-phenyl-10H-[1,2,6][3,4-b]quinoxaline (22)
3.7. Reactivity of 4-Chloro-10H-[1,2,6]thiadiazino[3,4-b]quinoxaline (19)
3.7.1. Reaction of 4-Chloro-10H-[1,2,6]thiadiazino[3,4-b]quinoxaline (19) with MnO2
3.7.2. Reaction of 4-Chloro-10H-[1,2,6]thiadiazino[3,4-b]quinoxaline (19) with HCl
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. Mono-5-substituted-3-chloro-4H-1,2,6-thiadiazin-4-one Antifungal Agents. U.S. Patent 4097594, 27 June 1978. [Google Scholar]
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. Mono-5-substituted-thio-3-chloro-4H-1,2,6-thiadiazin-4-one Antifungal Agents. U.S. Patent 4100281, 11 July 1978. [Google Scholar]
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. 3-Chloro-5-(optionally substituted heterocycloxy)-4H-1,2,6-thiadiazin-4-one Antifungal Agents. U.S. Patent 4143138, 6 March 1979. [Google Scholar]
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. Mono-5-substituted-3-chloro-4H-1,2,6-thiadiazin-4-one Antifungal Agents. U.S. Patent 4201780, 6 May 1980. [Google Scholar]
- Portnoy, R.C. Thiadiazinone Plant Disease Control Agents. U.S. Patent 4497807, 5 February 1985. [Google Scholar]
- Haddon, R.C.; Kaplan, M.L.; Marshall, J.H. Naphtho[1,8-cd:4,5-c′d′]bis[1,2,6]thiadiazine. A compound of ambiguous aromatic character. J. Am. Chem. Soc. 1978, 100, 1235–1239. [Google Scholar] [CrossRef]
- Gómez, T.; Macho, S.; Miguel, D.; Neo, A.G.; Rodríguez, T.; Torroba, T. Cyclopentathiadiazines, Cyclohepta- and Cyclopentadithiazoles: New Materials and a Rich Heterocyclic Chemistry of Cyclic Enaminonitriles. Eur. J. Org. Chem. 2005, 2005, 5055–5066. [Google Scholar] [CrossRef]
- Macho, S.; Miguel, D.; Neo, A.G.; Rodríguez, T.; Torroba, T. Cyclopentathiadiazines, new heterocyclic materials from cyclic enaminonitriles. Chem. Commun. 2005, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Lonchakov, A.V.; Rakitin, O.A.; Gritsan, N.P.; Zibarev, A.V. Breathing Some New Life into an Old Topic: Chalcogen-Nitrogen π-Heterocycles as Electron Acceptors. Molecules 2013, 18, 9850–9900. [Google Scholar] [CrossRef] [PubMed]
- Hermerschmidt, F.; Kalogirou, A.S.; Min, J.; Zissimou, G.A.; Tuladhar, S.M.; Ameri, T.; Faber, H.; Itskos, G.; Choulis, S.A.; Anthopoulos, T.D.; et al. 4H-1,2,6-Thiadiazin-4-one-containing small molecule donors and additive effects on their performance in solution-processed organic solar cells. J. Mater. Chem. C 2015, 3, 2358–2365. [Google Scholar] [CrossRef]
- Theodorou, E.; Ioannidou, H.A.; Ioannou, T.A.; Kalogirou, A.S.; Constantinides, C.P.; Manoli, M.; Koutentis, P.A.; Hayes, S.C. Spectroscopic characterization of C-4 substituted 3,5-dichloro-4H-1,2,6-thiadiazines. RSC Adv. 2015, 51, 18471–18481. [Google Scholar] [CrossRef]
- Ioannidou, H.A.; Koutentis, P.A. Substitution at C-4 in 3,5-disubstituted 4H-1,2,6-thiadiazin-4-ones. Tetrahedron 2012, 68, 2590–2597. [Google Scholar] [CrossRef]
- Ioannidou, H.A.; Kizas, C.; Koutentis, P.A. Palladium Catalyzed C–C Coupling Reactions of 3,5-Dichloro-4H-1,2,6-thiadiazin-4-one. Org. Lett. 2011, 13, 3466–3469. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, H.A.; Kizas, C.; Koutentis, P.A. Selective Stille Coupling Reactions of 3-Chloro-5-halo(pseudohalo)-4H-1,2,6-thiadiazin-4-ones. Org. Lett. 2011, 13, 5886–5889. [Google Scholar] [CrossRef] [PubMed]
- Geevers, J.; Trompen, W.P. Synthesis and reactions of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one. Recl. Trav. Chim. Pays Bas 1974, 93, 270–272. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W. Cyclisation chemistry of 4H-1,2,6-thiadiazines. J. Chem. Soc. Perkin Trans. 1 2000, 2601–2607. [Google Scholar] [CrossRef]
- Appel, R.; Janssen, H.; Siray, M.; Knoch, F. Synthese und Reaktionen des 4,5-Dichlor-1,2,3-dithiazolium-chlorids. Chem. Ber. 1985, 118, 1632–1643. [Google Scholar] [CrossRef]
- Rakitin, O.A. 1,2-Oxa/thia-3-azoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 6, Chapter 6.01; p. 1. [Google Scholar]
- Konstantinova, L.S.; Rakitin, O.A. Synthesis and properties of 1,2,3-dithiazoles. Russ. Chem. Rev. 2008, 77, 521–546. [Google Scholar] [CrossRef]
- Kim, K. Synthesis and Reactions of 1,2,3-Dithiazoles. Sulfur Rep. 1998, 21, 147–207. [Google Scholar] [CrossRef]
- Kim, K. Recent Advances in 1,2,3-Dithiazole Chemistry. Phosphorus Sulfur Silicon Relat. Elem. 1997, 120, 229–244. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Manoli, M.; Koutentis, P.A. Synthesis of N-aryl-3,5-dichloro-4H-1,2,6-thiadiazin-4-imines from 3,4,4,5-tetrachloro-4H-1,2,6-thiadiazine. Org. Lett. 2015. [Google Scholar] [CrossRef] [PubMed]
- Koutentis, P.A.; Koyioni, M.; Michaelidou, S.S. Synthesis of [(4-Chloro-5H-1,2,3-dithiazol-5-ylidene)amino]azines. Molecules 2011, 16, 8992–9002. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, W.R.; Armstrong, P. N-1,1-Trichlorocyanoacetimidoyl Chloride. J. Org. Chem. 1964, 29, 2772–2773. [Google Scholar] [CrossRef]
- Kristinnson, H. Addition of sulfenyl chlorides to the cyanogen bond in activated nitriles. Tetrahedron Lett. 1973, 14, 4489–4490. [Google Scholar] [CrossRef]
- Geevers, J.; Trompen, W.P. Reactions of N-chloro formimidoyl chlorides with sulfur. Tetrahedron Lett. 1974, 15, 1687–1690. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. The reaction of 4,5-dichloro-1,2,3-dithiazolium chloride with DMSO: An improved synthesis of 4-chloro-1,2,3-dithiazol-5H-one. Tetrahedron 2009, 65, 6855–6858. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W. Chemistry of 4-dicyanomethylene-1,2,6-thiadiazines. J. Chem. Soc. Perkin Trans. 1 2000, 1081–1088. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A.; Rikkou, M.D. The synthesis of pyrrolo[2,3-c][1,2,6]-thiadiazine-5-carbonitriles from (4H-1,2,6-thiadiazin-4-ylidene)malononitriles. Tetrahedron 2010, 66, 1817–1820. [Google Scholar] [CrossRef]
- Economopoulos, S.P.; Koutentis, P.A.; Ioannidou, H.A.; Choulis, S.A. Identifying potential candidates for donor-acceptor copolymers on a series of 4H-1,2,6-thiadiazines: An electrochemical approach. Electrochimica Acta 2013, 107, 448–453. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W. Reaction of tetracyanoethylene with SCl2; new molecular rearrangements. J. Chem. Soc. Perkin Trans. 1 2000, 1089–1094. [Google Scholar] [CrossRef]
- Emayan, K.; English, R.F.; Koutentis, P.A.; Rees, C.W. New routes to benzothiophenes, isothiazoles and 1,2,3-dithiazoles. J. Chem. Soc. Perkin Trans. 1 1997, 3345–3349. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Christoforou, I.C.; Ioannidou, H.A.; Manos, M.J.; Koutentis, P.A. Ring transformation of (4-chloro-5H-1,2,3-dithiazol-5-ylidene)acetonitriles to 3-haloisothiazole-5-carbonitriles. RSC Adv. 2014, 4, 7735–7748. [Google Scholar] [CrossRef]
- Christoforou, I.C.; Koutentis, P.A.; Rees, C.W. Reactions of 1,2,3-dithiazoles with halogenated malononitriles. J. Chem. Soc. Perkin Trans. 1 2002, 1236–1241. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W. Reactions of tetracyanoethylene oxide with 1,2,3-dithiazoles. J. Chem. Soc. Perkin Trans. 1 1998, 2505–2509. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. The reaction of 4,5-dichloro-1,2,3-dithiazolium chloride with dimethylsulfonium dicyanomethylide: An improved synthesis of (4-chloro-1,2,3-dithiazolylidene)-malononitrile. Tetrahedron 2009, 65, 6850–6854. [Google Scholar] [CrossRef]
- Xie, A.; Cao, M.; Liu, Y.; Feng, L.; Hu, X.; Dong, W. The Synthesis of Tetrazoles in Nanometer Aqueous Micelles at Room Temperature. Eur. J. Org. Chem. 2014, 2014, 436–441. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W. Chemistry of 4-chloro-5-cyano-1,2,3-dithiazolium chloride. J. Chem. Soc. Perkin Trans. 1 1999, 111–117. [Google Scholar] [CrossRef]
- Harwood, L.M. “Dry-Column” Flash Chromatography. Aldrichimica Acta 1985, 18, 25–25. [Google Scholar]
- Middleton, W.J.; Buhle, E.L.; McNally, J.G.; Zanger, M. S,S-Disubstituted Sulfonium Dicyanomethylides. J. Org. Chem. 1965, 30, 2384–2386. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalogirou, A.S.; Koutentis, P.A. A Qualitative Comparison of the Reactivities of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine and 4,5-Dichloro-1,2,3-dithiazolium Chloride. Molecules 2015, 20, 14576-14594. https://doi.org/10.3390/molecules200814576
Kalogirou AS, Koutentis PA. A Qualitative Comparison of the Reactivities of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine and 4,5-Dichloro-1,2,3-dithiazolium Chloride. Molecules. 2015; 20(8):14576-14594. https://doi.org/10.3390/molecules200814576
Chicago/Turabian StyleKalogirou, Andreas S., and Panayiotis A. Koutentis. 2015. "A Qualitative Comparison of the Reactivities of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine and 4,5-Dichloro-1,2,3-dithiazolium Chloride" Molecules 20, no. 8: 14576-14594. https://doi.org/10.3390/molecules200814576