
Comparative Immunogenicity of a Cytotoxic T Cell Epitope Delivered by Penetratin and TAT Cell Penetrating Peptides

Abstract
:1. Introduction
2. Results
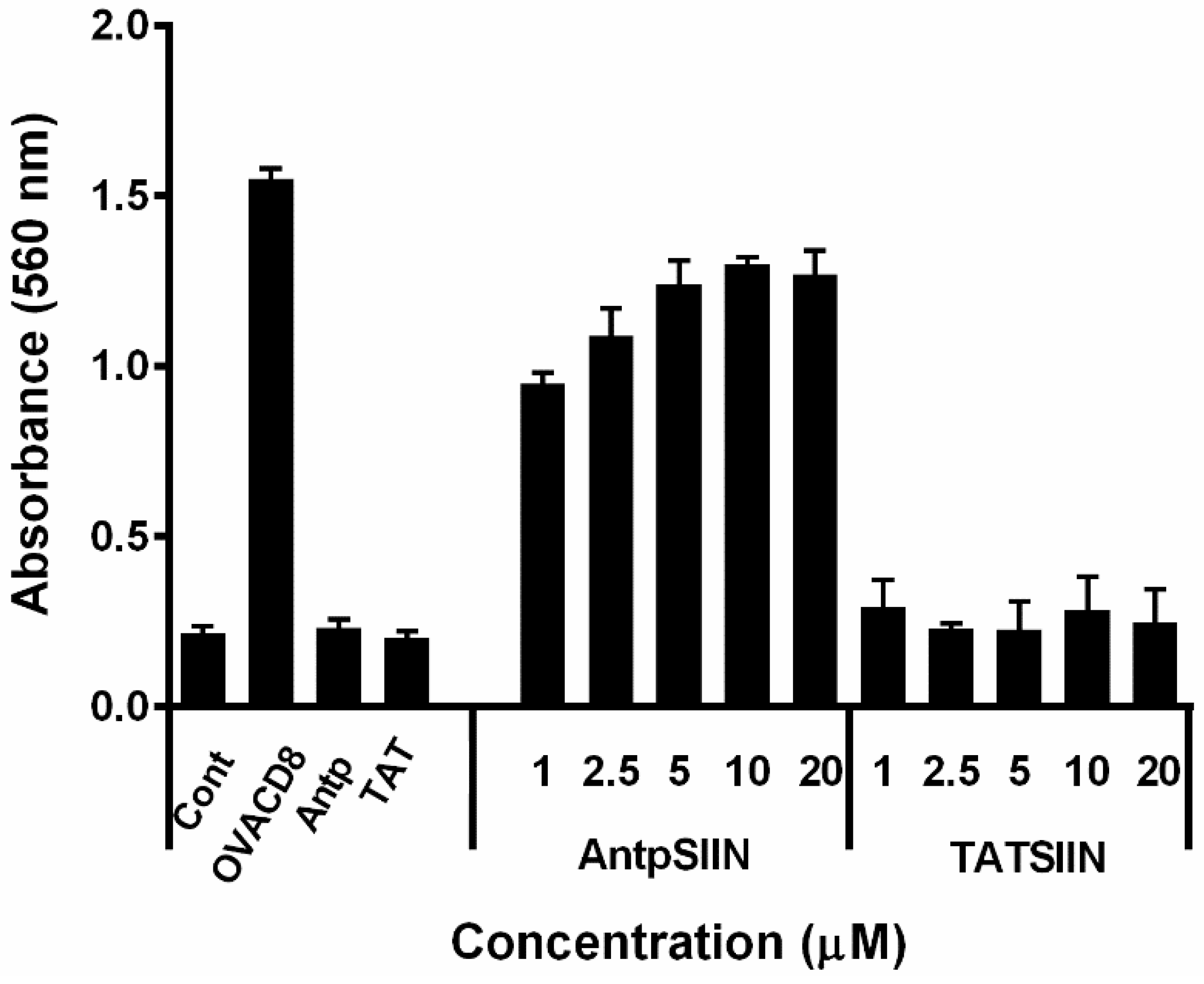
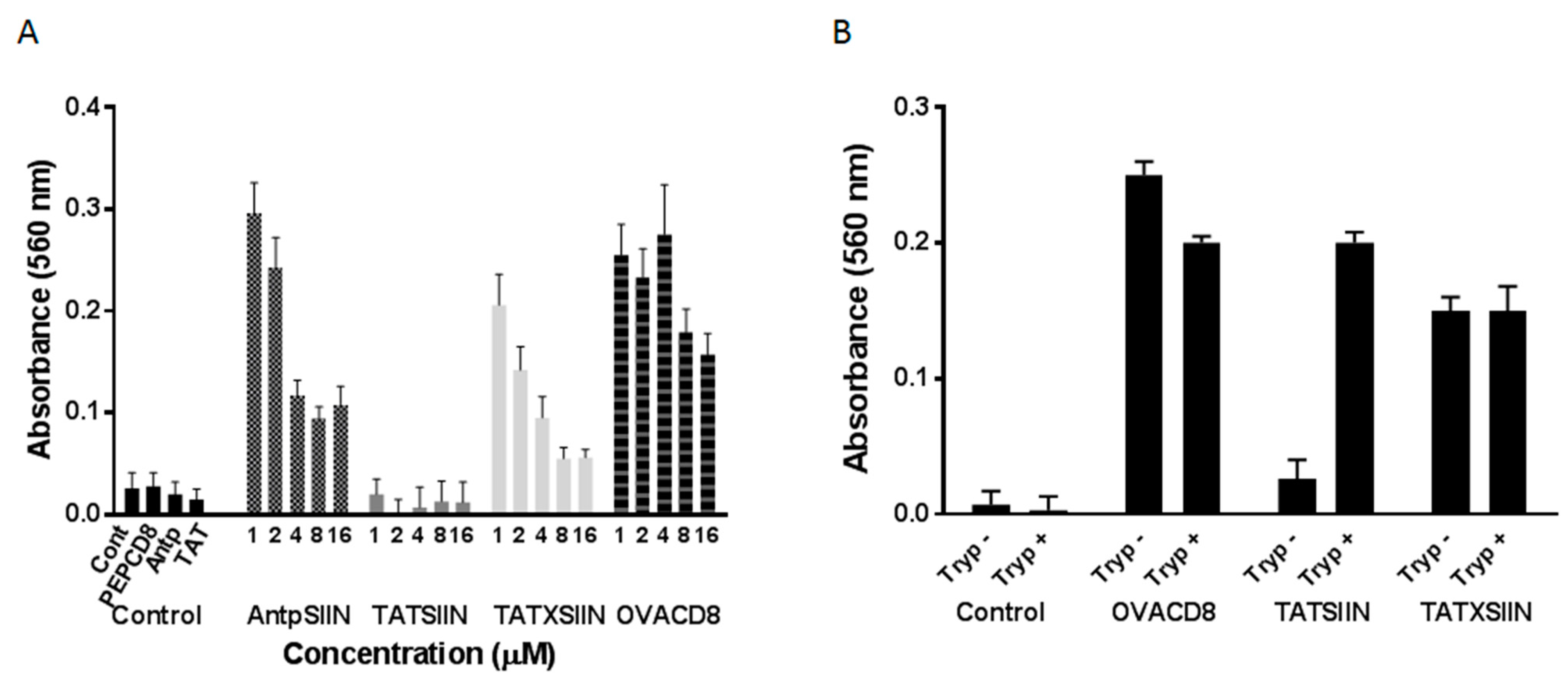
2.1. Stimulation of B3Z T Cells in Vitro by AntpSIIN and TATSIIN Pulsed DC
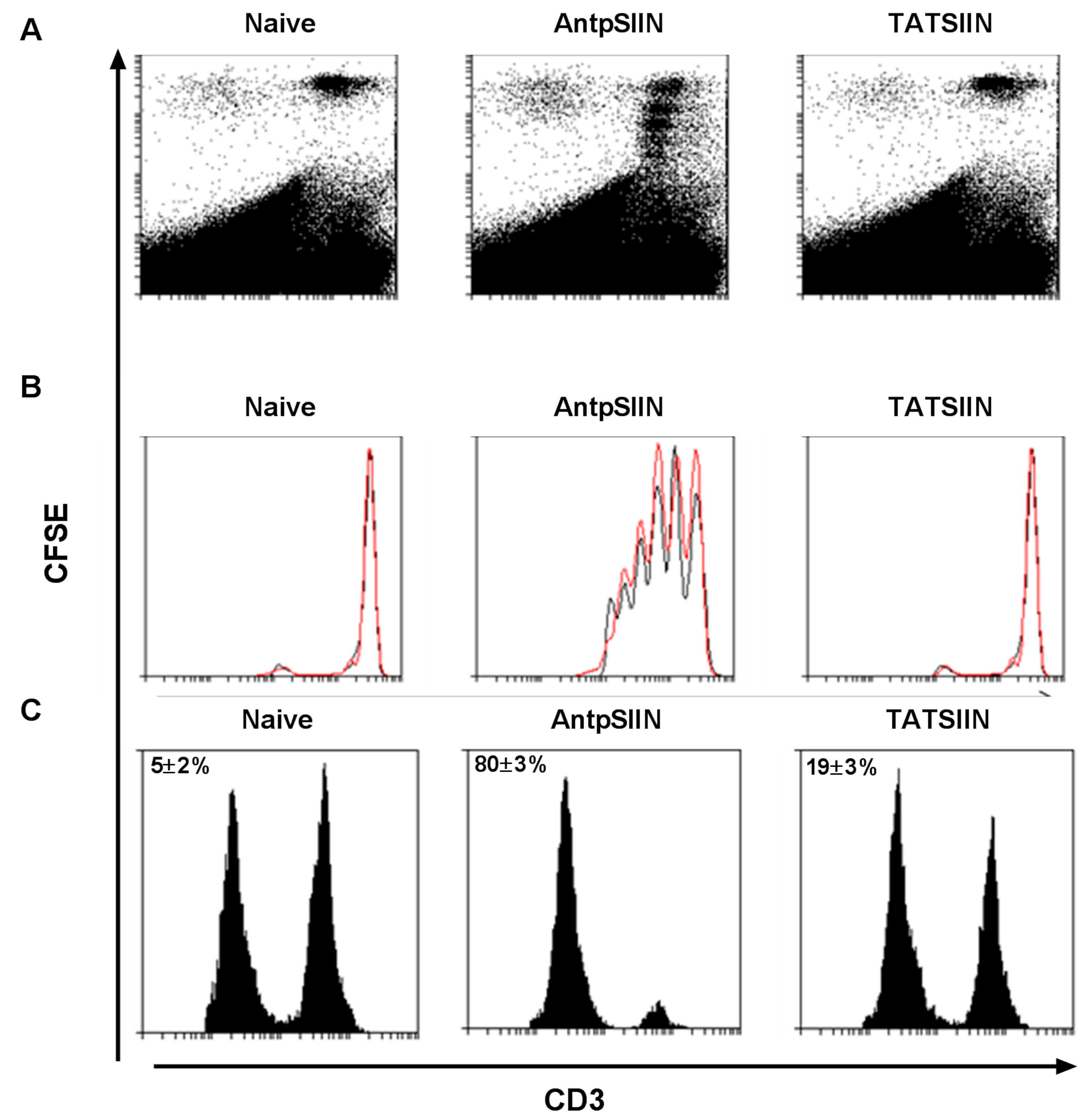
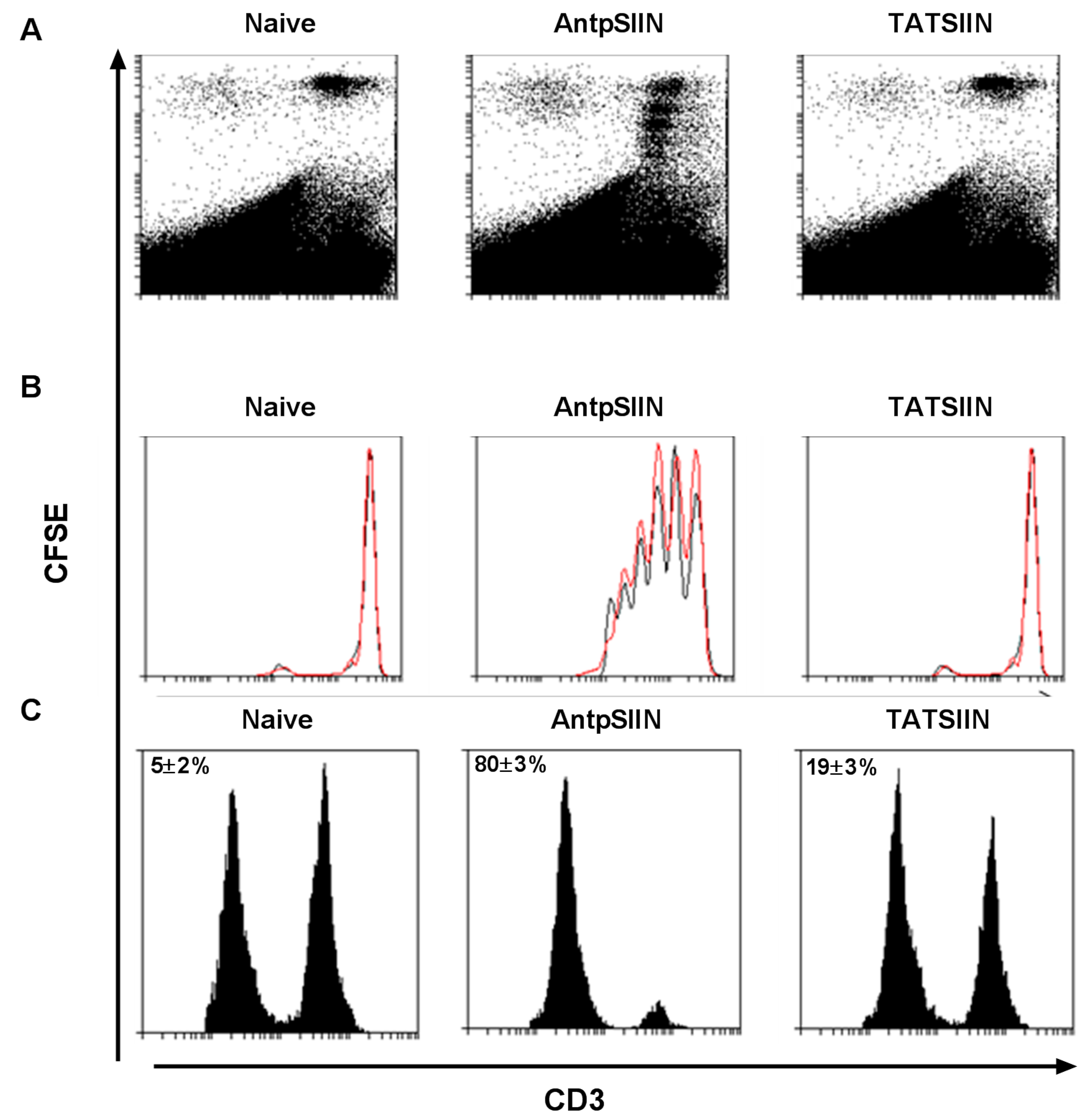
2.2. AntpSIIN but not TATSIIN Induce Potent in Vivo Proliferation and Killing
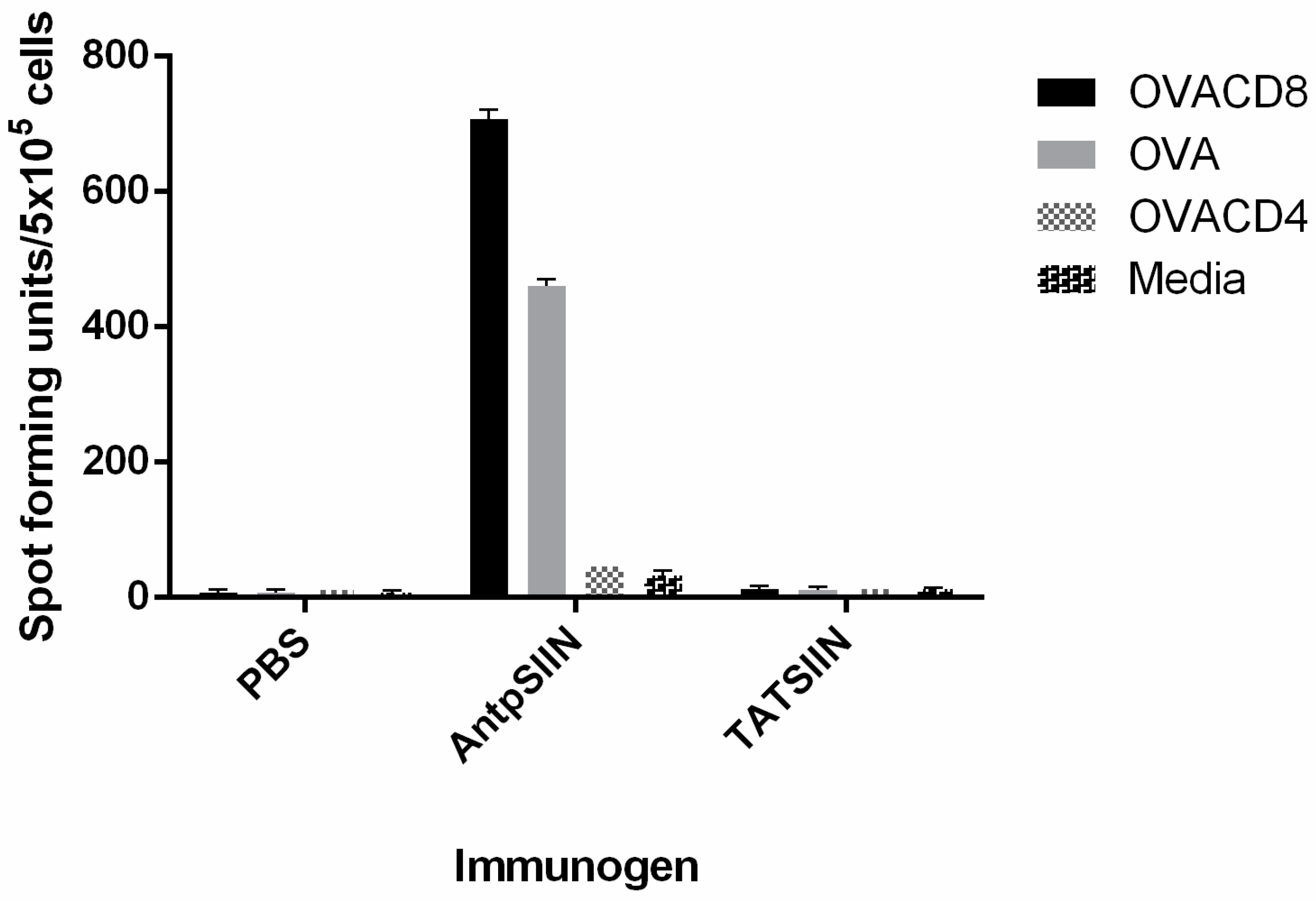
2.3. AntpSIIN but not TATSIIN Induce Strong OVA-Specific IFNγ T Cell Responses in Vivo


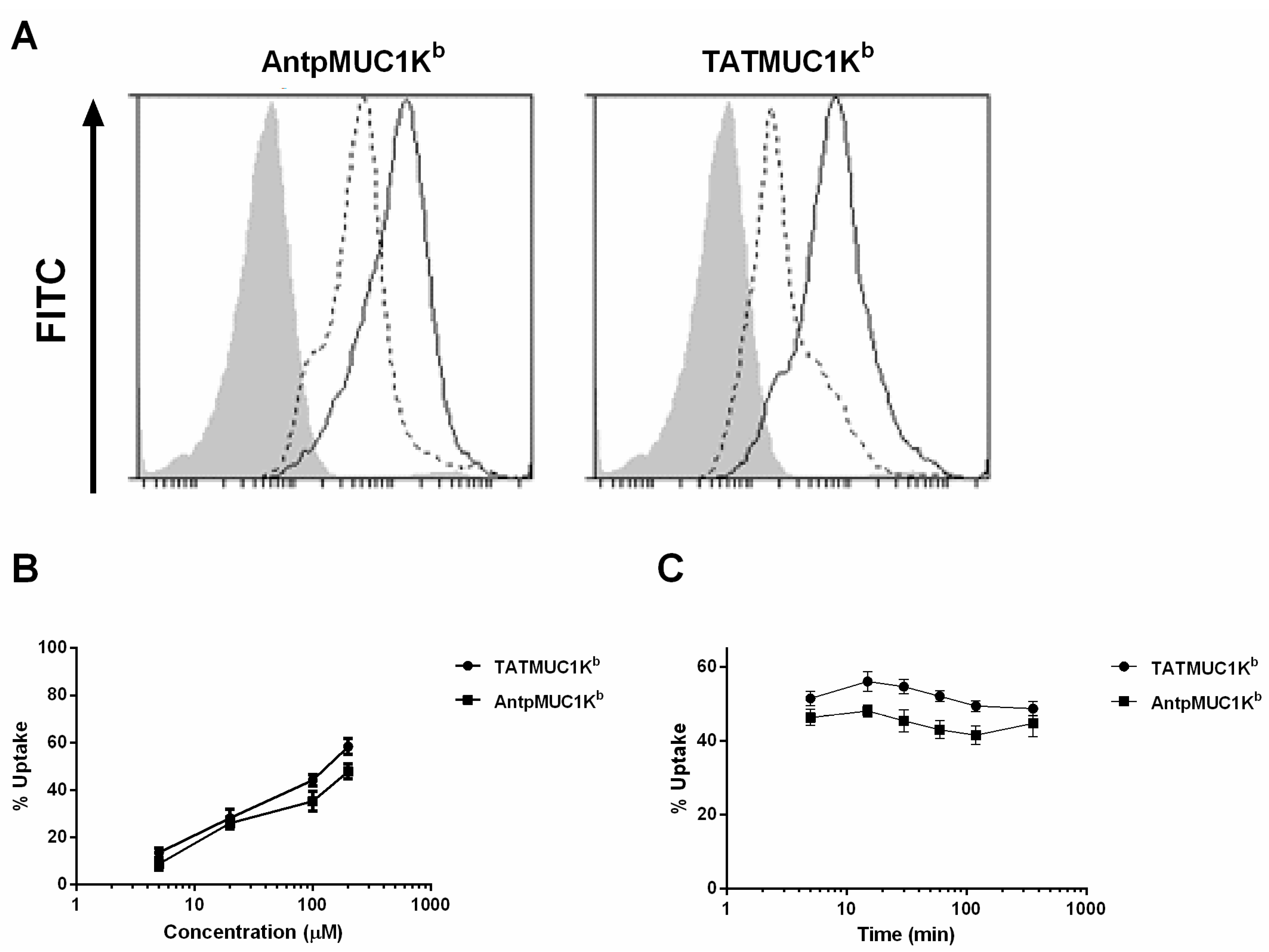
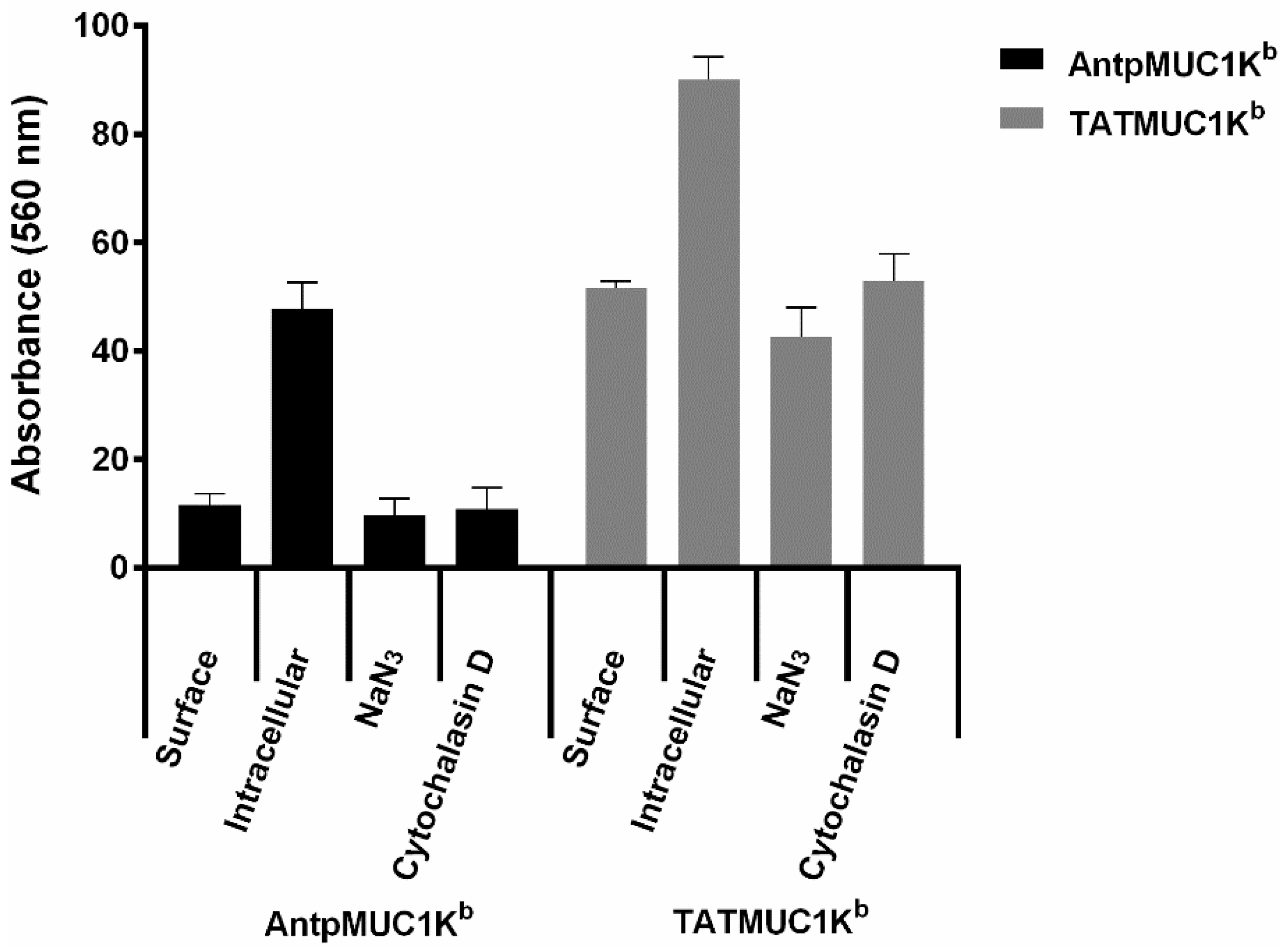
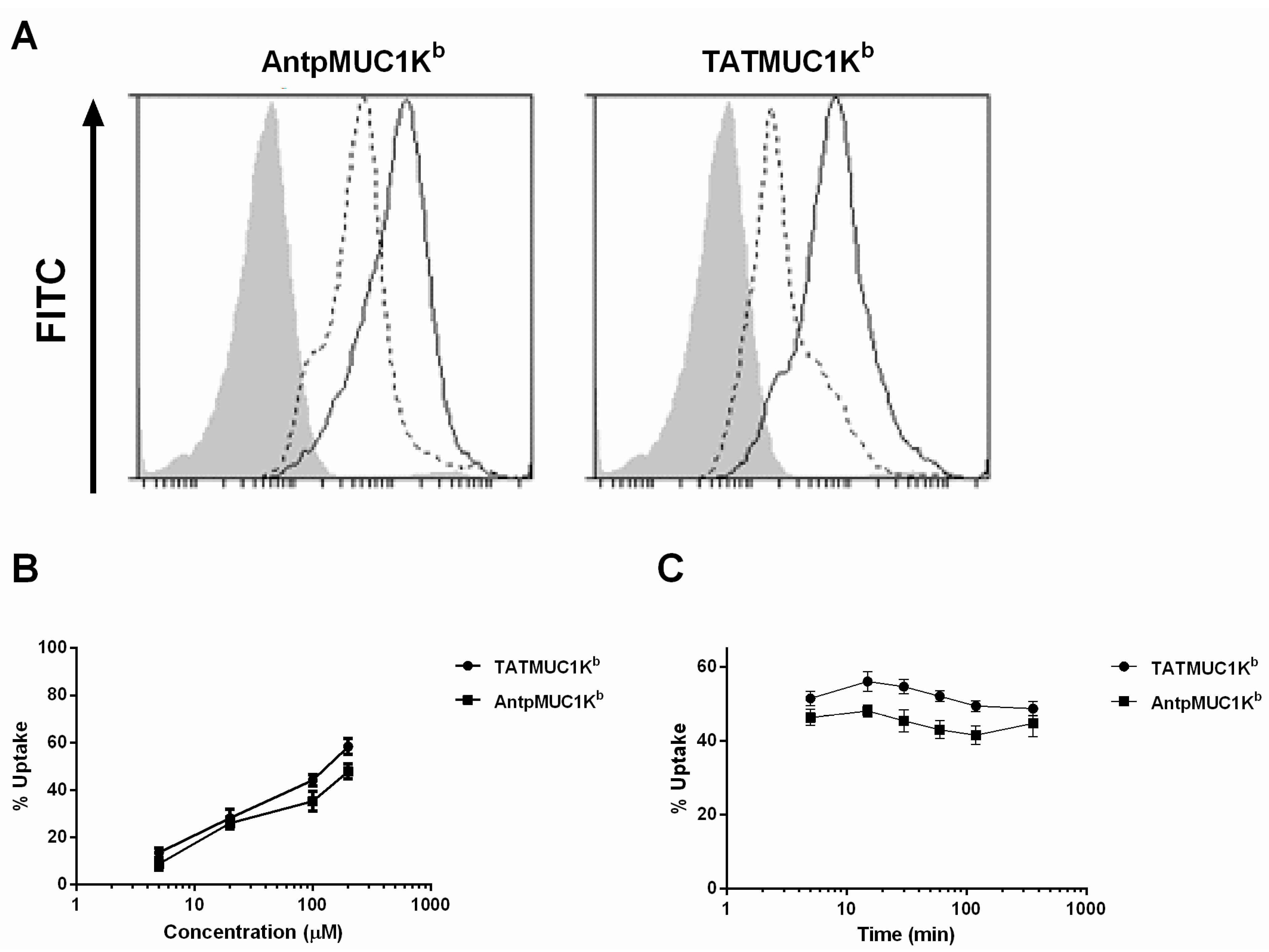
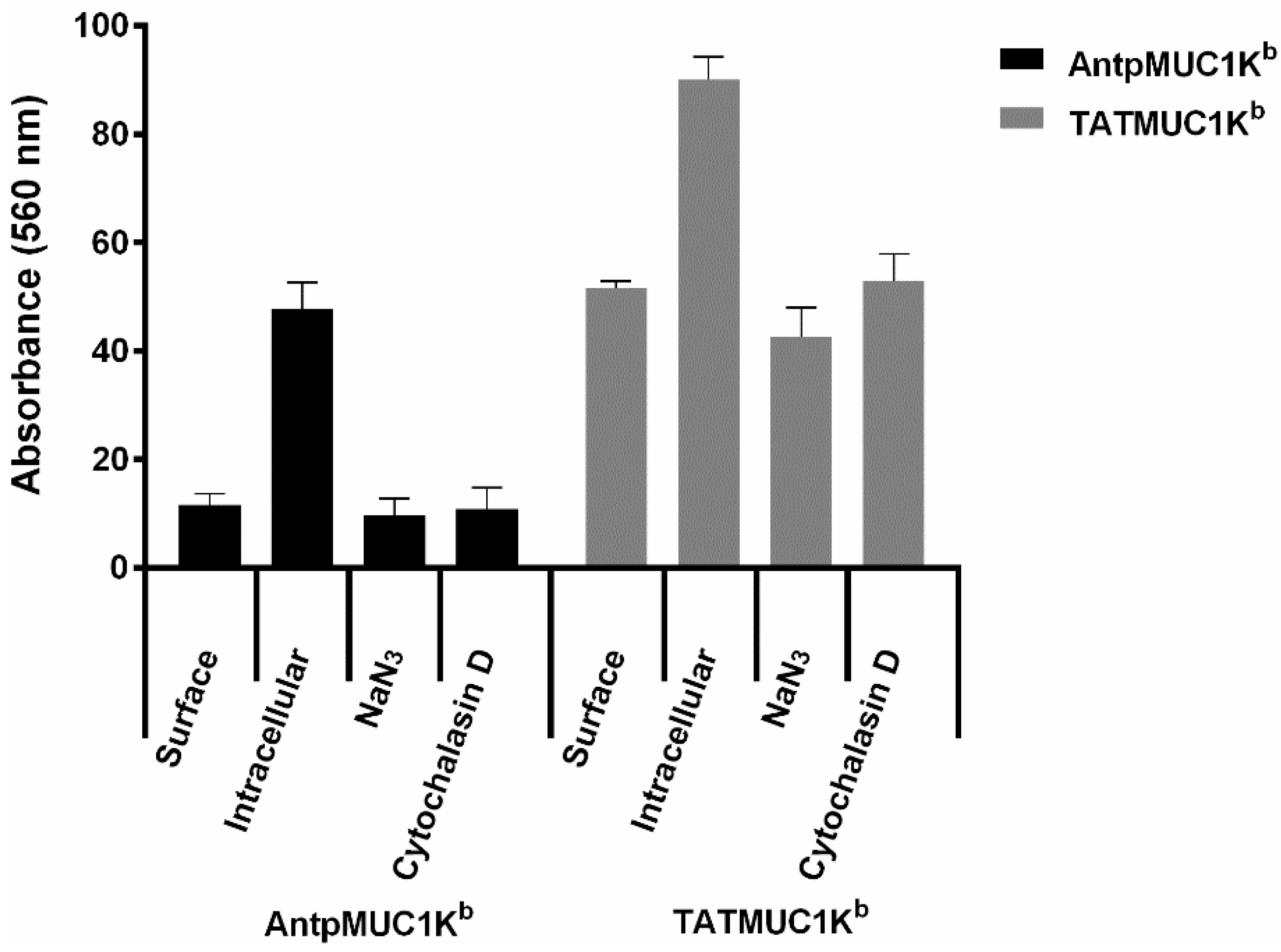
2.4. Rapid Internalisation of Antp and TAT Peptides Incorporating MUC1-H-2Kb Epitope
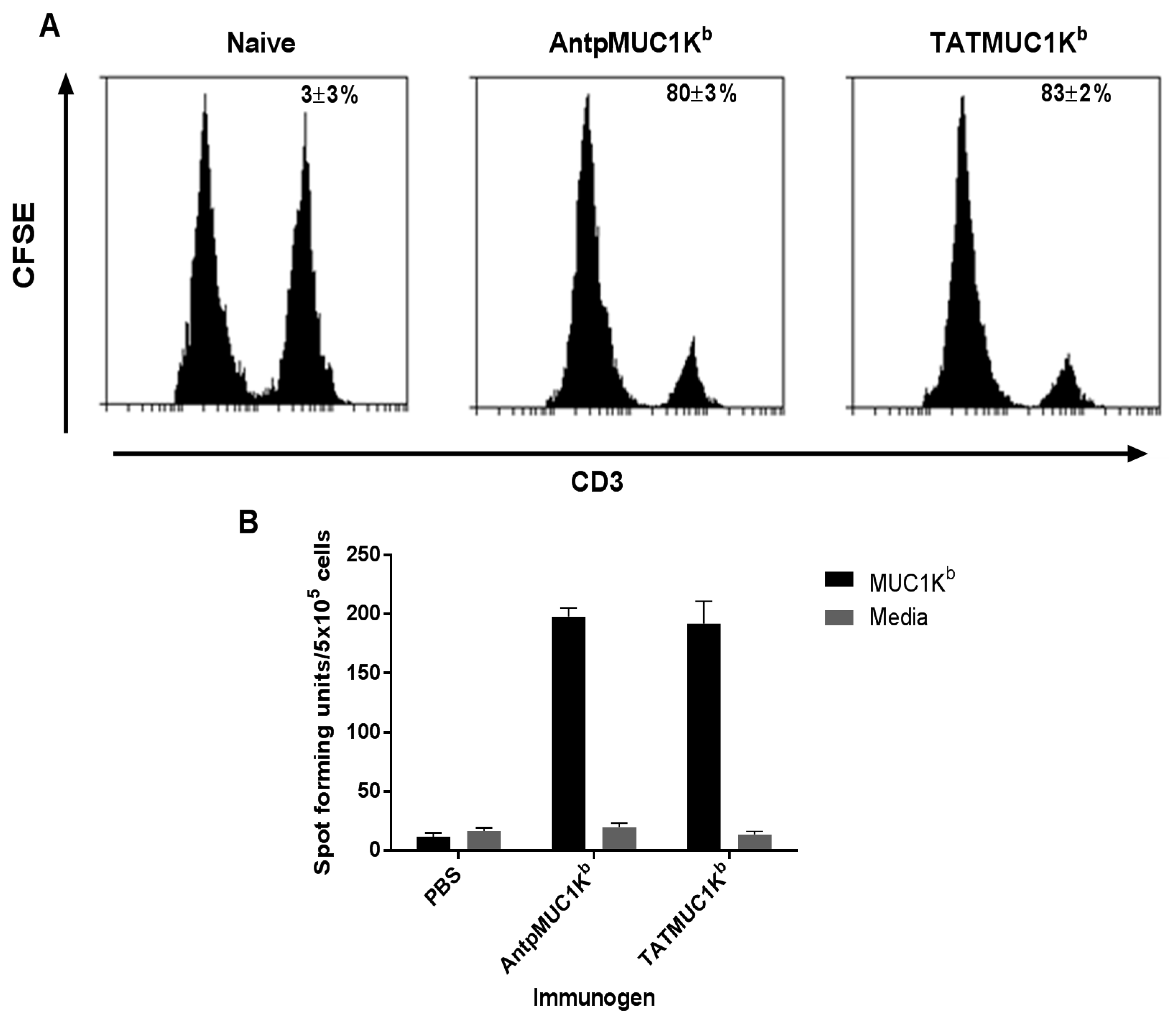
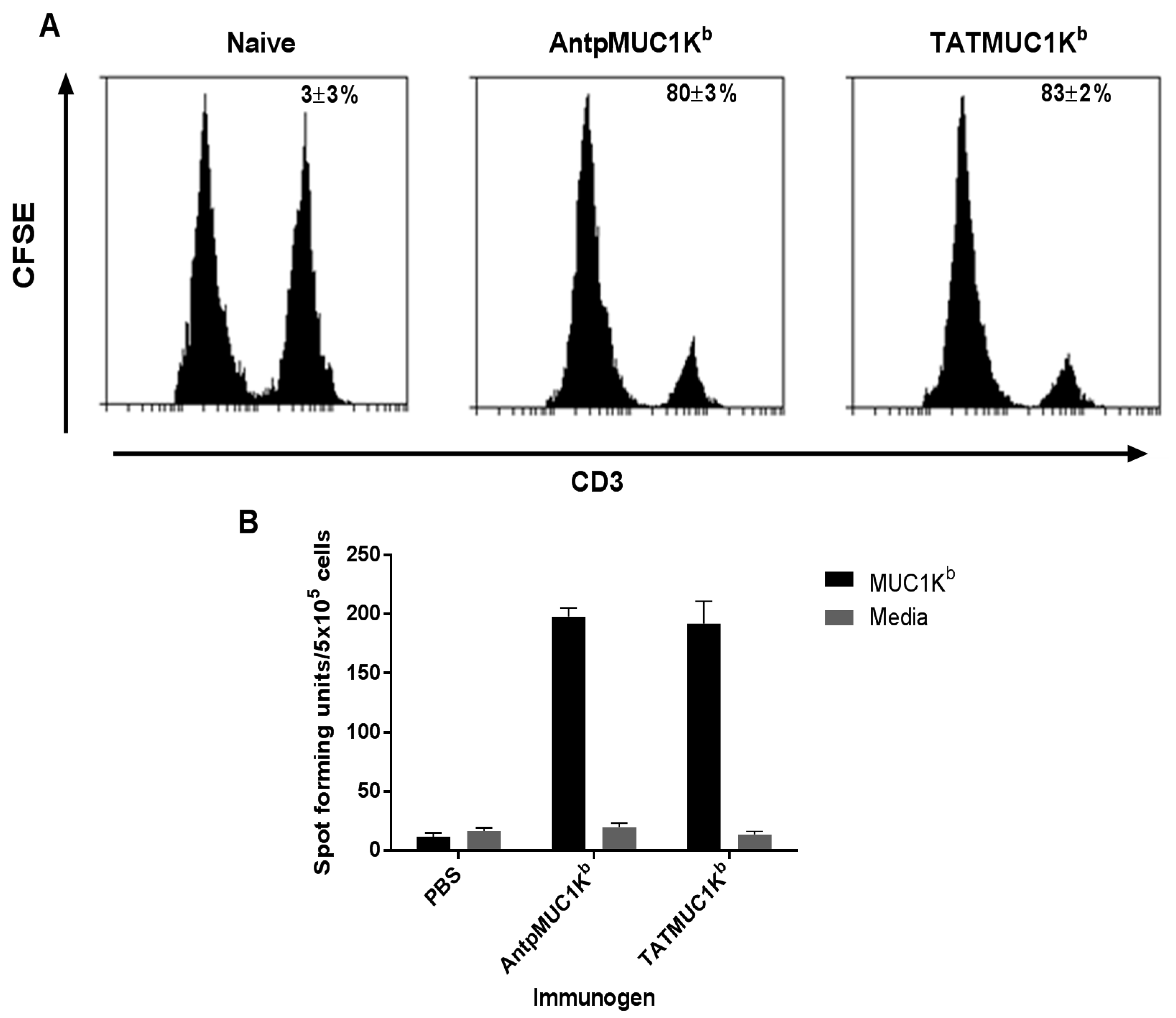
2.5. AntpMUC1Kb and TATMUC1Kb Induce Potent in vivo Cytotoxic T Cell Killing
2.6. AntpMUC1Kb and TATMUC1Kb Immunisation Induces Strong SAPDTRPAP T Cell Specific IFN-γ
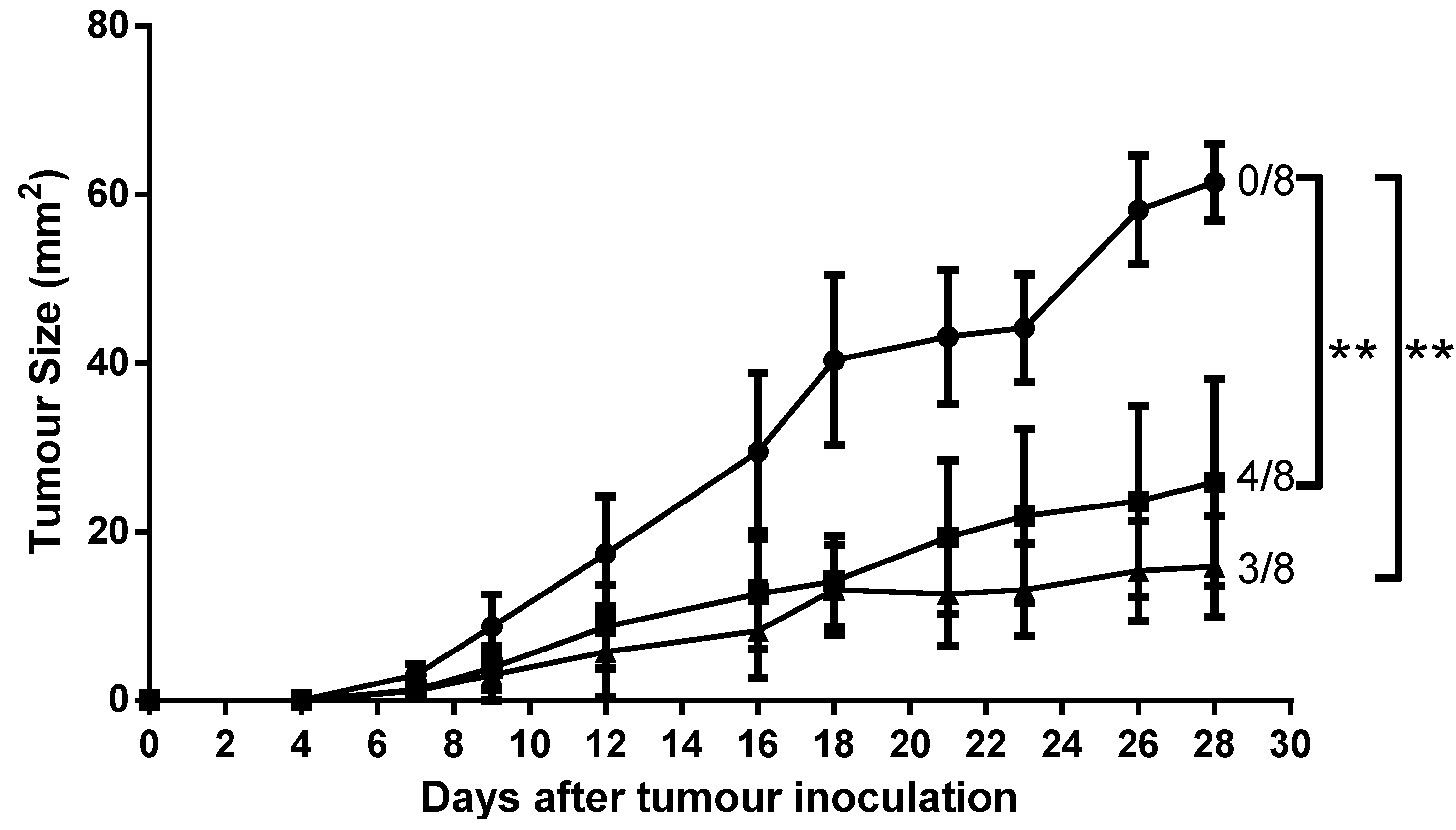
2.7. Mice Immunised with AntpMUC1Kb and TATMUC1Kb Inhibited MUC1+ Tumour Growth in C57BL/6 Mice
3. Discussion
4. Experimental Section
4.1. Peptides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Sequence | Description |
|---|---|---|
| Antp | RQIKIWFQNRRMKWKK | 16 amino acid Antennapedia peptide |
| TAT | RKKRRQRRR | 9-mer HIV TAT protein transduction domain |
| OVACD8 | SIINFEKL | ovalbumin H-2Kb CTL epitope 8-mer peptide. |
| OVACD4 | ISQAVHAAHAEINEAGR | ovalbumin IAb CD4 epitope 16-mer peptide (OVA323-339) |
| MUC1Kb | SAPDTRPAP | Mucin 1 H-2Kb epitope from the VNTR region. |
| AntpSIIN | RQIKIWFQNRRMKWKKSIINFEKL | C-terminal fusion peptide of Antp and OVACD8 |
| TATSIIN | RKKRRQRRRSIINFEKL | C-terminal fusion peptide of Tat and OVACD8 |
| AntpMUC1Kb | RQIKIWFQNRRMKWKKSAPDTRPAP | C-terminal fusion peptide of Antp and MUC1Kb |
| TATMUC1Kb | RKKRRQRRRSAPDTRPAP | C-terminal fusion peptide of Tat and MUC1Kb |
| PEPCD8 | LLPDEVSGLEQLESIINFEKL | Non-internalising control peptide including 13 amino acids N-terminal to SIINFEKL in native OVA sequence |
| TATXSIIN | RKKRRQRRREQLESIINFEKL | N-terminal fusion peptide of Tat and OVACD8 including 4 aa native sequence |
4.2. Mice
4.3. Cytotoxicity of Conjugates
4.4. Generation of Bone-Marrow Derived Dendritic Cells (BMDC)
4.5. Internalisation into BMDC
4.6. Internalisation Inhibition Studies
4.7. Stimulation of lacZ-Inducible Ovalbumin-Specific T-cell Hybrid Cells
4.8. In vivo Proliferation
4.9. In vivo Cytotoxicity Assay
4.10. Enzyme-Linked Immunosorbent Spot-Forming Cell Assay (ELISpot)
4.11. Antibody ELISA
4.12. Prophylactic Tumour Protection
4.13. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [PubMed]
- Delamarre, L.; Mellman, I. Harnessing dendritic cells for immunotherapy. Semin. Immunol. 2011, 23, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Nierkens, S.; Tel, J.; Janssen, E.; Adema, G.J. Antigen cross-presentation by dendritic cell subsets: One general or all sergeants? Trends Immunol. 2013, 34, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Thalhammer, T.; Tzakos, A.G.; Stojanovska, L. Targeting antigens to dendritic cell receptors for vaccine development. J. Drug Deliv. 2013, 2013, 869718. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Delamarre, L. Dendritic cell-targeted vaccines. Front. Immunol. 2014, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting ny-eso-1 to the dendritic cell receptor dec-205. Sci. Transl. Med. 2014, 6, 232ra251. [Google Scholar]
- Kastenmuller, W.; Kastenmuller, K.; Kurts, C.; Seder, R.A. Dendritic cell-targeted vaccines--hope or hype? Nat. Rev. Immunol. 2014, 14, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Chapman, R.; Powderly, J.; Blackwell, K.; Keler, T.; Green, J.; Riggs, R.; He, L.Z.; Ramakrishna, V.; Vitale, L.; et al. Phase i study utilizing a novel antigen-presenting cell-targeted vaccine with toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin. Cancer Res. 2011, 17, 4844–4853. [Google Scholar] [CrossRef] [PubMed]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot phase iii immunotherapy study in stage ii breast cancer patients using oxidized mannan-muc1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Pouniotis, D.S.; van Maanen, P.J.; Andriessen, R.W.; Lodding, J.; Xing, P.X.; McKenzie, I.F.; Loveland, B.E.; Pietersz, G.A. Delivery of tumor associated antigens to antigen presenting cells using penetratin induces potent immune responses. Vaccine 2006, 24, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.A.; Pouniotis, D.S.; Sheng, K.C.; Apostolopoulos, V.; Pietersz, G.A. A membrane penetrating multiple antigen peptide (map) incorporating ovalbumin cd8 epitope induces potent immune responses in mice. Biochim. Biophys. Acta 2010, 1798, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Chikh, G.G.; Kong, S.; Bally, M.B.; Meunier, J.C.; Schutze-Redelmeier, M.P. Efficient delivery of antennapedia homeodomain fused to ctl epitope with liposomes into dendritic cells results in the activation of cd8+ t cells. J. Immunol. 2001, 167, 6462–6470. [Google Scholar] [CrossRef] [PubMed]
- Pietersz, G.A.; Li, W.; Apostolopoulos, V. A 16-mer peptide (rqikiwfqnrrmkwkk) from antennapedia preferentially targets the class i pathway. Vaccine 2001, 19, 1397–1405. [Google Scholar] [CrossRef]
- Pouniotis, D.S.; Apostolopoulos, V.; Pietersz, G.A. Penetratin tandemly linked to a ctl peptide induces anti-tumour t-cell responses via a cross-presentation pathway. Immunology 2006, 117, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Pouniotis, D.S.; Esparon, S.; Apostolopoulos, V.; Pietersz, G.A. Whole protein and defined cd8(+) and cd4(+) peptides linked to penetratin targets both mhc class i and ii antigen presentation pathways. Immunol. Cell Boil. 2011, 89, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, L.; Wang, H.; Shang, X.; Niu, W.; Li, J.; Wu, Y. A novel mimovirus vaccine containing survivin epitope with adjuvant il-15 induces long-lasting cellular immunity and high antitumor efficiency. Mol. Immunol. 2008, 45, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.A.; Pouniotis, D.S.; Tang, C.K.; Apostolopoulos, V.; Pietersz, G.A. Cell-penetrating peptides: Application in vaccine delivery. Biochim. Biophys. Acta 2010, 1805, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Fu, T.; Wang, G.; Zeng, G.; Perry-Lalley, D.M.; Yang, J.C.; Restifo, N.P.; Hwu, P.; Wang, R.F. Induction of cd4(+) t cell-dependent antitumor immunity by tat-mediated tumor antigen delivery into dendritic cells. J. Clin. Investig. 2002, 109, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, H.S.; Park, H.M.; Kim, C.H.; Kim, T.G. Efficient induction of anti-tumor immunity by a tat-cea fusion protein vaccine with poly(i:C) in a murine colorectal tumor model. Vaccine 2011, 29, 8642–8648. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Cho, N.H.; Seong, S.Y. The tat-conjugated n-terminal region of mucin antigen 1 (muc1) induces protective immunity against muc1-expressing tumours. Clin. Exp. Immunol. 2009, 158, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Schutze-Redelmeier, M.P.; Kong, S.; Bally, M.B.; Dutz, J.P. Antennapedia transduction sequence promotes anti tumour immunity to epicutaneously administered ctl epitopes. Vaccine 2004, 22, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Haurum, J.S.; McKenzie, I.F. Muc1 peptide epitopes associated with five different h-2 class i molecules. Eur. J. Immunol. 1997, 27, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Karanikas, V.; Haurum, J.S.; McKenzie, I.F. Induction of hla-a2-restricted ctls to the mucin 1 human breast cancer antigen. J. Immunol. 1997, 159, 5211–5218. [Google Scholar] [PubMed]
- Apostolopoulos, V.; Loveland, B.E.; Pietersz, G.A.; McKenzie, I.F. Ctl in mice immunized with human mucin 1 are mhc-restricted. J. Immunol. 1995, 155, 5089–5094. [Google Scholar] [PubMed]
- Pietersz, G.A.; Li, W.; Osinski, C.; Apostolopoulos, V.; McKenzie, I.F. Definition of mhc-restricted ctl epitopes from non-variable number of tandem repeat sequence of muc1. Vaccine 2000, 18, 2059–2071. [Google Scholar] [CrossRef]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Farkhani, S.M.; Valizadeh, A.; Karami, H.; Mohammadi, S.; Sohrabi, N.; Badrzadeh, F. Cell penetrating peptides: Efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 2014, 57, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release 2014, 174, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.T.; Mitchell, D.J.; Brockstedt, D.G.; Fong, L.; Nolan, G.P.; Fathman, C.G.; Engleman, E.G.; Rothbard, J.B. Introduction of soluble proteins into the mhc class i pathway by conjugation to an hiv tat peptide. J. Immunol. 1997, 159, 1666–1668. [Google Scholar] [PubMed]
- Lu, J.; Higashimoto, Y.; Appella, E.; Celis, E. Multiepitope trojan antigen peptide vaccines for the induction of antitumor ctl and th immune responses. J. Immunol. 2004, 172, 4575–4582. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wettstein, P.J.; Higashimoto, Y.; Appella, E.; Celis, E. Tap-independent presentation of ctl epitopes by trojan antigens. J. Immunol. 2001, 166, 7063–7071. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Xing, P.X.; McKenzie, I.F. Murine immune response to cells transfected with human muc1: Immunization with cellular and synthetic antigens. Cancer Res. 1994, 54, 5186–5193. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brooks, N.; Esparon, S.; Pouniotis, D.; Pietersz, G.A. Comparative Immunogenicity of a Cytotoxic T Cell Epitope Delivered by Penetratin and TAT Cell Penetrating Peptides. Molecules 2015, 20, 14033-14050. https://doi.org/10.3390/molecules200814033
Brooks N, Esparon S, Pouniotis D, Pietersz GA. Comparative Immunogenicity of a Cytotoxic T Cell Epitope Delivered by Penetratin and TAT Cell Penetrating Peptides. Molecules. 2015; 20(8):14033-14050. https://doi.org/10.3390/molecules200814033
Chicago/Turabian StyleBrooks, Nicole, Sandra Esparon, Dodie Pouniotis, and Geoffrey A. Pietersz. 2015. "Comparative Immunogenicity of a Cytotoxic T Cell Epitope Delivered by Penetratin and TAT Cell Penetrating Peptides" Molecules 20, no. 8: 14033-14050. https://doi.org/10.3390/molecules200814033