
Novel Fluorinated Phosphorus–Sulfur Heteroatom Compounds: Synthesis and Characterization of Ferrocenyl- and Aryl-Phosphonofluorodithioic Salts, Adducts, and Esters


Abstract
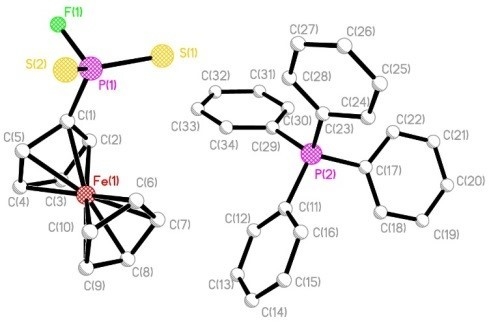
:
1. Introduction
2. Results and Discussion

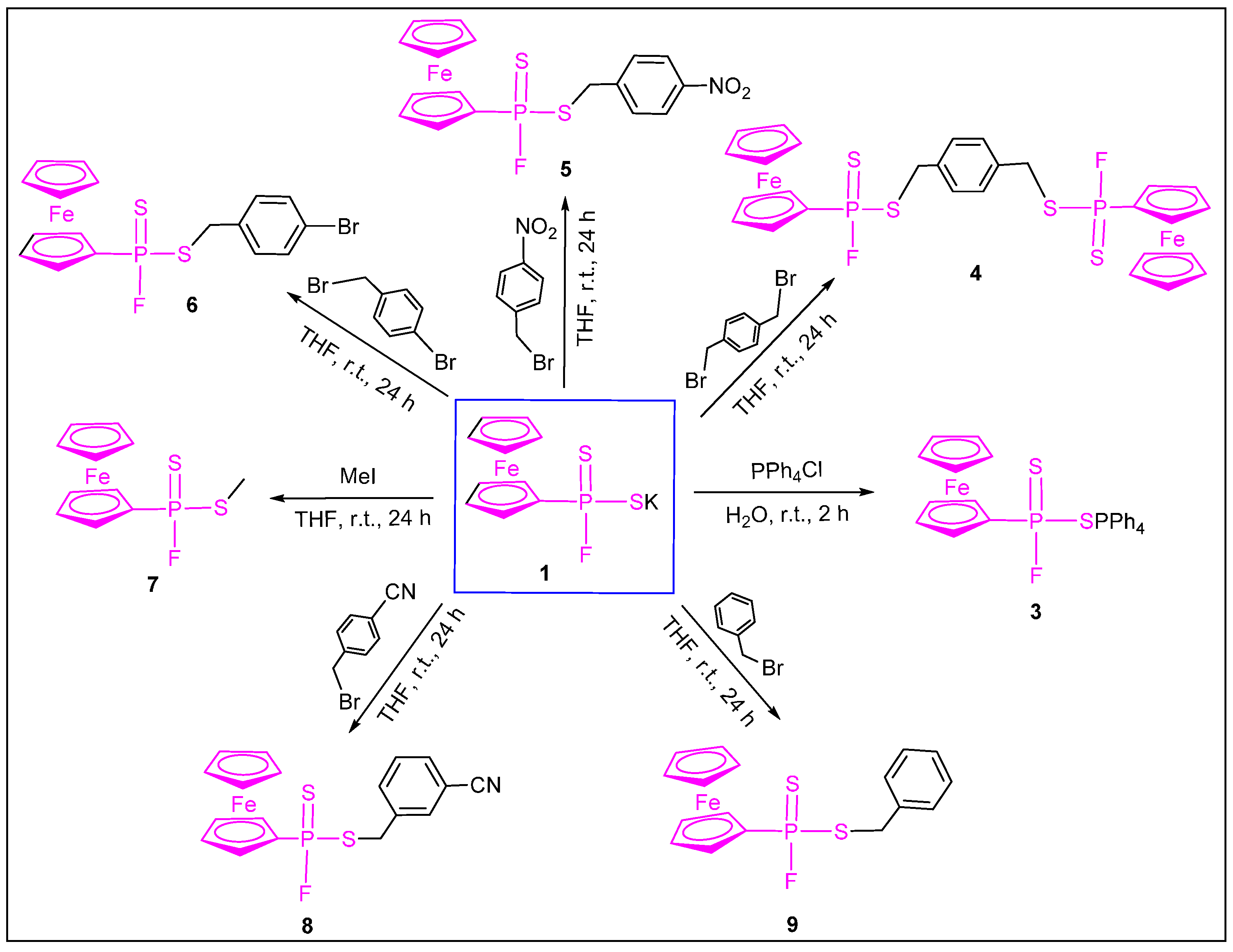
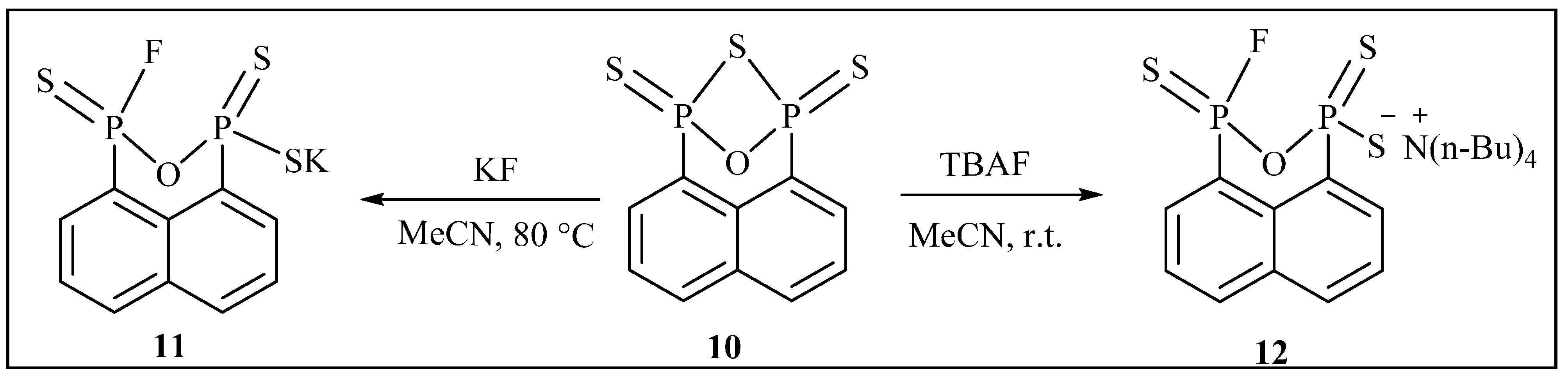
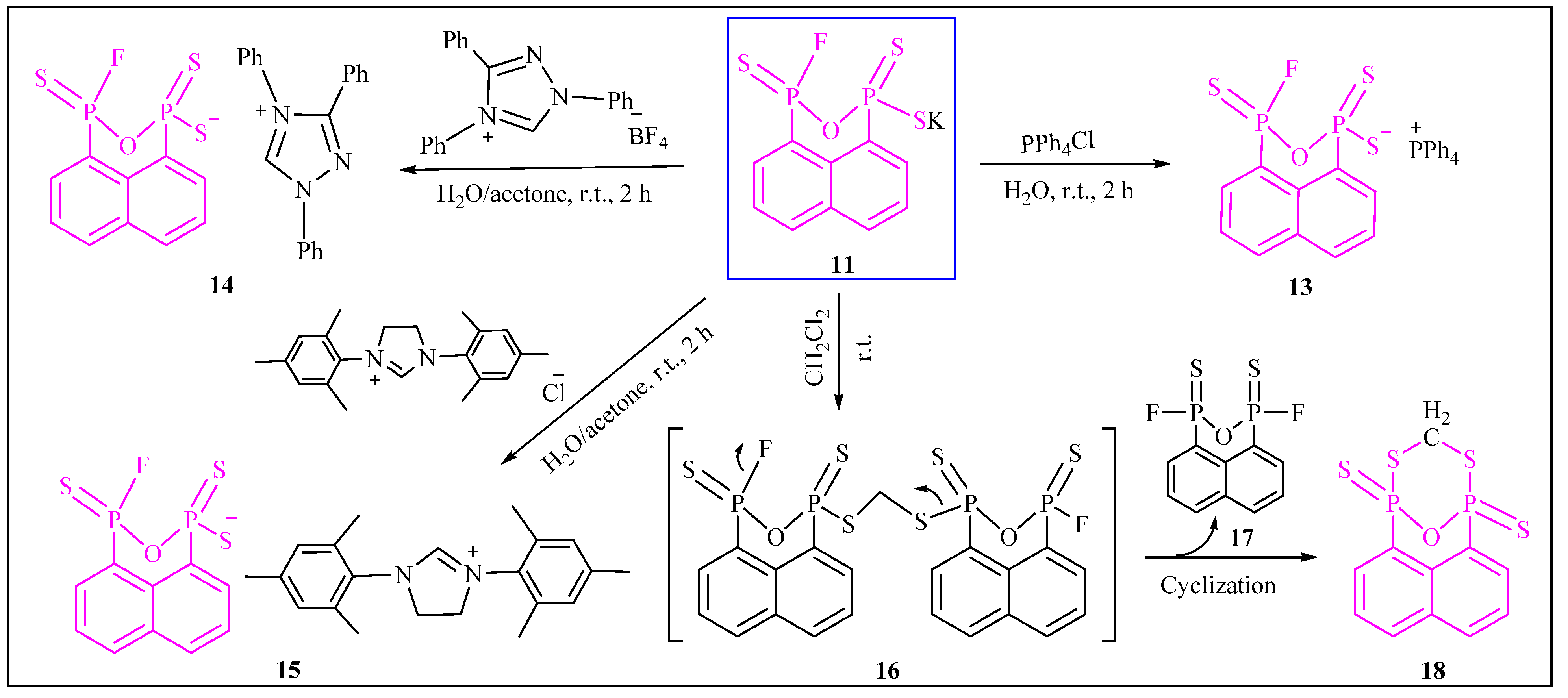

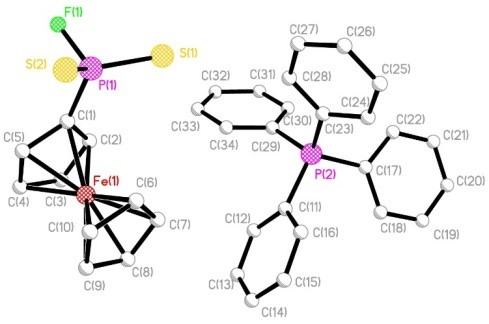{kind=link}
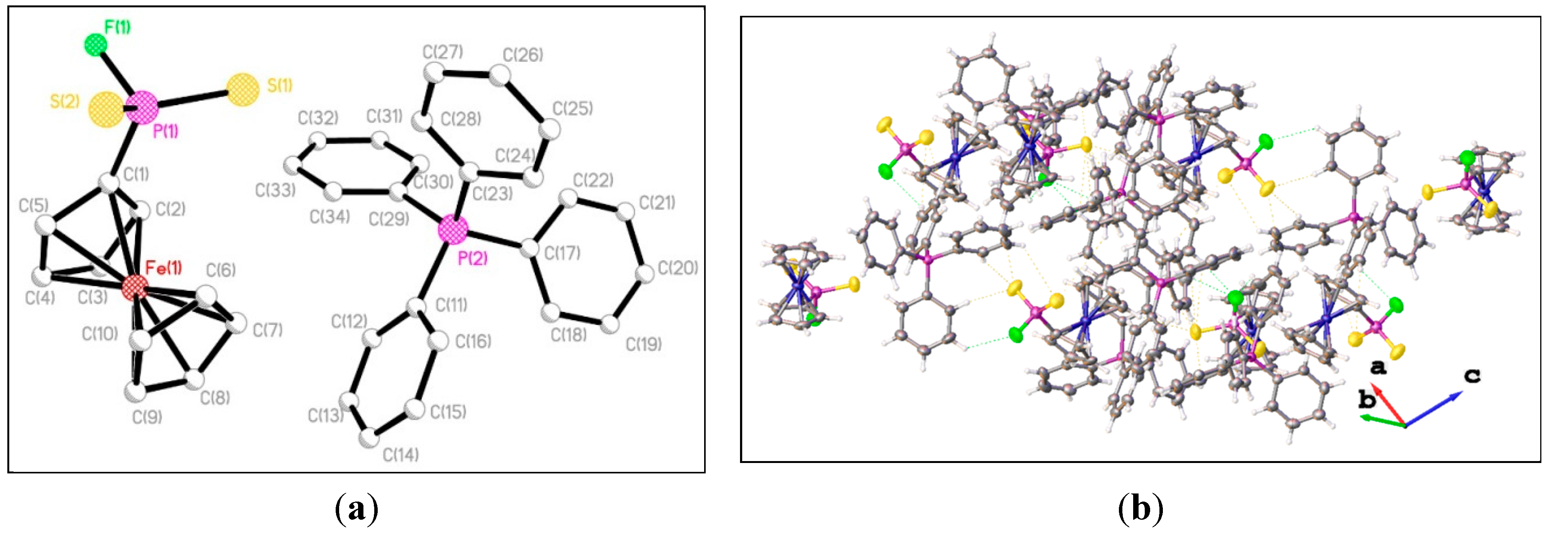{kind=link}
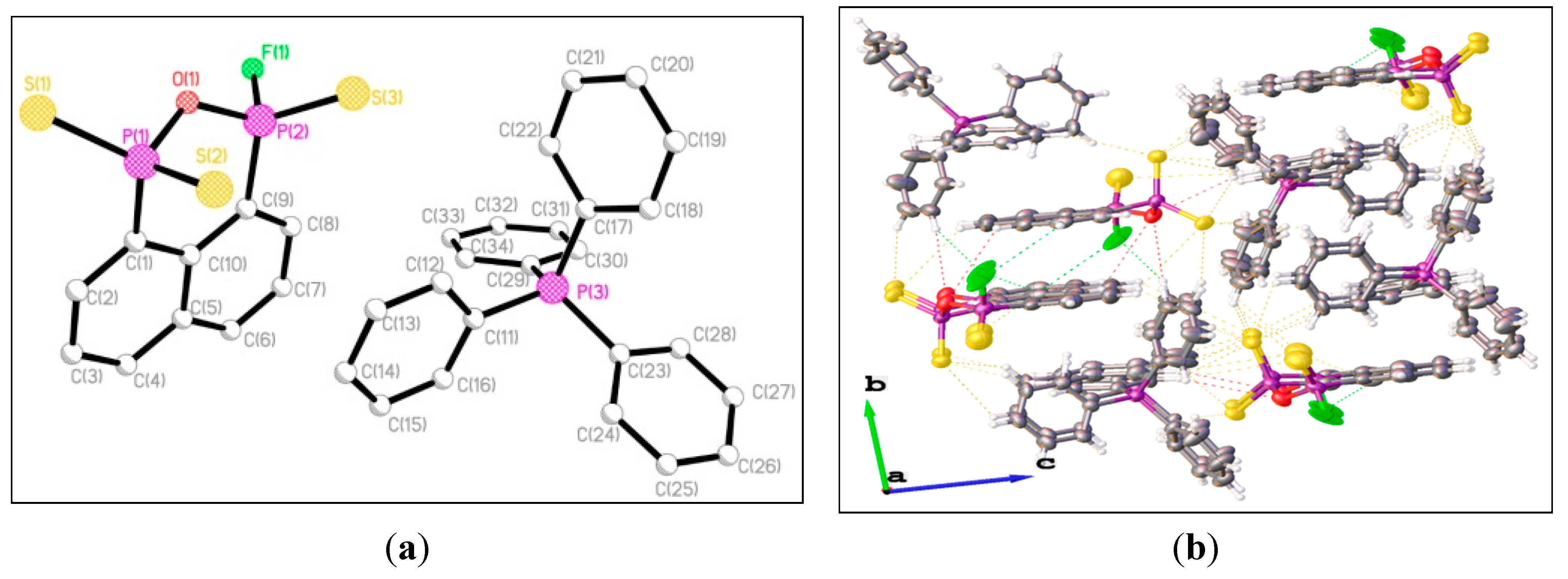{kind=link}
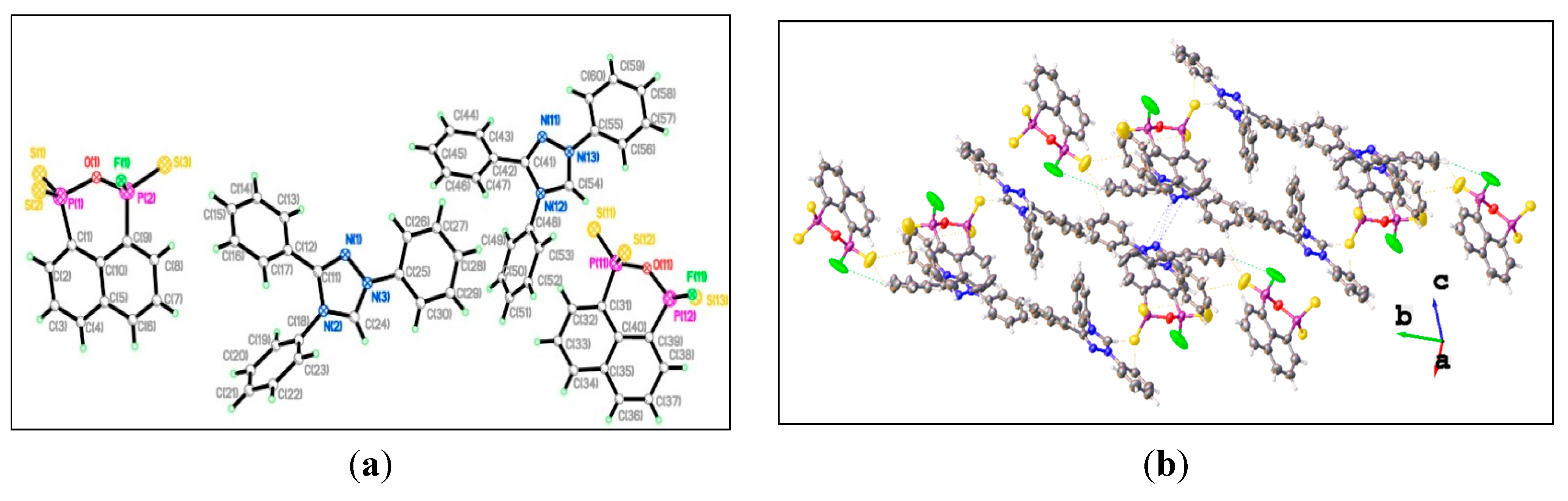{kind=link}
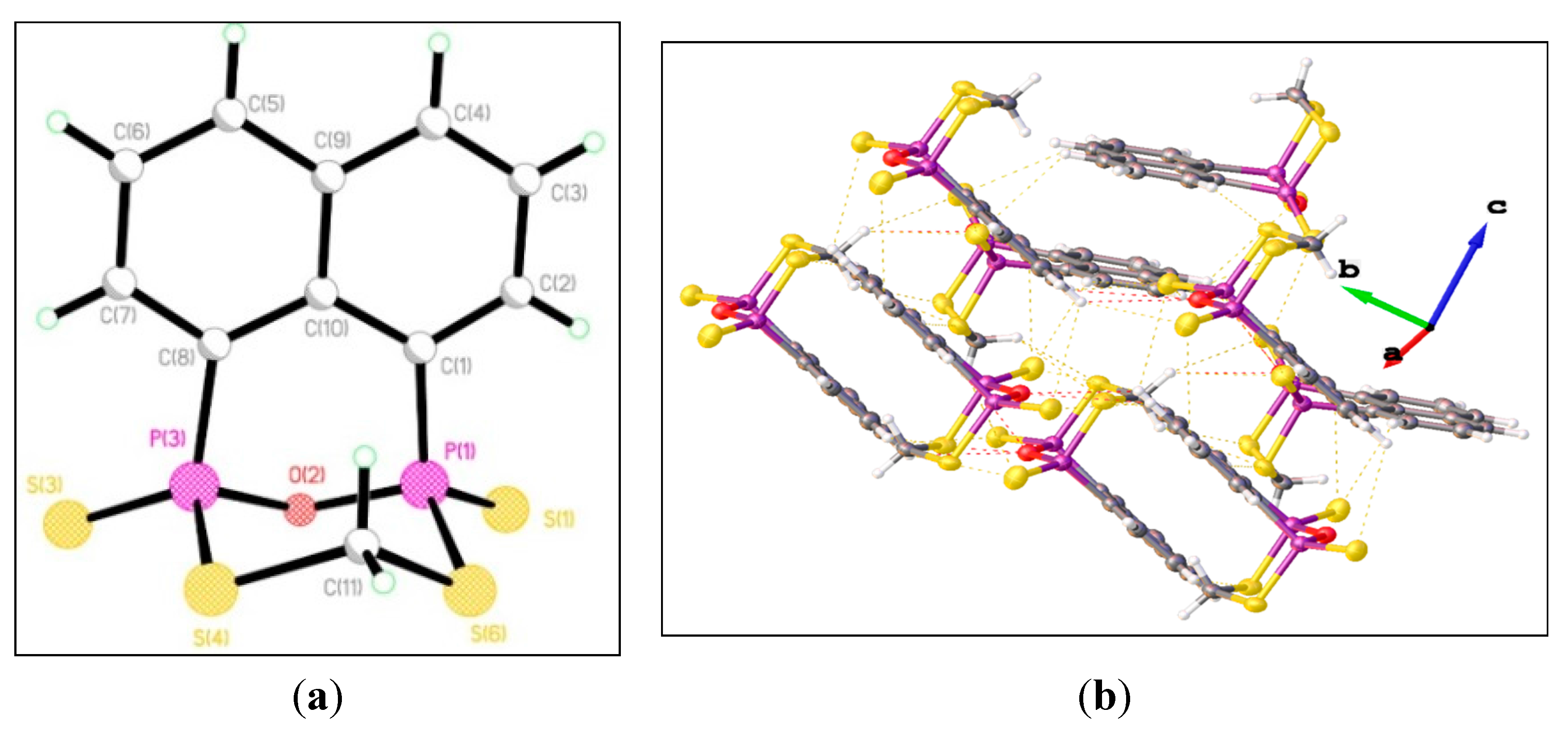{kind=link}
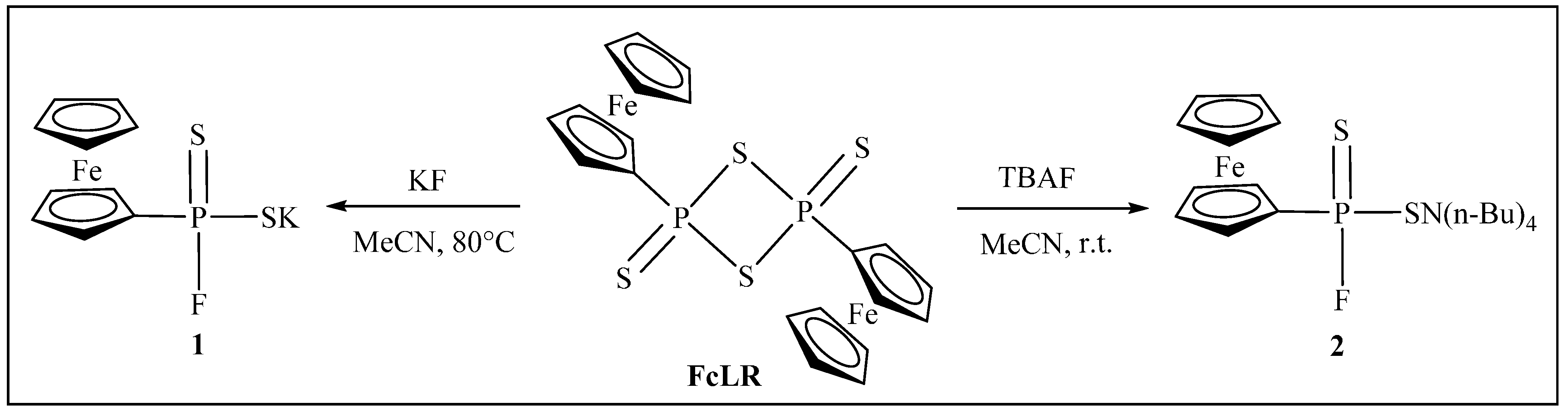{kind=link}
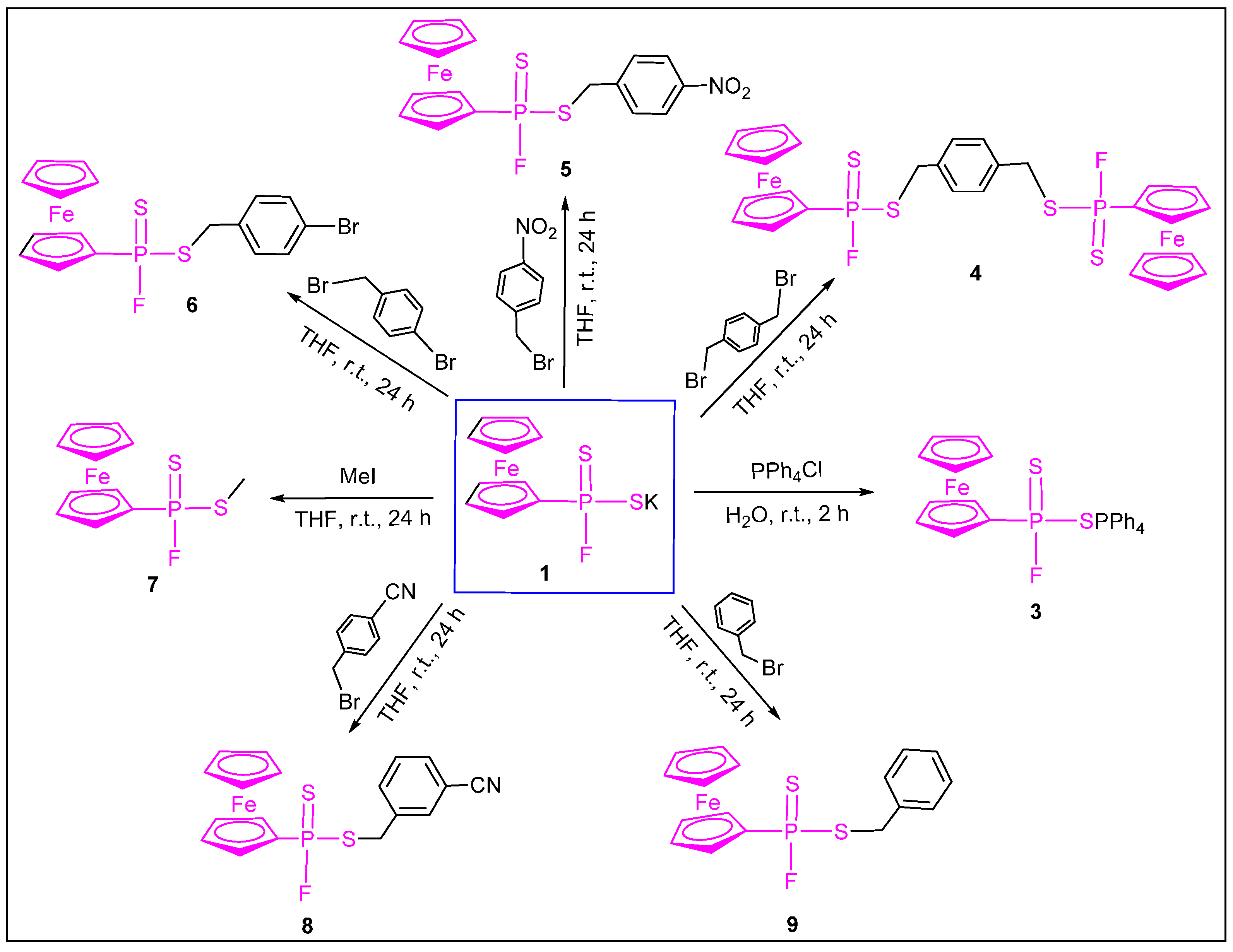{kind=link}
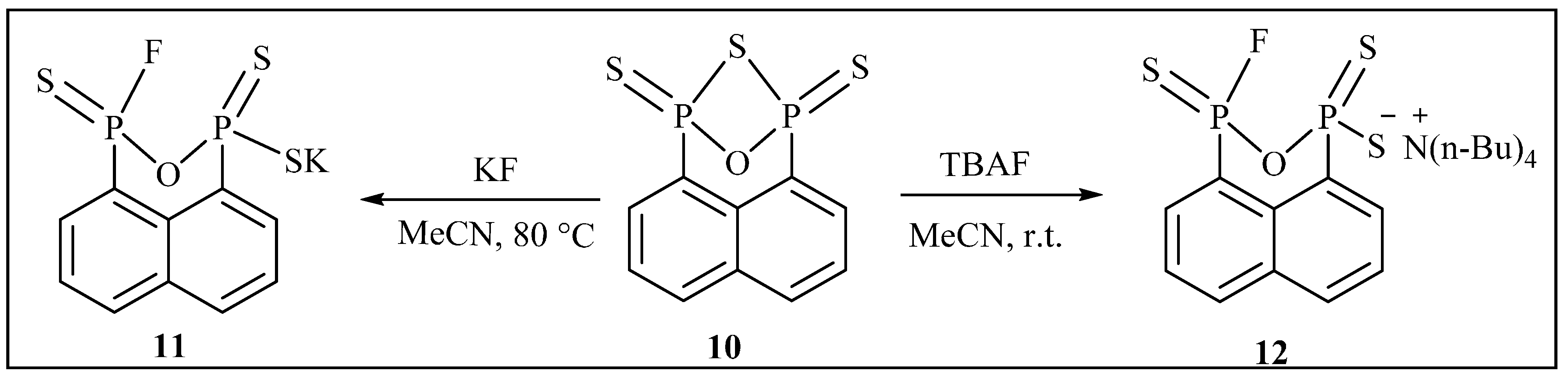{kind=link}
{kind=link}
{kind=link}
| Compound | 3 | 13 | 14 | 18 |
|---|---|---|---|---|
| Formula | C34H29FFeP2S2 | C34H26FOP3S3 | C30H22FN3OP3S3 | C10H6O2P2S4 |
| M | 638.52 | 658.68 | 617.65 | 348.34 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/n | P21/c | P-1 | P21/n |
| a/Å | 11.704(2) | 9.563(3) | 9.992(4) | 11.171(3) |
| b/Å | 17.994(2) | 16.876(5) | 16.808(8) | 8.618(2) |
| c/Å | 14.981(3) | 19.258(6) | 18.117(8) | 14.122(4) |
| α | 90 | 90 | 104.020(11) | 90 |
| β | 106.902(9) | 93.820(7) | 90.270(10) | 99.002(6) |
| γ | 90 | 90 | 104.404(11) | 90 |
| U/A3 | 3018.6(9) | 3101.1(15) | 2852(3) | 1342.8(6) |
| Z | 4 | 4 | 4 | 4 |
| µ/mm−1 | 7.726 | 4.277 | 4.090 | 9.32 |
| Reflections collected | 25,726 | 23,431 | 21,800 | 9765 |
| Independent reflections | 5310 | 5430 | 9960 | 2346 |
| Rint | 0.1707 | 0.1124 | 0.1052 | 0.0696 |
| R1 | 0.0824 | 0.1037 | 0.0988 | 0.0526 |
| wR2 [I > 2σ(I)] | 0.0912 | 0.2408 | 0.2433 | 0.1339 |
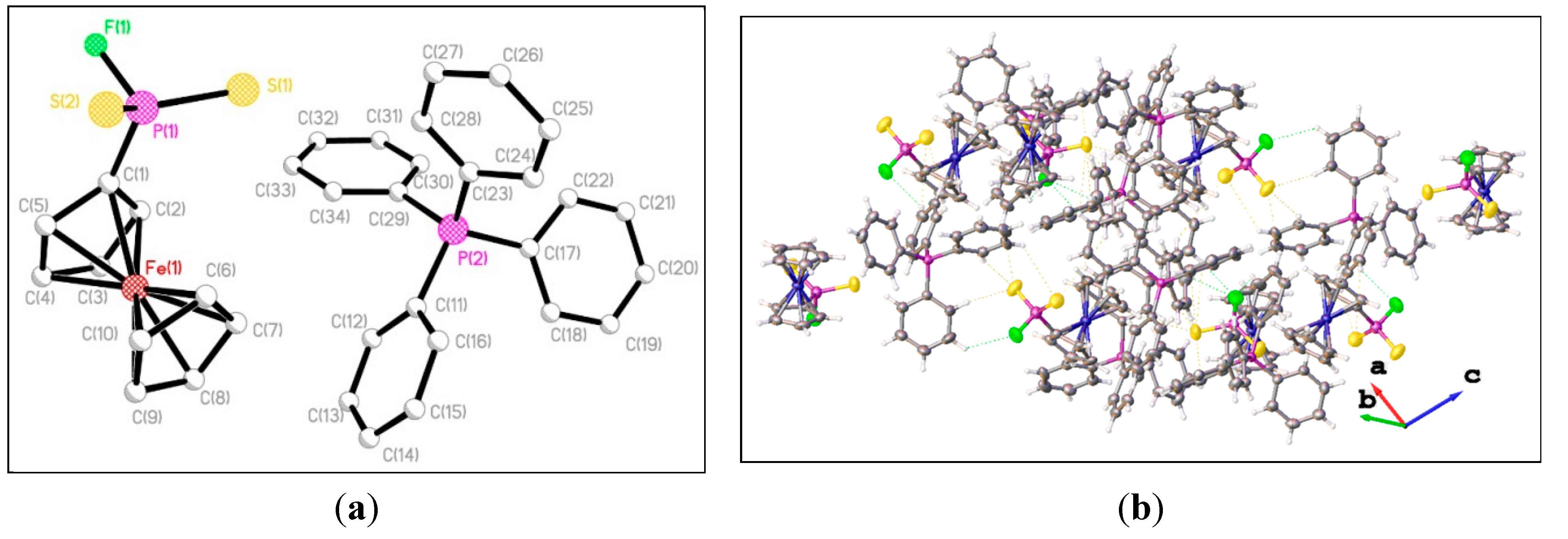
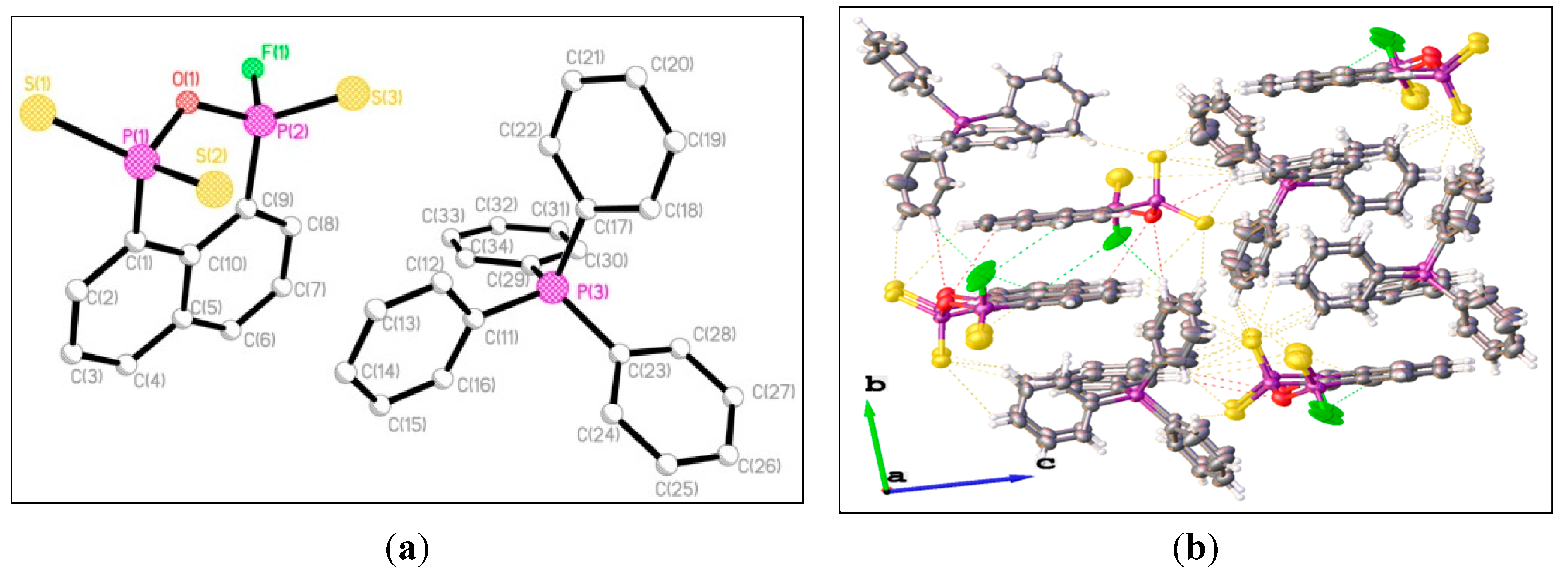
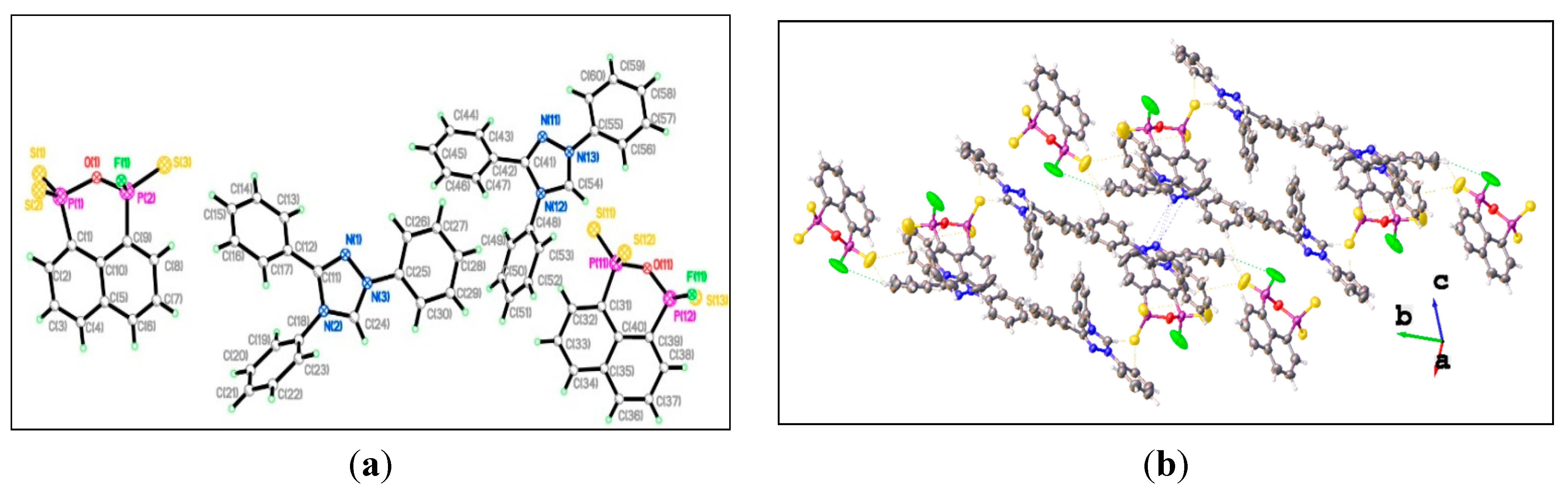

3. Experimental Section
3.1. General
3.2. Synthesis
3.2.1. Synthesis of Potassium Ferrocenylphosphonofluoridodithioate (1)
3.2.2. Synthesis of Tetrabutylammonium Ferrocenylphosphonofluoridodithioate (2)
3.2.3. Synthesis of Tetraphenylphosphonium Ferrocenylphosphonofluoridodithioate (3)
3.2.4. Synthesis of 1,4-Phenylenebis(methylene) Bis(ferrocenylphosphonofluoridodithioate) (4)
3.2.5. Synthesis of 4-Nitrobenzyl Ferrocenylphosphonofluoridodithioate (5)
3.2.6. Synthesis of 4-Bromobenzyl Ferrocenylphosphonofluoridodithioate (6)
3.2.7. Synthesis of Methylyl Ferrocenylphosphonofluoridodithioate (7)
3.2.8. Synthesis of 4-Cyanobenzyl Ferrocenylphosphonofluoridodithioate (8)
3.2.9. Synthesis of Benzyl Ferrocenylphosphonofluoridodithioate (9)
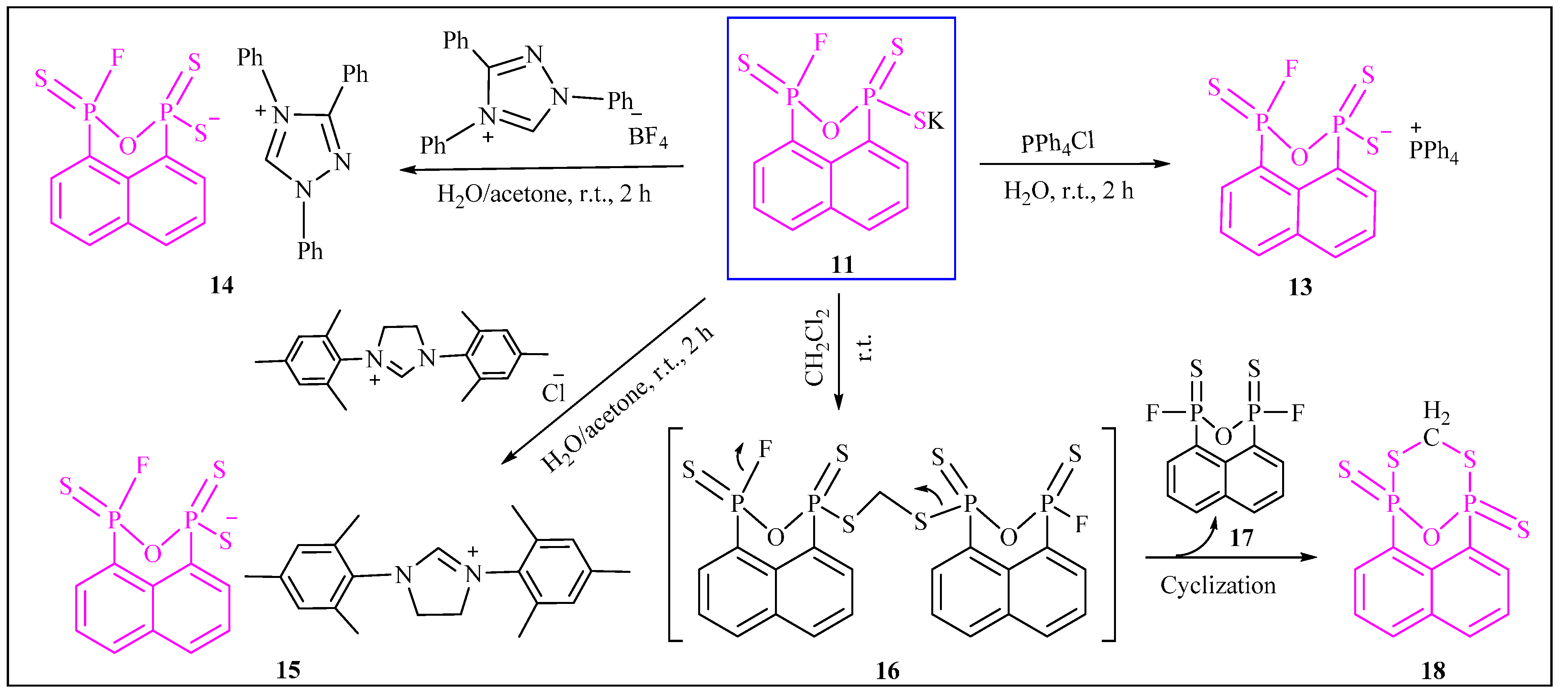
3.2.10. Synthesis of Potassium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (11)
3.2.11. Synthesis of Tetrabutylammonium 3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (12)
3.2.12. Synthesis of Tetraphenylphosphonium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (13)
3.2.13. Synthesis of 1,3,4-Triphenyl-1H-1,2,4-triazol-4-ium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (14)
3.2.14. Synthesis of 1,3-Dimesityl-4,5-dihydro-1H-imidazol-3-ium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (15)
3.2.15. Synthesis of 1,5-Epoxynaphtho[1,8-cd][1,7,2,6]dithiadiphosphocine 1,5-disulfide (18)
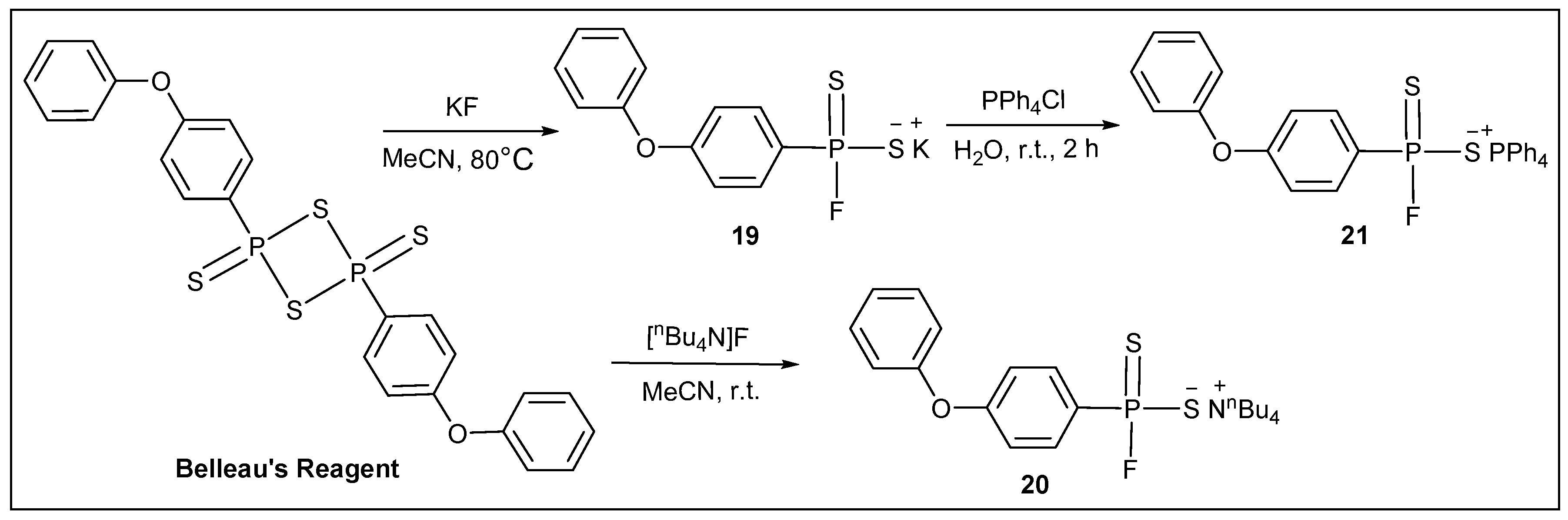
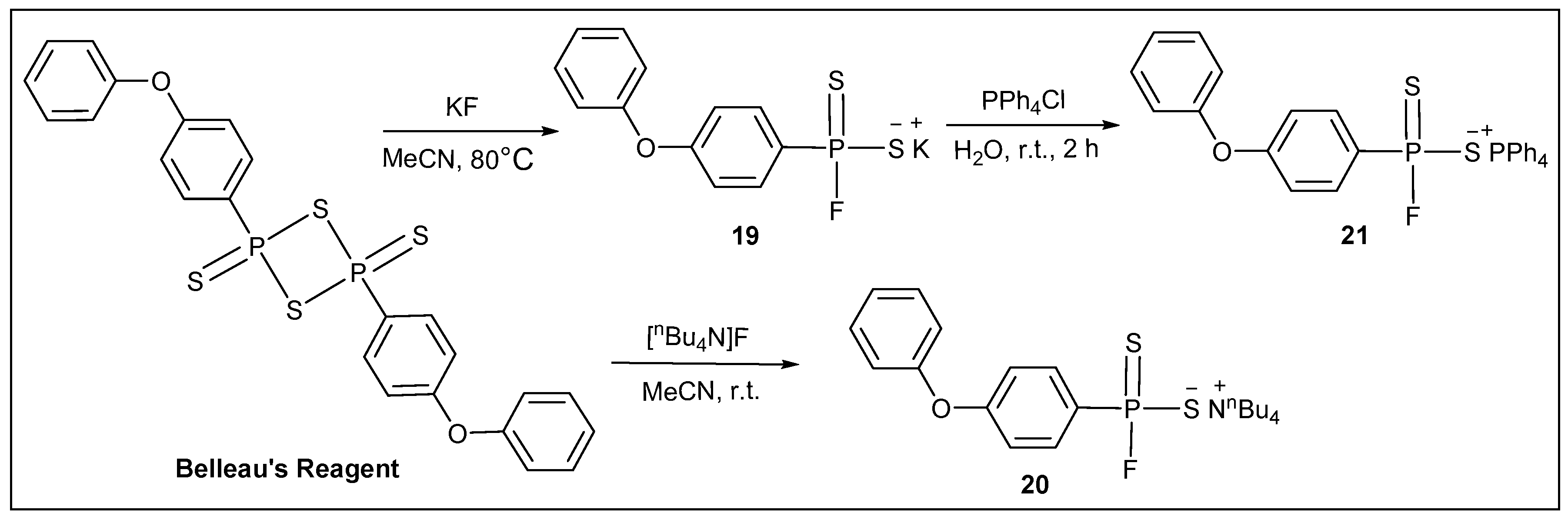
3.2.16. Synthesis of Potassium (4-phenoxyphenyl)phosphonofluoridodithioate (19)
3.2.17. Synthesis of Tetrabutylammonium (4-phenoxyphenyl)phosphonofluoridodithioate (20)
3.2.18. Synthesis of Tetraphenylphosphonium (4-phenoxyphenyl)phosphonofluoridodithioate (21)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wilson, B.W.; Walker, C.R. Regulation of newly synthesized acetyleholinesterase in muscle cultures treated with diisopropylfluorophosphate. Proc. Natl. Acad. Sci. USA 1974, 71, 3194–3198. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, P.A.; Lamdem, L.A. Inhibition of chymotrypsin by phosphonate and phosphonamidate peptide analogs. Bioorg. Chem. 1986, 14, 356–377. [Google Scholar] [CrossRef]
- Engel, R. Phosphonates as analogues of natural phosphates. Chem. Rev. 1977, 77, 349–367. [Google Scholar] [CrossRef]
- Camps, F.; Coll, J.; Fabrias, G.; Guerrero, A. Synthesis of dientic fluorinated analogs of insect sex pheromones. Tetrahedron 1984, 40, 2871–2878. [Google Scholar] [CrossRef]
- Sikder, A.K.; Ghosh, A.K.; Jaiswal, D.K. Quaternary salts of 3,3′-bis-pyridinium monooximes: Synthesis and biological activity. J. Pharm. Sci. 1993, 82, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Marjit, D.N.; Sharma, U.S. Hydrolysis of diisopropyl phosphorofluoridate catalysed by copper(II)-diamine complexes. Indian J. Chem. 1989, 28A, 958–960. [Google Scholar]
- Sikder, A.K.; Pandey, K.S.; Jaiswal, D.K.; Dube, S.N.; Kumar, D.; Hussain, K.; Bhattacharya, R.; Das Gupta, S. The 3.3′-bispyridinium monooximes as antidotes agaist organophosphorous intoxication. J. Pharm. Pharmacol. 1992, 44, 1038–1040. [Google Scholar] [PubMed]
- Anderson, C.; Freeman, J.; Lucas, L.H.; Farley, M.; Dalhoumi, H.; Widlanski, T.S. Estrone sulfatase: Probing structural requirements for substrate and inhibitor. Biochemsitry 1997, 36, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- Ashani, Y.; Leader, H.; Rothschild, N.; Dosoretz, C. Combined effect of organophosphorus hydrolase and oxime on the reactivation rate of diethylphosphoryl-acetylcholinesterase conjugates. Biochem. Pharmacol. 1998, 55, 159–168. [Google Scholar] [CrossRef]
- Bollmark, M.; Stawinski, J. H-phosphonates-chemistry and applications. Nucleosides Nucleotides 1998, 17, 663–680. [Google Scholar] [CrossRef]
- Eyer, P. The role of oximes in the management of organophosphorus pesticide poisoning. Toxicol. Rev. 2003, 22, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Oh, K.A.; Park, N.J.; Park, N.S.; Kim, Y.J.; Yum, E.K.; Jung, Y.S. Reactivation study of pyridinium oximes for acetylcholinesterases inhibited by paraoxon or DFP. J. Appl. Biomed. 2006, 4, 67–72. [Google Scholar]
- Roesky, H.W.; Tebbe, F.N.; Muetterties, E.L. New phosphorus-sulfur chemistry. J. Am. Chem. Soc. 1967, 89, 1272–1274. [Google Scholar] [CrossRef]
- Roesky, H.W.; Tebbe, F.N.; Muetteries, E.L. Thiophosphate chemistry: Anion set X2PS2−, (XPS2)2S2−, and (XPS2)2S22−. Inorg. Chem. 1970, 9, 831–836. [Google Scholar] [CrossRef]
- Roesky, H.W. Phosphorus compounds. XIX. Preparation and reactions of thiophosphoramidic dihalides and alkyldithiophosphonic acid fluorides. Chem. Ber. 1968, 101, 3679–3687. [Google Scholar] [CrossRef]
- Roesky, H.W.; Beyer, H. Phosphorus compounds. XXX. Substitution reactions in thiophosphinyl halide compounds. Chem. Ber. 1969, 102, 2588–2594. [Google Scholar] [CrossRef]
- Roesky, H.W.; Grimm, L.F. Phosphorus compounds. XXVIII. Preparation and characterization of thiophosphoryl compounds containing a phosphorus-nitrogen double bond. Chem. Ber. 1969, 102, 2319–2329. [Google Scholar] [CrossRef]
- Roesky, H.W.; Grimm, L.F. Phosphorus compounds. 50. Reactions with N-halophosphoranylidenethiophosphorylhalidde amides. Chem. Ber. 1970, 103, 1664–1673. [Google Scholar] [CrossRef]
- Roesky, H.W.; Grimm, L.F. N,N′-Sulfonylbis(sulffur difluoride diimide). Angew. Chem. Int. Ed. 1970, 9, 244–245. [Google Scholar]
- Harris, R.K.; Woplin, J.R.; Murruy, M.; Schmutzler, R. Preparation and nuclear magnetic resonance spectra of symmetrical spin systems containing phosphorus: Bis(fluorophosphinothioyl) sulfphides. J. Chem. Soc. Dalton Trans. 1972, 1590–1596. [Google Scholar] [CrossRef]
- Fluck, E.; Schimdt, R.; Haubold, W. Reaktionen des pyridinium-fluordithiophosphorsaure-betains. Phosphorus Sulfur Silicon Relat. Elem. 1978, 5, 141–146. [Google Scholar] [CrossRef]
- Stawinski, J.; Bollmark, M. A facile access to nucleoside phosphorofluoridate, nucleoside phosphorofluoridothioate, and nucleoside phosphorofluoridethioate monoesters. Tetrahedron Lett. 1996, 37, 5739–5741. [Google Scholar]
- Dabkowski, W.; Michalska, M.; Tworowska, I. Highly efficient synthesis of phosphorodithioates derived from 3′-thiothymidine by anhydro-ring opening of 2,3′-anhydro5′-tritylthylidine with O,O-disubstituted phosphorodithioic acids. Chem. Commun. 1998, 427–428. [Google Scholar] [CrossRef]
- Tworowska, I.; Dabkowski, W. New efficient synthesis of phosphonofluorodithioates ROP(S)(S−)F and their structural analogues. Chem. Commun. 1998, 2611–2612. [Google Scholar] [CrossRef]
- Hua, G.; Du, J.; Slawin, A.M.Z.; Woollins, J.D. Novel fluorinated phosphorus-selenium heteroatom compounds: Phenylphosphonofluorodislenoic salts, adducts and esters. Inorg. Chem. 2013, 52, 8214–8217. [Google Scholar] [CrossRef] [PubMed]
- St. J. Foreman, M.R.; Slawin, A.M.Z.; Woollins, J.D. 2,4-Diferrocenyl-1,3-dithiadiphosphetane 2,4-disulfide; structure and reactions with catechols and [PtCl2(PR3)2](R = Et or Bun). J. Chem. Soc. Dalton Trans. 1996, 3653–3657. [Google Scholar] [CrossRef]
- Parveen, S.; Kilian, P.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and characterisation of four- and six-membered P-Se heterocycles. Dalton Trans. 2006, 2586–2590. [Google Scholar] [CrossRef] [PubMed]
- Sağlam, E.G.; Çelik, Ö.; Yılmaz, H.; Acar, N. Synthesis and spectroscopic characterization of novel aryl-dithiofluorophosphonate derivatives and X-ray studies of [(4-CH3OC6H4)(F)P(S)S−][PH4P+]. Phosphorus Sulfur Silicon Relat. Elem. 2012, 187, 1339–1346. [Google Scholar] [CrossRef]
- Roesky, H.W. Über die darstellung von alkyldithiofluorophosphaten. Z. Naturforschung 1967, 22, 716–718. [Google Scholar]
- Takeda, M.; Hallenga, K.; Shigezane, M.; Waelchli, M.; Löhr, F.; Markley, J.L.; Kainosho, M. Construction and performance of an NMR tube with a sample cavity formed within magnetic susceptibility-matched glass. J. Magn. Reson. 2011, 209, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Slawin, A.M.Z.; Williams, D.J.; Wood, P.T.; Woollins, J.D. The preparation and X-ray structure of naphthalenedithiadiphosphetanedisulphide. J. Chem. Soc. Chem. Commun. 1987, 1741–1741. [Google Scholar] [CrossRef]
- Gray, I.P.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and structure of [Fc(EtO)PS2]-complexes. Z. Anorg. Allg. Chem. 2004, 630, 1851–1857. [Google Scholar] [CrossRef]
- Fei, Z.; Slawin, A.M.Z.; Woollins, J.D. Nucleophilic reactions of 2,4-(naphthalene-1,8-diyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide. Polyhedron 2001, 20, 2577–2581. [Google Scholar] [CrossRef]
- Liang, C.X.; Allen, L.C. Sulfur does not form double bonds in phosphorothioate anions. J. Am. Chem. Soc. 1987, 109, 6449–6453. [Google Scholar] [CrossRef]
- Wong, C.Y.; McDonald, R.; Cavell, R.G. Hexacoordinate phosphorus. 7. Synthesis and characterization of neutral phosphorus(V) compounds containing divalent tridentate diphenol imine, azo, and thio ligants. Inorg. Chem. 1996, 35, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.; Goycoolea, F.M.; Rivera, D.Z.; Onofre, J.J.; Martinez, K.; Lizardi, J.; Reyes, M.S.; Gordillo, B.; Contreras, C.V.; Barradas, O.G.; et al. Substituent effects on the 31P-NMR chemical shifts of arylphosphorothionates. Tetrahedron 2006, 62, 2520–2528. [Google Scholar] [CrossRef]
- Kilian, P.; Marek, J.; Marek, R.; Tousek, J.; Humpa, O.; Slawin, A.M.Z.; Touzin, J.; Novosad, J.; Woollins, J.D. New P–S–N containing ring systems. Reaction of 2,4-(naphthalene-1,8-diyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide with methylbis(trimethylsily)amine. Dalton Trans. 1999, 2231–2236. [Google Scholar] [CrossRef]
- Kilian, P.; Marek, J.; Marek, R.; Touin, J.; Novosad, J.; Woollins, J.D. New P–S–N containing ring systems. Reaction of 2,4-(naphthalene-1,8-diyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide and its 4-methoxynaphthalene derivative with hexamethyldisilazane. Dalton Trans. 1998, 1175–1180. [Google Scholar] [CrossRef]
- Eleftheriou, M.E.; Novosad, J.; Williams, D.J.; Woollins, D.J. Reaction of 1,3-epithionaphtho[1,8-cd][1,2λ5,6λ5]thiadiphosphinine-1,3-dithione; the preparation and X-ray structure of Np(S)(SMe)SP(S)(OMe), the first C3P2S ring. J. Chem. Soc. Chem. Commun. 1991, 116–117. [Google Scholar] [CrossRef]
- Fuller, A.L.; Scott-Hayward, L.A.S.; Li, Y.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Automated chemical crystallography. J. Am. Chem. Soc. 2010, 132, 5799–5802. [Google Scholar] [CrossRef] [PubMed]
- Flugrath, J.W.P. The finer things in X-ray diffraction data collection. Acta Crystallogr. 1999, D55, 1718–1725. [Google Scholar]
- Altomare, A.; Burla, M.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Beurskens, P.T.; Admiraal, G.; Beurskens, G.; Bosman, W.P.; Israel, R.; Smits, J.M.M. The DIRDIF-99 Program System; Technical Report of the Crystallography Laboratory, University of Nijmegen: Nijmegen, The Netherlands, 1999. [Google Scholar]
- Crystal Structure; Version 3.8 Single Crystal Structure Analysis Software; Rigaku/MSC: The Woodlands, TX, USA, 2006.
- Sheldrick, G.M. A short history of SHELXL. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of all compounds are not available except compounds 1 and 2 from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, G.; Du, J.; Surgenor, B.A.; Slawin, A.M.Z.; Woollins, J.D. Novel Fluorinated Phosphorus–Sulfur Heteroatom Compounds: Synthesis and Characterization of Ferrocenyl- and Aryl-Phosphonofluorodithioic Salts, Adducts, and Esters. Molecules 2015, 20, 12175-12197. https://doi.org/10.3390/molecules200712175
Hua G, Du J, Surgenor BA, Slawin AMZ, Woollins JD. Novel Fluorinated Phosphorus–Sulfur Heteroatom Compounds: Synthesis and Characterization of Ferrocenyl- and Aryl-Phosphonofluorodithioic Salts, Adducts, and Esters. Molecules. 2015; 20(7):12175-12197. https://doi.org/10.3390/molecules200712175
Chicago/Turabian StyleHua, Guoxiong, Junyi Du, Brian A. Surgenor, Alexandra M. Z. Slawin, and J. Derek Woollins. 2015. "Novel Fluorinated Phosphorus–Sulfur Heteroatom Compounds: Synthesis and Characterization of Ferrocenyl- and Aryl-Phosphonofluorodithioic Salts, Adducts, and Esters" Molecules 20, no. 7: 12175-12197. https://doi.org/10.3390/molecules200712175