
Iridium-Catalysed ortho-Directed Deuterium Labelling of Aromatic Esters—An Experimental and Theoretical Study on Directing Group Chemoselectivity

Abstract
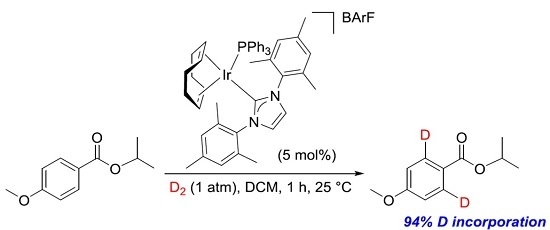
:
1. Introduction
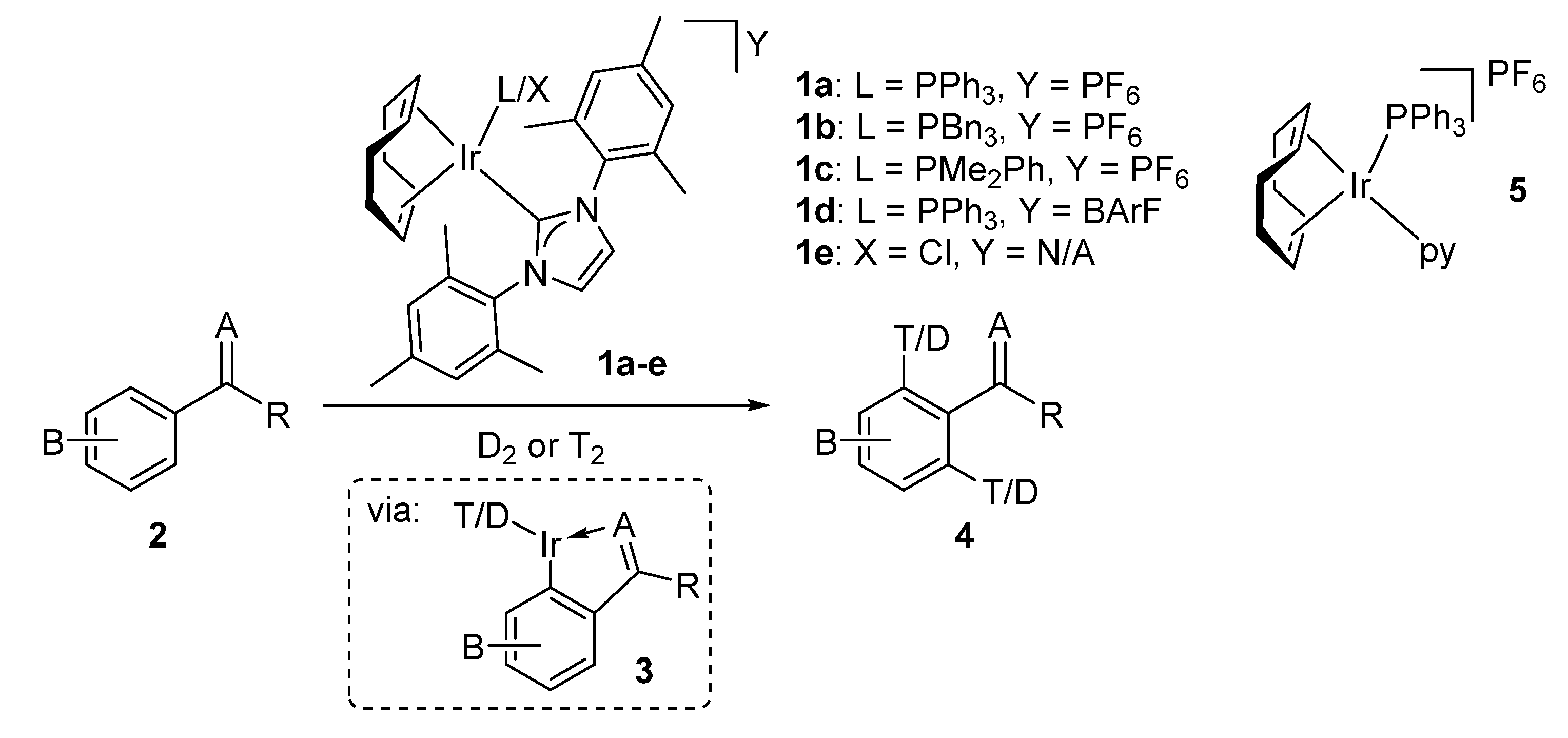
2. Results and Discussion
2.1. Catalyst Screening and Comparisons with Crabtree’s Catalyst

2.2. Applicable Substrate Scope with Catalyst 1a
2.2.1. Para-Substituted Methyl Benzoates


| Entry | X | Substrate | %D a |
|---|---|---|---|
| 1 | H | 8a | 52 |
| 2 | CH3 | 8b | 42 |
| 3 | CF3 | 8c | 32 |
| 4 | Cl | 8d | 93 |
| 5 | OMe | 8e | 89 |
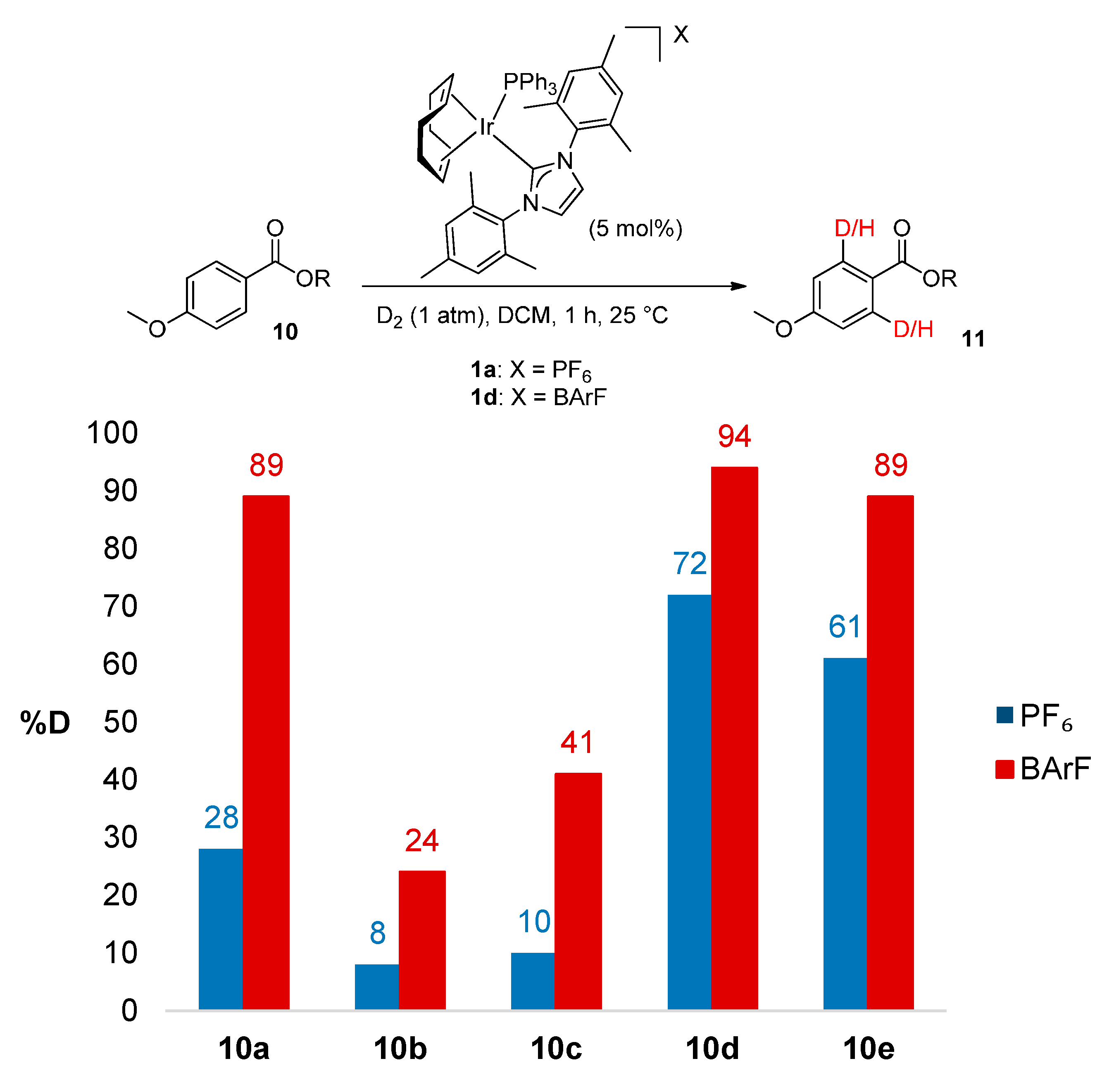
2.2.2. Scope of O-Alkyl Substitution with Electron-Rich Benzoate Esters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
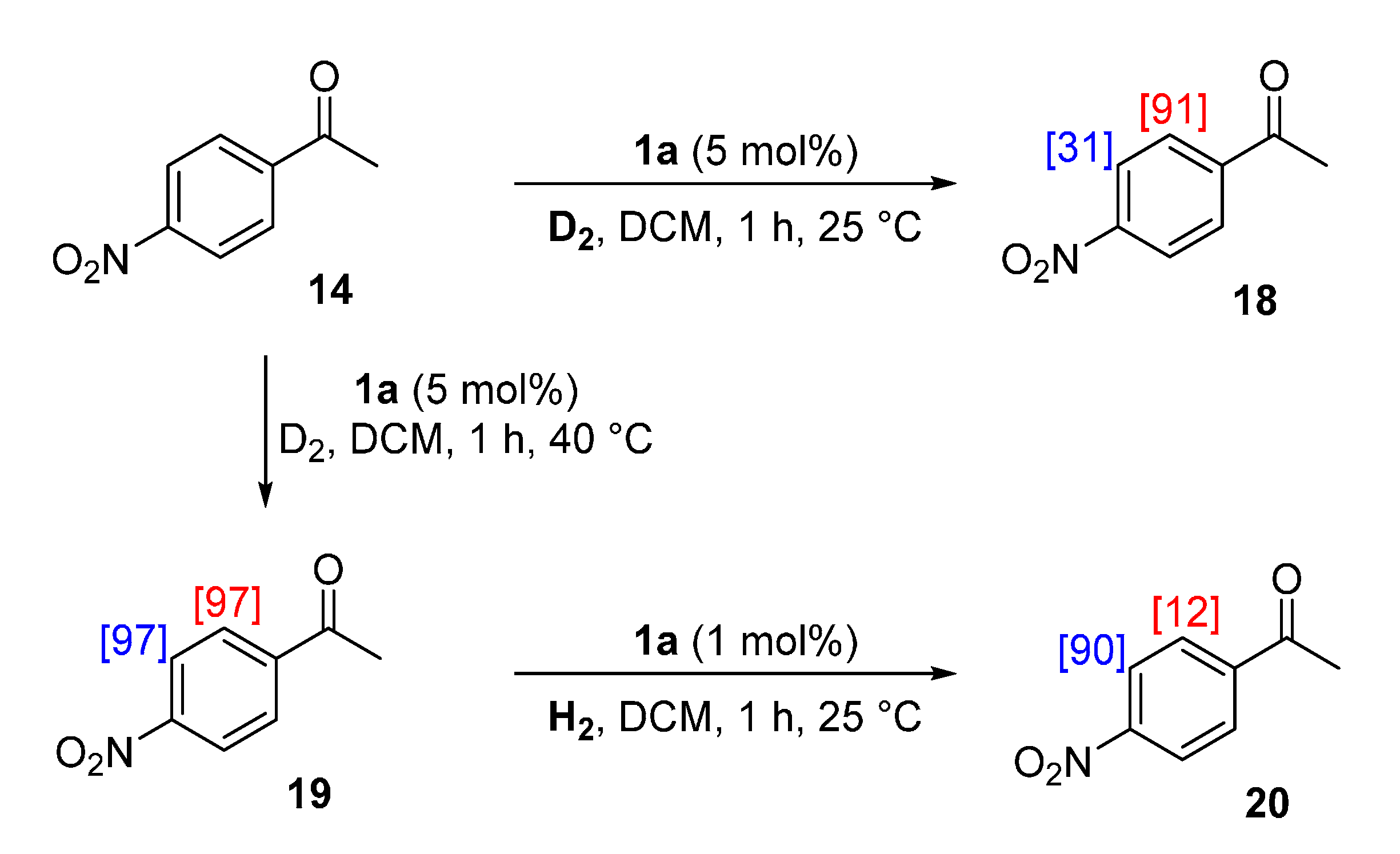{kind=link}
| Entry | R | Substrate | %D a |
|---|---|---|---|
| 1 | n-Pr | 10a | 28 |
| 2 | CH2CF3 | 10b | 8 |
| 3 | t-Bu | 10c | 10 |
| 4 | i-Pr | 10d | 73 |
| 5 | Bn | 10e | 62 |
2.3. Reaction Optimisation for Efficient Labelling of Challenging Benzoate Ester Substrates
2.3.1. Temperature Effects
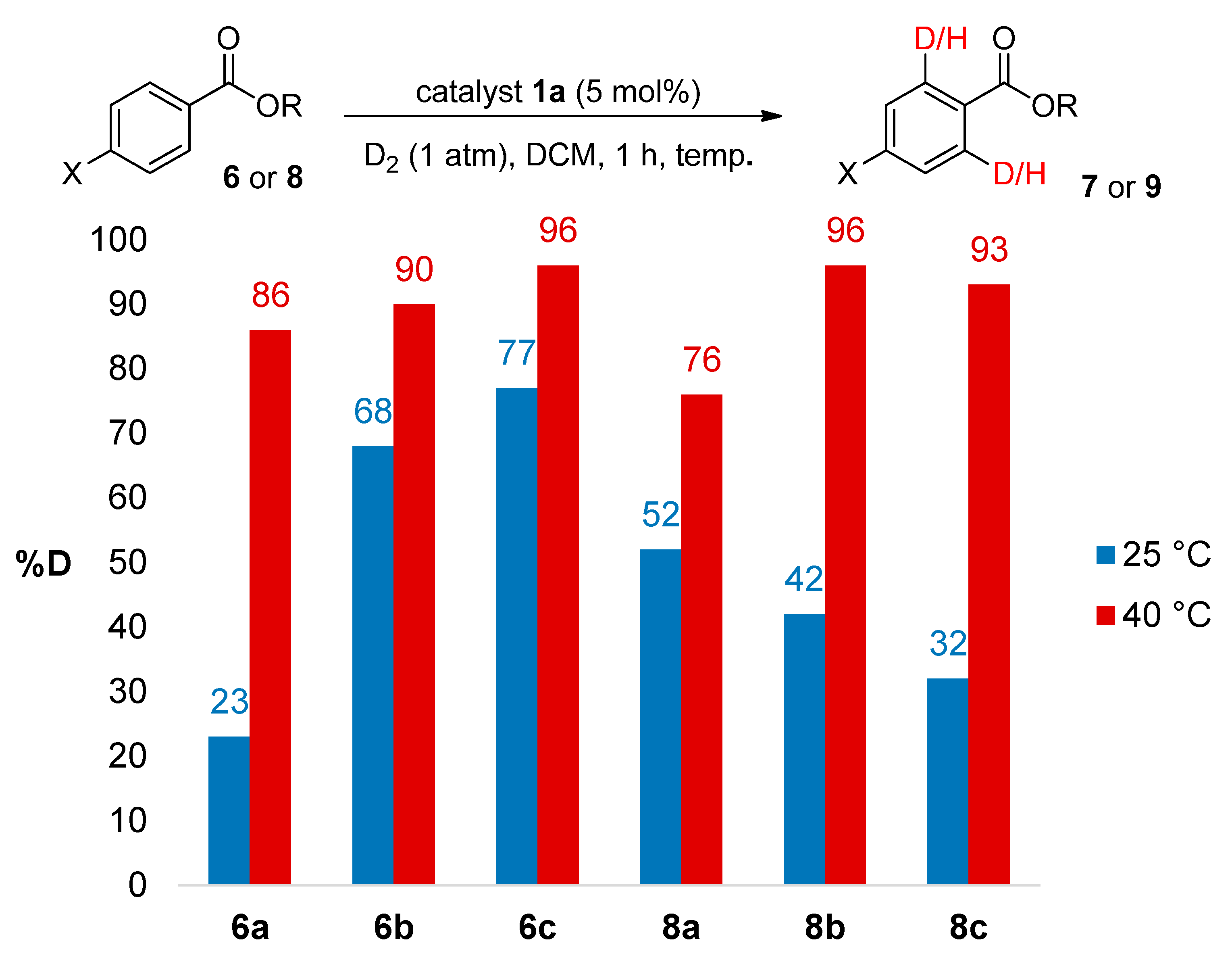
2.3.2. Catalyst Counterion Effects

2.4. Exploring Chemoselectivity Issues in Ester Labelling
2.4.1. Experimental Observations
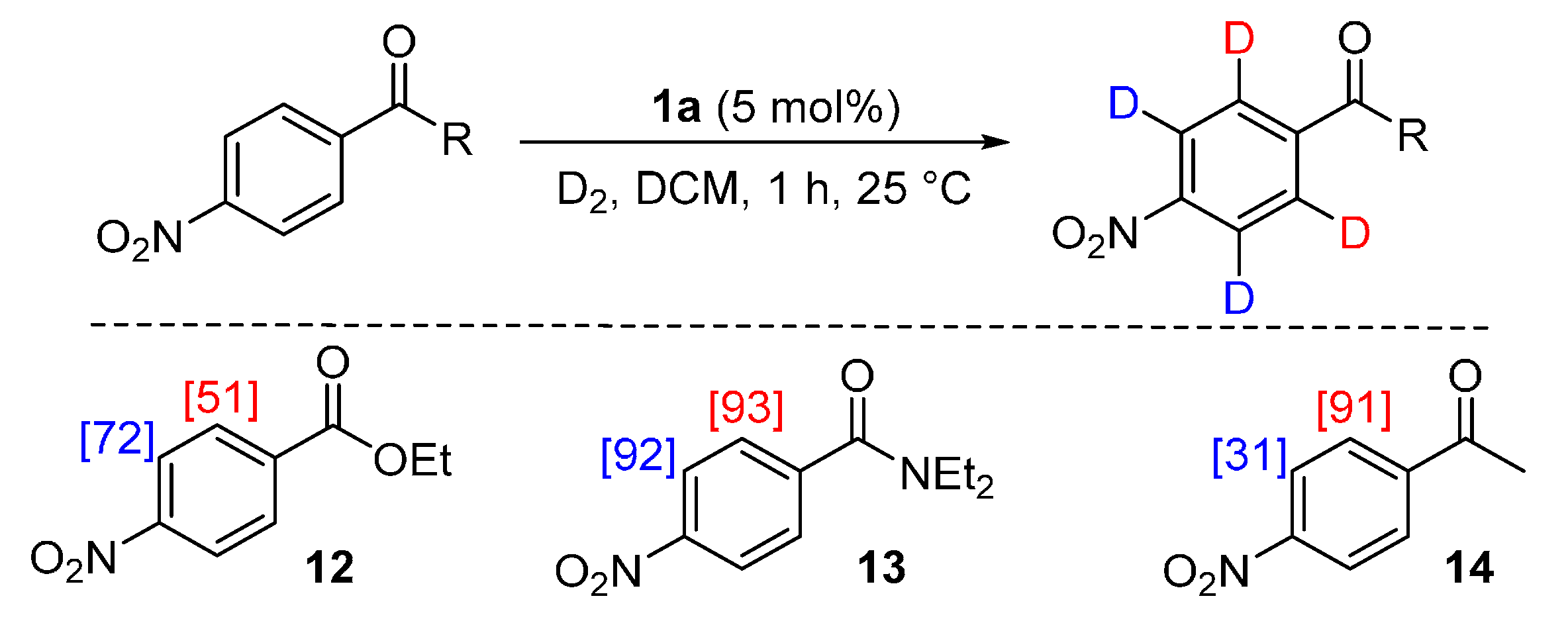

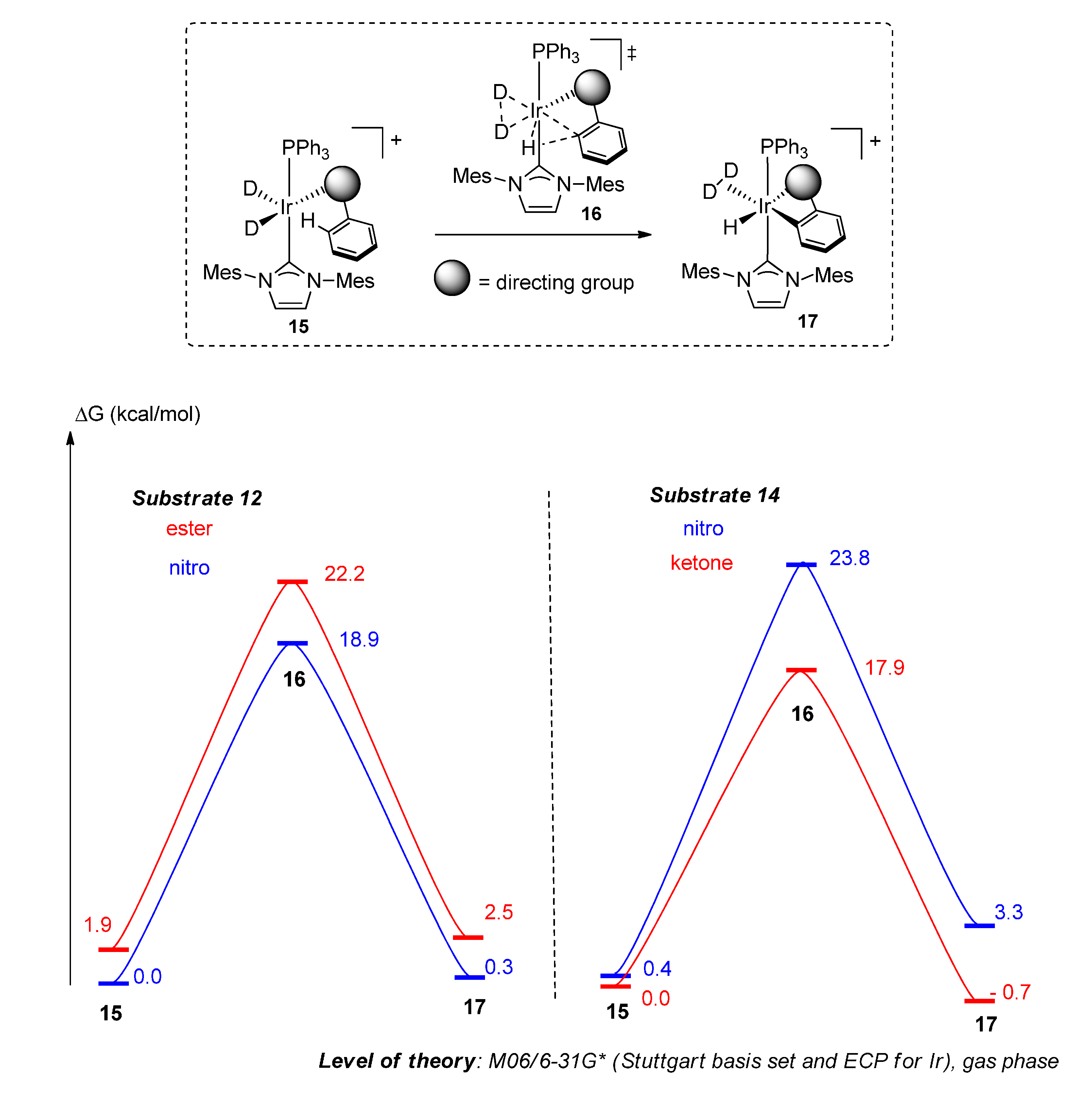
2.4.2. Theoretical Analysis
2.4.3. Practical Exploitation of Directing Group Chemoselectivity
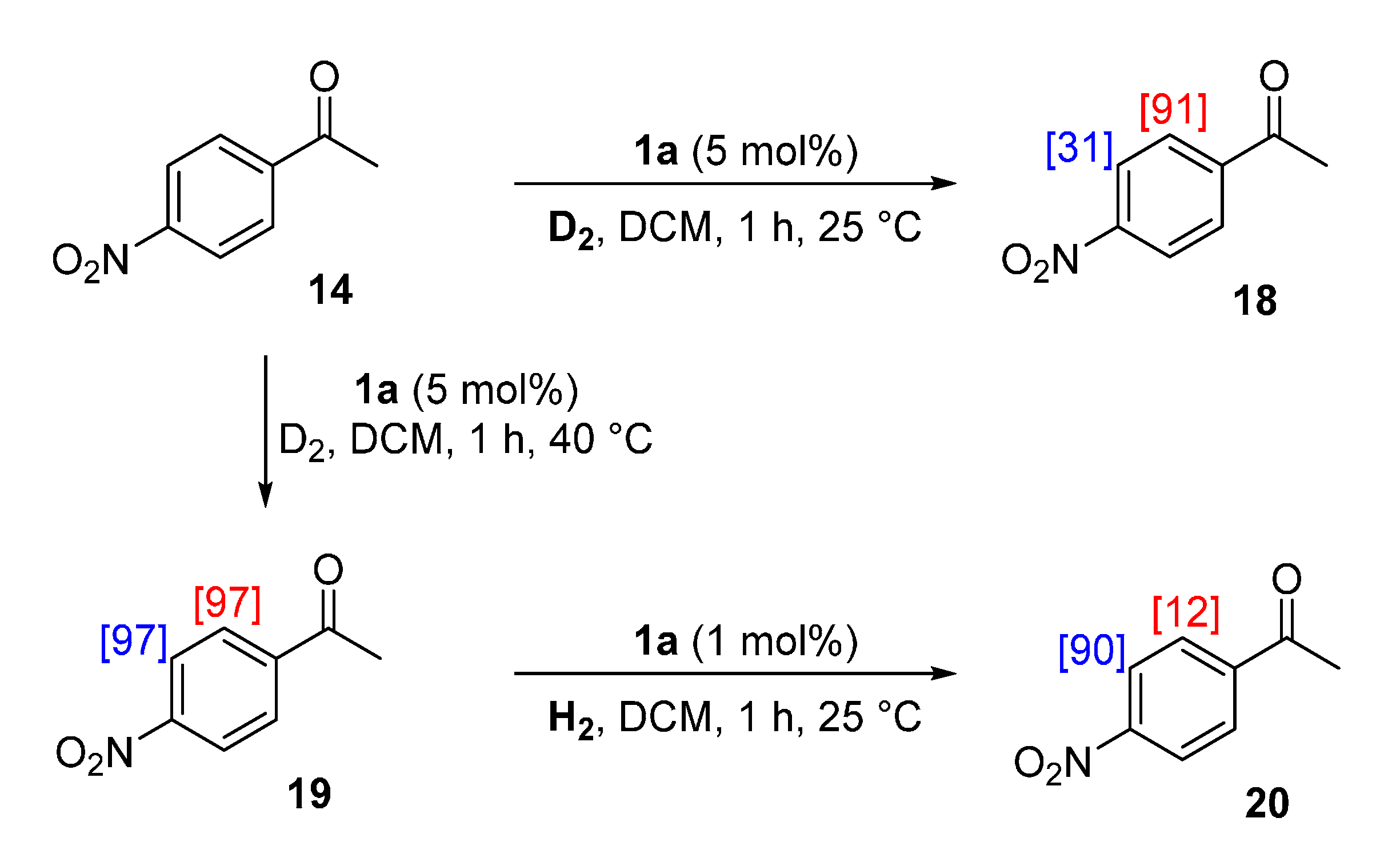
3. Experimental Section
3.1. General Considerations
3.2. General Procedures
3.2.1. Deuteration of Substrates Using Iridium(I) Complexes 1a, 1b, 1d and 5
3.2.2. Deuteration of Substrates for Rate Studies
3.3. Labelling Studies
3.3.1. Deuteration of Ethyl benzoate 6a with Complex 5

3.3.2. Deuteration of Ethyl 4-methylbenzoate 6b with Complex 5

3.3.3. Deuteration of Ethyl 4-(trifluoromethyl)benzoate 6c with Complex 5

3.3.4. Deuteration of Ethyl 4-chlorobenzoate 6d with Complex 5

3.3.5. Deuteration of Ethyl 4-methoxybenzoate 6e with Complex 5

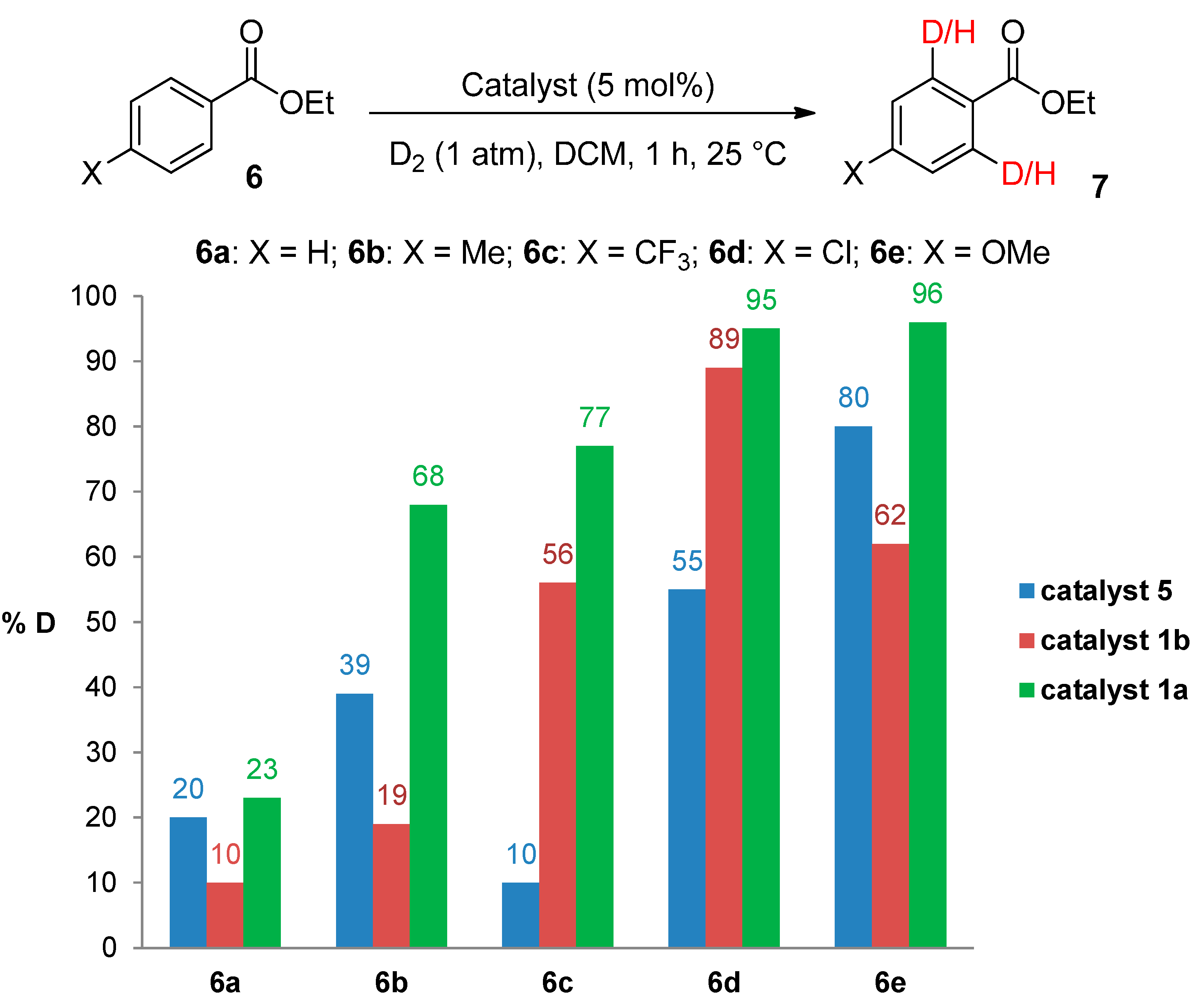
3.3.6. Deuteration of Esters 6a–e Using Catalysts 1b and 1a
3.3.7. Deuteration of Methyl Benzoate 8a with Complex 1a

| Entry | Substrate | Catalyst | %D (run 1) | %D (run 2) |
|---|---|---|---|---|
| 1 | 6a | 1b (10.5 mg) | 10 | 10 |
| 2 | 6b | 15 | 23 | |
| 3 | 6c | 62 | 50 | |
| 4 | 6d | 85 | 93 | |
| 5 | 6e | 65 | 59 | |
| 6 | 6a | 1a (10.1 mg) | 6 | 40 |
| 7 | 6b | 53 | 83 | |
| 8 | 6c | 74 | 79 | |
| 9 | 6d | 95 | 95 | |
| 10 | 6e | 96 | 96 |
3.3.8. Deuteration of Methyl 4-methylbenzoate 8b with Complex 1a

3.3.9. Deuteration of Methyl 4-(trifluoromethyl)benzoate 8c with Complex 1a

3.3.10. Deuteration of Methyl 4-chlorobenzoate 8d with Complex 1a

3.3.11. Deuteration of Methyl 4-methoxybenzoate 8e with Complex 1a

3.3.12. Deuteration of n-Propyl 4-methoxybenzoate 10a with Complex 1a

3.3.13. Deuteration of 2,2,2-Trifluoroethyl 4-methoxybenzoate 10b with Complex 1a

3.3.14. Deuteration of t-Butyl 4-methoxybenzoate 10c with Complex 1a

3.3.15. Deuteration of Iso-propyl 4-methoxybenzoate 10d with Complex 1a

3.3.16. Deuteration of Benzyl 4-methoxybenzoate 10e with Complex 1a

3.4. Temperature Effects
| Entry | Substrate | %D (run 1) | %D (run 2) |
|---|---|---|---|
| 1 | 6a | 85 | 86 |
| 2 | 6b | 89 | 91 |
| 3 | 6c | 95 | 96 |
| 4 | 8a | 75 | 76 |
| 5 | 8b | 96 | 95 |
| 6 | 8c | 92 | 93 |
3.5. Catalyst Counterion Effects
3.5.1. Deuteration of n-Propyl 4-methoxybenzoate 10a with Complex 1d
3.5.2. Deuteration of 2,2,2-Trifluoroethyl 4-methoxybenzoate 10b with Complex 1d
3.5.3. Deuteration of t-Butyl 4-methoxybenzoate 10c with Complex 1d
3.5.4. Deuteration of iso-Propyl 4-methoxybenzoate 10d with Complex 1d
3.5.5. Deuteration of Benzyl 4-methoxybenzoate 10e with Complex 1d
3.6. Chemoselectivity Studies
3.6.1. Deuteration of Ethyl 4-nitrobenzoate 12 with Complex 1a

3.6.2. Deuteration of N,N-Diethyl 4-nitrobenzamide 13 with Complex 1a

3.6.3. Deuteration of 4-Nitroacetophenone 14 with Complex 1a

3.7. Rate Studies
3.7.1. Deuteration of Ethyl 4-nitrobenzoate 12 with Complex 1a

3.7.2. Deuteration of 4-Nitroacetophenone 14 with Complex 1a

3.8. Practical Exploitation of Directing Group Chemoselectivity

3.8.1. Deuteration of 4-Nitroacetophenone 14 with Complex 1a
3.8.2. Deuteration of 4-Nitroacetophenone 14 with Complex 1a
3.8.3. Hydrogenation of 4-Nitroacetophenone-d4 19 with Complex 1a
4. Computational Details
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Isin, E.M.; Elmore, C.S.; Nilsson, G.N.; Thompson, R.A.; Weidolf, L. Use of radiolabeled compounds in drug metabolism and pharmacokinetic studies. Chem. Res. Toxicol. 2012, 25, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.H.; Hickey, M.J.; Kingston, L.P.; Wilkinson, D.J. Metal-catalysed isotopic exchange labelling: 30 years of experience in pharmaceutical R&D. J. Label. Compd. Radiopharm. 2010, 53, 731–738. [Google Scholar]
- Hesk, D.; Lavey, C.F.; McNamara, P. Tritium labelling of pharmaceuticals by metal-catalysed exchange methods. J. Label. Compd. Radiopharm. 2010, 53, 722–730. [Google Scholar] [CrossRef]
- Heys, J.R. Organoiridium complexes for hydrogen isotope exchange labeling. J. Label. Compd. Radiopharm. 2007, 50, 770–778. [Google Scholar] [CrossRef]
- Lockley, W.J.S.; McEwen, A.; Cooke, R. Tritium: A coming of age for drug discovery and development ADME studies. J. Label. Compd. Radiopharm. 2012, 55, 235–257. [Google Scholar] [CrossRef]
- Nilsson, G.N.; Kerr, W.J. The development and use of novel iridium complexes as catalysts for ortho-directed hydrogen isotope exchange reactions. J. Label. Compd. Radiopharm. 2010, 53, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Sawama, Y.; Monguchi, Y.; Sajiki, H. Efficient H-D Exchange Reactions Using Heterogeneous Platinum-Group Metal on Carbon-H2-D2O System. Synlett 2012, 23, 959–972. [Google Scholar] [CrossRef]
- Atzrodt, J.; Derdau, V.; Fey, T.; Zimmermann, J. The renaissance of H/D exchange. Angew. Chem. Int. Ed. 2007, 46, 7744–7765. [Google Scholar] [CrossRef] [PubMed]
- Lockley, W.J.S. 30 Years with ortho-directed hydrogen isotope exchange labelling. J. Label. Compd. Radiopharm. 2007, 50, 779–788. [Google Scholar] [CrossRef]
- Hesk, D.; McNamara, P. Synthesis of isotopically labelled compounds at Schering-Plough, an historical perspective. J. Label. Compd. Radiopharm. 2007, 50, 875–887. [Google Scholar] [CrossRef]
- Lin, D.; Chang, W. Chemical derivatization and the selection of deuterated internal standard for quantitative determination—Methamphetamine example. J. Anal. Toxicol. 2000, 24, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Atzrodt, J.; Derdau, V. Pd- and Pt-catalyzed H/D exchange methods and their application for internal MS standard preparation from a Sanofi-Aventis perspective. J. Label. Compd. Radiopharm. 2010, 53, 674–685. [Google Scholar] [CrossRef]
- Parkin, G. Applications of deuterium isotope effects for probing aspects of reactions involving oxidative addition and reductive elimination of H–H and C–H bonds. J. Label. Compd. Radiopharm. 2007, 50, 1088–1114. [Google Scholar] [CrossRef]
- Heinekey, D.M. Transition metal dihydrogen complexes: Isotope effects on reactivity and structure. J. Label. Compd. Radiopharm. 2007, 50, 1063–1071. [Google Scholar] [CrossRef]
- Quasdorf, K.W.; Huters, A.D.; Lodewyk, M.W.; Tantillo, D.J.; Garg, N.K. Total synthesis of oxidized welwitindolinones and (−)-N-methylwelwitindolinone C isonitrile. J. Am. Chem. Soc. 2012, 134, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Irvine, S.; Kennedy, A.R.; Kerr, W.J.; Andersson, S.; Nilsson, G.N. Highly active iridium(I) complexes for catalytic hydrogen isotope exchange. Chem. Commun. 2008, 1115–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochrane, A.R.; Idziak, C.; Kerr, W.J.; Mondal, B.; Paterson, L.C.; Tuttle, T.; Andersson, S.; Nilsson, G.N. Practically convenient and industrially-aligned methods for iridium-catalysed hydrogen isotope exchange processes. Org. Biomol. Chem. 2014, 12, 3598–3603. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R.; Kerr, W.J.; Moir, R.; Reid, M. Anion effects to deliver enhanced iridium catalysts for hydrogen isotope exchange processes. Org. Biomol. Chem. 2014, 12, 7927–7931. [Google Scholar] [CrossRef] [PubMed]
- Atzrodt, J.; Derdau, V.; Kerr, W.J.; Reid, M.; Rojahn, P.; Weck, R. Expanded applicability of iridium(I) NHC/phosphine catalysts in hydrogen isotope exchange processes with pharmaceutically-relevant heterocycles. Tetrahedron 2015, 71, 1924–1929. [Google Scholar] [CrossRef]
- Kerr, W.J.; Reid, M.; Tuttle, T. Iridium-Catalyzed C–H Activation and Deuteration of Primary Sulfonamides: An Experimental and Computational Study. ACS Catal. 2015, 5, 402–410. [Google Scholar] [CrossRef]
- Cochrane, A.R.; Irvine, S.; Kerr, W.J.; Reid, M.; Andersson, S.; Nilsson, G.N. Application of Neutral Iridium(I) N-Heterocyclic Carbene Complexes in ortho-Directed Hydrogen Isotope Exchange. J. Label. Compd. Radiopharm. 2013, 56, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.J.; Mudd, R.J.; Paterson, L.C.; Brown, J.A. Iridium(I)-Catalyzed Regioselective C–H Activation and Hydrogen-Isotope Exchange of Non-aromatic Unsaturated Functionality. Chem. Eur. J. 2014, 20, 14604–14607. [Google Scholar] [CrossRef] [PubMed]
- Cross, P.W.C.; Ellames, G.J.; Gibson, J.S.; Herbert, J.M.; Kerr, W.J.; McNeill, A.H.; Mathers, T.W. Conditions for deuterium exchange mediated by iridium complexes formed in situ. Tetrahedron 2003, 59, 3349–3358. [Google Scholar] [CrossRef]
- Ellames, G.; Gibson, J.; Herbert, J.; McNeill, A. The scope and limitations of deuteration mediated by Crabtree’s catalyst. Tetrahedron 2001, 57, 9487–9497. [Google Scholar] [CrossRef]
- Heys, J.R.; Shu, A.Y.L.; Senderoff, S.G.; Phillips, N.M. Deuterium exchange labelling of substituted aromatics using [IrH2(Me2CO)2(PPh3)2BF4. J. Label. Compd. Radiopharm. 1993, 33, 431–438. [Google Scholar] [CrossRef]
- Shu, A.; Chen, W.; Heys, J. Organoiridium catalyzed hydrogen isotope exchange: ligand effects on catalyst activity and regioselectivity. J. Organomet. Chem. 1996, 524, 87–93. [Google Scholar] [CrossRef]
- Piola, L.; Fernández-Salas, J.A.; Manzini, S.; Nolan, S.P. Regioselective ruthenium catalysed H–D exchange using D2O as the deuterium source. Org. Biomol. Chem. 2014, 12, 8683–8688. [Google Scholar] [CrossRef] [PubMed]
- Hesk, D.; Das, P.R.; Evans, B. Deuteration of Acetanilides and Other Substituted Aromatics Using [Ir(COD)(Cy3P)Py)PF6 as Catalyst. J. Label. Compd. Radiopharm. 1995, 36, 497–502. [Google Scholar] [CrossRef]
- Brown, J.A.; Cochrane, A.R.; Irvine, S.; Kerr, W.J.; Mondal, B.; Parkinson, J.A.; Paterson, L.C.; Reid, M.; Tuttle, T.; Andersson, S.; et al. The Synthesis of Highly Active Iridium(I) Complexes and their Application in Catalytic Hydrogen Isotope Exchange. Adv. Synth. Catal. 2014, 356, 3551–3562. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Seeman, J.I. Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin-Hammett Winstein-Holness kinetics. Chem. Rev. 1983, 83, 83–134. [Google Scholar] [CrossRef]
- Shang, R.; Fu, Y.; Li, J.B.; Zhang, S.L.; Guo, Q.X.; Liu, L. Synthesis of Aromatic Esters via Pd-Catalyzed Decarboxylative Coupling of Potassium Oxalate Monoesters with Aryl Bromides and Chlorides. J. Am. Chem. Soc. 2009, 131, 5738–5379. [Google Scholar]
- Moller, H.; Thimm, H.J. Cosmetic Preparations with Alkoxybenzoic Acid Esters as Inflammation Inhibitors and Method. U.S. Patent 4,136,165; filed 22 April 1977, and issued 23 January 1979,
- Mori, N.; Togo, H. Facile oxidative conversion of alcohols to esters usingmolecular iodine. Tetrahedron 2005, 61, 5915–5925. [Google Scholar] [CrossRef]
- Jackson, R.W. A mild and selective method for the cleavage of tert-butyl esters. Tetrahedron Lett. 2001, 42, 5163–5165. [Google Scholar] [CrossRef]
- Kuh, L.P. Ionization of Organic Esters in Sulfuric Acid. II. Alkyl Oxygen Fission. J. Am. Chem. Soc. 1949, 71, 1575–1577. [Google Scholar]
- Rivero-Cruz, B.; Rivero-Cruz, I.; Rodríguez-Sotres, R.; Mata, R. Effect of natural and synthetic benzyl benzoates on calmodulin. Phytochemistry 2007, 68, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Narayanan, V.L.; Risbood, P.A.; Hollingshead, M.G.; Stinson, S.F.; Varma, R.K.; Sausville, E.A. Synthesis, structure–activity relationship, and p210bcr−abl protein tyrosine kinase activity of novel AG 957 analogs. Bioorg. Med. Chem. 2005, 13, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Andrae, D.; Häußerman, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devlin, J.; Kerr, W.J.; Lindsay, D.M.; McCabe, T.J.D.; Reid, M.; Tuttle, T. Iridium-Catalysed ortho-Directed Deuterium Labelling of Aromatic Esters—An Experimental and Theoretical Study on Directing Group Chemoselectivity. Molecules 2015, 20, 11676-11698. https://doi.org/10.3390/molecules200711676
Devlin J, Kerr WJ, Lindsay DM, McCabe TJD, Reid M, Tuttle T. Iridium-Catalysed ortho-Directed Deuterium Labelling of Aromatic Esters—An Experimental and Theoretical Study on Directing Group Chemoselectivity. Molecules. 2015; 20(7):11676-11698. https://doi.org/10.3390/molecules200711676
Chicago/Turabian StyleDevlin, Jennifer, William. J. Kerr, David M. Lindsay, Timothy J. D. McCabe, Marc Reid, and Tell Tuttle. 2015. "Iridium-Catalysed ortho-Directed Deuterium Labelling of Aromatic Esters—An Experimental and Theoretical Study on Directing Group Chemoselectivity" Molecules 20, no. 7: 11676-11698. https://doi.org/10.3390/molecules200711676