
Practical Radiosynthesis and Preclinical Neuroimaging of [11C]isradipine, a Calcium Channel Antagonist

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
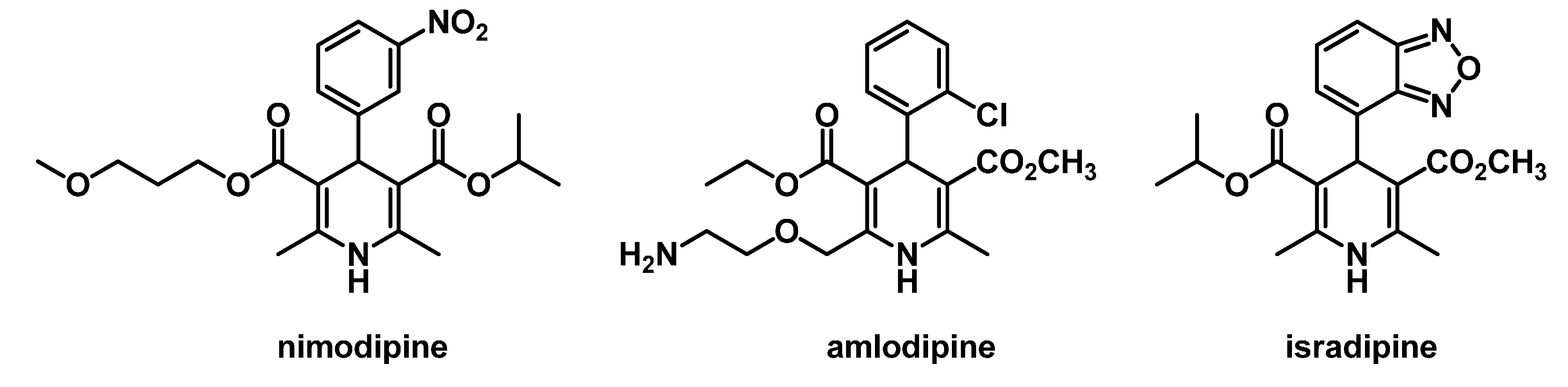
:1. Introduction
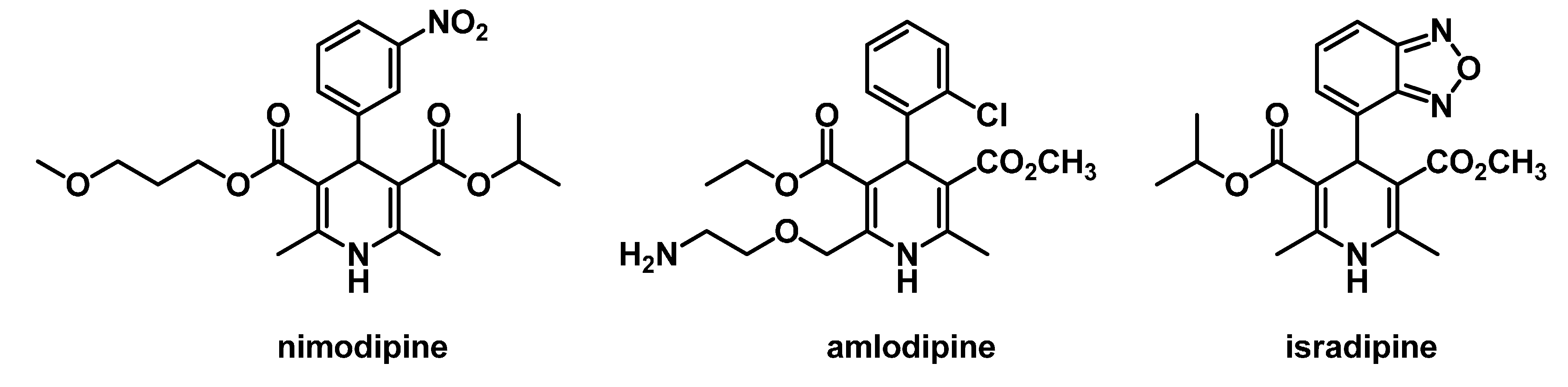
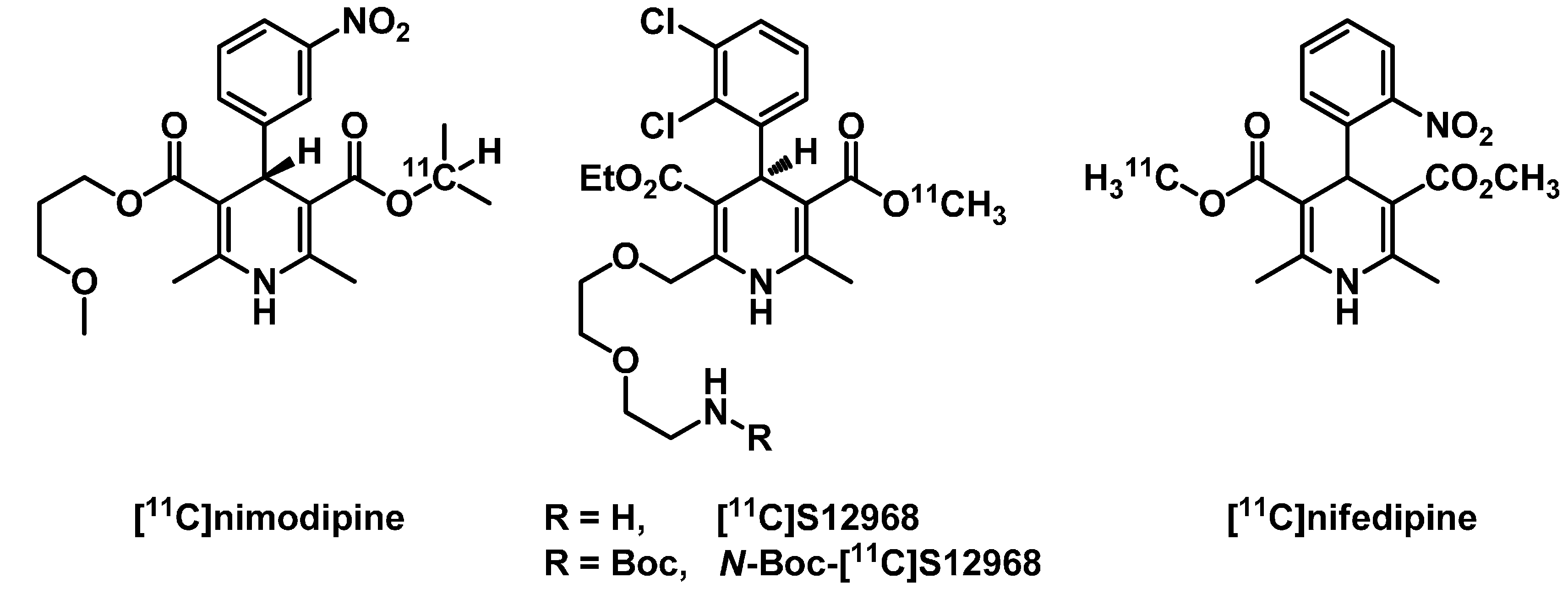
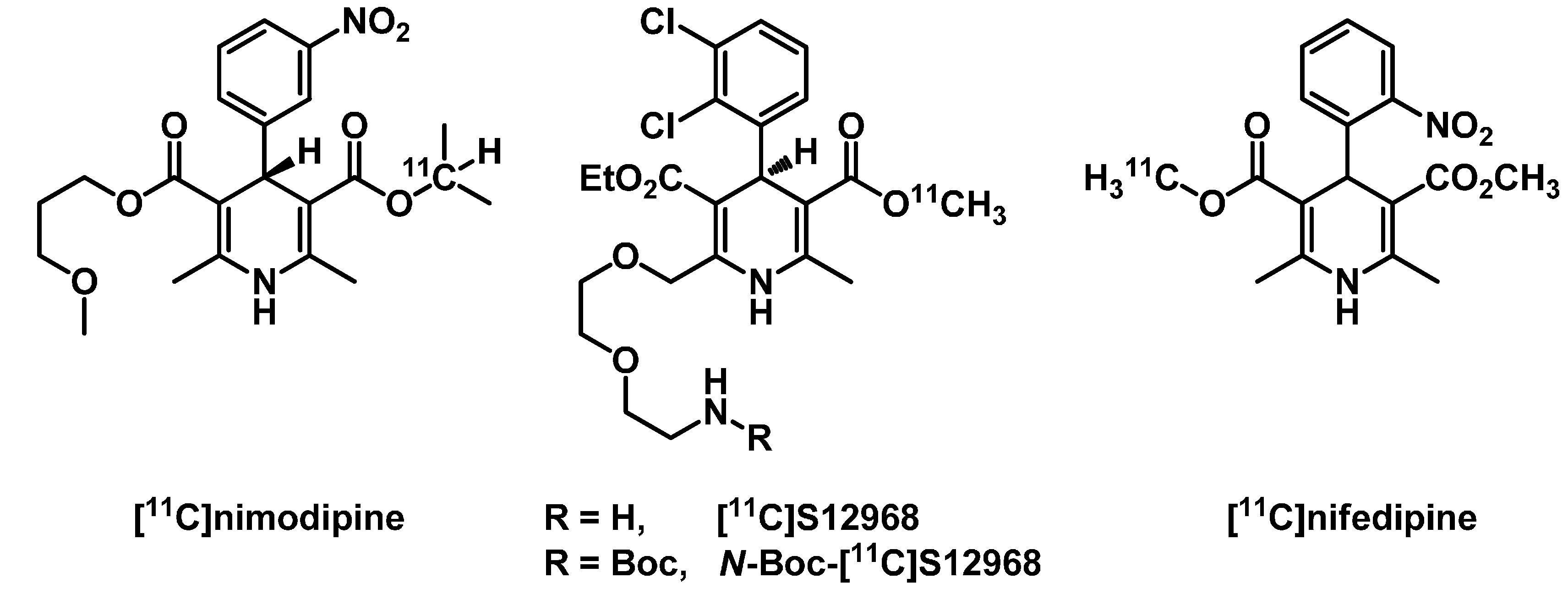
2. Results and Discussion
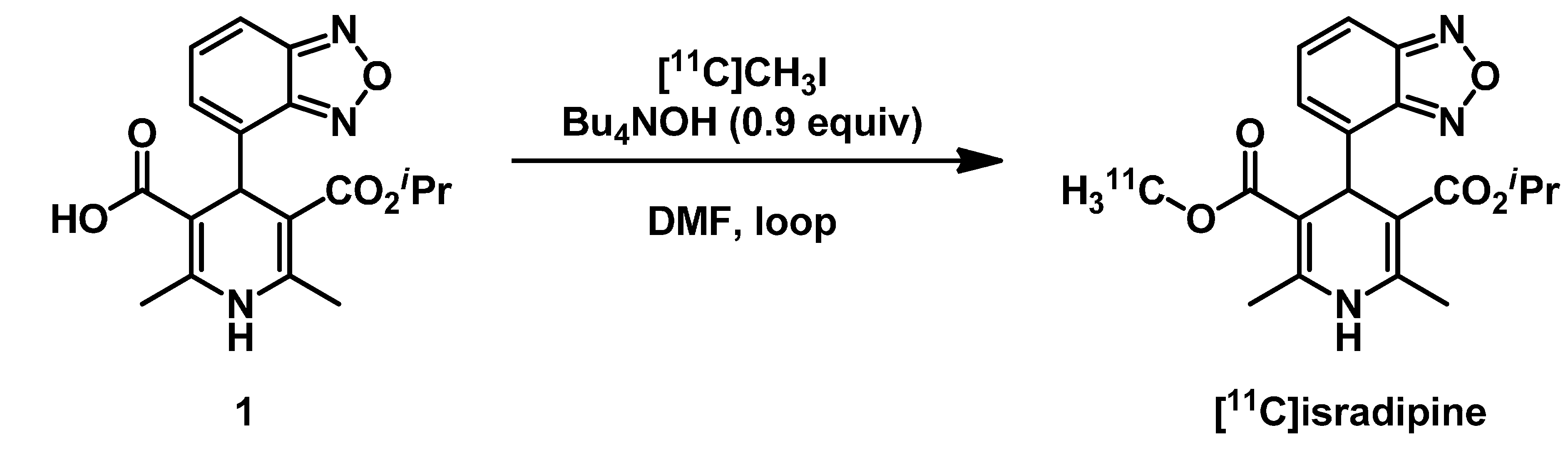
2.1. Radiosynthesis of [11C]Isradipine

2.2. Partition Coefficient of [11C]isradipine
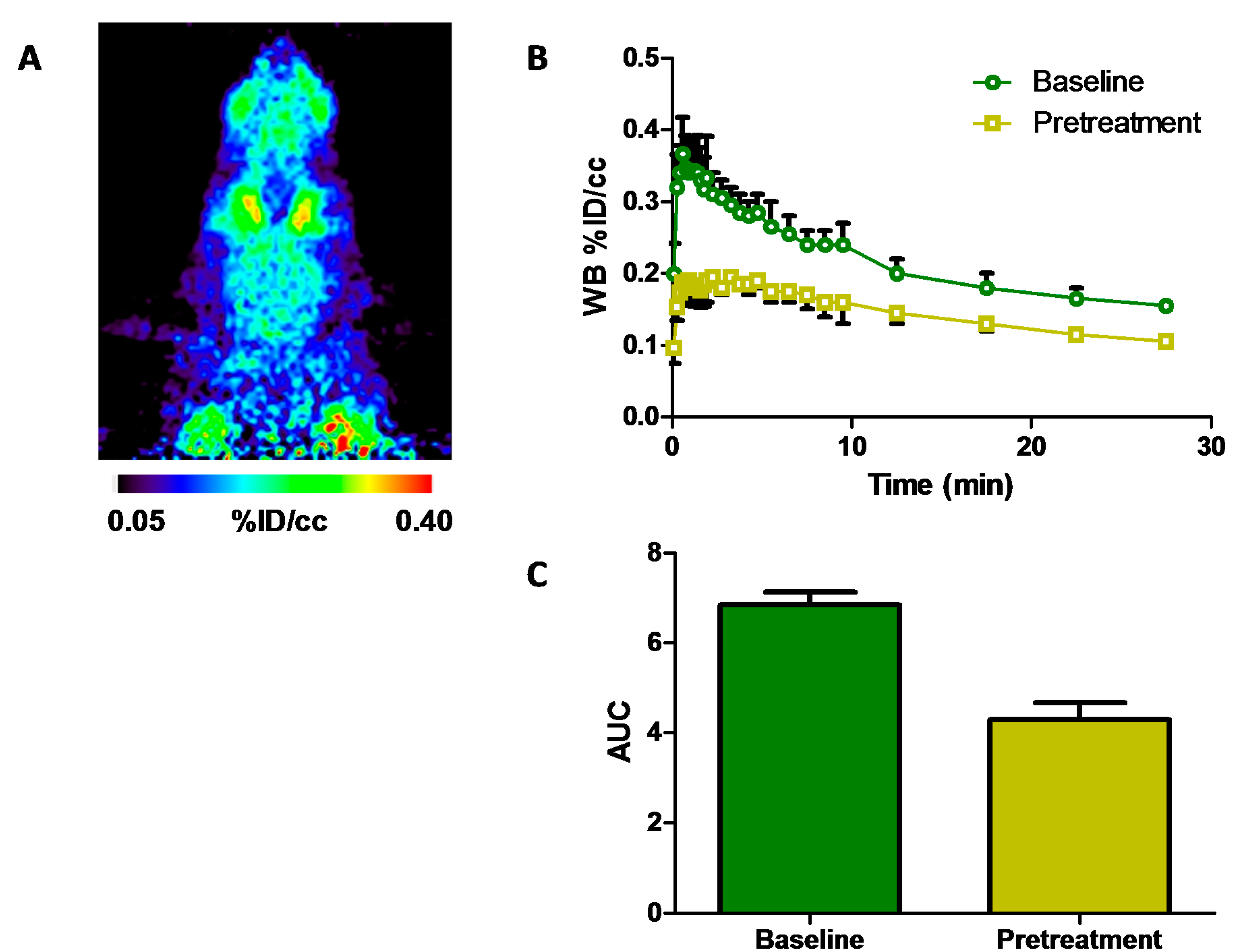
2.3. Positron Emission Tomography Neuroimaging of [11C]Isradipine
 ) and after pretreatment (
) and after pretreatment (  ) with isradipine (2 mg·kg−1 i.p., 30 min prior to time-of-injection (TOI)); (C) area-under-the-curve analysis of whole brain uptake 0–5 min after tracer injection at baseline and after pretreatment, paired t-test, p < 0.05; WB, whole brain; % ID/cc, percent of injected dose per cubic centimeter; AUC, area-under-the-curve.
) and after pretreatment ( ) with isradipine (2 mg·kg−1 i.p., 30 min prior to time-of-injection (TOI)); (C) area-under-the-curve analysis of whole brain uptake 0–5 min after tracer injection at baseline and after pretreatment, paired t-test, p < 0.05; WB, whole brain; % ID/cc, percent of injected dose per cubic centimeter; AUC, area-under-the-curve.
) with isradipine (2 mg·kg−1 i.p., 30 min prior to time-of-injection (TOI)); (C) area-under-the-curve analysis of whole brain uptake 0–5 min after tracer injection at baseline and after pretreatment, paired t-test, p < 0.05; WB, whole brain; % ID/cc, percent of injected dose per cubic centimeter; AUC, area-under-the-curve.
) and after pretreatment ( ) with isradipine (2 mg·kg−1 i.p., 30 min prior to time-of-injection (TOI)); (C) area-under-the-curve analysis of whole brain uptake 0–5 min after tracer injection at baseline and after pretreatment, paired t-test, p < 0.05; WB, whole brain; % ID/cc, percent of injected dose per cubic centimeter; AUC, area-under-the-curve.
3. Experimental Section
3.1. Radiosynthesis of [11C]Isradipine
3.2. PET Imaging
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar]
- Striessnig, J.; Koschak, A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene-knockout models. Channels 2008, 2, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.R.; O’Donovan, M.C.; Meng, Y.A.; Jones, I.R.; Ruderfer, D.M.; Jones, L.; Fan, J.; Kirov, G.; Perlis, R.H.; Green, E.K.; et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008, 40, 1056–1058. [Google Scholar]
- Sklar, P.; Ripke, S.; Scott, L.J.; Andreassen, O.A.; Cichon, S.; Craddock, N.; Mahon, P.B. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 2011, 43, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Triggle, D.J.; Hawthorn, M.; Gopalakrishnan, M.; Minarini, A.; Avery, S.; Rutledge, A.; Bangalore, R.; Zheng, W. Synthetic Organic Ligands active at Voltage-Gated Calcium Channels. Ann. N. Y. Acad. Sci. 1991, 635, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Deyo, R.A.; Straube, K.T.; Disterhoft, J.F. Nimodipine facilitates associative learning in aging rabbits. Science 1989, 243, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Scriabine, A.; Schuurman, T.; Traber, J. Pharmacological basis for the use of nimodipine in central nervous system disorders. FASEB J. 1989, 3, 1799–1806. [Google Scholar] [PubMed]
- Pucilowski, O.; Plaźnik, A.; Overstreet, D.H. Isradipine Suppresses Amphetamine-Induced Conditioned Place Preference and Locomotor Stimulation in the Rat. Neuropsychopharmacology 1995, 12, 239–244. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. “Rejuvenation” protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Link, M.C.; Wiemann, M.; Bingmann, D. Organic and inorganic calcium antagonists inhibit veratridine-induced epileptiform activity in CA3 neurons of the guinea pig. Epilepsy Res. 2008, 78, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Casamassima, F.; Huang, J.; Fava, M.; Sachs, G.S.; Smoller, J.W.; Cassano, G.B.; Lattanzi, L.; Fagerness, J.; Stange, J.P.; Perlis, R.H. Phenotypic effects of a bipolar liability gene among individuals with major depressive disorder. Am. J. Med. Genet. 2010, 153B, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Casamassima, F.; Hay, A.C.; Benedetti, A.; Lattanzi, L.; Cassano, G.B.; Perlis, R.H. L-type calcium channels and psychiatric disorders: A brief review. Am. J. Med. Genet. 2010, 153B, 1373–1390. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Cooper, G.; Dunne, S.F.; Dusel, B.; Luan, C.H.; Surmeier, D.J.; Silverman, R.B. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat. Commun. 2012, 3, 1146. [Google Scholar] [CrossRef]
- Pourbadie, H.G.; Naderi, N.; Mehranfard, N.; Janahmadi, M.; Khodagholi, F.; Motamedi, F. Preventing Effect of L-Type Calcium Channel Blockade on Electrophysiological Alterations in Dentate Gyrus Granule Cells Induced by Entorhinal Amyloid Pathology. PLoS ONE 2015, 10, e0117555. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, E.; Poetschke, C.; Duda, J.; Schlaudraff, F.; Lammel, S.; Schiemann, J.; Fauler, M.; Hetzel, A.; Watanabe, M.; Lujan, R.; et al. CaV1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain 2014, 137, 2287–2302. [Google Scholar]
- Triggle, D.J. 1,4-Dihydropyridines as calcium channel ligands and privileged structures. Cell. Mol. Neurobiol. 2003, 23, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson’s disease (STEADY-PD). Mov. Disord. 2013, 28, 1823–1831. [Google Scholar]
- Ostacher, M.J.; Iosifescu, D.V.; Hay, A.; Blumenthal, S.R.; Sklar, P.; Perlis, R.H. Pilot investigation of isradipine in the treatment of bipolar depression motivated by genome-wide association. Bipolar Disord. 2014, 16, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.D.; Halldin, C. PET radioligands targeting the brain GABAA/benzodiazepine receptor complex. J. Label. Compd. Radiopharm. 2013, 56, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.X.; Yin, Y.F.; Li, Y.M. Neural nAChRs PET imaging probes. Nucl. Med. Commun. 2014, 35, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Horti, A.G.; Kuwabara, H.; Holt, D.P.; Dannals, R.F.; Wong, D.F. Recent PET radioligands with optimal brain kinetics for imaging nicotinic acetylcholine receptors. J. Label. Compd. Radiopharm. 2013, 56, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Sobrio, F. Radiosynthesis of carbon-11 and fluorine-18 labelled radiotracers to image the ionotropic and metabotropic glutamate receptors: 11C and 18 F chemistry to image the glutamate receptors. J. Label. Compd. Radiopharm. 2013, 56, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Dollé, F.; Valette, H.; Hinnen, F.; Fuseau, C.; Péglion, J.L.; Crouzel, C. Synthesis and Characterization of a 11C-Labelled Derivative of S12968: An Attempt to Image in Vivo Brain Calcium Channels. Nucl. Med. Biol. 1998, 25, 339–342. [Google Scholar] [CrossRef]
- Wilson, A.A.; Dannals, R.F.; Ravert, H.T.; Burns, H.D.; Lever, S.Z.; Wagner, H.N. Radiosynthesis of [11C]nifedipine and [11C]nicardipine. J. Label. Compd. Radiopharm. 1989, 27, 589–598. [Google Scholar] [CrossRef]
- Stone-Elander, S.; Roland, P.; Schwenner, E.; Halldin, C.; Widén, L. Synthesis of [isopropyl-11C]nimodipine for in vivo studies of dihydropyridine binding in man using positron emission tomography. Int. J. Radiat. Appl. Instrum. Part A 1991, 42, 871–875. [Google Scholar] [CrossRef]
- Holschbach, M.; Roden, W.; Hamkens, W. Synthesis of carbon-11 labelled calcium channel antagonists. J. Label. Compd. Radiopharm. 1991, 29, 431–442. [Google Scholar] [CrossRef]
- Pleiss, U. 1,4-Dihydropyridines (DHPs)-a class of very potent drugs: Syntheses of isotopically labeled DHP derivatives during the last four decades. J. Label. Compd. Radiopharm. 2007, 50, 818–830. [Google Scholar] [CrossRef]
- Sadeghpour, H.; Jalilian, A.R.; Shafiee, A.; Akhlaghi, M.; Miri, R.; Mirzaei, M. Radiosynthesis of dimethyl-2-[18F]-(fluoromethyl)-6-methyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate for L-type calcium channel imaging. Radiochim. Acta 2008, 96, 849–854. [Google Scholar] [CrossRef]
- Valette, H.; Crouzel, C.; Syrota, A.; Fuseau, C.; Bourachot, M.L. Canine myocardial dihydropyridine binding sites: A positron emission tomographic study with the calcium channel inhibitor 11C-S11568. Life Sci. 1994, 55, 1471–1477. [Google Scholar] [CrossRef]
- Dollé, F.; Hinnen, F.; Valette, H.; Fuseau, C.; Duval, R.; Péglion, J.L.; Crouzel, C. Synthesis of two optically active calcium channel antagonists labelled with carbon-11 for in vivo cardiac PET imaging. Bioorg. Med. Chem. 1997, 5, 749–764. [Google Scholar] [CrossRef]
- Dollé, F.; Valette, H.; Hinnen, F.; Demphel, S.; Bramoulle, Y.; Peglion, J.L.; Crouzel, C. Highly efficient synthesis of [11C]S12968 and [11C]S12967, for the in vivo imaging of the cardiac calcium channels using PET. J. Label. Compd. Radiopharm. 2001, 44, 481–499. [Google Scholar] [CrossRef]
- Valette, H.; Dollé, F.; Guenther, I.; Hinnen, F.; Fuseau, C.; Coulon, C.; Péglion, J.L.; Crouzel, C. Myocardial Kinetics of the 11C-Labeled Enantiomers of the Ca2+ Channel Inhibitor S11568: An in Vivo Study. J. Nucl. Med. 2001, 42, 932–937. [Google Scholar] [PubMed]
- Valette, H.; Dollé, F.; Guenther, I.; Fuseau, C.; Coulon, C.; Hinnen, F.; Péglion, J.L.; Crouzel, C. In vivo quantification of myocardial dihydropyridine binding sites: A PET study in dogs. J. Nucl. Med. 2002, 43, 1227–1233. [Google Scholar] [PubMed]
- Maan, A.C.; Ptasienski, J.; Hosey, M.M. Influence of Mg++ on the effect of diltiazem to increase dihydropyridine binding to receptors on Ca++-channels in chick cardiac and skeletal muscle membranes. J. Pharmacol. Exp. Ther. 1986, 239, 768–774. [Google Scholar] [PubMed]
- Uchida, S.; Yamada, S.; Nagai, K.; Deguchi, Y.; Kimura, R. Brain pharmacokinetics and in vivo receptor binding of 1,4-dihydropyridine calcium channel antagonists. Life Sci. 1997, 61, 2083–2090. [Google Scholar] [CrossRef]
- Crouzel, C.; Syrota, A. The use of [11C]diazomethane for labelling a calcium channel antagonist: PN 200-110 (isradipine). Int. J. Radiat. Appl. Instrum. Part A 1990, 41, 241–242. [Google Scholar] [CrossRef]
- Wilson, A.A.; Garcia, A.; Jin, L.; Houle, S. Radiotracer synthesis from [11C]-iodomethane: A remarkably simple captive solvent method. Nucl. Med. Biol. 2000, 27, 529–532. [Google Scholar] [CrossRef]
- Wilson, A.A.; Garcia, A.; Houle, S.; Vasdev, N. Utility of commercial radiosynthetic modules in captive solvent [11C]-methylation reactions. J. Label. Compd. Radiopharm. 2009, 52, 490–492. [Google Scholar] [CrossRef]
- Waterhouse, R. Determination of lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Herbette, L.G.; Vant Erve, Y.M.; Rhodes, D.G. Interaction of 1,4 dihydropyridine calcium channel antagonists with biological membranes: Lipid bilayer partitioning could occur before drug binding to receptors. J. Mol. Cell. Cardiol. 1989, 21, 187–201. [Google Scholar] [CrossRef]
- Wilson, A.A.; Jin, L.; Garcia, A.; DaSilva, J.N.; Houle, S. An admonition when measuring the lipophilicity of radiotracers using counting techniques. Appl. Radiat. Isot. 2001, 54, 203–208. [Google Scholar] [CrossRef]
- Samples Availability: Samples of the compound 1 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotstein, B.H.; Liang, S.H.; Belov, V.V.; Livni, E.; Levine, D.B.; Bonab, A.A.; Papisov, M.I.; Perlis, R.H.; Vasdev, N. Practical Radiosynthesis and Preclinical Neuroimaging of [11C]isradipine, a Calcium Channel Antagonist. Molecules 2015, 20, 9550-9559. https://doi.org/10.3390/molecules20069550
Rotstein BH, Liang SH, Belov VV, Livni E, Levine DB, Bonab AA, Papisov MI, Perlis RH, Vasdev N. Practical Radiosynthesis and Preclinical Neuroimaging of [11C]isradipine, a Calcium Channel Antagonist. Molecules. 2015; 20(6):9550-9559. https://doi.org/10.3390/molecules20069550
Chicago/Turabian StyleRotstein, Benjamin H., Steven H. Liang, Vasily V. Belov, Eli Livni, Dylan B. Levine, Ali A. Bonab, Mikhail I. Papisov, Roy H. Perlis, and Neil Vasdev. 2015. "Practical Radiosynthesis and Preclinical Neuroimaging of [11C]isradipine, a Calcium Channel Antagonist" Molecules 20, no. 6: 9550-9559. https://doi.org/10.3390/molecules20069550