1. Introduction
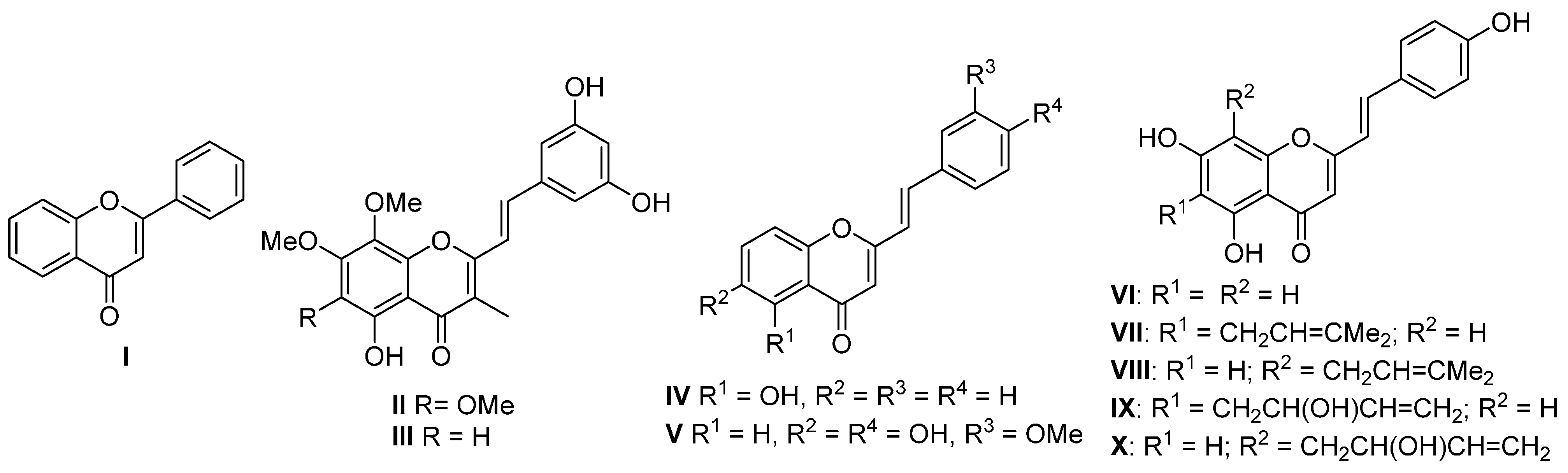
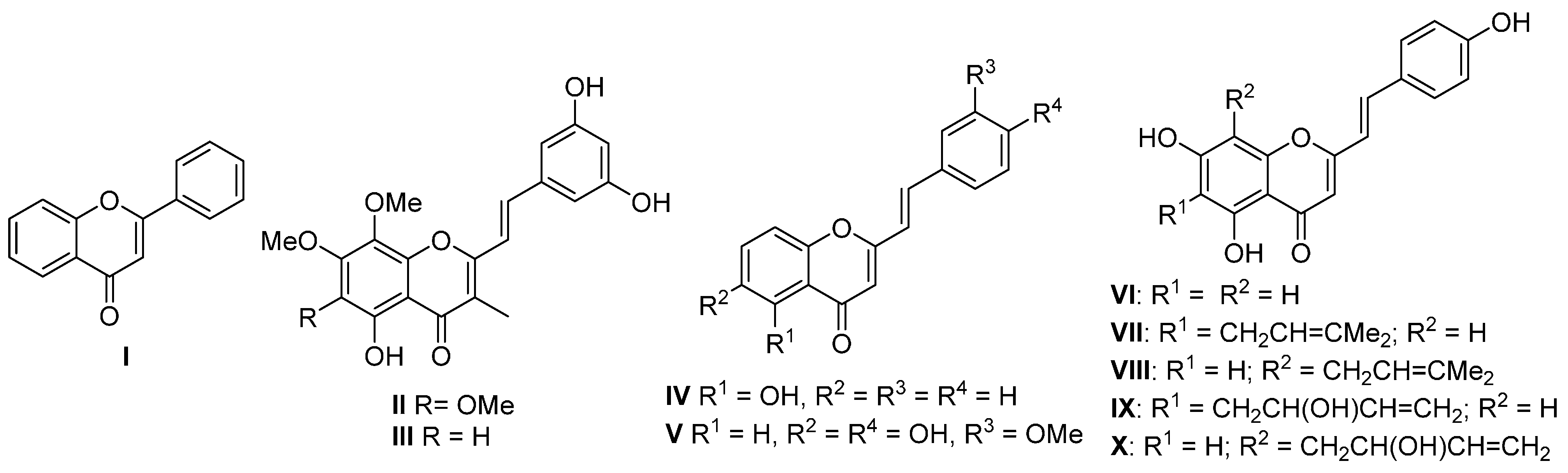
Flavones (
I), the most prominent group of naturally occurring chromones, are present in a wide variety of plants [
1] and are well-known by their broad range of biological properties, such as antibacterial, antifungal [
2,
3], antiviral [
4], antiinflammatory [
5], antioxidant [
6], antiallergic [
7], hepatoprotective [
8], antithrombotic and antitumoral [
9,
10] activities (
Figure 1). In contrast to flavones, there are only nine natural (
E)-2-styrylchromones
II–
X [
11,
12,
13,
14,
15], a group of oxygen heterocyclic compounds that although being scarce in Nature have shown significant biological activities [
16,
17,
18] (
Figure 1). 2-Styrylchromones have potential therapeutic applications in the treatment of cancer [
19], allergies [
20], viral infections [
21], gout [
22] and oxidative stress related damage [
23]. These compounds have demonstrated strong protective effects against pro-oxidant agents observed in cellular [
23] and in non-cellular systems [
24], making them good antioxidant compound candidates.
Figure 1.
Structures of flavone I and naturally occurring (E)-2-styrylchromones II–X.
Figure 1.
Structures of flavone I and naturally occurring (E)-2-styrylchromones II–X.
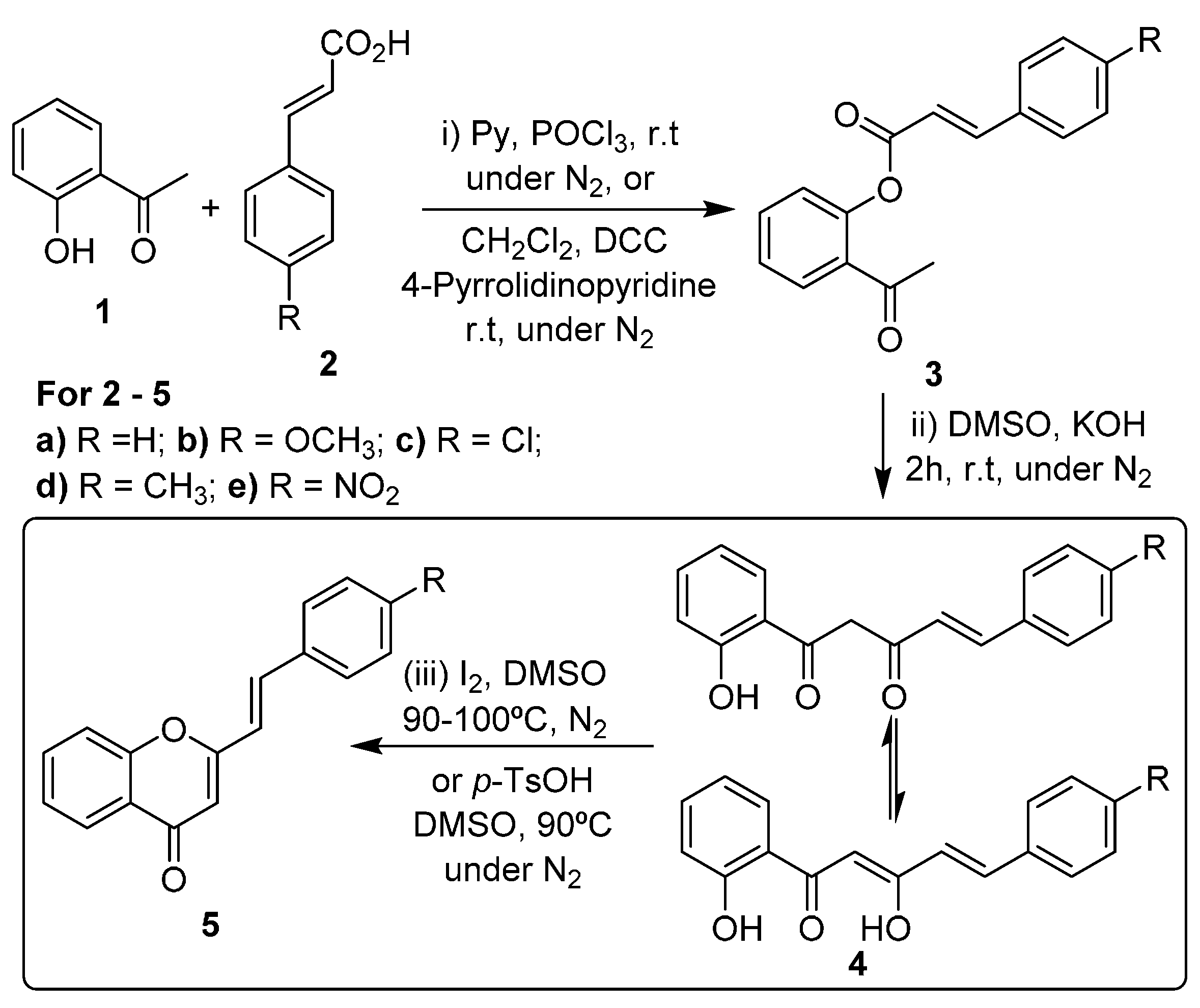
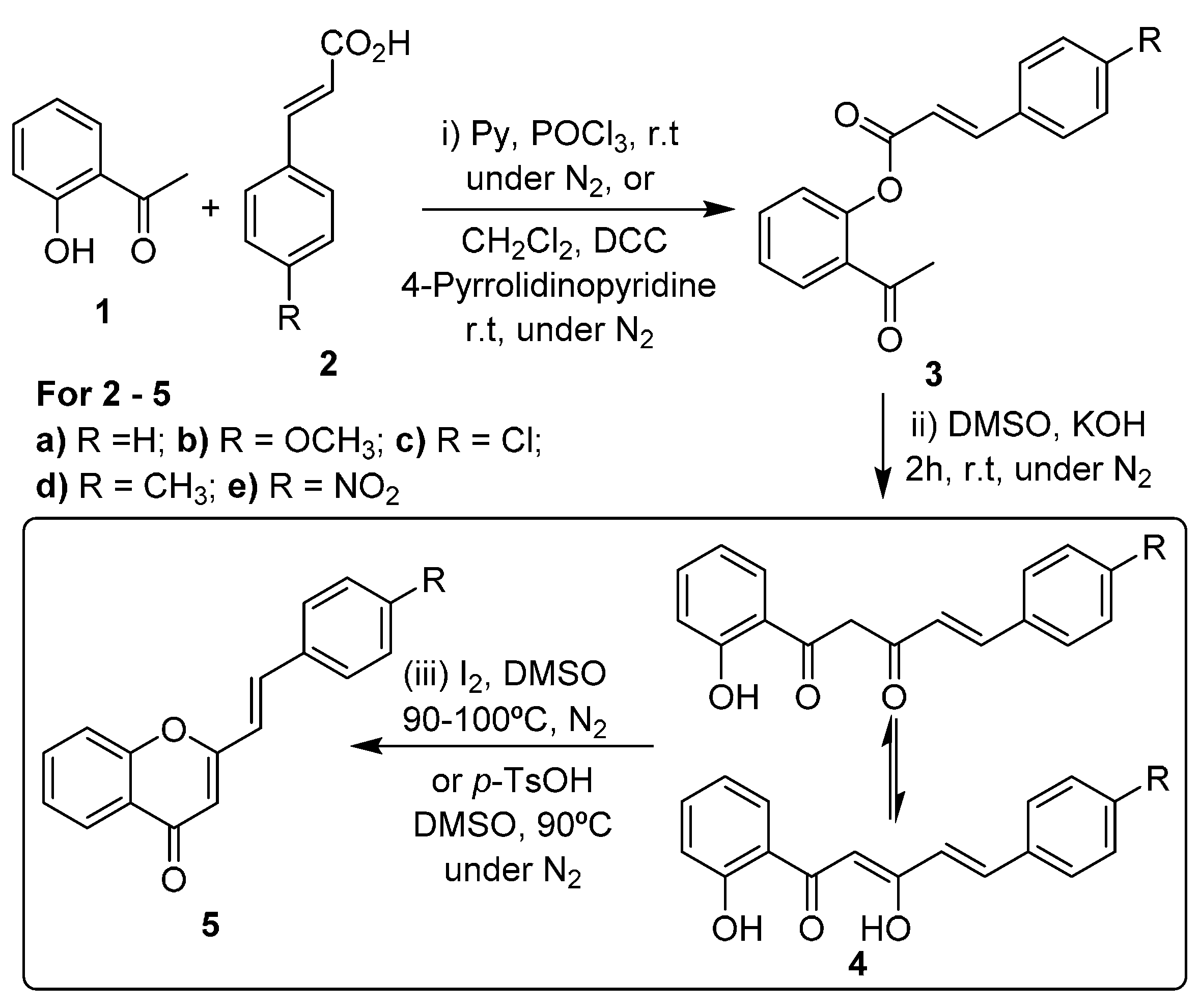
The most common strategies for the synthesis of (
E)-2-styrylchromones are the aldol condensation of 2′-hydroxyacetophenone derivatives with cinnamaldehydes, followed by cyclodehydrogenation of the formed 2′-hydroxycinnamylideneacetophenones, and the Baker-Venkataraman method [
16,
17,
18,
25]. This second strategy involves the
O-acylation of 2′-hydroxyacetophenone (
1,
Scheme 1, step i) followed by base-catalyzed rearrangement of the formed esters
3 to give 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones
4 (which exist in equilibrium with their enolic form;
Scheme 1, step ii). This rearrangement can be performed in high yield under solvent-free conditions, using grinding techniques [
26]. The last step of this method consists in the cyclodehydration of compounds
4 into (
E)-2-styrylchromones
5, using strong acidic conditions,
p-toluenesulfonic acid or a catalytic amount of iodine in DMSO at 90–100 °C (
Scheme 1, step iii) [
16,
17,
18,
25].
A similar procedure is also well-established for the synthesis of flavones. Also in this case the cyclodehydration reaction of 1-(2-hydroxyphenyl)propane-1,3-diones (which exist also in equilibrium with their enolic forms) is an important step in the synthesis of flavones using the Baker-Venkataraman approach (the same sequence presented in
Scheme 1, but using benzoic acids instead of cinnamic acids) [
27,
28,
29]. This process usually is a catalytic transformation performed in different media. Some reaction conditions employed an excess of sulfuric acid in glacial acetic acid [
30], cationic exchange resins in isopropyl alcohol [
31], CuCl
2 in ethanol [
32], ionic liquids under microwave irradiation [
33], heteropolyacids, carbon supported triflic acid [
34,
35,
36,
37] and also grinding techniques in the presence of phosphorus pentoxide [
38]. Some of these methods require high temperatures or long times to complete the reactions [
30,
31,
37], other require the preparation of the catalysts, involving non-green and time consuming procedures [
34,
35,
36,
37], and almost all of them were used to prepare flavones or simple chromones, but were not used in the synthesis of 2-styrylchromones [
30,
31,
32,
33,
34,
35,
36,
37]. Pawar and co-workers [
33] described the conversion of 1-(2-hydroxyphenyl)-3-phenylpropane-1,3-diones to the corresponding flavones under microwave irradiation using the ionic liquid [EtNH
3]NO
3·(EAN). Despite the structural differences, EAN shares many properties with water [
39]. However, while water is a safe, easily available and relatively low cost and environmentally friendly solvent, the ionic liquid has to be prepared. EAN is usually synthesized by heating ethyl nitrate with an alcoholic solution of ammonia or by reacting ethylamine with concentrated nitric acid [
40] which cannot be considered green processes. In addition organic nitrates are potentially explosive, especially when rigorously dried. The recovery of the ionic liquid requires evaporation of the aqueous layer at 80 °C that represents additional time and energy consumption.
Scheme 1.
Synthesis of (E)-2-styrylchromones by the Baker-Venkataraman method.
Scheme 1.
Synthesis of (E)-2-styrylchromones by the Baker-Venkataraman method.
The synthesis of flavones using trifluoromethanesulfonic acid supported on carbon as catalyst [
37] was performed using toluene as solvent and requires 5 h for reaction completion. Makrandi and co-workers [
38] described a green synthesis of flavones and 2-styrylchromones via cyclodehydration of the corresponding 1-(2-hydroxyaryl)-3-(aryl/styryl)propane-1,3-diones under solvent-free conditions in the presence of phosphorus pentoxide using grinding techniques. However the scope of the reaction was poorly checked since only substrates containing electron-donating substituents were used.
A different and eco-friendly methodology for the direct synthesis of flavones, from phloroglucinol and β-ketoesters, was reported by Seijas and co-workers [
41]. The reaction involves the cycloaddition of an α-oxo ketene intermediate followed by an uncatalyzed thermal Fries rearrangement. The flavones were obtained in very good yields (68%–96%) after 3 min of microwave irradiation using solvent-free conditions, but the methodology was not extended to the synthesis of 2-styrylchromones. Due to the formation of polar transition states, the reaction benefits from microwave activation, however 800 W output power and 240 °C are required to achieve short reaction times and high reaction yields. Furthermore, when the melting point of the reactants is higher a longer reaction time is required.
Our interest in organic reactions using exclusively water as solvent prompted us to investigate the aqueous cyclodehydration of appropriate β-diketones to prepare (
E)-2-styrylchromones and flavones. The use of water in organic synthesis, without the presence of any organic solvent, can be beneficial because water is an available, cheap, safe and environmentally benign solvent. So far, extensive work revealed that a variety of organic reactions including dehydration reactions can be performed using water as solvent [
42,
43,
44]. Here we present a protocol for the base-catalyzed cyclodehydration reaction of compounds
4 and
6 into the corresponding (
E)-2-styrylchromones
5 and flavones
7 in water, in the absence of any organic solvent, and using potassium carbonate as base (
Scheme 1 and
Scheme 2). The desired compounds
5 and
7 were obtained in yields over 60%, except for compound
7b. This method represents a cheap, easy and suitable protocol for the cyclodehydration of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones
4 and 3-aryl-1-(2-hydroxyphenyl)propane-1,3-diones
6 (which exist in equilibrium with their enolic forms) which contributes to the establishment of environmentally friendly organic synthesis.
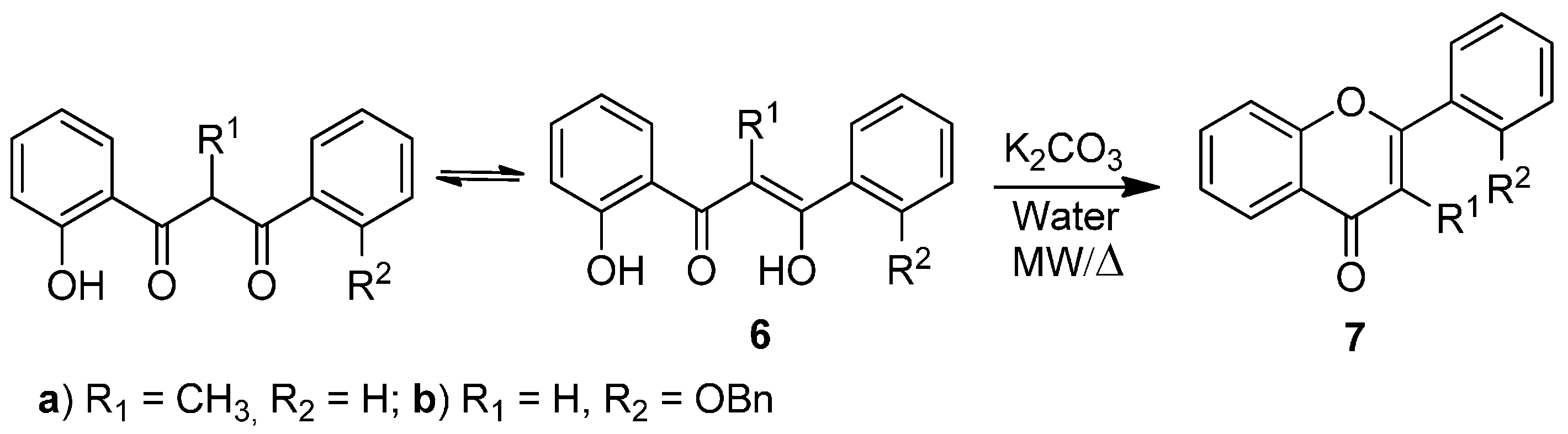
Scheme 2.
Cyclodehydration reaction of 3-aryl-1-(2-hydroxyphenyl)propane-1,3-diones 6a,b to flavones 7a,b.
Scheme 2.
Cyclodehydration reaction of 3-aryl-1-(2-hydroxyphenyl)propane-1,3-diones 6a,b to flavones 7a,b.
2. Results and Discussion
The typical established methods for the cyclodehydration reaction of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones
4 involve the use of strong acidic conditions,
p-toluenesulfonic acid, or a catalytic amount of iodine in DMSO at 90–100 °C [
16,
17,
18,
25]. In order to establish a more environmental friendly procedure for this cyclodehydration reaction commonly used in our research group, we performed the base-catalyzed (potassium carbonate) cyclodehydration reaction of
4a in water. To the best of our knowledge, the cyclodehydration reaction of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones
4 in aqueous basic conditions has not been previously reported. Firstly, we performed the reactions in a small scale (50–200 mg), in order to establish the optimal reaction conditions and to fully characterize the obtained compounds.
Our results indicate that 0.5 molar equiv of base are enough to perform the expected cyclodehydration reaction under classical reflux heating (
Table 1, Entries 1 and 2). Under these conditions (
E)-2-styrylchromone
5a was obtained in 59% yield. In order to increase the yield, and since the used β-diketone is not soluble in water, we tried the addition of a phase transfer catalyst (PTC) (tetrabutylammonium bromide, TBAB) to improve the solubility of
4a. However in the presence of this catalyst the yield was only 12% (
Table 1, Entry 3). We also tried to replace potassium carbonate by another base, tetramethylammonium hydroxide (TMAOH) as a bifunctional catalyst, acting as base and as PTC (
Table 1, Entries 4 and 5). Since the product was obtained in low yields, we concluded that the reaction works better in the first conditions, using potassium carbonate as base. Under these conditions (
E)-2-styrylchromones
5b–
e were obtained as main compounds in 61%, 70%, 70% and 20% yields, respectively (
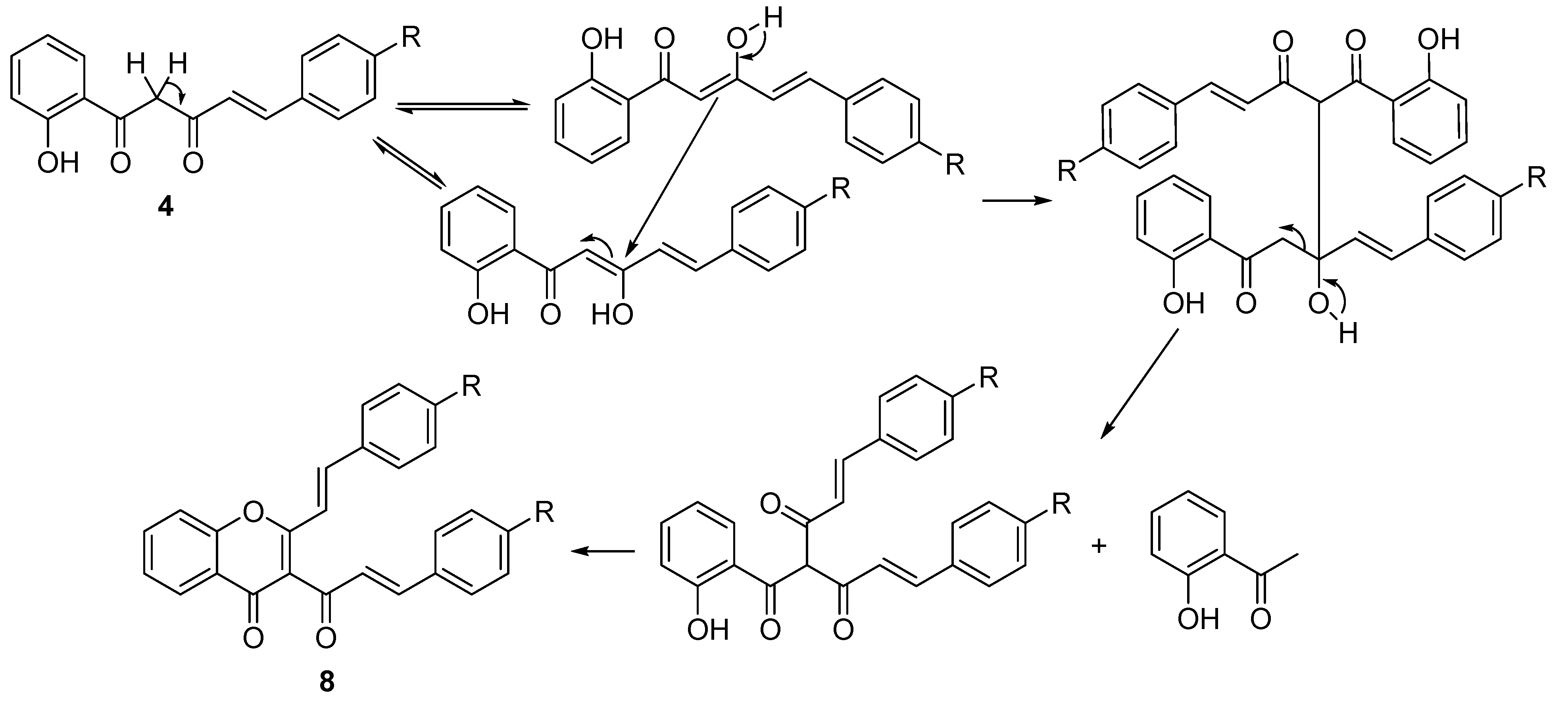
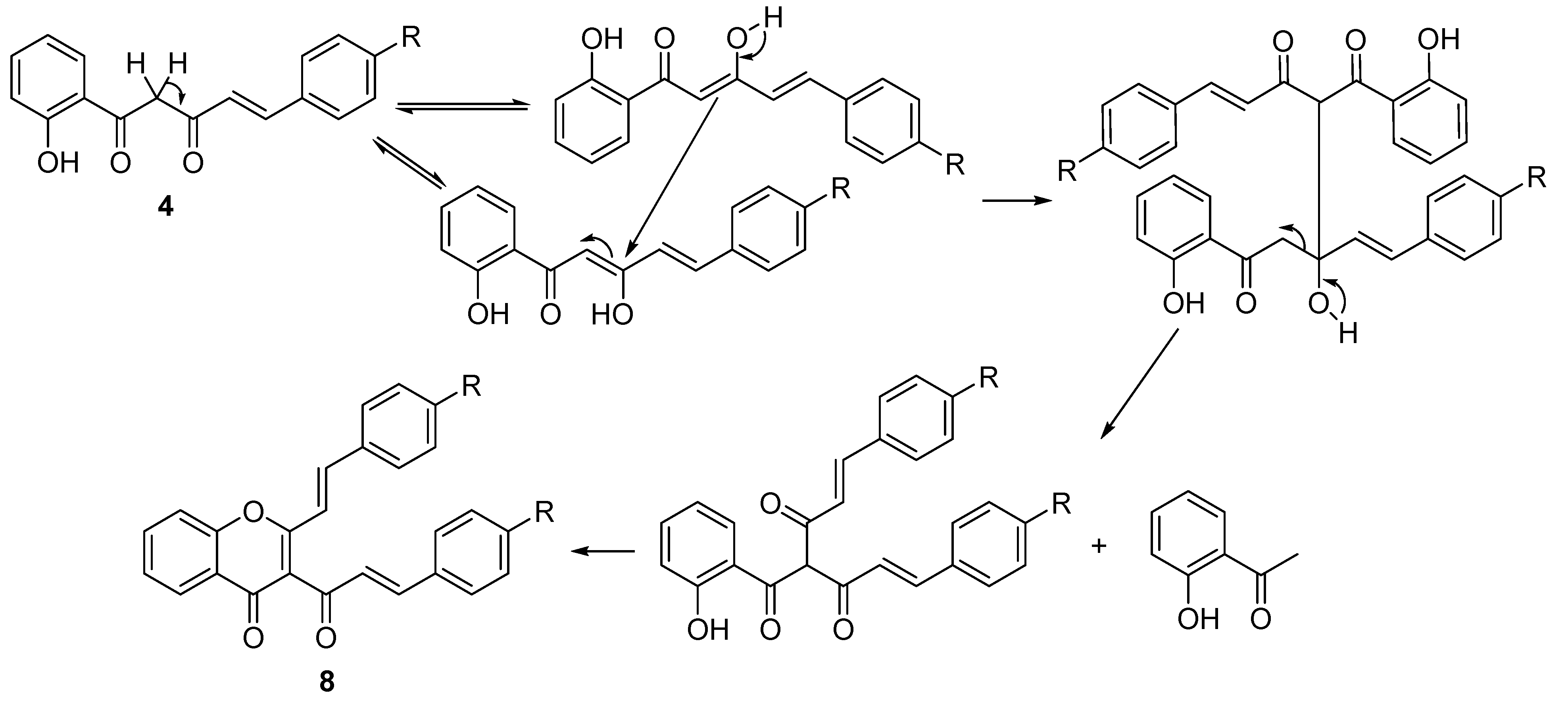
Table 1, Entries 6–9) and two by-products were also identified in each case. The NMR spectra of the by-product with higher Rf value, obtained in very low yield (less than 10%), is consistent with a benzalacetone structure, resulting from the hydrolysis of the starting material
4 in alkaline medium [
45]. The other by-product with lower Rf value was identified as the (
E,E)-3-cinnamoyl-2-styrylchromone
8 (see NMR spectra in
Supporting Information,
Figure S14) and was obtained in very low yield (less than 5%). A plausible mechanism for the formation of this compound is similar to the mechanism proposed for the formation of the (
E,E)-3-cinnamoyl-2-styrylchromone in the cyclodehydration of the corresponding 5-aryl-3-hydroxy-1-(2-hydroxyaryl)-2,4-pentadien-1-one, when performed with a mixture of DMSO and a catalytic amount of iodine at 90 °C [
46] (
Scheme 3).
Scheme 3.
Proposed mechanism for the formation of (E,E)-3-cinnamoyl-2-styrylchromones 8.
Scheme 3.
Proposed mechanism for the formation of (E,E)-3-cinnamoyl-2-styrylchromones 8.
Table 1.
Cyclodehydration of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones 4a–e to (E)-2-styrylchromones 5a–e in water, using classical reflux heating conditions.
Table 1.
Cyclodehydration of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones 4a–e to (E)-2-styrylchromones 5a–e in water, using classical reflux heating conditions.
| Entry | Compound | Base/molar equiv | Time (h) | Yield of 5 (%) a |
|---|
| 1 | 4a R = H | K2CO3/1 | 4 b | 58 |
| 2 | 4a R = H | K2CO3/0.5 | 4 | 59 |
| 3 | 4a R = H | K2CO3/0.5 + TBAB/0.5 | 3 | 12 |
| 4 | 4a R = H | TMAOH/1 | 2 | 19 |
| 5 | 4a R = H | TMAOH/0.5 | 2 | 47 |
| 6 | 4b R = OCH3 | K2CO3/0.5 | 4 | 61 |
| 7 | 4c R = Cl | K2CO3/0.5 | 4 | 70 |
| 8 | 4d R = CH3 | K2CO3/0.5 | 4 b | 70 |
| 9 | 4e R = NO2 | K2CO3/0.5 | 4 | 20 |
| 10 | 4a R = H | K2CO3/0.5 | 4 | 55 c |
In order to isolate the product by simple filtration, avoiding the use of the expensive and time-consuming chromatographic techniques, the cyclodehydration of
4a was performed on a bigger scale (800 mg) thus establishing an easy scalable protocol. In this case, the yield obtained of
5a was 55%, which was determined through precipitation (after adjustment of pH to 3–4), filtration and recrystallization of the product
5a (
Table 1, Entry 10). Since 2-styrylchromones
5a–
e are water insoluble the scalability of the reaction facilitates the precipitation and product isolation.
To evaluate the effect of microwave radiation in this reaction we performed the base-catalyzed cyclodehydration reaction of 1-(2-hydroxyphenyl)-5-phenylpent-4-ene-1,3-dione
4a under open and closed vessel conditions, using water as solvent. Water is a good solvent for microwave assisted synthesis due to its high dielectric constant [
47]. So, we studied the cyclodehydration reaction of
4a under different temperatures, reaction time and amount of base (results summarized in
Table 2). Using open vessel conditions (reflux, 30 min), the yields for (
E)-2-styrylchromones
5a,
b,
d were 66%, 56% and 55%, respectively (
Table 2, Entries 3, 5 and 9), but this reaction time is not suitable for the synthesis of (
E)-2-styrylchromones with electron-accepting substituents such as
5c,
e (
Table 2, Entries 7 and 11). In these cases better yields were obtained (58% and 56%, respectively) after 10 min of irradiation (
Table 2, Entries 6 and 10).
Table 2.
Cyclodehydration of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones 4a–e to (E)-2-styrylchromones 5a–e in water, under microwave irradiation, using open vessel reflux and closed vessel (120 °C) conditions.
Table 2.
Cyclodehydration of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones 4a–e to (E)-2-styrylchromones 5a–e in water, under microwave irradiation, using open vessel reflux and closed vessel (120 °C) conditions.
| Entry | Compound | Method | Base/molar equiv | Time (min) | Yield of 5 (%) a |
|---|
| 1 | 4a R =H | Open | K2CO3/1 | 10 | 35 |
| 2 | 4a R =H | Open | K2CO3/0.5 | 5 | 54 |
| 3 | 4a R = H | Open | K2CO3/0.5 | 30 | 66 |
| 4 | 4b R = OCH3 | Open | K2CO3/0.5 | 10 | 47 |
| 5 | 4b R = OCH3 | Open | K2CO3/0.5 | 30 | 56 |
| 6 | 4c R = Cl | Open | K2CO3/0.5 | 10 | 58 |
| 7 | 4c R = Cl | Open | K2CO3/0.5 | 30 | 47 |
| 8 | 4d R = CH3 | Open | K2CO3/0.5 | 10 | 45 |
| 9 | 4d R = CH3 | Open | K2CO3/0.5 | 30 | 55 |
| 10 | 4e R = NO2 | Open | K2CO3/0.5 | 10 | 56 |
| 11 | 4e R = NO2 | Open | K2CO3/0.5 | 30 | 38 |
| 12 | 4a R =H | Closed | K2CO3/0.5 | 5 | 50 |
| 13 | 4a R = H | Closed | K2CO3/0.5 | 30 | 67 |
| 14 | 4b R = OCH3 | Closed | K2CO3/0.5 | 30 | 77 |
| 15 | 4c R = Cl | Closed | K2CO3/0.5 | 10 | 66 |
| 16 | 4c R = Cl | Closed | K2CO3/0.5 | 15 | 38 |
| 17 | 4c R = Cl | Closed | K2CO3/0.5 | 30 | 41 |
| 18 | 4d R = CH3 | Closed | K2CO3/0.5 | 30 | 57 |
| 19 | 4e R = NO2 | Closed | K2CO3/0.5 | 30 | 27 |
| 20 | 4e R = NO2 | Closed | TMAOH/0.5 | 10 | 60 |
Employing closed vessel conditions (120 °C, 30 min) compounds
5a,
b,
d were obtained in 67%, 77% and 57% yields, respectively (
Table 2, Entries 13, 14 and 18). For compounds
5c,
e we observed a high degradation in the reaction mixture after 30 min of closed vessel microwave conditions (
Table 2, Entries 17 and 19), but good yields (66% and 60%, respectively) were obtained only with 10 min of irradiation (
Table 2, Entries 15 and 20). In the case of the cyclodehydration of derivative
4e, TMAOH was used as base and as PTC in order to improve the solubility of this compound in water which led to the formation of compound
5e in 60% yield (
Table 2, Entry 20).
We also performed the reaction with a lower amount of K
2CO
3 (0.05 equiv) using closed vessel microwave conditions. In a first attempt the cyclodehydration of
4a was performed at 120 °C for 30 min; however the 2-styrylchromone
5a was obtained in 16% yield and 26% of
4a was recovered. In order to improve the yield, we repeated the reaction at 200 °C (for 30 min). Under these conditions, compounds
5a–
e were obtained in 64%–75% yield (
Table 3).
Table 3.
Cyclodehydration of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones 4a–e to (E)-2-styrylchromones 5a–e in water and in the presence of a catalytic amount of K2CO3, using closed vessel microwave conditions, at 200 °C.
Table 3.
Cyclodehydration of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones 4a–e to (E)-2-styrylchromones 5a–e in water and in the presence of a catalytic amount of K2CO3, using closed vessel microwave conditions, at 200 °C.
| Entry | Compound | Base/molar equiv | Time (min) | Yield of 5 (%) a |
|---|
| 1 | 4a R = H | K2CO3/0.05 | 30 | 65 |
| 2 | 4b R = OCH3 | K2CO3/0.05 | 30 | 75 |
| 3 | 4c R = Cl | K2CO3/0.05 | 30 | 64 |
| 4 | 4d R = CH3 | K2CO3/0.05 | 30 | 68 |
| 5 | 4e R = NO2 | K2CO3/0.05 | 30 | 69 |
Comparing the results obtained in classical and microwave heating methods, a significant reduction of the reaction time was achieved from 2–4 h (classical heating) to 10–30 min (microwave heating). In addition, we demonstrated that it is possible to use a catalytic amount of base (0.05 equiv) using rapid microwave heating at a substantially higher temperature (at 200 °C), taking advantage of the use of water under closed vessel microwave conditions.
Recently, a new synthetic route of flavones was reported, consisting in a one-pot procedure by treatment of 2′-hydroxyacetophenones with 3 molar equiv of aroyl chloride in net K
2CO
3/acetone (1% w/w water) [
48]. Under these conditions flavones were obtained in 51%–65% yields together with 3-aroylflavones (11%–23%); this study was not extended to the synthesis of 2-styrylchromones.
After establishing the best conditions for the base-catalyzed cyclodehydration reaction of 5-aryl-1-(2-hydroxyphenyl)pent-4-ene-1,3-diones
4a–
e under classical reflux heating and microwave irradiation, we extended our study to other 3-aryl-1-(2-hydroxyphenyl)propane-1,3-diones
6a,
b in order to prepare flavones
7a,
b (
Scheme 2,
Table 4). Cyclodehydration of compound
6a occurs in excellent yield (quantitative yield) in both classical reflux heating for 1 h and under microwave irradiation for 30 min (
Table 4, Entries 1 and 3). In the case of compound
6b the reaction was performed only under closed vessel microwave conditions. After 15 min of irradiation some unreacted starting material was observed while after 30 min the expected flavone
7b was obtained in 45% yield (
Table 4, Entry 4). However after longer reaction time (45 min) the yield was not improved (46% yield) due to high degradation in the reaction mixture (
Table 4, Entry 5). These results indicate that the substituents on the diketone structure have a great effect on the yield of the cyclodehydration reaction.
Table 4.
Cyclodehydration reaction of 3-aryl-1-(2-hydroxyphenyl)propane-1,3-diones 6a,b into flavones 7a,b, under classical reflux heating and closed vessel microwave conditions, at 200 °C.
Table 4.
Cyclodehydration reaction of 3-aryl-1-(2-hydroxyphenyl)propane-1,3-diones 6a,b into flavones 7a,b, under classical reflux heating and closed vessel microwave conditions, at 200 °C.
| Entry | Compound | Method | Reaction Time (min) | Yield of 7 (%) a |
|---|
| 1 | 6a R1 =CH3, R2 = H | Oil bath | 60 | Quantitative |
| 2 | 6a R1 =CH3, R2 = H | Microwave | 15 | 70 |
| 3 | 6a R1 =CH3, R2 = H | Microwave | 30 | Quantitative |
| 4 | 6b R1 =H, R2 = OBn | Microwave | 30 | 45 |
| 5 | 6b R1 =H, R2 = OBn | Microwave | 45 | 46 |
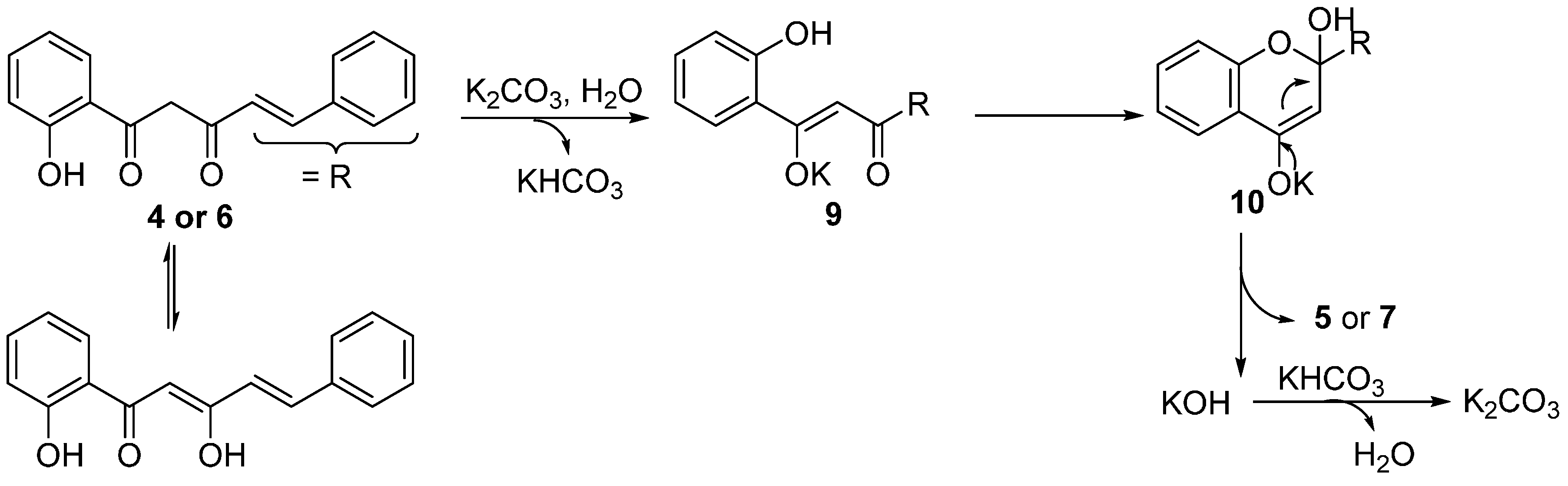
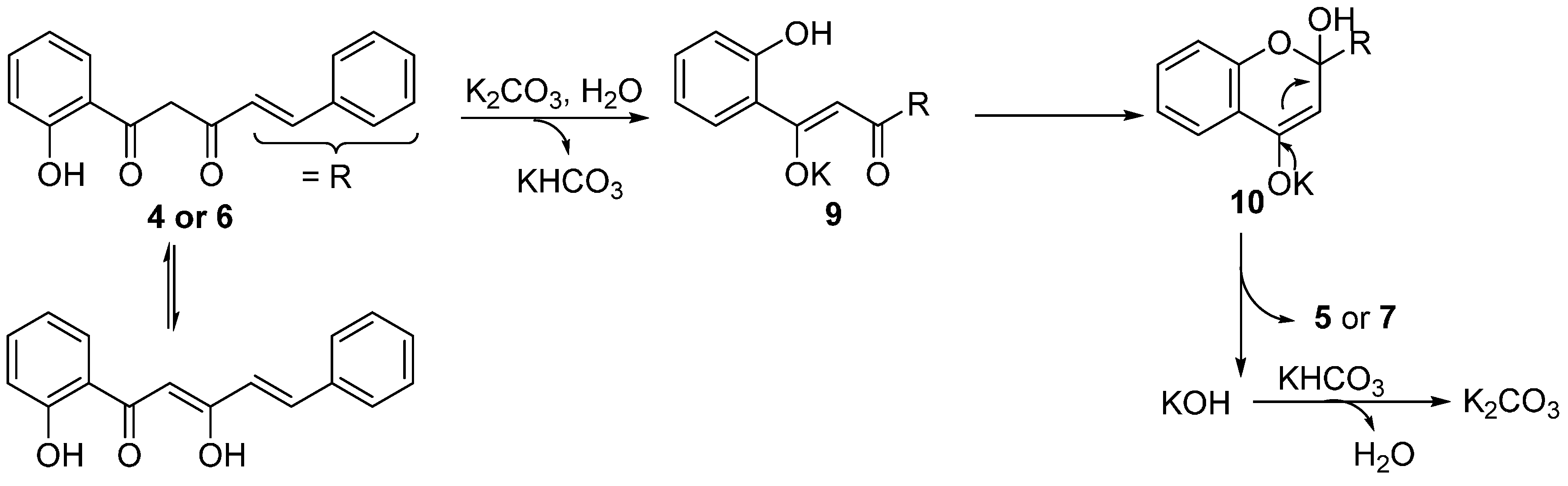
A plausible mechanism for the formation of 2-styrylchromones
5a–
e and flavones
7a,
b under the described experimental conditions is proposed in
Scheme 4. Enolization of the α-carbonyl group in
4 or
6, leads to intermediate
9 in the presence of a base (K
2CO
3), and intramolecular cycloaddition of
9 gives
10. Rearomatization of
10 by elimination of KOH provides the corresponding products
5 or
7. A similar mechanism was proposed by Fu and coworkers for the synthesis of chromones by K
2CO
3-catalysed (0.20 equiv) intramolecular cyclization of the corresponding diketones in DMF [
49].
Scheme 4.
Possible mechanism on base-catalyzed cyclodehydration of β-diketones 4a–e and 6a,b in water.
Scheme 4.
Possible mechanism on base-catalyzed cyclodehydration of β-diketones 4a–e and 6a,b in water.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}