
Phytochemical Profiling and Evaluation of Pharmacological Activities of Hypericum scabrum L.


 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Total Polyphenolic Compounds
2.2. Total Flavonoids contents
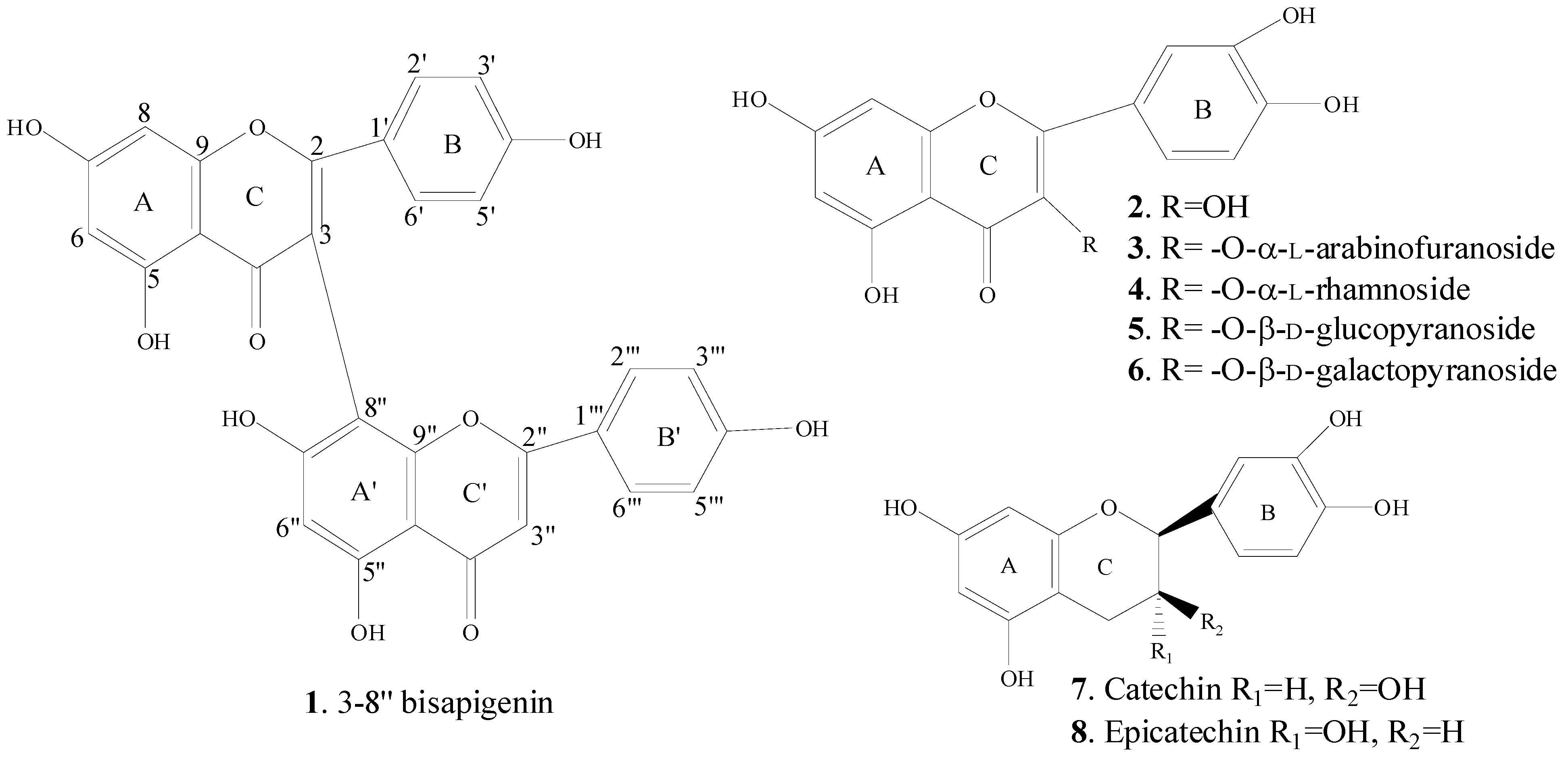
2.3. Spectral Identification of Isolated Compounds
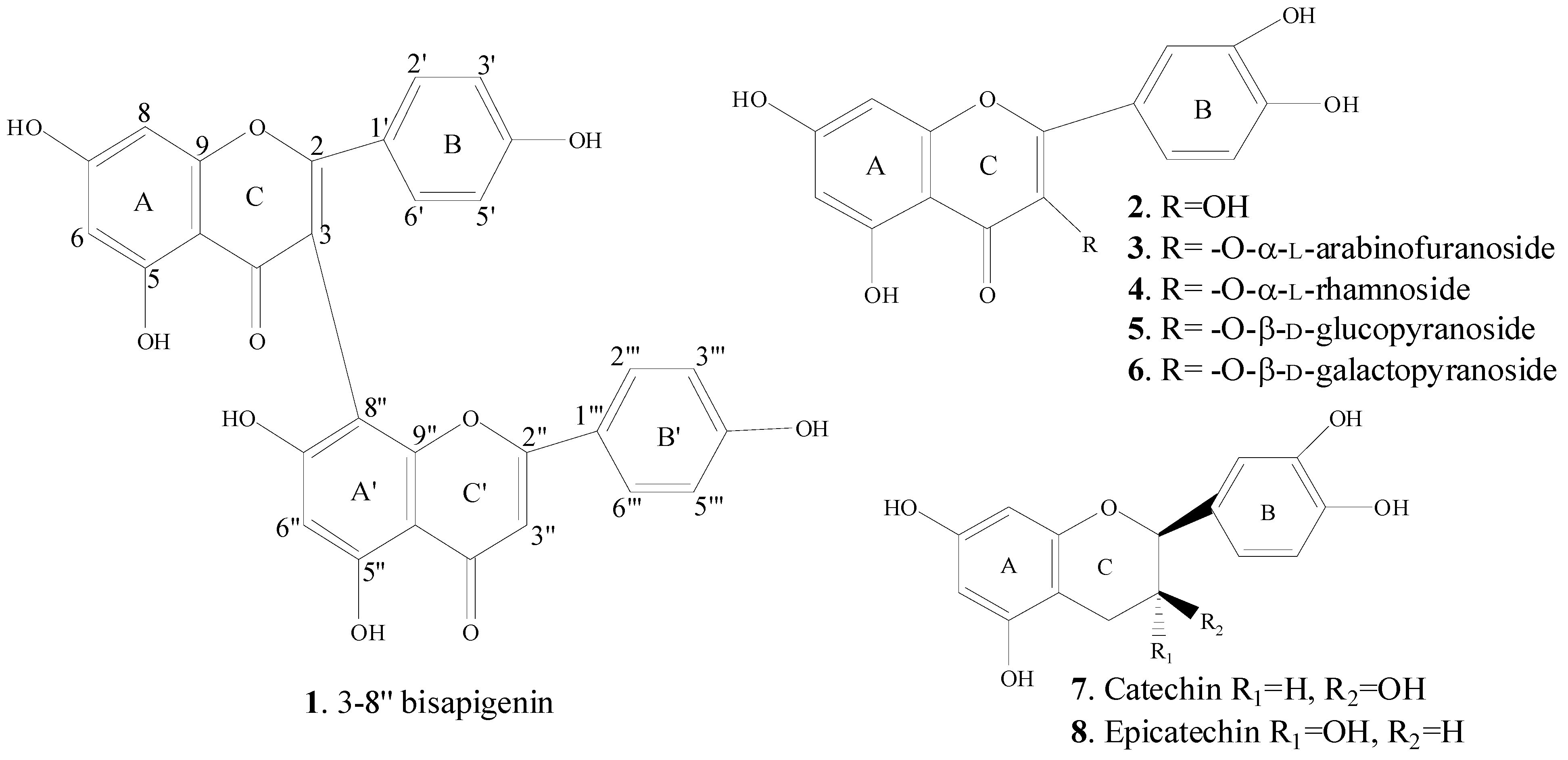
2.4. Pharmacological Activities
2.4.1. PTP-1B Inhibition Screening
2.4.2. In Vitro Antimicrobial Screening
2.4.3. Antioxidant Activity

{kind=link}
{kind=link}
| Samples | Inhibition Zone Diameter (mm) | PTP-1B Inhibition; IC50 Values (µM) | DPPH Scavenging Effects; IC50 Values (µM) | DPPH Scavenging Effects; IC50 Values (μg∙mL−1) | ||
|---|---|---|---|---|---|---|
| SA | EC | CA | ||||
| 1 | 5.5 | n.e. | n.e. | >50 | >500 | - |
| 2 | 12 | n.e. | 6.5 | 2.19 ± 0.2 | 24.88 ± 1.4 | - |
| 3 | n.e. | 5.5 | n.e. | >50 | 38.29 ± 2 | - |
| 4 | n.e. | 8 | 5.5 | >50 | 32.10 ± 1.6 | - |
| 5 | 11 | n.e. | n.e. | >50 | 24.78 ± 1.3 | - |
| 6 | 11 | 6.5 | n.e. | >50 | 29.52 ± 1.5 | - |
| 7a | 7 | 10 | 7 | 2.03 ± 0.1 | - | >500 |
| 8a | n.e. | n.e. | n.e. | 2.16 ± 0.1 | - | 13.86 ± 0.7 |
| 9a | n.e. | n.e. | n.e. | 4.91 ± 0.3 | - | 24.62 ± 1.3 |
| 10 | n.e. | 12 | n.e. | 2.91 ± 0.2 | - | 1.19 ± 0.1 |
| 11 | n.e. | n.e. | n.e. | 2.1 ± 0.1 | - | 6.18 ± 0.3 |
| 12 | n.e. | n.e. | n.e. | 1.57 ± 0.1 | - | 10.81 ± 0.6 |
| 13 | n.e. | n.e. | n.e. | 14.29 ± 0.8 | - | 16.29 ± 0.8 |
| Vitamin C | - | - | - | - | 30.32 ± 2.4 | 5.34 ± 0.4 |
| PTP-1B | - | - | - | 1.97 ± 0.5 | - | - |
| Ampicillin | 19 | 14 | - | - | - | - |
| Amphotericin B | - | - | 15 | - | - | - |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Determination of Total Polyphenolic Compounds and Total Contents of Flavonoids
3.3.1. Preparation of Sample
3.3.2. Total Polyphenolic Compounds
3.3.3. Determination of Total Flavonoids
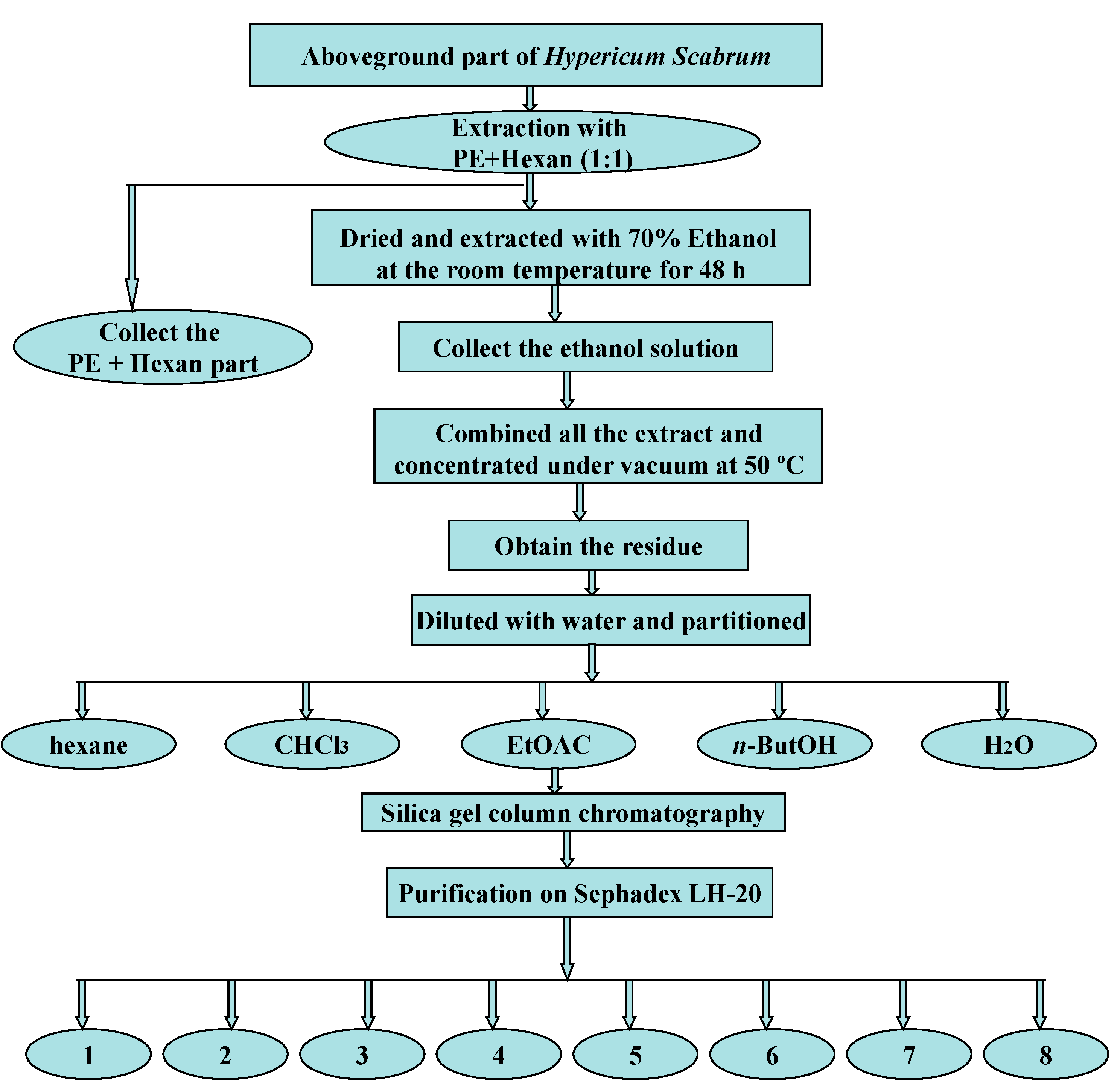
3.4. Extraction, Isolation and Purification Procedures
3.5. Mass Spectrometry
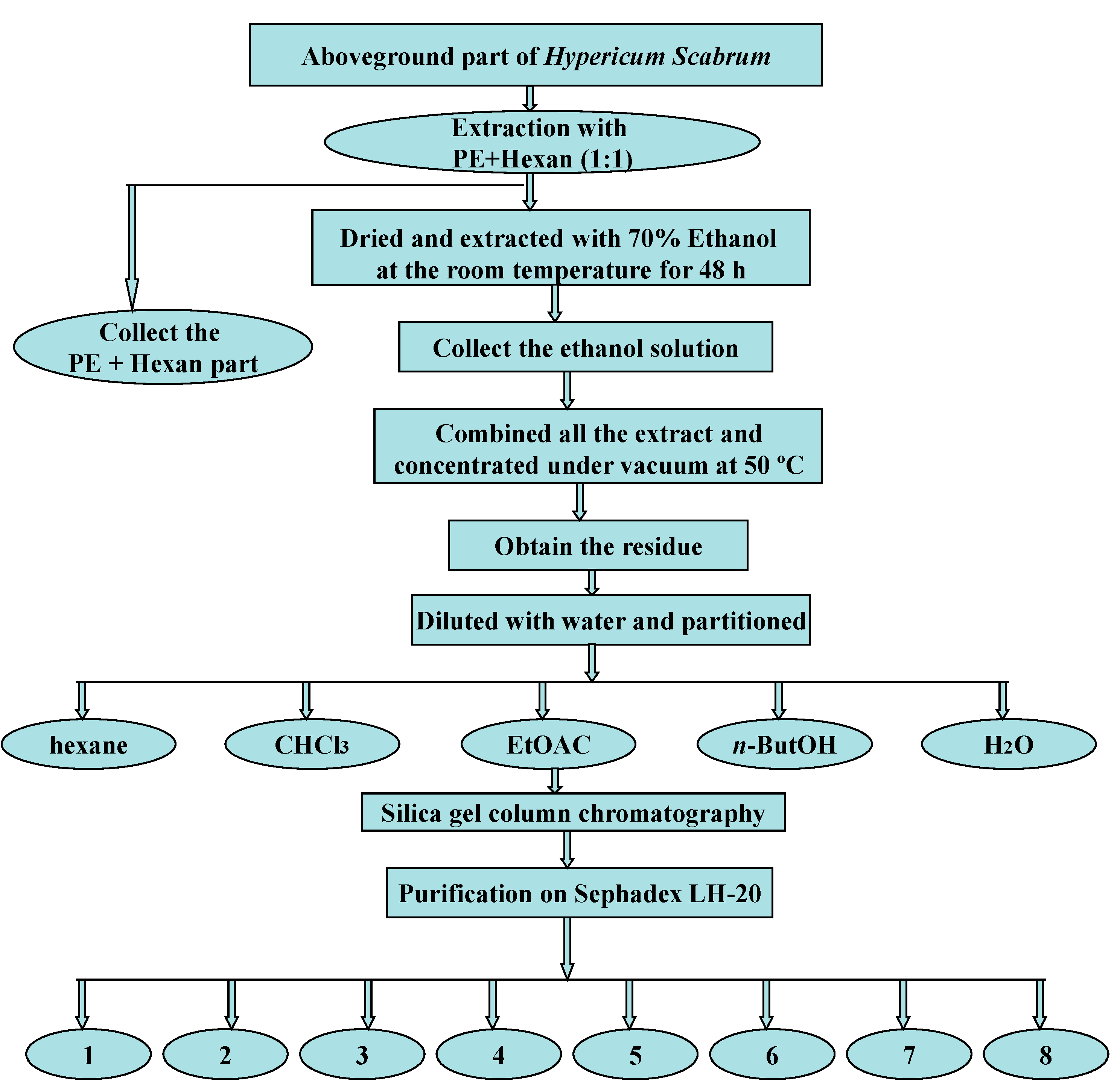
3.6. Analytical Data of the Identified Compounds
3.7. Protein Tyrosine Phosphatase 1B (PTP-1B) Inhibition Assay
3.8. Antimicrobial Activity
3.9. DPPH Radical Scavenging Assay
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Raskin, I.; Ribnicky, D.M.; Komarnytsky, S.; Ilic, N.; Poulev, A.; Borisjuk, N.; Brinker, A.; Moreno, D.A.; Ripoll, C.; Yakoby, N. Plants and human health in the twenty-first century. Trends Biotechnol. 2002, 20, 522–531. [Google Scholar] [CrossRef]
- Middleton, E., Jr. Effect of plant flavonoids on immune and inflammatory cell function. Adv. Exp. Med. Biol. 1998, 439, 175–182. [Google Scholar] [PubMed]
- Liu, E.H.; Qi, L.W.; Cao, J.; Li, P.; Li, C.Y.; Peng, Y.B. Advances of modern chromatographic and electrophoretic methods in separation and analysis of flavonoids. Molecules 2008, 13, 2521–2544. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.N.; Stecher, G.; Bonn, G.K. Determination of total polyphenolic compounds and flavonoids in Juglans regia leaves. Pak. J. Pharm. Sci. 2014, 27, 865–869. [Google Scholar] [PubMed]
- Sultana, T.; Stecher, G.; Mayer, R.; Trojer, L.; Qureshi, M.N.; Abel, G.; Popp, M.; Bonn, G.K. Quality assessment and quantitative analysis of flavonoids from tea samples of different origins by HPLC-DAD-ESI-MS. J. Agric. Food Chem. 2008, 56, 3444–3453. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T. Novel natural antioxidants for utilization in food and biological systems. In Postharvest Biochemistry of Plant Food-Materials in the Tropics; Uritani, I., Garcia, V.V., Mendoza, E.M., Eds.; Japan Scientific Society Press: Tokyo, Janpan, 1994; pp. 241–251. [Google Scholar]
- Numonov, S.; Usmanova, S.; Aisa, H. A triterpenoid and flavonoids from Dracocephalum heterophyllum. Chem. Nat. Compd. 2013, 48, 1109–1110. [Google Scholar] [CrossRef]
- Numonov, S.; Usmanova, S.; Aisa, H. Chemical composition of Dracocephalum heterophyllum. Chem. Nat. Compd. 2013, 49, 511–513. [Google Scholar] [CrossRef]
- Sharopov, F.; Gulmurodov, I.; Setzer, W. Essential oil composition of Hypericum perforatum L. and Hypericum scabrum L. growing wild in Tajikistan. J. Chem. Pharm. Res. 2010, 2, 284–290. [Google Scholar]
- Erdoğrul, Ö.; Azirak, S.; Tosyali, C. Antimicrobial activities of Hypericum scabrum L. extracts. KSU J. Sci. Eng. 2004, 7, 38–42. [Google Scholar]
- Chang, C.C.; Yang, M.H.; Wen, H.M.; Chern, J.C. Estimation of total flavonoid content in propolis by two complementary colorimetric methods. J. Food Drug Anal. 2002, 10, 178–182. [Google Scholar]
- Zheng, J.; Wang, N.; Fan, M.; Chen, H.; Liu, H.; Yao, X. A new biflavonoid from Selaginella uncinata. Asian J. Tradit. Med. 2007, 2, 92–97. [Google Scholar]
- Barbosa, F.G.; Lima, M.A.S.; Silveira, E.R. Total NMR assignments of new [C7-O-C7′′]-biflavones from leaves of the limonene–carvone chemotype of Lippia alba (Mill) NE Brown. Magn. Reson. Chem. 2005, 43, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, S.; Kumar, H.; Verma, D. Chemical Examination of Bergenia stracheyi (Hk.) for antioxidative flavonoids. Nat. Sci. 2009, 7, 29–34. [Google Scholar]
- Nowak, S.; Wolbis, M. Flavonoids from some species of genus Scopolia Jacq. Acta Pol. Pharm. 2002, 59, 275–280. [Google Scholar] [PubMed]
- Sidana, J.; Neeradi, D.; Choudhary, A.; Singh, S.; Foley, W.J.; Singh, I.P. Antileishmanial polyphenols from Corymbia maculata. J. Chem. Sci. 2013, 125, 765–775. [Google Scholar] [CrossRef]
- Foo, L.; Newman, R.; Waghorn, G.; McNabb, W.; Ulyatt, M. Proanthocyanidins from Lotus corniculatus. Phytochemistry 1996, 41, 617–624. [Google Scholar] [CrossRef]
- Hye, M.; Taher, M.; Ali, M.; Ali, M.; Zaman, S. Isolation of (+)-catechin from Acacia catechu (Cutch Tree) by a convenient method. J. Sci. Res. 2009, 1, 300–305. [Google Scholar]
- Ivanov, S.A.; Nomura, K.; Malfanov, I.L.; Sklyar, I.V.; Ptitsyn, L.R. Isolation of a novel catechin from Bergenia rhizomes that has pronounced lipase-inhibiting and antioxidative properties. Fitoterapia 2011, 82, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, J.; Liu, H.; Zhou, L.; Liu, Z.; Wang, J.; Han, J.; Yu, Z.; Yang, F. Chemical analysis and biological activity of the essential oils of two valerianaceous species from China: Nardostachys chinensis and Valeriana officinalis. Molecules 2010, 15, 6411–6422. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Numonov, S.; Bobakulov, K.; Qureshi, M.N.; Zhao, H.; Aisa, H.A. Phytochemical Profiling and Evaluation of Pharmacological Activities of Hypericum scabrum L. Molecules 2015, 20, 11257-11271. https://doi.org/10.3390/molecules200611257
Jiang L, Numonov S, Bobakulov K, Qureshi MN, Zhao H, Aisa HA. Phytochemical Profiling and Evaluation of Pharmacological Activities of Hypericum scabrum L. Molecules. 2015; 20(6):11257-11271. https://doi.org/10.3390/molecules200611257
Chicago/Turabian StyleJiang, Lan, Sodik Numonov, Khayrulla Bobakulov, Muhammad Nasimullah Qureshi, Haiqing Zhao, and Haji Akber Aisa. 2015. "Phytochemical Profiling and Evaluation of Pharmacological Activities of Hypericum scabrum L." Molecules 20, no. 6: 11257-11271. https://doi.org/10.3390/molecules200611257