
Non-Invasive Biomarkers for Duchenne Muscular Dystrophy and Carrier Detection

 ,
, 
Abstract
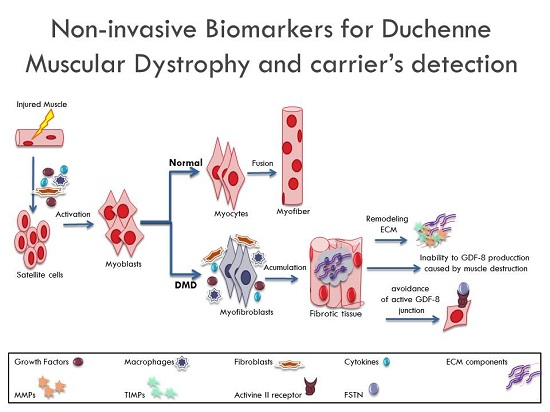
: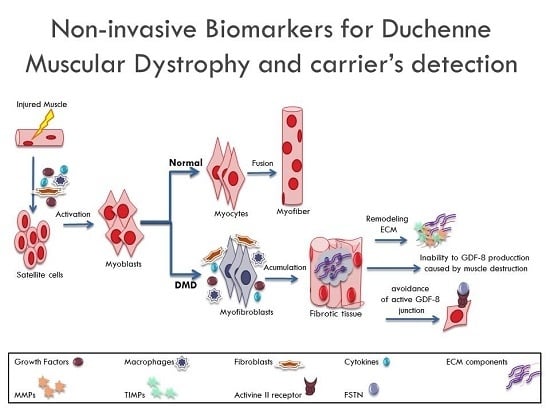
1. Introduction
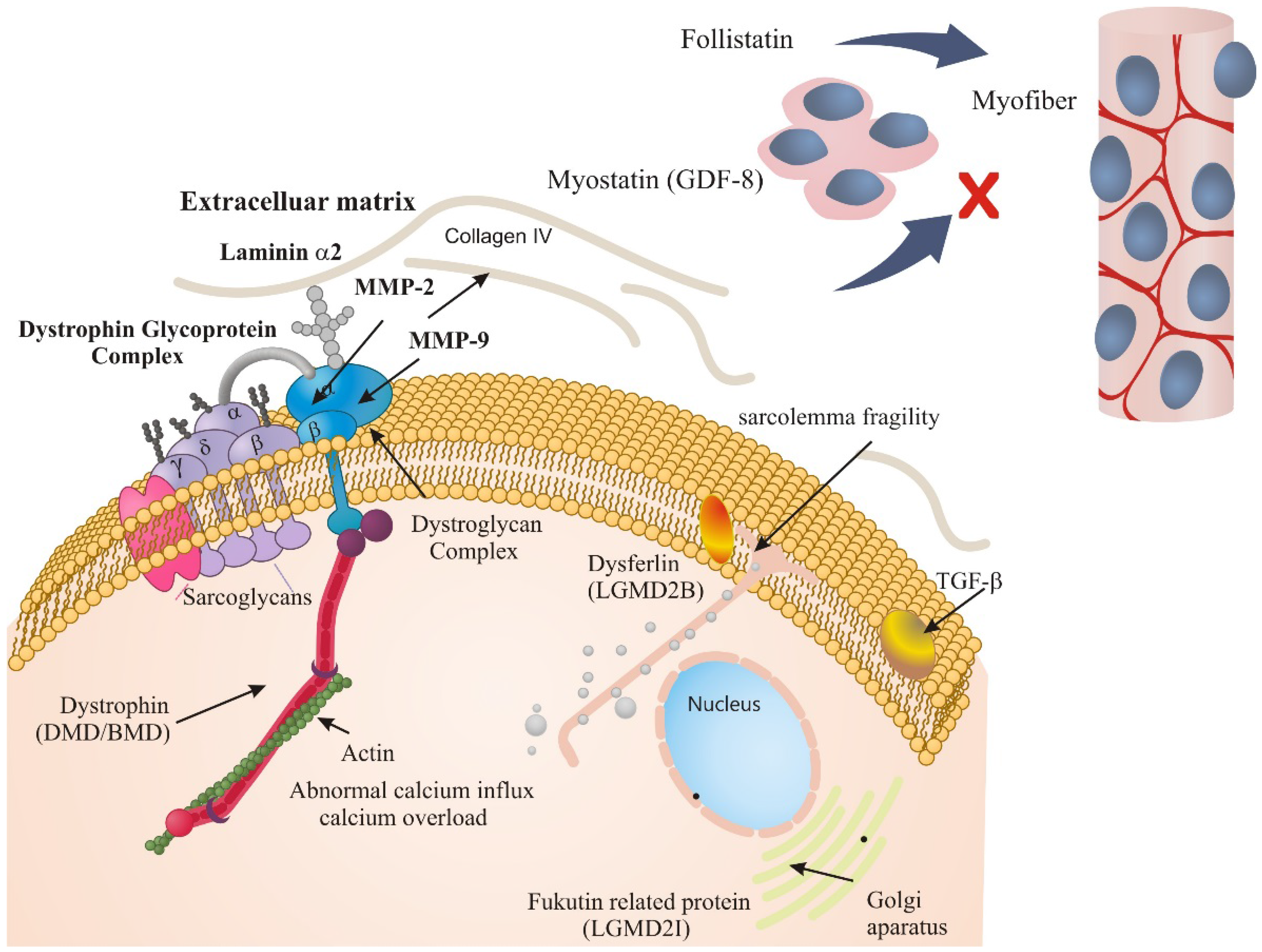
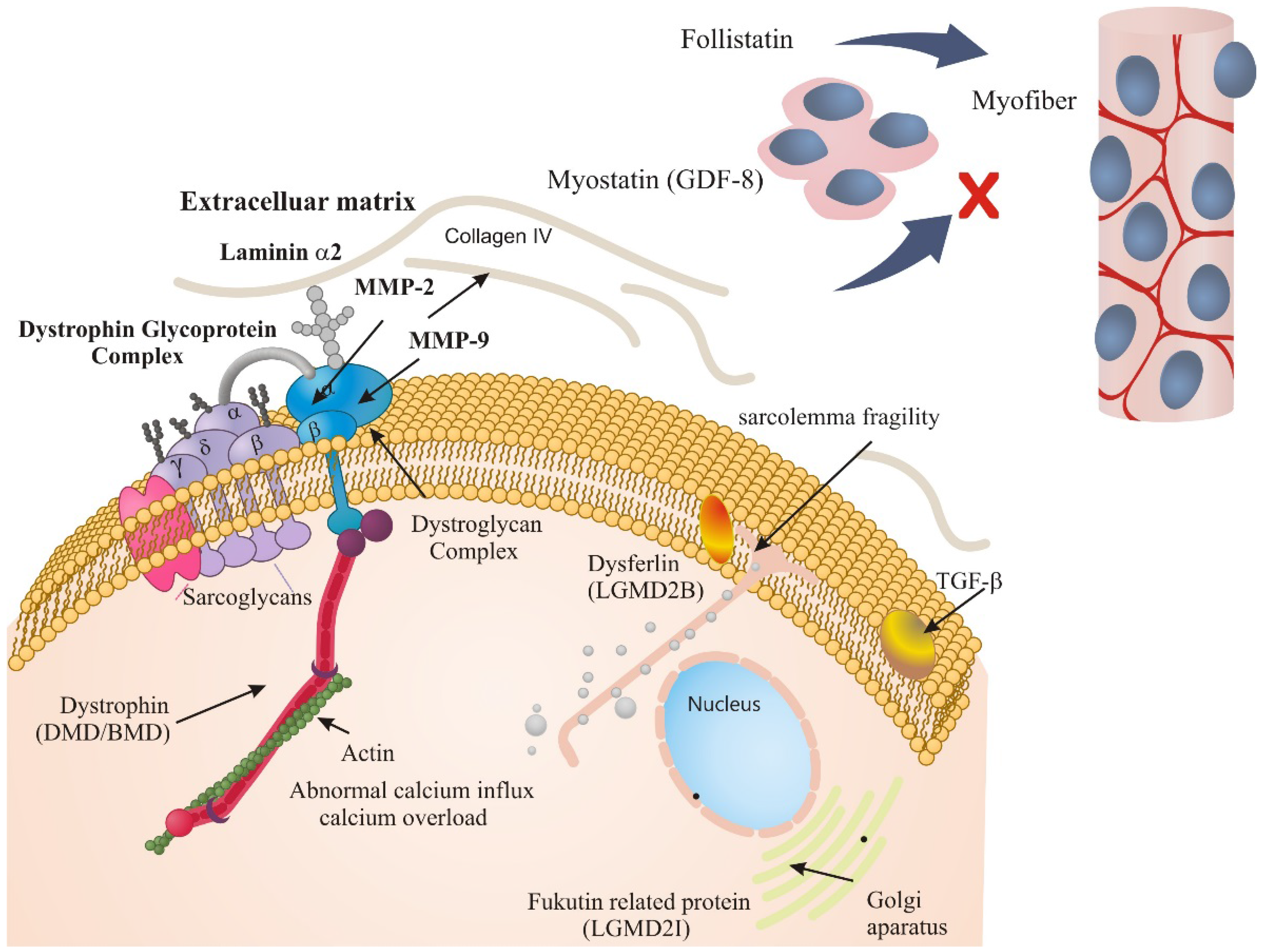
2. Results and Discussion
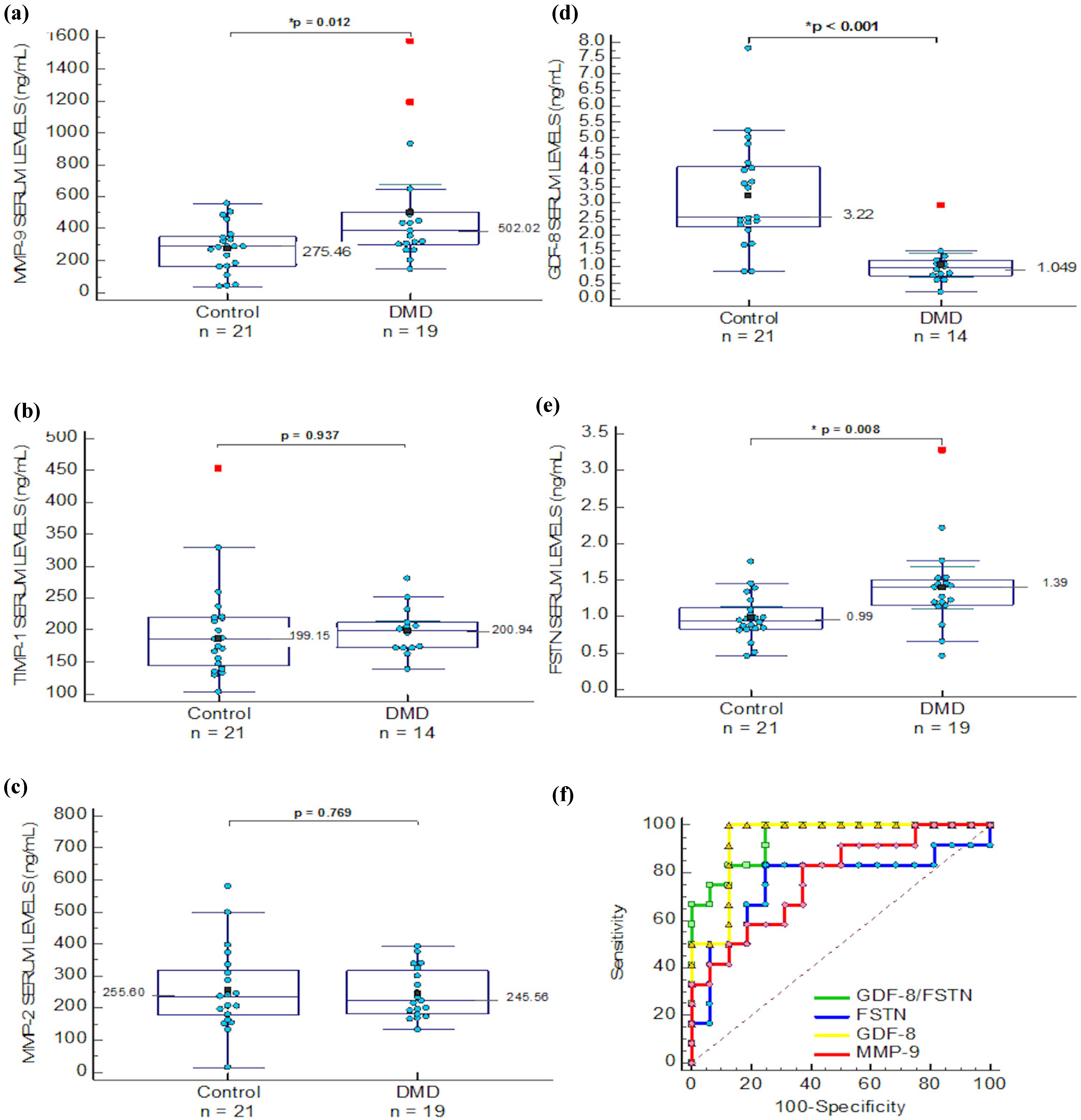
2.1. ECM Regulators in Duchenne Muscular Dystrophy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | DMD | BMD | Carriers | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patients n = 19 Mean (±SD) | Controls n = 21 Mean (±SD) | p Value | Patients n = 4 Mean (±SD) | Controls n = 4 Mean (±SD) | p Value | Carriers n = 17 Mean (±SD)/Median (Range) | Controls n = 17 Mean (±SD)/Median (Range) | p Value | |
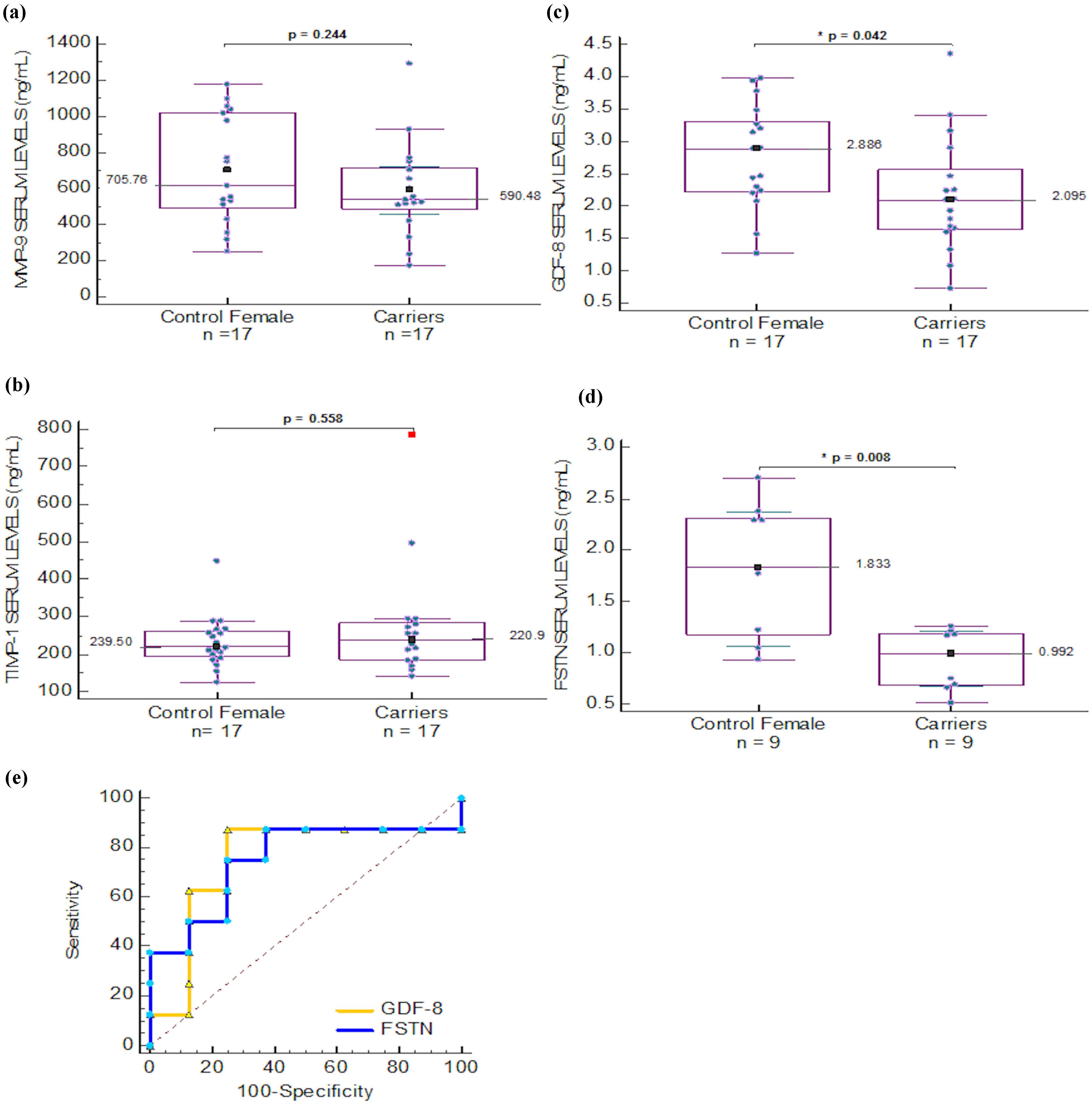
| MMP-9 | 502.021 (174.297) | 275.46 (68.62) | 0.012 * | 785.05 (180.9) | 165.3 (114.18) | 0.001 * | 590.48 (134.79) | 705.76 (155.62) | 0.244 |
| MMP-2 | 245.56 (37.69) | 255.60 (58.40) | 0.769 | 254.76 (32.98) | 519.83 (112.93) | 0.004 * | ND | ND | ND |
| FSTN | 1.39 (0.28) | 0.99 (0.14) | 0.008 * | 1.02 (0.152) | 0.715 (0.152) | 0.028 * | ¶ 0.992 (0.516–1.259) | ¶ 1.833 (0.934–2.702) | 0.008 * |
| GDF-8 | # 1.049 (0.364) | 3.22 (0.752) | ≤0.01 * | ND | ND | ND | 2.095 (0.726–4.354) | 2.886 (1.272–3.974) | 0.042 * |
| TIMP-1 | # 200.94 (22.3) | 199.15 (35.5) | 0.937 | ND | ND | ND | 239.5 (142.29–785.1) | 220.93 (124.7–448.7) | 0.558 |
 ), FSTN (
), FSTN (  ), MMP-9 (
), MMP-9 (  ) and GDF-8/FSTN ratio (
) and GDF-8/FSTN ratio (  ) levels ROC analysis.
), FSTN ( ), MMP-9 ( ) and GDF-8/FSTN ratio ( ) levels ROC analysis.
) levels ROC analysis.
), FSTN ( ), MMP-9 ( ) and GDF-8/FSTN ratio ( ) levels ROC analysis.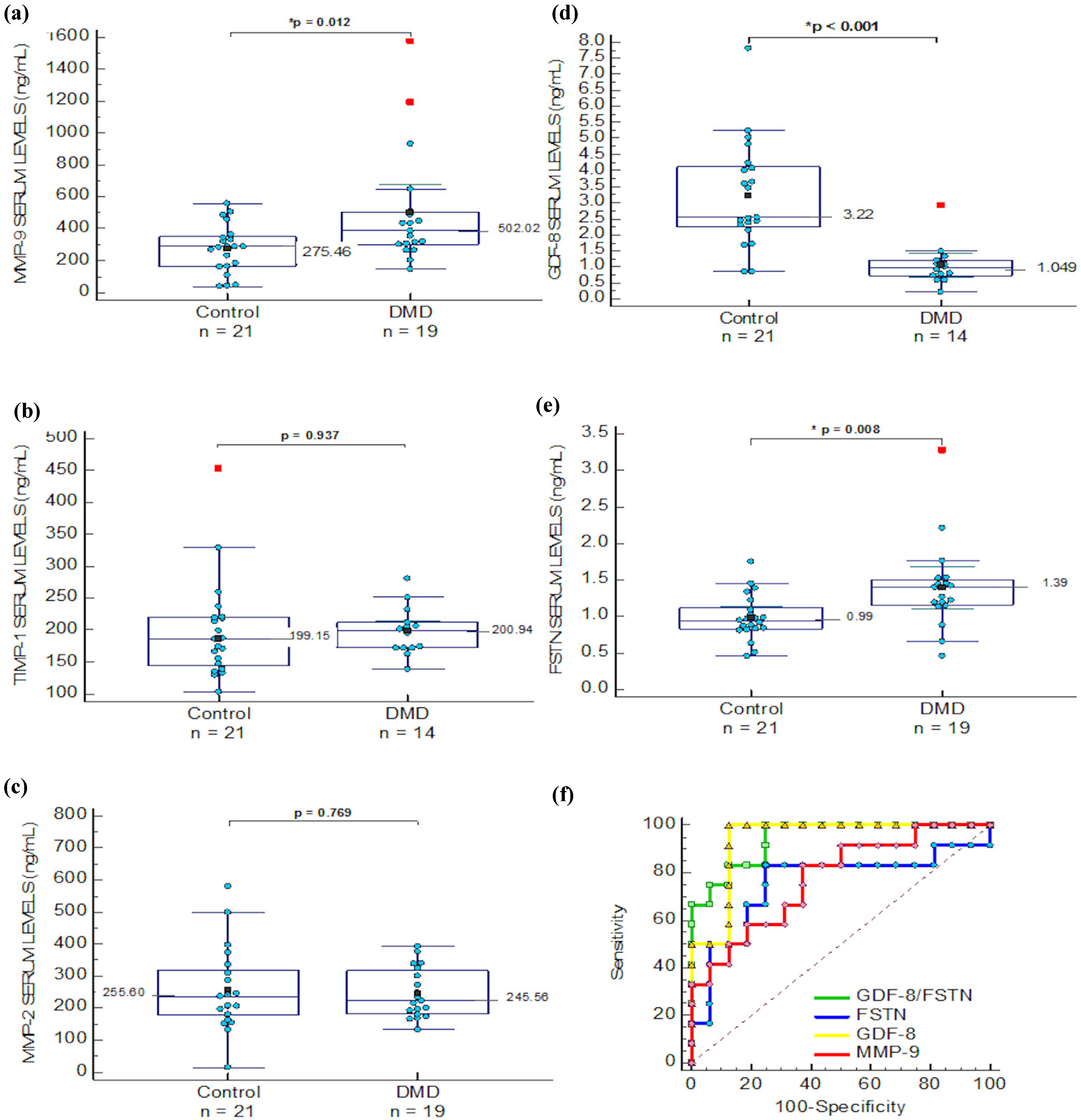
| Biomarker | Age (Years) | NSAA | 6 MW | Barthel | Brook L | Vignos | T10mw | T10mr | Gowers | Stair | Chair | Shirt | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MMP-9 | Correlation coefficient | 0.13 | −0.34 | 0.16 | 0.05 | 0.29 | 0.39 | 0.2 | 0.650 ** | 0.514 * | 0.21 | −0.12 | 0.39 |
| p-Value | 0.58 | 0.25 | 0.61 | 0.85 | 0.21 | 0.09 | 0.56 | 0.01 | 0.04 | 0.43 | 0.62 | 0.11 | |
| TIMP-1 | Correlation coefficient | −0.24 | 0.68 | −0.23 | 0.07 | −0.27 | −0.07 | 0.09 | 0.05 | 0.09 | 0.39 | −0.37 | −0.09 |
| p-Value | 0.44 | 0.06 | 0.62 | 0.86 | 0.38 | 0.82 | 0.82 | 0.89 | 0.81 | 0.29 | 0.32 | 0.81 | |
| GDF-8 | Correlation coefficient | 0.02 | −0.13 | -0.3 | −0.33 | −0.22 | −0.16 | −0.27 | −0.36 | −0.13 | −0.09 | 0.29 | −0.07 |
| p-Value | 0.95 | 0.75 | 0.4 | 0.32 | 0.47 | 0.61 | 0.42 | 0.25 | 0.7 | 0.8 | 0.39 | 0.84 | |
| MMP-2 | Correlation coefficient | −0.1 | 0.41 | 0.13 | 0.3 | −0.3 | −0.37 | 0.29 | 0.03 | −0.25 | −0.17 | −0.586 * | −0.41 |
| p-Value | 0.68 | 0.16 | 0.67 | 0.37 | 0.22 | 0.13 | 0.41 | 0.9 | 0.39 | 0.55 | 0.02 | 0.11 | |
| FSTN | Correlation coefficient | −0.22 | −0.14 | −0.02 | 0.02 | 0.30 | 0.03 | 0.19 | 0.17 | 0.00 | 0.1 | −0.07 | 0.16 |
| p-Value | 0.38 | 0.64 | 0.94 | 0.94 | 0.21 | 0.91 | 0.59 | 0.51 | 0.99 | 0.73 | 0.79 | 0.56 | |
2.2. Matrix Metalloproteinases in Other Muscular Dystrophies
2.3. Muscle Growth Regulators in Dystrophinopathies and Other Muscular Dystrophies
2.4. Serum Biomarkers for Carrier Detection in Duchenne Muscular Dystrophy
2.5. Sensibility and Specificity of Serum Biomarkers in DMD Patients and Carriers
) and FSTN ( ) levels in female serum samples.
) and FSTN ( ) levels in female serum samples.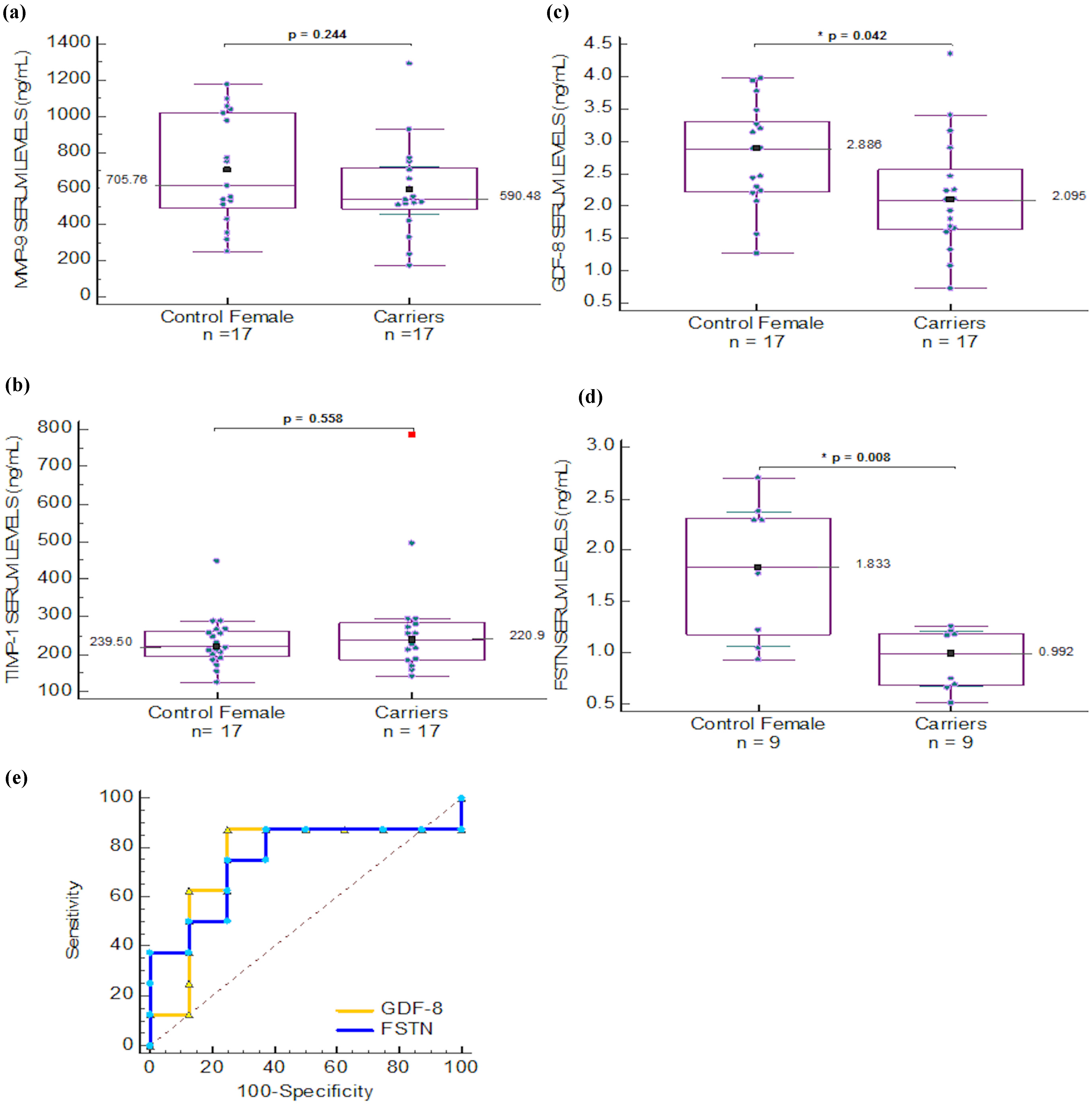
2.6. Discussion
3. Experimental Section
3.1. Study Participants
3.2. Muscular Dystrophy Patients and Healthy Controls
3.3. Neuromuscular Assessments for Patients with Duchenne Muscular Dystrophy
3.4. Carrier Detection in DMD Families
3.5. DNA Analysis
3.6. Multiplex Ligation Dependent Probe Amplification
3.7. STR Segregation Analysis
3.8. Immunodetection Analysis
3.9. Serum Samples
3.10. Determination of Proteins in Human Serum Samples
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhattacharya, S.; Das, A.; Dasgupta, R.; Bagchi, A. Analyses of the presence of mutations in dystrophin protein to predict their relative influences in the onset of duchenne muscular dystrophy. Cell. Signal. 2014, 26, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Van Westering, T.L.; Betts, C.A.; Wood, M.J. Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef] [PubMed]
- Piko, H.; Vancso, V.; Nagy, B.; Ban, Z.; Herczegfalvi, A.; Karcagi, V. Dystrophin gene analysis in hungarian duchenne/becker muscular dystrophy families—Detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul. Disord. 2009, 19, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Mercier, S.; Toutain, A.; Toussaint, A.; Raynaud, M.; de Barace, C.; Marcorelles, P.; Pasquier, L.; Blayau, M.; Espil, C.; Parent, P.; et al. Genetic and clinical specificity of 26 symptomatic carriers for dystrophinopathies at pediatric age. Eur. J. Hum. Genet. 2013, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 2008, 265, 191–225. [Google Scholar] [PubMed]
- Nadarajah, V.D.; Van Putten, M.; Chaouch, A.; Garrood, P.; Straub, V.; Lochmuller, H.; Ginjaar, H.B.; Aartsma-Rus, A.M.; van Ommen, G.J.; den Dunnen, J.T.; et al. Serum matrix metalloproteinase-9 (mmp-9) as a biomarker for monitoring disease progression in duchenne muscular dystrophy (dmd). Neuromuscul. Disord. 2011, 21, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Hurnaus, S.; Mueller-Felber, W.; Pongratz, D.; Schoser, B.G. Serum levels of matrix metalloproteinases-2 and -9 and their tissue inhibitors in inflammatory neuromuscular disorders. Eur. Neurol. 2006, 55, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindi, S.M.; Shin, J.; Ogura, Y.; Li, H.; Kumar, A. Matrix metalloproteinase-9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS ONE 2013, 8, e72121. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, M.; Sciandra, F.; Brancaccio, A. Role of gelatinases in pathological and physiological processes involving the dystrophin-glycoprotein complex. J. Int. Soc. Matrix Biol. 2015, 44–46, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, D.; Sciandra, F.; Gioia, M.; Marini, S.; Gori, A.; Giardina, B.; Tarantino, U.; Coletta, M.; Brancaccio, A.; Bozzi, M. Alpha-dystroglycan is a potential target of matrix metalloproteinase mmp-2. J. Int. Soc. Matrix Biol. 2015, 41, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Perez, V.; Siachoque, N.; Miksztowicz, V.; Berg, G.; Rodriguez, M.; Donato, M.; Gelpi, R.J. Dystrophin proteolysis: A potential target for mmp-2 and its prevention by ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H88–H96. [Google Scholar] [CrossRef] [PubMed]
- Kherif, S.; Lafuma, C.; Dehaupas, M.; Lachkar, S.; Fournier, J.G.; Verdiere-Sahuque, M.; Fardeau, M.; Alameddine, H.S. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: A study in experimentally injured and mdx muscles. Dev. Biol. 1999, 205, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, D.; Nakamura, A.; Fukushima, K.; Yoshida, K.; Takeda, S.; Ikeda, S. Matrix metalloproteinase-2 ablation in dystrophin-deficient mdx muscles reduces angiogenesis resulting in impaired growth of regenerated muscle fibers. Hum. Mol. Genet. 2011, 20, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Zocevic, A.; Rouillon, J.; Wong, B.; Servais, L.; Voit, T.; Svinartchouk, F. Evaluation of the serum matrix metalloproteinase-9 as a biomarker for monitoring disease progression in duchenne muscular dystrophy. Neuromuscul. Disord. 2015, 25, 444–446. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.E.; Boxhorn, L.K. Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J. Cell. Physiol. 1987, 133, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Foster, W.; Deasy, B.M.; Chan, Y.; Prisk, V.; Tang, Y.; Cummins, J.; Huard, J. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: A key event in muscle fibrogenesis. Am. J. Pathol. 2004, 164, 1007–1019. [Google Scholar] [CrossRef]
- Ishitobi, M.; Haginoya, K.; Zhao, Y.; Ohnuma, A.; Minato, J.; Yanagisawa, T.; Tanabu, M.; Kikuchi, M.; Iinuma, K. Elevated plasma levels of transforming growth factor beta1 in patients with muscular dystrophy. Neuroreport 2000, 11, 4033–4035. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.; Yaworsky, P.J.; Walsh, F.S. Myostatin as a therapeutic target for musculoskeletal disease. Cell. Mol. Life Sci. 2008, 65, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.T.; Ryall, J.G.; Snell, S.M.; Nair, L.; Koopman, R.; Krasney, P.A.; Ibebunjo, C.; Holden, K.S.; Loria, P.M.; Salatto, C.T.; et al. Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am. J. Pathol. 2010, 176, 2425–2434. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.J. Ataluren: First global approval. Drugs 2014, 74, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of duchenne muscular dystrophy (demand ii): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ferlini, A.; Vroom, E. Biomarkers and surrogate endpoints in duchenne: Meeting report. Neuromuscul. Disord. 2014, 24, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Ayoglu, B.; Chaouch, A.; Lochmuller, H.; Politano, L.; Bertini, E.; Spitali, P.; Hiller, M.; Niks, E.H.; Gualandi, F.; Ponten, F.; et al. Affinity proteomics within rare diseases: A bio-nmd study for blood biomarkers of muscular dystrophies. EMBO Mol. Med. 2014, 6, 918–936. [Google Scholar] [CrossRef] [PubMed]
- Hashim, R.; Shaheen, S.; Ahmad, S.; Sattar, A.; Khan, F.A. Comparison of serum creatine kinase estimation with short tandem repeats based linkage analysis in carriers and affected children of duchenne muscular dystrophy. J. Ayub Med. Coll. Abbottabad 2011, 23, 125–128. [Google Scholar] [PubMed]
- Rouillon, J.; Zocevic, A.; Leger, T.; Garcia, C.; Camadro, J.M.; Udd, B.; Wong, B.; Servais, L.; Voit, T.; Svinartchouk, F. Proteomics profiling of urine reveals specific titin fragments as biomarkers of duchenne muscular dystrophy. Neuromuscul. Disord. 2014, 24, 563–573. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new tgf-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Soejima, M.; Iwanuma, O.; Saka, H.; Matsunaga, S.; Sakiyama, K.; Ide, Y. Expression of myostatin and follistatin in mdx mice, an animal model for muscular dystrophy. Zool. Sci. 2009, 26, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, K.; Nakamura, A.; Ueda, H.; Yuasa, K.; Yoshida, K.; Takeda, S.; Ikeda, S. Activation and localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of the muscular dystrophy dog (cxmdj). BMC Musculoskelet. Disord. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.E.; Alonso, D.F.; Yoshiji, H.; Thorgeirsson, U.P. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur. J. Cell Biol. 1997, 74, 111–122. [Google Scholar] [PubMed]
- Kolkenbrock, H.; Orgel, D.; Hecker-Kia, A.; Zimmermann, J.; Ulbrich, N. Generation and activity of the ternary gelatinase b/timp-1/lmw-stromelysin-1 complex. Biol. Chem. Hoppe Seyler 1995, 376, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Borden, P.; Heller, R.A. Transcriptional control of matrix metalloproteinases and the tissue inhibitors of matrix metalloproteinases. Crit. Rev. Eukaryot. Gene Expr. 1997, 7, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Cynthia Martin, F.; Hiller, M.; Spitali, P.; Oonk, S.; Dalebout, H.; Palmblad, M.; Chaouch, A.; Guglieri, M.; Straub, V.; Lochmuller, H.; et al. Fibronectin is a serum biomarker for duchenne muscular dystrophy. Proteomics Clin. Appl. 2014, 8, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kong, M.; Ye, Y.; Hong, S.; Cheng, L.; Jiang, L. Serum mir-206 and other muscle-specific micrornas as non-invasive biomarkers for duchenne muscular dystrophy. J. Neurochem. 2014, 129, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. Mirnas as serum biomarkers for duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Haginoya, K.; Chiba, Y.; Uematsu, M.; Hino-Fukuyo, N.; Tanaka, S.; Onuma, A.; Iinuma, K.; Tsuchiya, S. Elevated plasma levels of tissue inhibitors of metalloproteinase-1 and their overexpression in muscle in human and mouse muscular dystrophy. J. Neurol. Sci. 2010, 297, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Brunherotti, M.A.; Sobreira, C.; Rodrigues-Junior, A.L.; de Assis, M.R.; Terra Filho, J.; Baddini Martinez, J.A. Correlations of egen klassifikation and barthel index scores with pulmonary function parameters in duchenne muscular dystrophy. Heart Lung J. Crit. Care 2007, 36, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Klapac, L.R.; Janssen, P.M.; Shontz, K.M.; Canan, B.; Montgomery, C.L.; Griffin, D.; Heller, K.; Schmelzer, L.; Handy, C.; Clark, K.R.; et al. Micro-dystrophin and follistatin co-delivery restores muscle function in aged dmd model. Hum. Mol. Genet. 2013, 22, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Camerino, G.M.; Cannone, M.; Giustino, A.; Massari, A.M.; Capogrosso, R.F.; Cozzoli, A.; De Luca, A. Gene expression in mdx mouse muscle in relation to age and exercise: Aberrant mechanical-metabolic coupling and implications for pre-clinical studies in duchenne muscular dystrophy. Hum. Mol. Genet. 2014, 23, 5720–5732. [Google Scholar] [CrossRef] [PubMed]
- Awano, H.; Takeshima, Y.; Okizuka, Y.; Saiki, K.; Yagi, M.; Matsuo, M. Wide ranges of serum myostatin concentrations in duchenne muscular dystrophy patients. Clin. Chim. Acta 2008, 391, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Han, D.S.; Hwu, W.L.; Thurberg, B.L.; Yang, W.S. Myostatin and insulin-like growth factor i: Potential therapeutic biomarkers for pompe disease. PLoS ONE 2013, 8, e71900. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Brandt, C.; Nielsen, A.R.; Hojman, P.; Whitham, M.; Febbraio, M.A.; Pedersen, B.K.; Plomgaard, P. Exercise induces a marked increase in plasma follistatin: Evidence that follistatin is a contraction-induced hepatokine. Endocrinology 2011, 152, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Mallidis, C.; Artaza, J.; Taylor, W.; Gonzalez-Cadavid, N.; Bhasin, S. Characterization of 5ʹ-regulatory region of human myostatin gene: Regulation by dexamethasone in vitro. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1128–E1136. [Google Scholar] [PubMed]
- Qin, J.; Du, R.; Yang, Y.Q.; Zhang, H.Q.; Li, Q.; Liu, L.; Guan, H.; Hou, J.; An, X.R. Dexamethasone-induced skeletal muscle atrophy was associated with upregulation of myostatin promoter activity. Res. Vet. Sci. 2013, 94, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Carrero, J.J.; Qureshi, A.R.; Anderstam, B.; Heimburger, O.; Barany, P.; Lindholm, B.; Stenvinkel, P. Circulating follistatin in patients with chronic kidney disease: Implications for muscle strength, bone mineral density, inflammation, and survival. Clin. J. Am. Soc. Nephrol. 2011, 6, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Mathews, K.D.; Cunniff, C.; Kantamneni, J.R.; Ciafaloni, E.; Miller, T.; Matthews, D.; Cwik, V.; Druschel, C.; Miller, L.; Meaney, F.J.; et al. Muscular dystrophy surveillance tracking and research network (md starnet): Case definition in surveillance for childhood-onset duchenne/becker muscular dystrophy. J. Child. Neurol. 2010, 25, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Cardenas, N.A.; Ibarra-Hernandez, F.; Lopez-Hernandez, L.B.; Escobar-Cedillo, R.E.; Ruano-Calderon, L.A.; Gomez-Diaz, B.; Garcia-Calderon, N.; Carriedo-Davila, M.F.; Rojas-Hurtado, L.G.; Luna-Padron, E.; et al. Diagnosis and treatment with steroids for patients with duchenne muscular dystrophy: Experience and recommendations for mexico. Rev. Neurol. 2013, 57, 455–462. [Google Scholar] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet. Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Gustincich, S.; Manfioletti, G.; Del Sal, G.; Schneider, C.; Carninci, P. A fast method for high-quality genomic DNA extraction from whole human blood. Biotechniques 1991, 11, 298–300, 302. [Google Scholar] [PubMed]
- Lopez-Hernandez, L.B.; Gomez-Diaz, B.; Luna-Angulo, A.B.; Anaya-Segura, M.; Bunyan, D.J.; Zuniga-Guzman, C.; Escobar-Cedillo, R.E.; Roque-Ramirez, B.; Ruano-Calderon, L.A.; Rangel-Villalobos, H.; et al. Comparison of mutation profiles in the duchenne muscular dystrophy gene among populations: Implications for potential molecular therapies. Int. J. Mol. Sci. 2015, 16, 5334–5346. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Diaz, B.; Rosas-Vargas, H.; Roque-Ramirez, B.; Meza-Espinoza, P.; Ruano-Calderon, L.A.; Fernandez-Valverde, F.; Escalante-Bautista, D.; Escobar-Cedillo, R.E.; Sanchez-Chapul, L.; Vargas-Canas, S.; et al. Immunodetection analysis of muscular dystrophies in mexico. Muscle Nerve 2012, 45, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.; Hartmann, C.; Scholz, V.; Jauch, A.; Zschocke, J. Mlpa analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics 2005, 6, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Most samples included in the study are still available for further analyses, stored at the Trasnlational Biomedicine Laboratory, in the Biomedical Research Division of the National Medical Centre “20 de Noviembre”, Institute for Social Security of State Workers, Mexico City.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anaya-Segura, M.A.; García-Martínez, F.A.; Montes-Almanza, L.Á.; Díaz, B.-G.; Ávila-Ramírez, G.; Alvarez-Maya, I.; Coral-Vázquez, R.M.; Mondragón-Terán, P.; Escobar-Cedillo, R.E.; García-Calderón, N.; et al. Non-Invasive Biomarkers for Duchenne Muscular Dystrophy and Carrier Detection. Molecules 2015, 20, 11154-11172. https://doi.org/10.3390/molecules200611154
Anaya-Segura MA, García-Martínez FA, Montes-Almanza LÁ, Díaz B-G, Ávila-Ramírez G, Alvarez-Maya I, Coral-Vázquez RM, Mondragón-Terán P, Escobar-Cedillo RE, García-Calderón N, et al. Non-Invasive Biomarkers for Duchenne Muscular Dystrophy and Carrier Detection. Molecules. 2015; 20(6):11154-11172. https://doi.org/10.3390/molecules200611154
Chicago/Turabian StyleAnaya-Segura, Mónica Alejandra, Froylan Arturo García-Martínez, Luis Ángel Montes-Almanza, Benjamín-Gómez Díaz, Guillermina Ávila-Ramírez, Ikuri Alvarez-Maya, Ramón Mauricio Coral-Vázquez, Paul Mondragón-Terán, Rosa Elena Escobar-Cedillo, Noemí García-Calderón, and et al. 2015. "Non-Invasive Biomarkers for Duchenne Muscular Dystrophy and Carrier Detection" Molecules 20, no. 6: 11154-11172. https://doi.org/10.3390/molecules200611154