
KMUP-1 Attenuates Endothelin-1-Induced Cardiomyocyte Hypertrophy through Activation of Heme Oxygenase-1 and Suppression of the Akt/GSK-3β, Calcineurin/NFATc4 and RhoA/ROCK Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
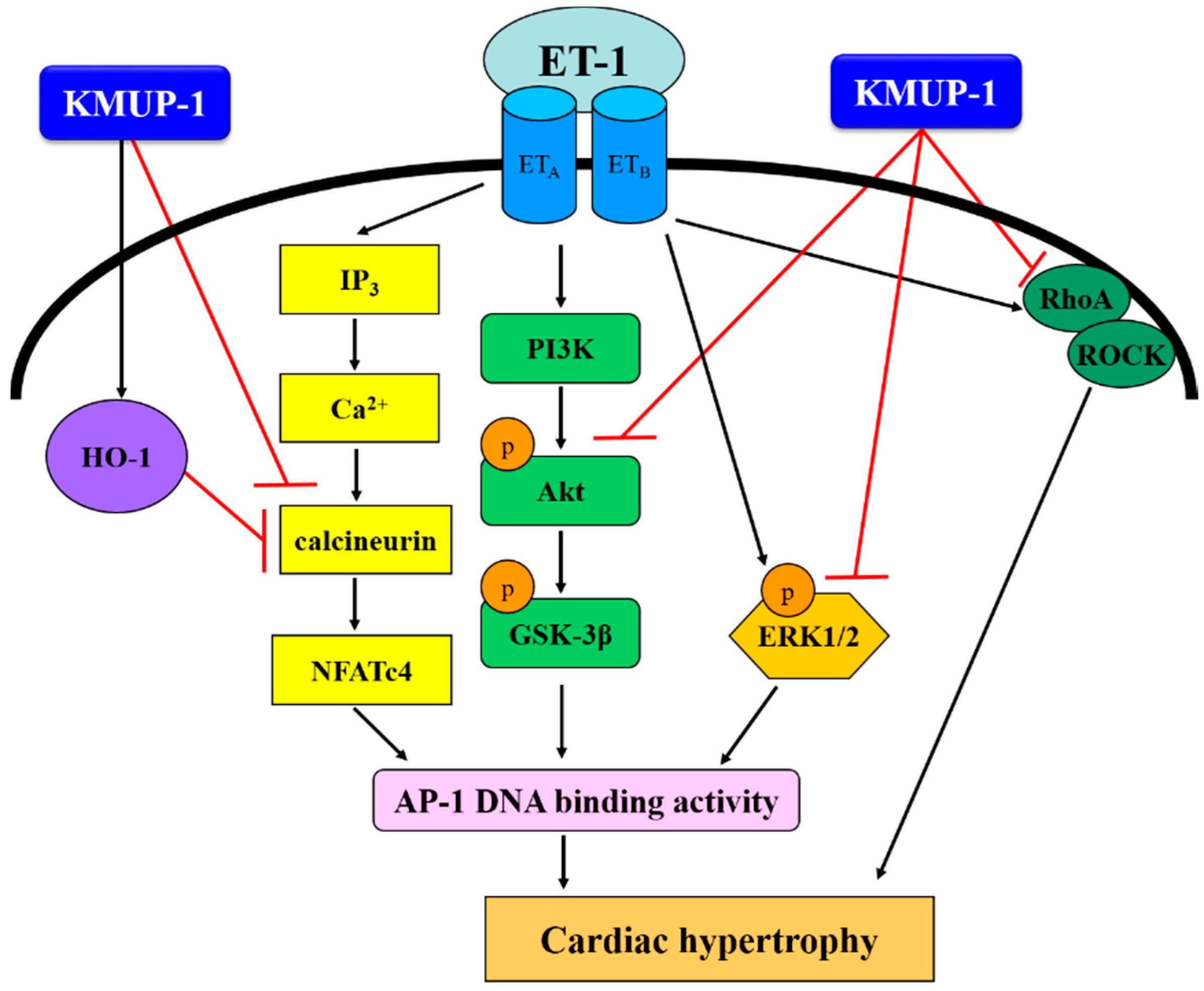
2. Results and Discussion
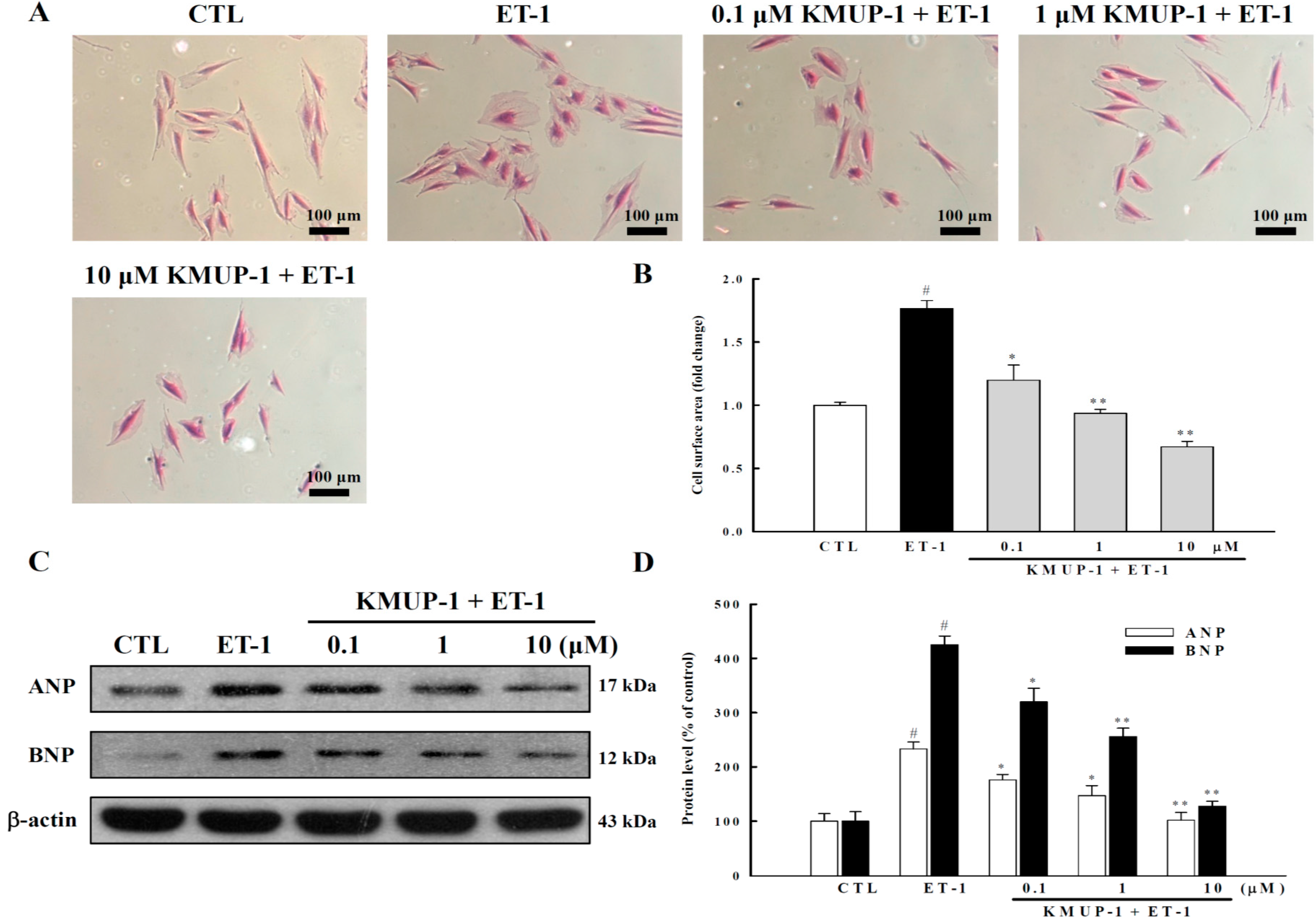
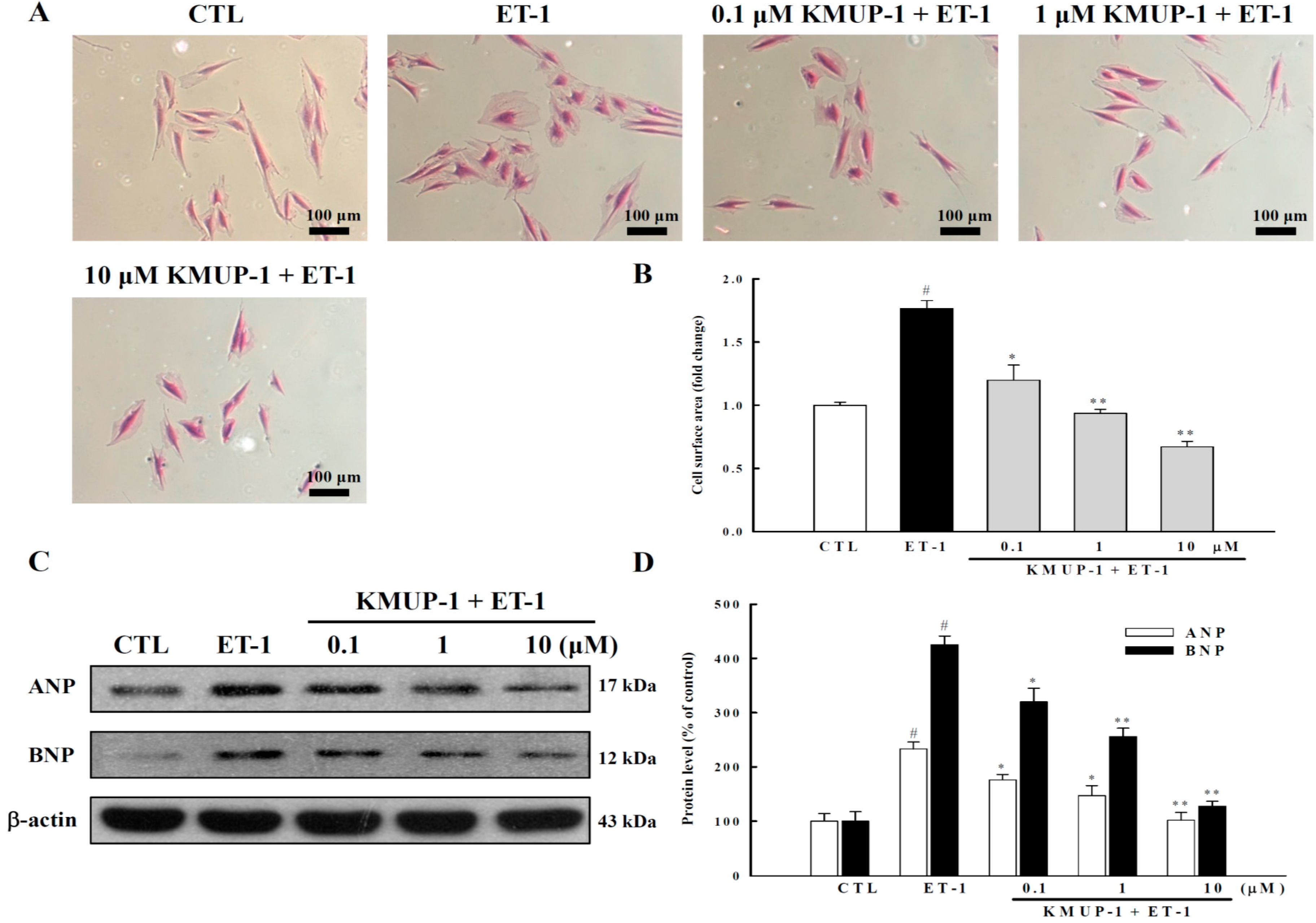
2.1. KMUP-1 Inhibited ET-1-Induced Cardiomyocyte Hypertrophy
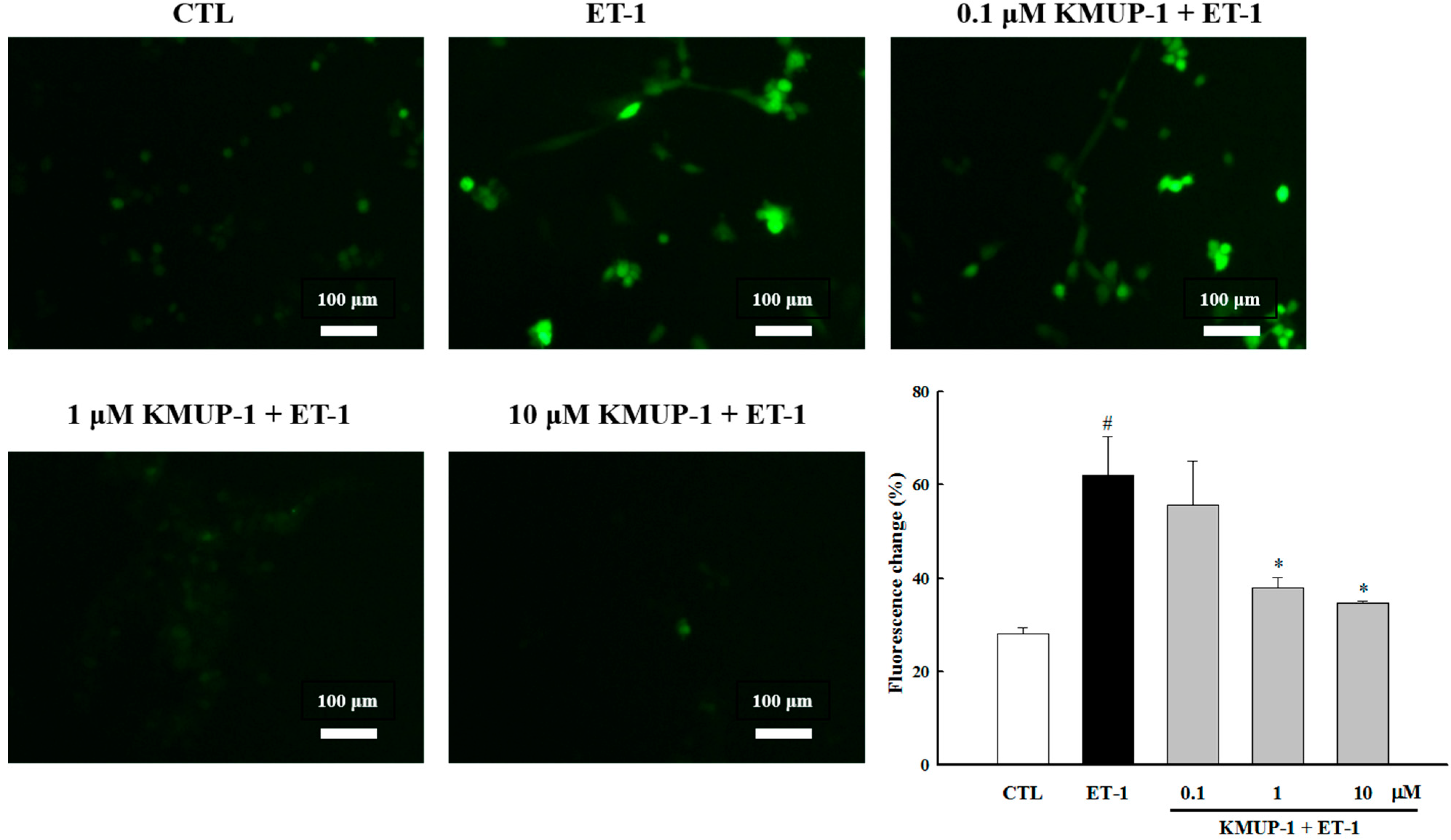
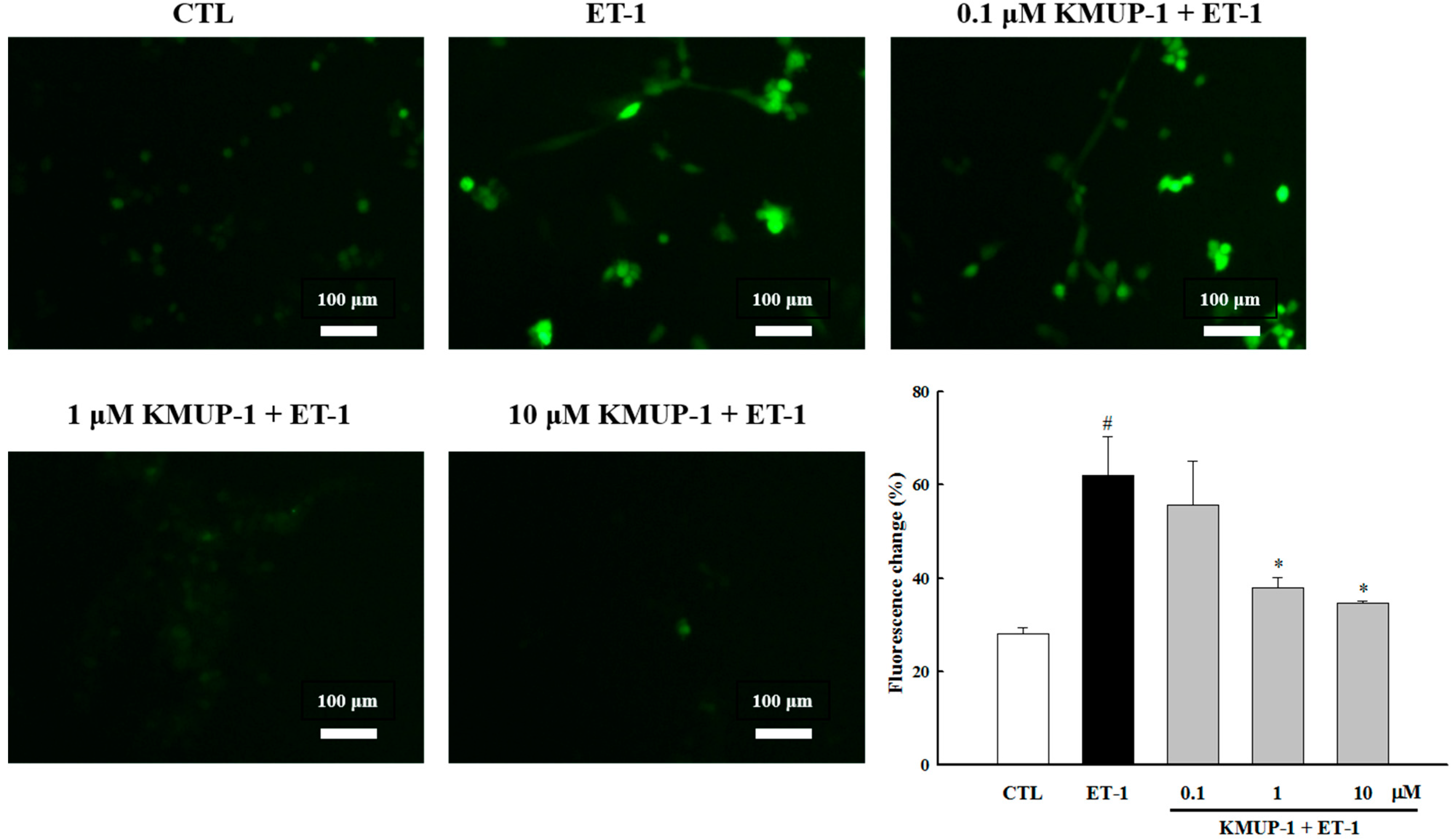
2.2. KMUP-1 Attenuated ET-1-Induced ROS Generation
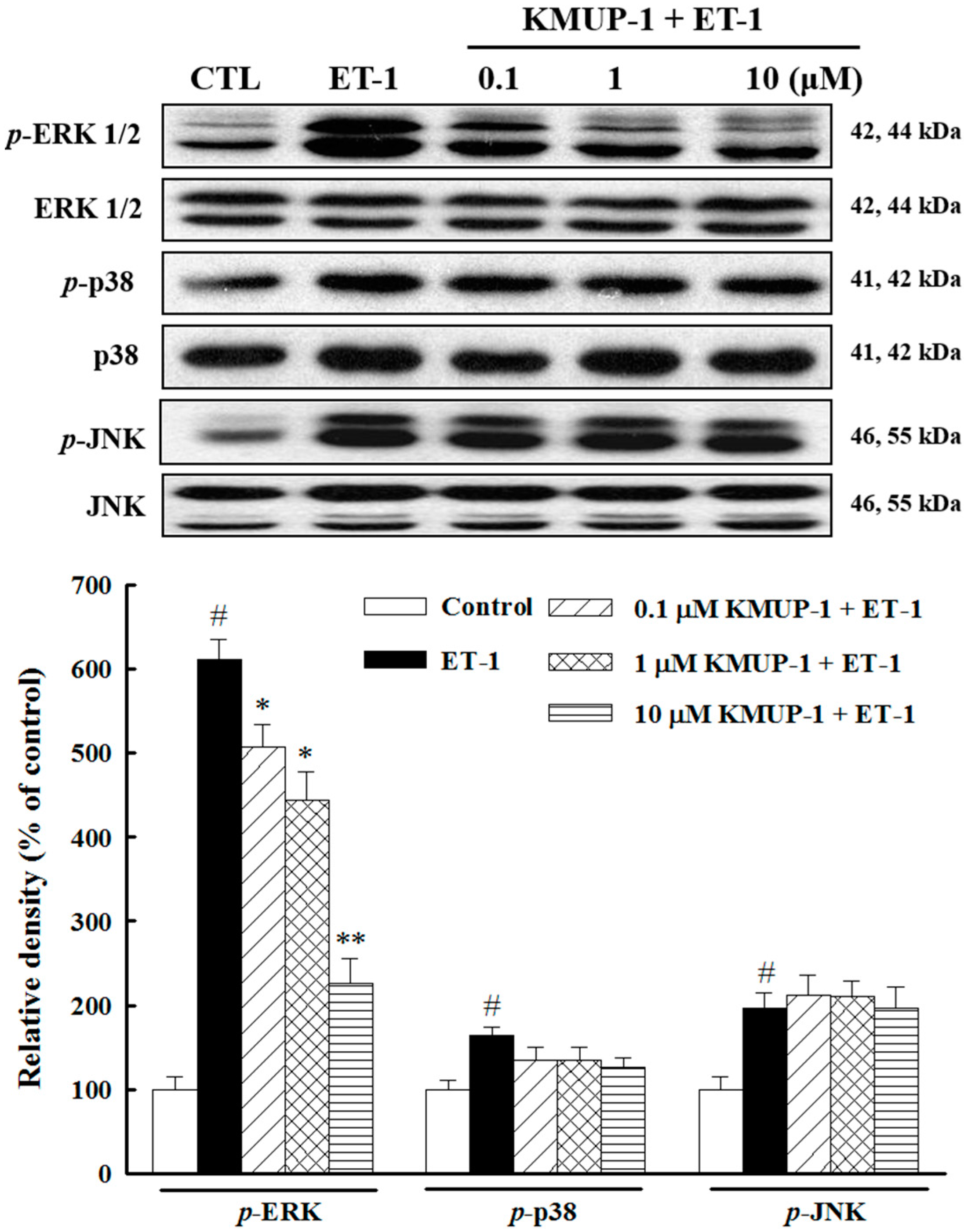
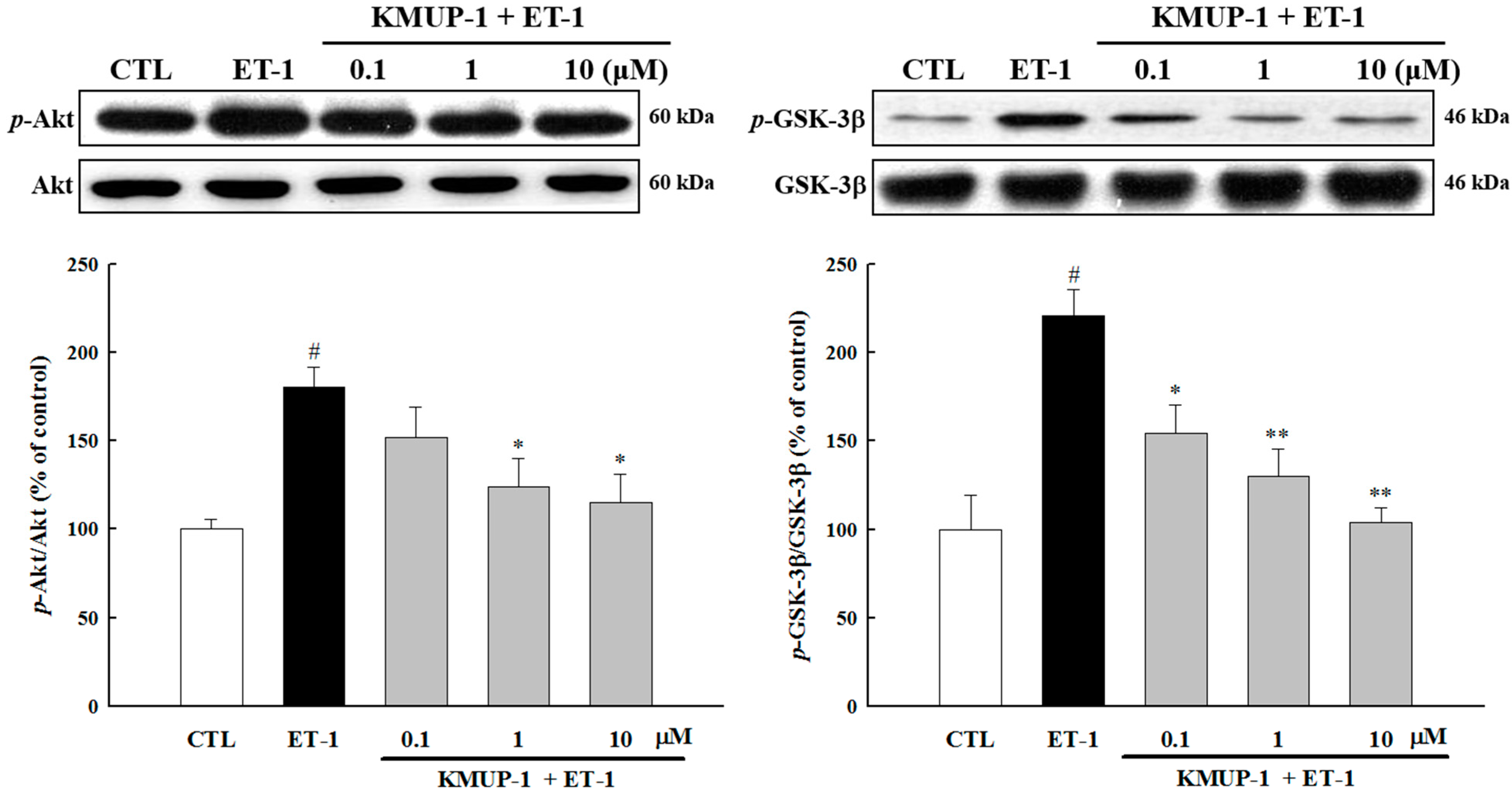
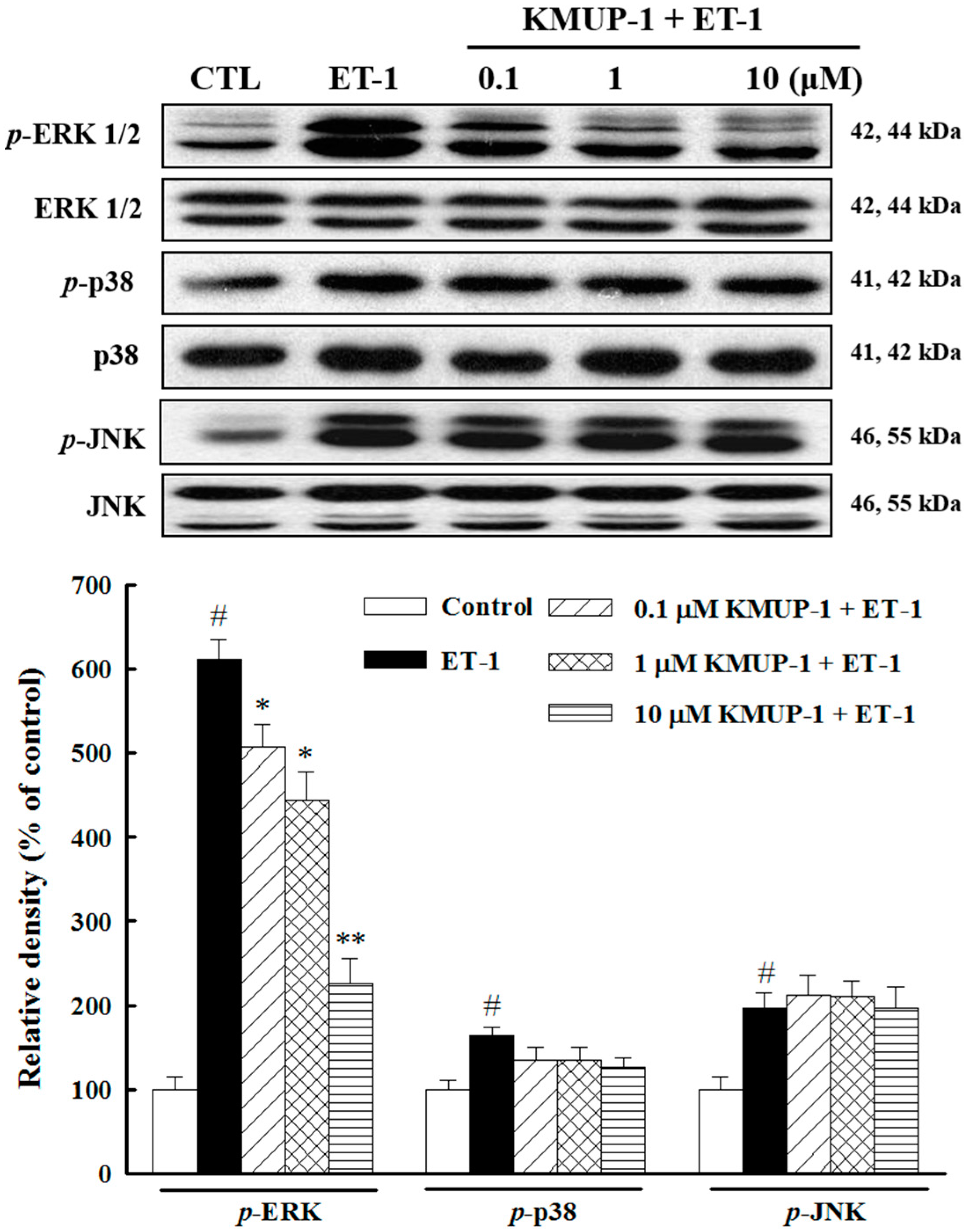
2.3. Effects of KMUP-1 on MAPKs and Akt/GSK-3β Signaling

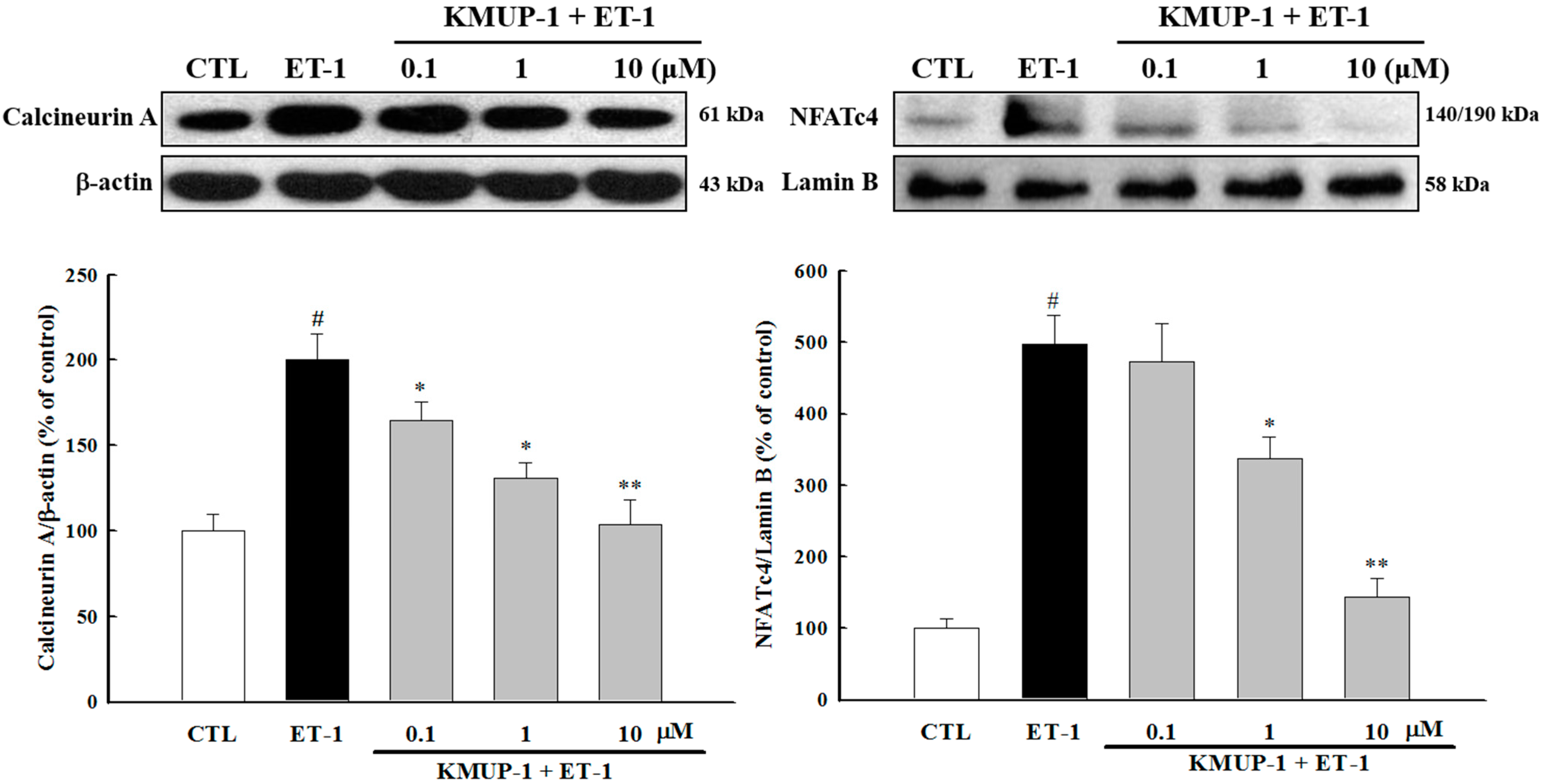
2.4. KMUP-1 Suppressed ET-1-Activated Calcineurin/NFATc4 Signaling Pathway

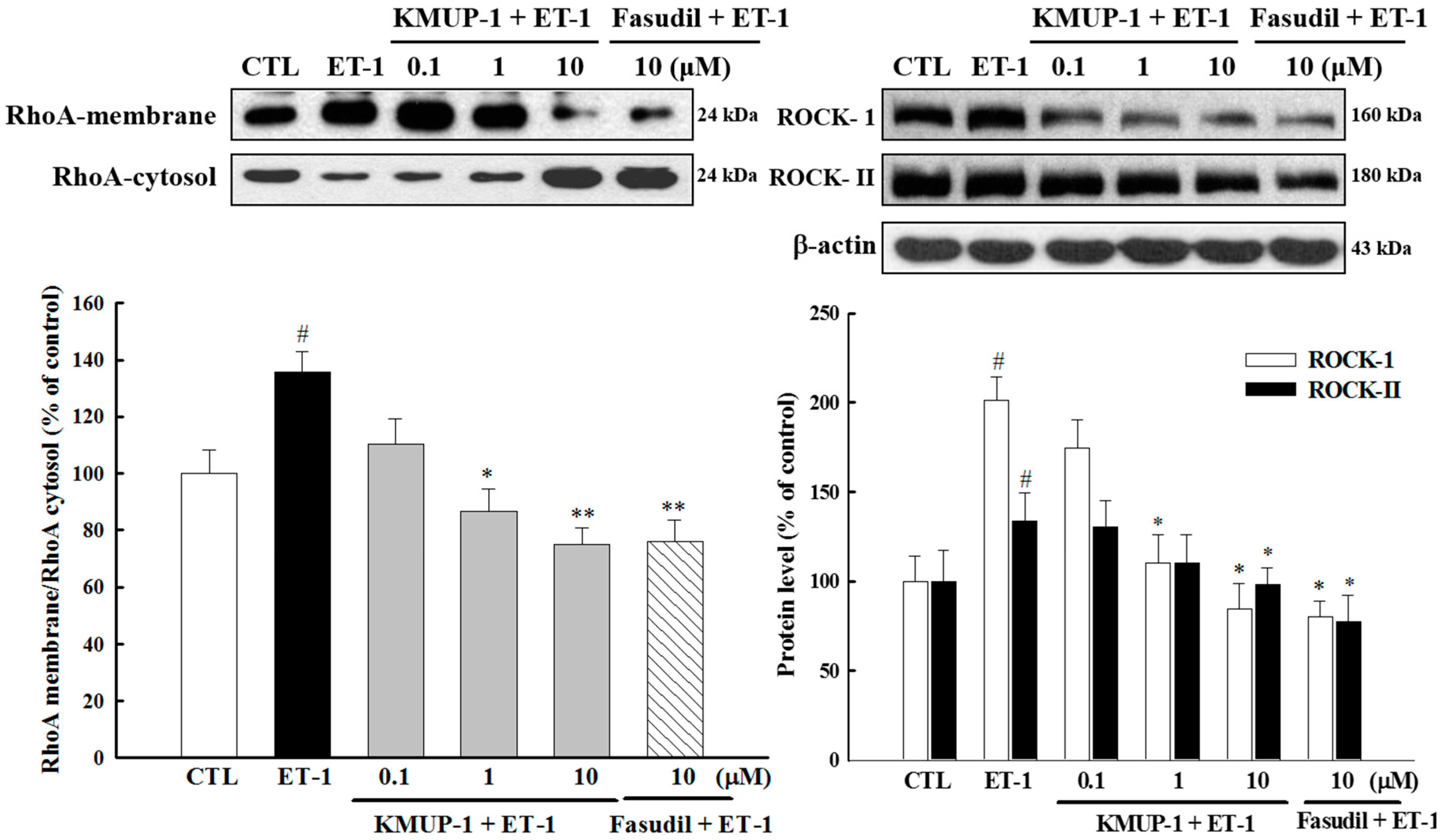
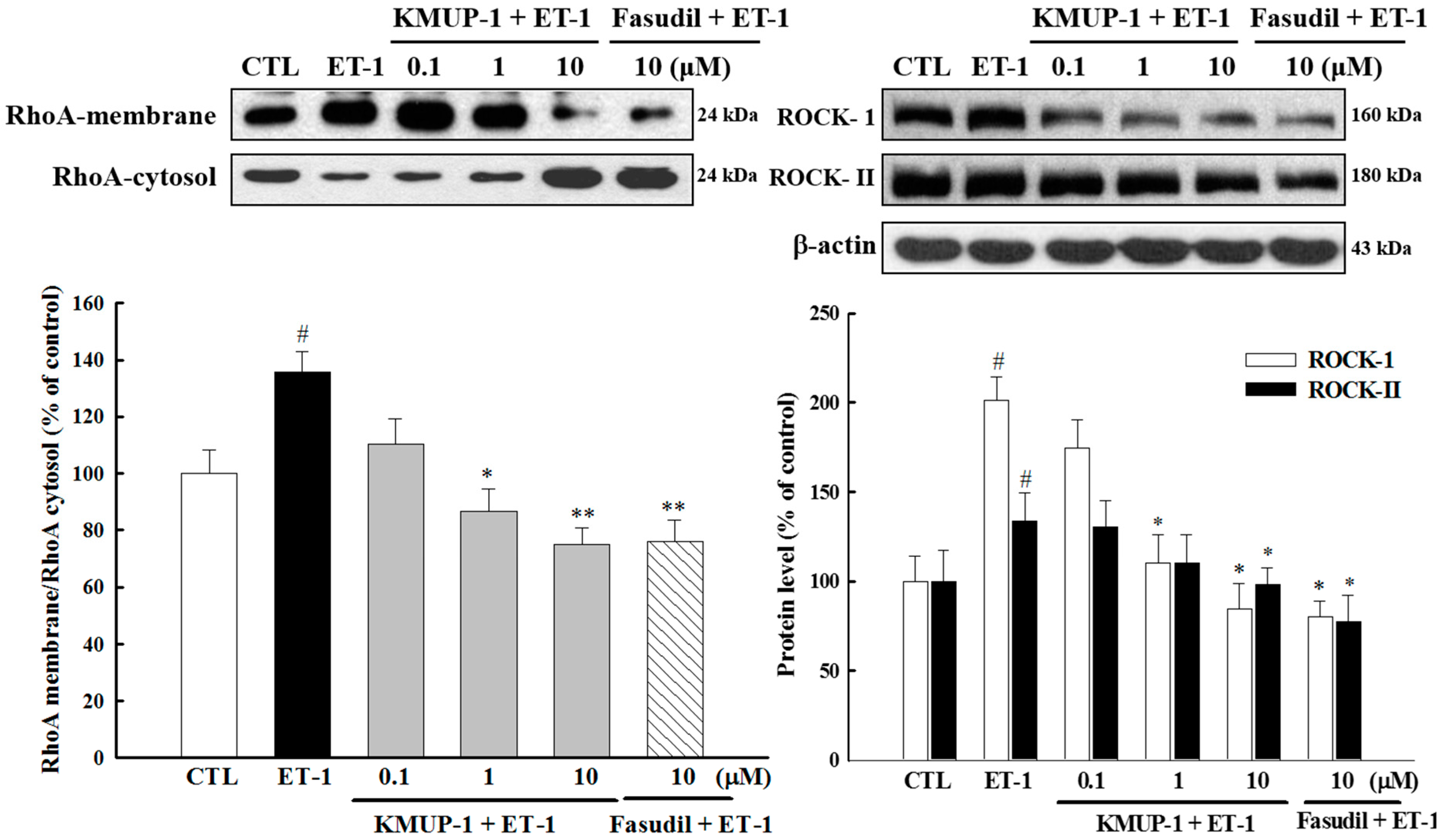
2.5. Effect of KMUP-1 on ET-1-Induced RhoA and ROCK Activation
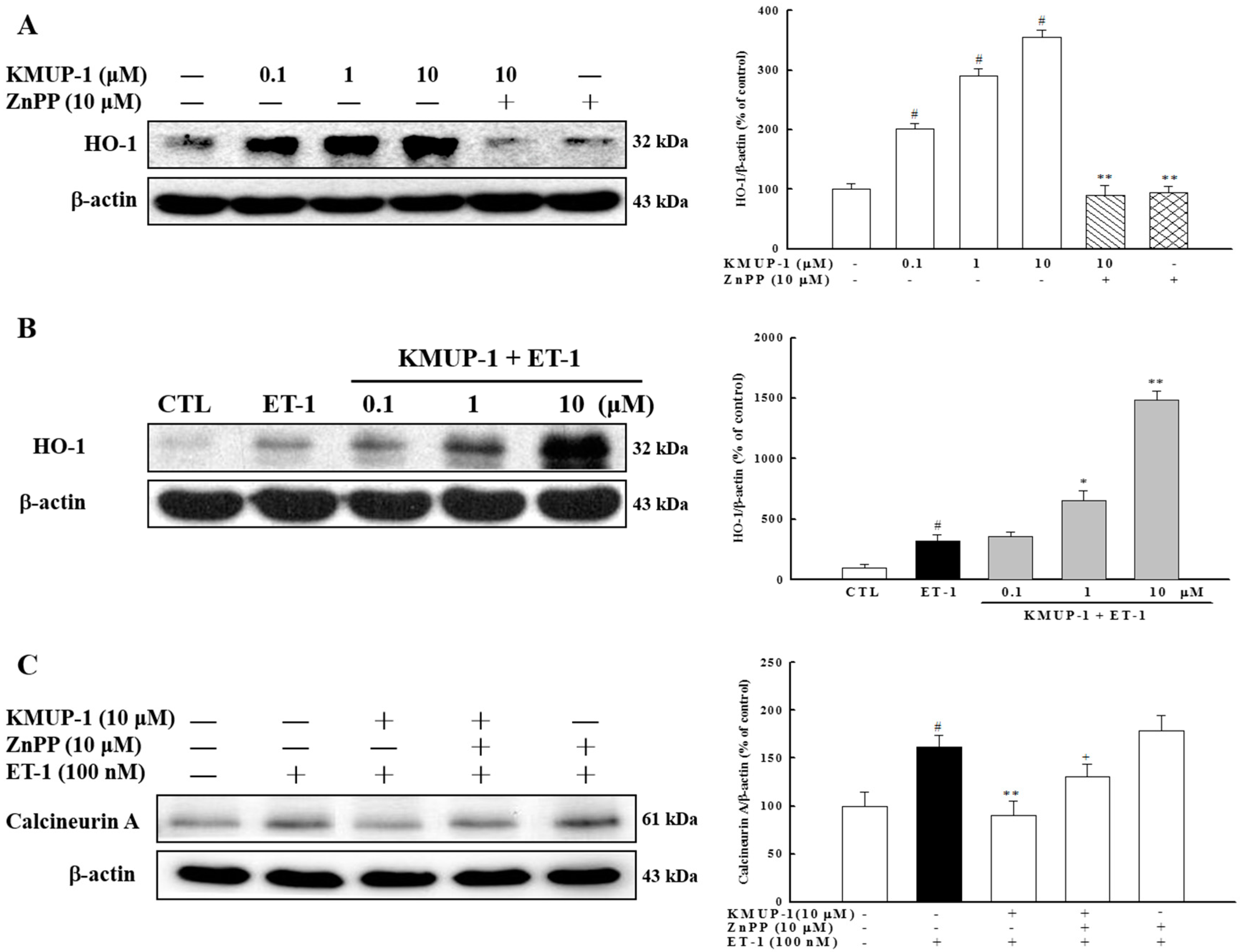
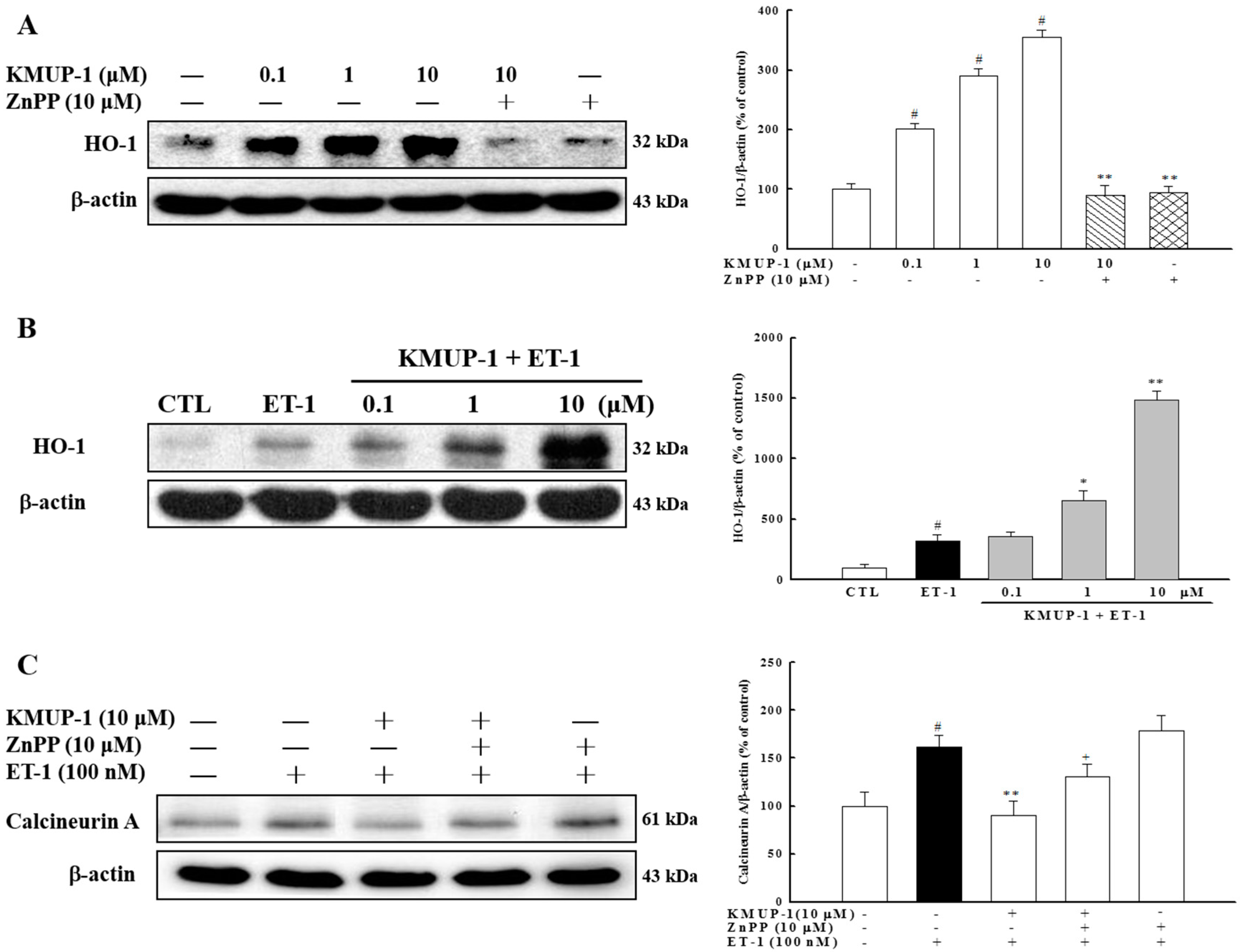
2.6. Effect of KMUP-1 on HO-1 Protein Expression
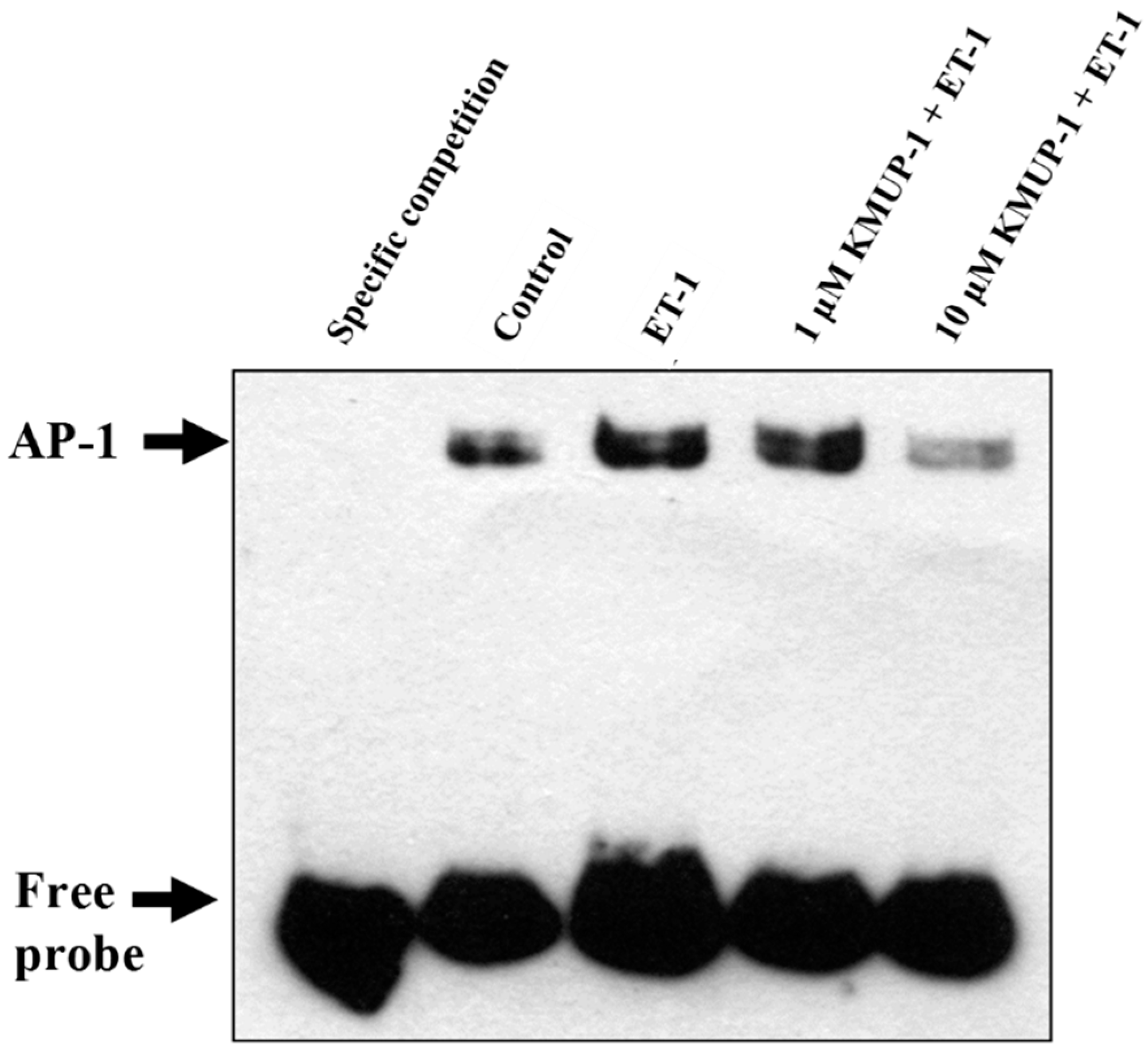
2.7. KMUP-1 Attenuated ET-1-Induced DNA Binding Activity of AP-1

3. Experimental Section
3.1. Materials
3.2. Cell Culture and Hypertrophy Induction
3.3. Measurement of Cell Surface Area
3.4. Determination of Intracellular Reactive Oxygen Species
3.5. Western Blot Analysis
3.6. Electrophoretic Mobility Shift Assay (EMSA)
3.7. Statistical Analysis
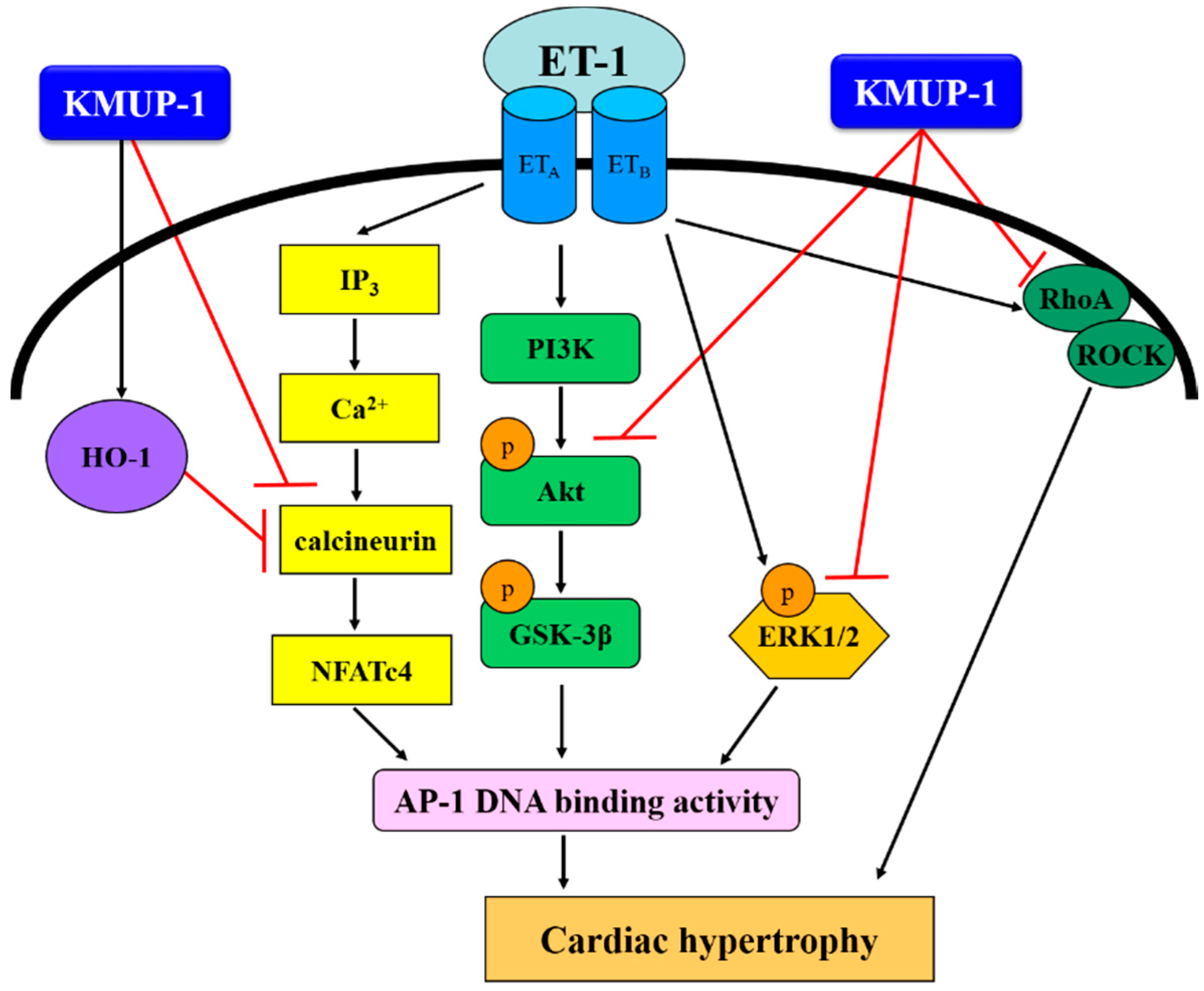
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Santulli, G. Epidemiology of Cardiovascular Disease in the 21st Century: Updated Numbers and Updated Facts. J. Cardiovasc. Dis. Res. 2013, 1, 1–2. [Google Scholar]
- Menaouar, A.; Florian, M.; Wang, D.; Danalache, B.; Jankowski, M.; Gutkowska, J. Anti-hypertrophic effects of oxytocin in rat ventricular myocytes. Int. J. Cardiol. 2014, 175, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Ling, H.; Grimm, M.; Zhang, T.; Bers, D.M.; Brown, J.H. Cardiac hypertrophy and heart failure development through Gq and CaM Kinase II signaling. J. Cardiovasc. Pharmacol. 2010, 56, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Cipolletta, E.; Sorriento, D.; del Giudice, C.; Anastasio, A.; Monaco, S.; Maione, A.S.; Condorelli, G.; Puca, A.; Trimarco, B.; et al. CaMK4 Gene Deletion Induces Hypertension. J. Am. Heart Assoc. 2012, 1, e001081. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.L.; Gu, J.L.; Wang, C.; Reith, A.D.; Lee, J.C.; Mirabile, R.C.; Kreutz, R.; Wang, Y.; Maleeff, B.; Parsons, A.A.; et al. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrineinduced cardiomyocyte hypertrophy. J. Biol. Chem. 2000, 275, 37895–37901. [Google Scholar] [CrossRef] [PubMed]
- Irukayama-Tomobe, Y.; Miyauchi, T.; Sakai, S.; Kasuya, Y.; Ogata, T.; Takanashi, M.; Iemitsu, M.; Sudo, T.; Goto, K.; Yamaguchi, I. Endothelin-1-induced cardiac hypertrophy is inhibited by activation of peroxisome proliferator-activated receptor-alpha partly via blockade of c-Jun NH2-terminal kinase pathway. Circulation 2004, 109, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, N.; Jesmin, S.; Zaedi, S.; Maeda, S.; Soma, M.; Aonuma, K.; Yamaguchi, I.; Miyauchi, T. Eicosapentanoic acid prevents endothelin-1-induced cardiomyocyte hypertrophy in vitro through the suppression of TGF-beta1 and phosphorylated JNK. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H835–H845. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Zeidan, A.; Javadov, S.; Kilić, A.; Rajapurohitam, V.; Karmazyn, M. Nitric oxide inhibits endothelin-1-induced neonatal cardiomyocyte hypertrophy via a RhoA-ROCK-dependent pathway. J. Mol. Cell. Cardiol. 2009, 47, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Guan, P.; Zhang, J.P.; Li, Y.Q.; Chang, Y.Z.; Shi, Z.H.; Wang, F.Y.; Chu, L. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses isoproterenol-induced heart failure in rats via JNK and ERK1/2 pathways. J. Cell. Biochem. 2011, 112, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.N.; Lin, R.J.; Lin, C.Y.; Shen, K.P.; Chiang, L.C.; Chen, I.J. A xanthine-based KMUP-1 with cyclic GMP enhancing and K+ channels opening activities in rat aortic smooth muscle. Br. J. Pharmacol. 2001, 134, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.N.; Chen, C.W.; Liou, S.F.; Yeh, J.L.; Chung, H.H.; Chen, I.J. Inhibition of proinflammatory tumor necrosis factor-{alpha}-induced inducible nitric-oxide synthase by xanthine-based 7-[2-[4-(2-chlorobenzene)piperazinyl]ethyl]-1,3-dimethylxanthine (KMUP-1) and 7-[2-[4-(4-nitrobenzene)piperazinyl]ethyl]-1,3-dimethylxanthine (KMUP-3) in rat trachea: The involvement of soluble guanylate cyclase and protein kinase G. Mol. Pharmacol. 2006, 70, 977–985. [Google Scholar] [PubMed]
- Liu, C.M.; Lo, Y.C.; Wu, B.N.; Wu, W.J.; Chou, Y.H.; Huang, C.H.; An, L.M.; Chen, I.J. cGMP-enhancing- and alpha1A/alpha1D-adrenoceptor blockade-derived inhibition of Rho-kinase by KMUP-1 provides optimal prostate relaxation and epithelial cell anti-proliferation efficacy. Prostate 2007, 67, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Lo, Y.C.; Tai, M.H.; Wu, B.N.; Wu, W.J.; Chou, Y.H.; Chai, C.Y.; Huang, C.H.; Chen, I.J. Piperazine-designed alpha 1A/alpha 1D-adrenoceptor blocker KMUP-1 and doxazosin provide down-regulation of androgen receptor and PSA in prostatic LNCaP cells growth and specifically in xenografts. Prostate 2009, 69, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.L; Hsu, J.H.; Wu, P.J; Liou, S.F.; Liu, C.P.; Chen, I.J.; Wu, B.N.; Dai, Z.K.; Wu, J.R. KMUP-1 attenuates isoprenaline-induced cardiac hypertrophy in rats through NO/cGMP/PKG and ERK1/2/calcineurin A pathways. Br. J. Pharmacol. 2010, 159, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.P.; Dai, Z.K.; Huang, C.H.; Yeh, J.L.; Wu, B.N.; Wu, J.R.; Chen, I.J. Endothelial nitric oxide synthase-enhancing G-protein coupled receptor antagonist inhibits pulmonary artery hypertension by endothelin-1-dependent and endothelin-1-independent pathways in a monocrotaline model. Kaohsiung J. Med. Sci. 2014, 30, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Bueno, O.F.; Molkentin, J.D. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ. Res. 2002, 91, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ev, I. ROS and RNS signaling in heart disorders: Could antioxidant treatment be successful? Oxid. Med. Cell. Longev. 2011, 2011, 293769. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.G.; Molinaro, M.; Angelone, T.; Lionetti, V.; Madonna, R.; Mangiacapra, F.; Moccia, F.; Penna, C.; Sartiani, L.; Quaini, F.; et al. Nitroso-Redox Balance and Modulation of Basal Myocardial Function: an Update from the Italian Society of Cardiovascular Research (SIRC). Curr. Drug Targets 2015, in press. [Google Scholar] [CrossRef]
- Hirotani, S.; Otsu, K.; Nishida, K.; Higuchi, Y.; Morita, T.; Nakayama, H.; Yamaguchi, O.; Mano, T.; Matsumura, Y.; Ueno, H.; et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist- induced cardiomyocyte hypertrophy. Circulation 2002, 105, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Liu, J.C.; Chen, Y.L.; Chen, C.H.; Lin, H.; Lin, J.W.; Chiu, W.T.; Chen, J.J.; Cheng, T.H. Inhibitory effect of trilinolein on endothelin-1-induced c-fos gene expression in cultured neonatal rat cardiomyocytes. Naunyn Schmiedeberg’s Arch. Pharmacol. 2005, 372, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, A.; Woodman, O.L.; Cao, A.H.; Drummond, G.R.; Marshall, T.; Kaye, D.M.; Ritchie, R.H. Antioxidant actions contribute to the antihypertrophic effects of atrial natriuretic peptide in neonatal rat cardiomyocytes. Cardiovasc. Res. 2006, 72, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Tongers, J.; Fiedler, B.; König, D.; Kempf, T.; Klein, G.; Heineke, J.; Kraft, T.; Gambaryan, S.; Lohmann, S.M.; Drexler, H.; et al. Heme oxygenase-1 inhibition of MAP kinases, calcineurin/NFAT signaling, and hypertrophy in cardiac myocytes. Cardiovasc. Res. 2004, 63, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zheng, W.; Pi, R.; Gao, J.; Zhang, H.; Wang, P.; Le, K.; Liu, P. Activation of peroxisome proliferator-activated receptor-alpha prevents glycogen synthase 3beta phosphorylation and inhibits cardiac hypertrophy. FEBS Lett. 2007, 581, 3311–3316. [Google Scholar] [CrossRef] [PubMed]
- Maillet, M; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell. Biol. 2013, 14, 38–48. [Google Scholar]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Marks, A.R. Dysfunctional ryanodine receptors in the heart: new insights into complex cardiovascular diseases. J. Mol. Cell. Cardiol. 2013, 58, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Chen, Z.; Santulli, G.; Gu, L.; Yang, Z.G.; Yuan, Z.Q.; Zhao, Y.T.; Xin, H.B.; Deng, K.Y.; Wang, S.Q.; et al. Functional role of Calstabin2 in age-related cardiac alterations. Sci. Rep. 2014, 4, 7425. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Ding, W.; Liao, Y.; Liu, Y.; Yan, D.; Zhang, Y.; Wang, R.; Zheng, N.; Liu, S.; Liu, J. Polydatin prevents hypertrophy in phenylephrine induced neonatal mouse cardiomyocytes and pressure-overload mouse models. Eur. J. Pharmacol. 2015, 746, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhang, J.D.; Wang, B.; Lv, Y.J.; Jiang, H.; Liu, G.L.; Qiao, Y.; Ren, M.; Guo, X.F. Quercetin inhibits left ventricular hypertrophy in spontaneously hypertensive rats and inhibits angiotensin II-induced H9C2 cells hypertrophy by enhancing PPAR-γ expression and suppressing AP-1 activity. PLoS ONE 2013, 8, e72548. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Castellazzi, M.; Wollert, K.C.; Trautwein, C.; Schunkert, H.; Drexler, H. JunD attenuates phenylephrine-mediated cardiomyocyte hypertrophy by negatively regulating AP-1 transcriptional activity. Cardiovasc. Res. 2006, 71, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Santulli, G.; Fusco, A.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension 2010, 56, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Shravah, J.; Luo, H.; Raedschelders, K.; Chen, D.D.; Ansley, D.M. Propofol protects against hydrogen peroxide-induced injury in cardiac H9c2 cells via Akt activation and Bcl-2 up-regulation. Biochem. Biophys. Res. Commun. 2009, 389, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liou, S.-F.; Hsu, J.-H.; Chen, Y.-T.; Chen, I.-J.; Yeh, J.-L. KMUP-1 Attenuates Endothelin-1-Induced Cardiomyocyte Hypertrophy through Activation of Heme Oxygenase-1 and Suppression of the Akt/GSK-3β, Calcineurin/NFATc4 and RhoA/ROCK Pathways. Molecules 2015, 20, 10435-10449. https://doi.org/10.3390/molecules200610435
Liou S-F, Hsu J-H, Chen Y-T, Chen I-J, Yeh J-L. KMUP-1 Attenuates Endothelin-1-Induced Cardiomyocyte Hypertrophy through Activation of Heme Oxygenase-1 and Suppression of the Akt/GSK-3β, Calcineurin/NFATc4 and RhoA/ROCK Pathways. Molecules. 2015; 20(6):10435-10449. https://doi.org/10.3390/molecules200610435
Chicago/Turabian StyleLiou, Shu-Fen, Jong-Hau Hsu, You-Ting Chen, Ing-Jun Chen, and Jwu-Lai Yeh. 2015. "KMUP-1 Attenuates Endothelin-1-Induced Cardiomyocyte Hypertrophy through Activation of Heme Oxygenase-1 and Suppression of the Akt/GSK-3β, Calcineurin/NFATc4 and RhoA/ROCK Pathways" Molecules 20, no. 6: 10435-10449. https://doi.org/10.3390/molecules200610435