
Glutathione Levels and Susceptibility to Chemically Induced Injury in Two Human Prostate Cancer Cell Lines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
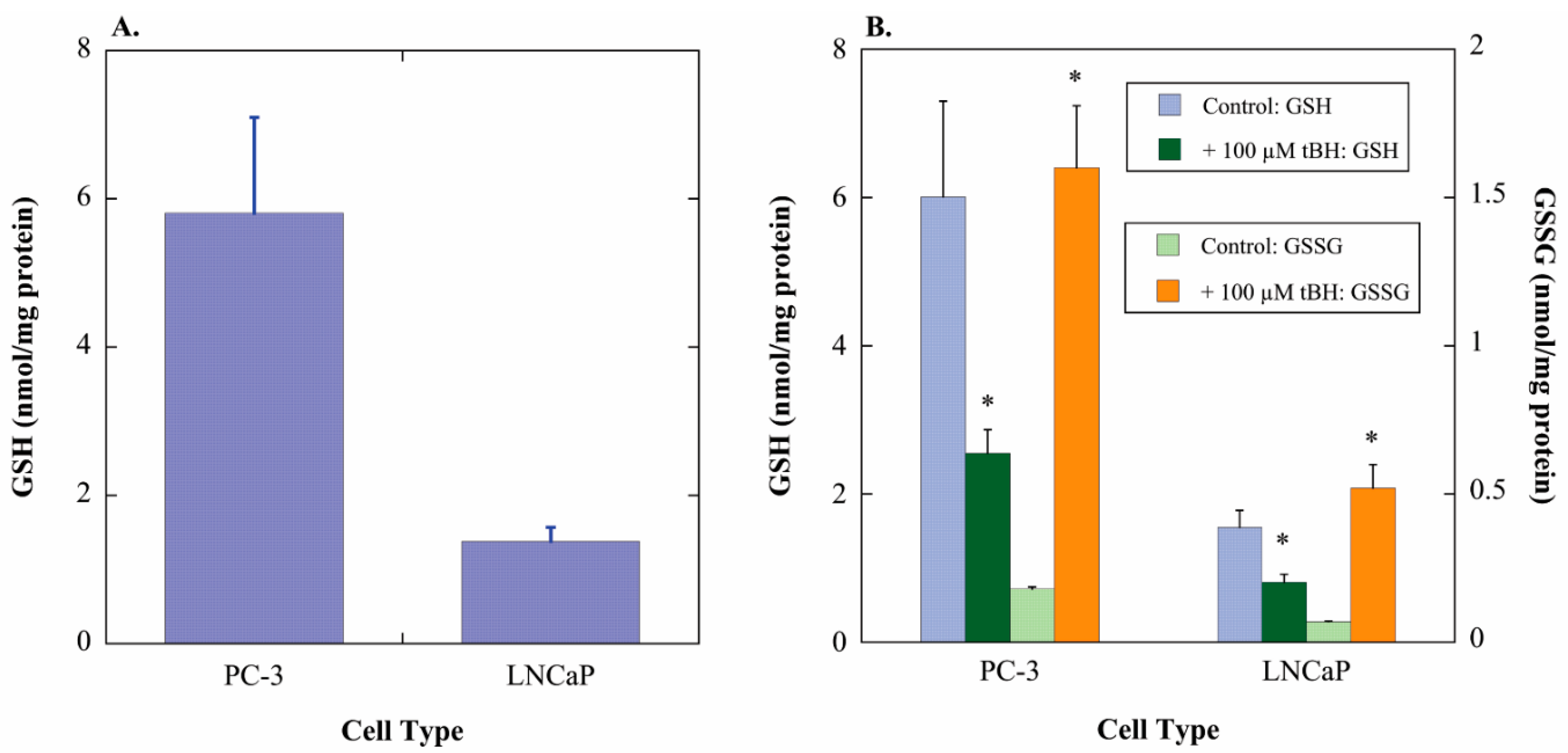
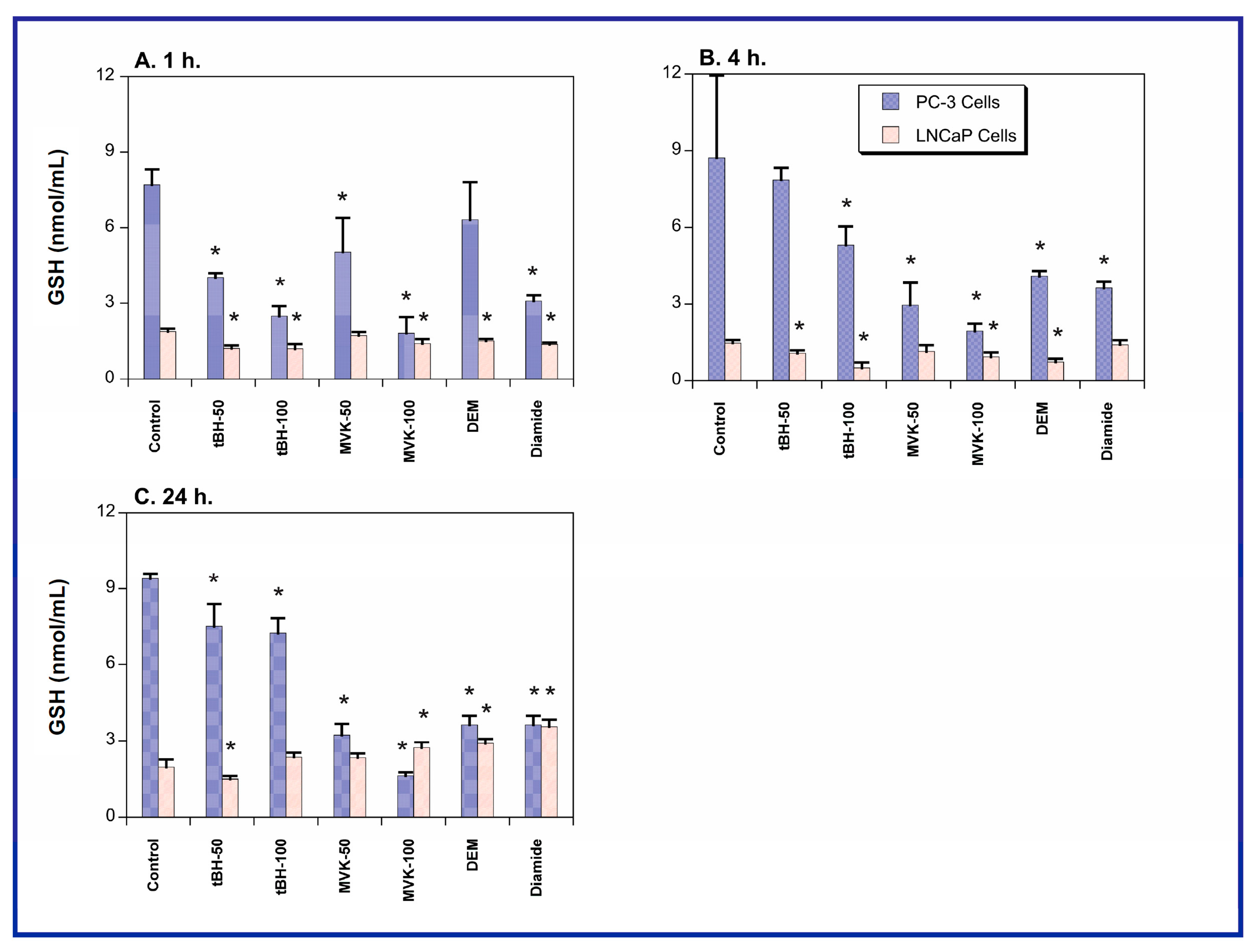
2.1. GSH Concentrations and Oxidant-Induced GSH Oxidation in PC-3 and LNCaP Cells
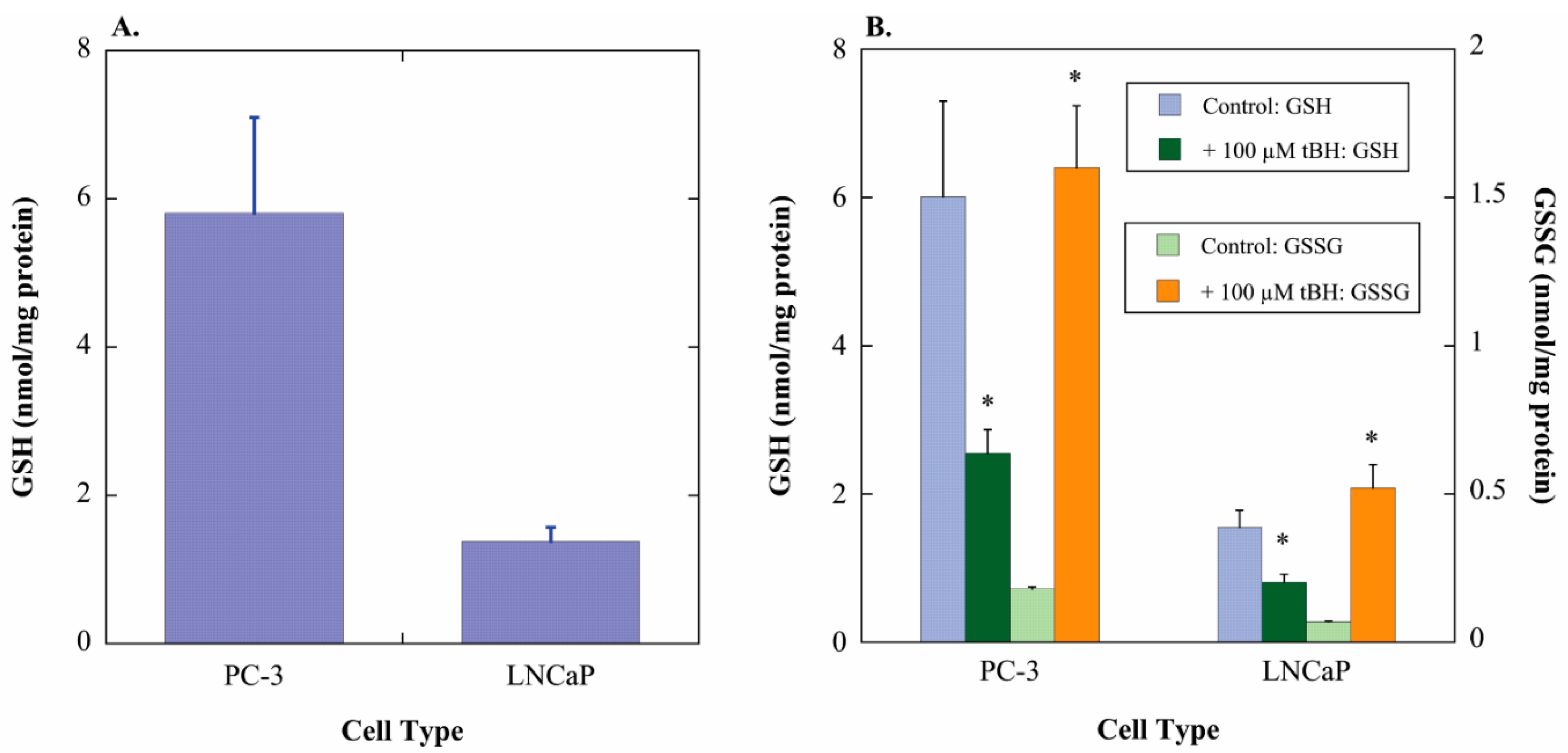
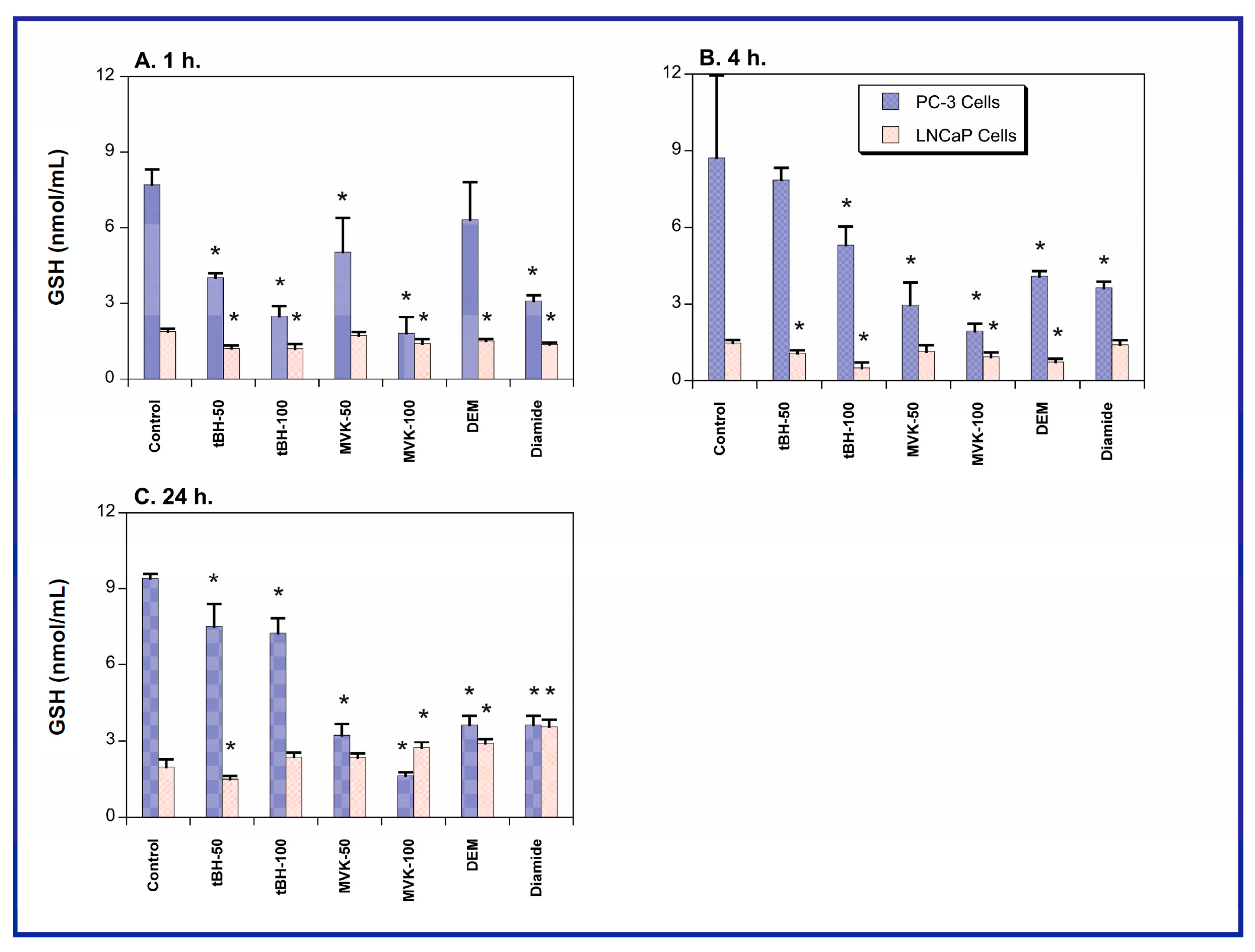
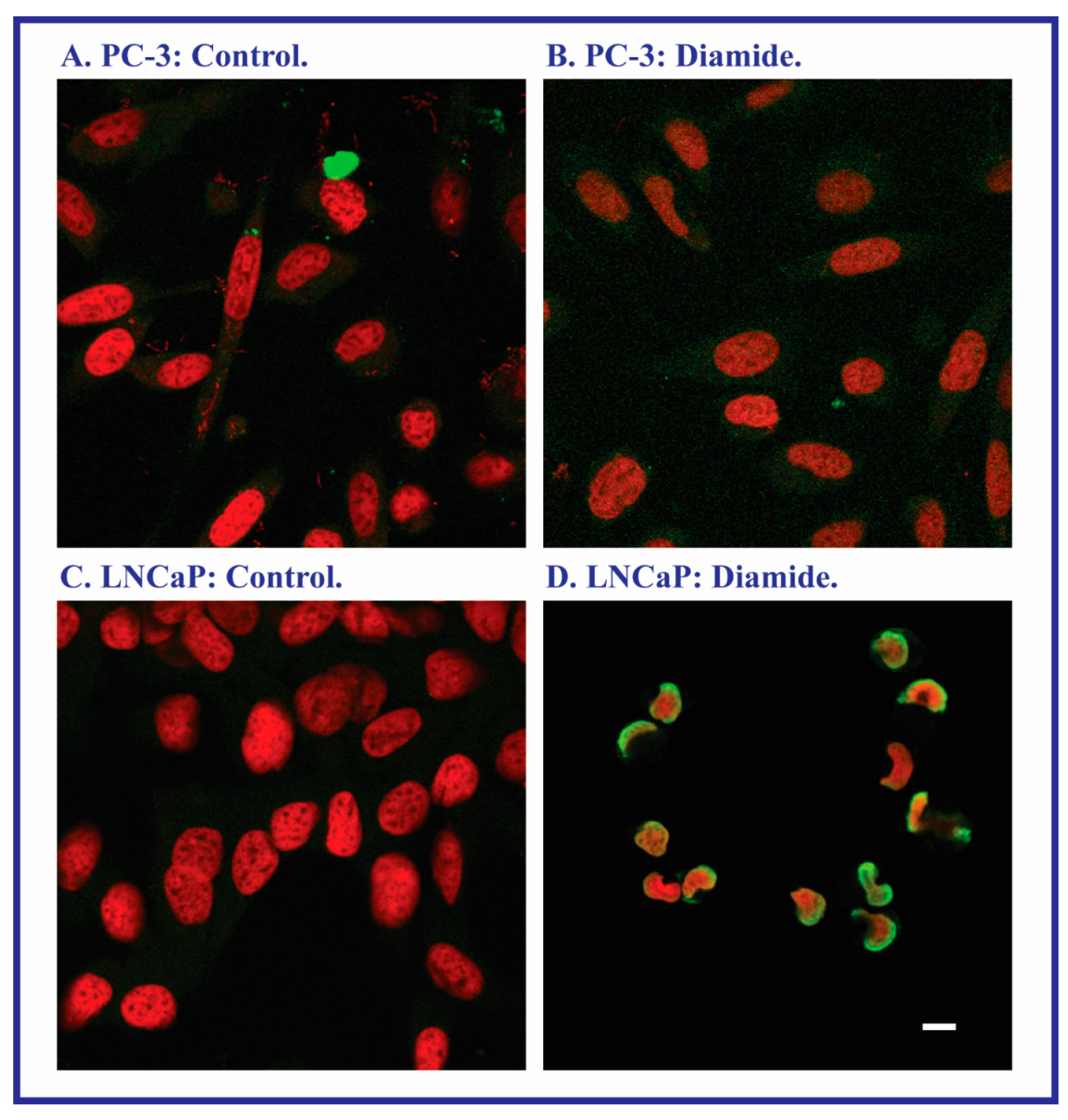
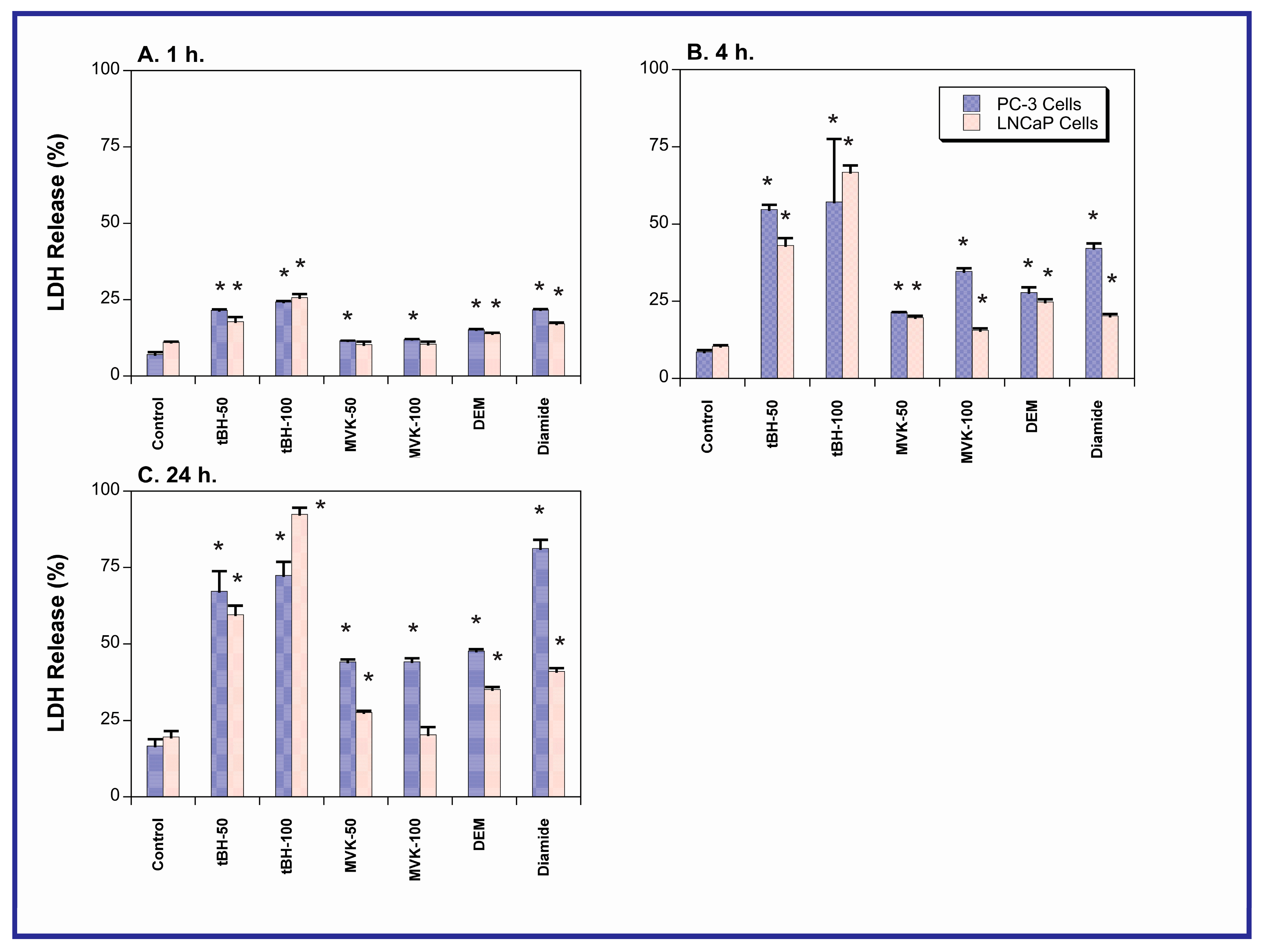
2.2. Acute Cellular Necrosis and Apoptosis Induced by Toxicants in PC-3 and LNCaP Cells
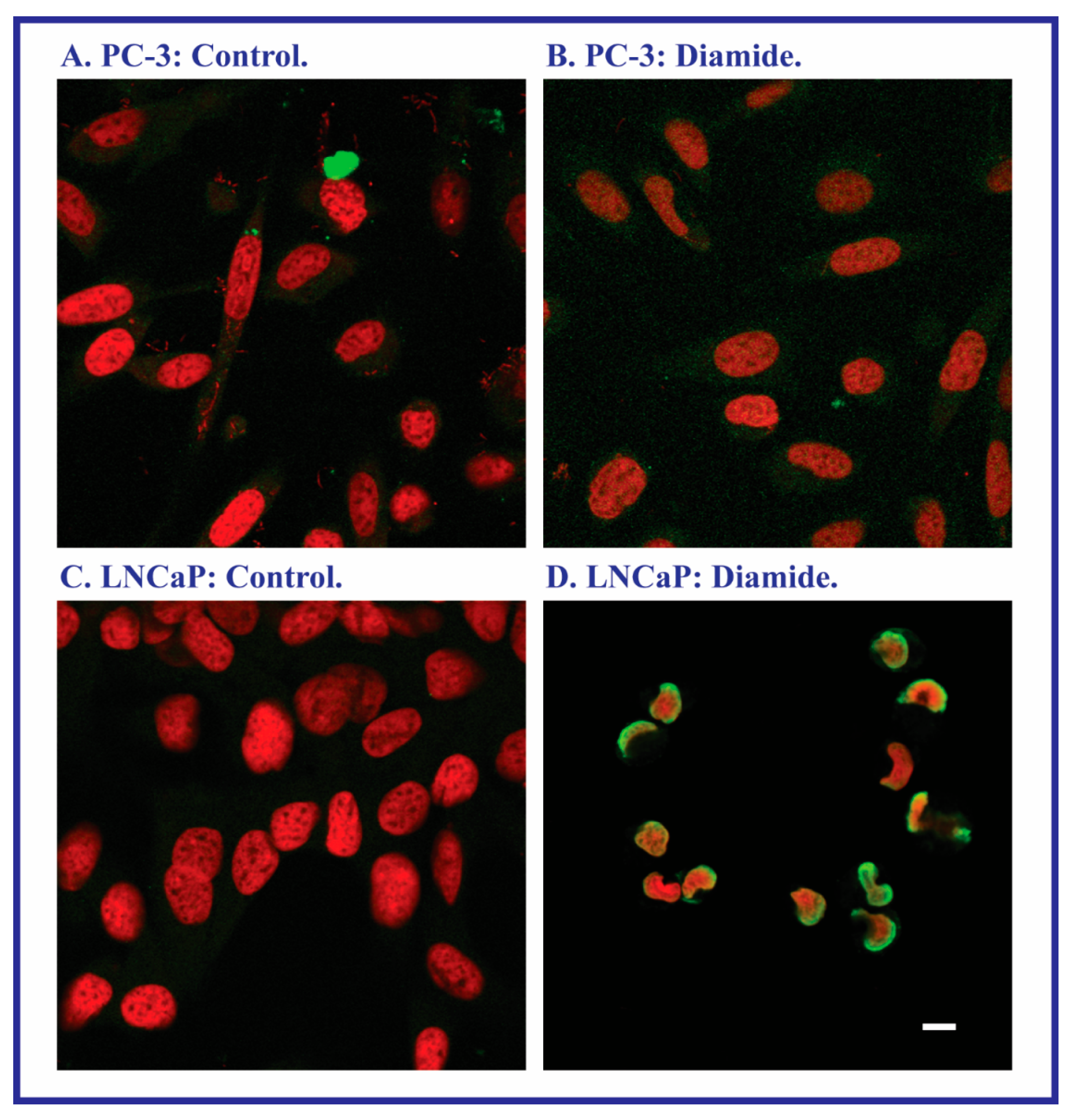
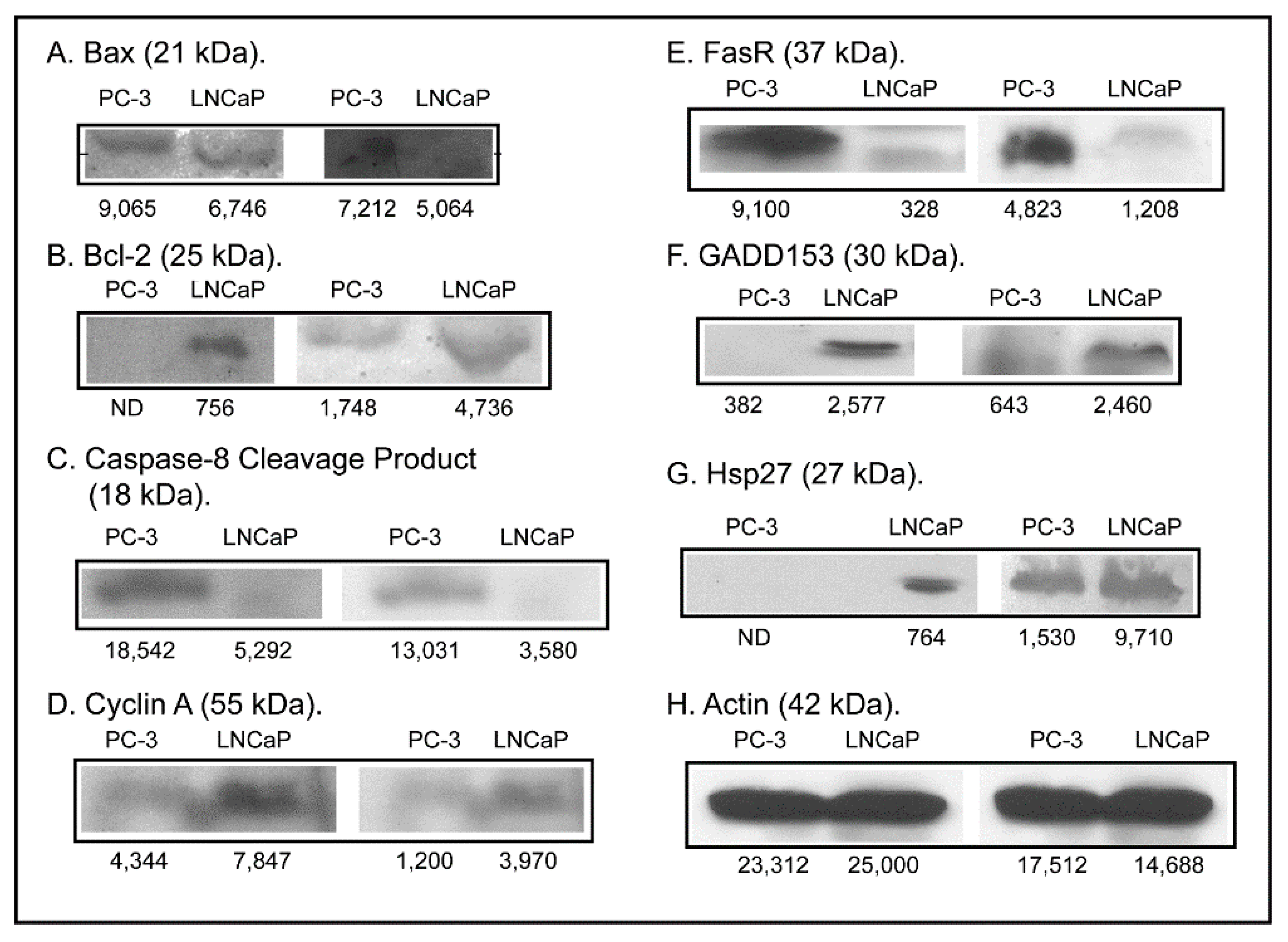
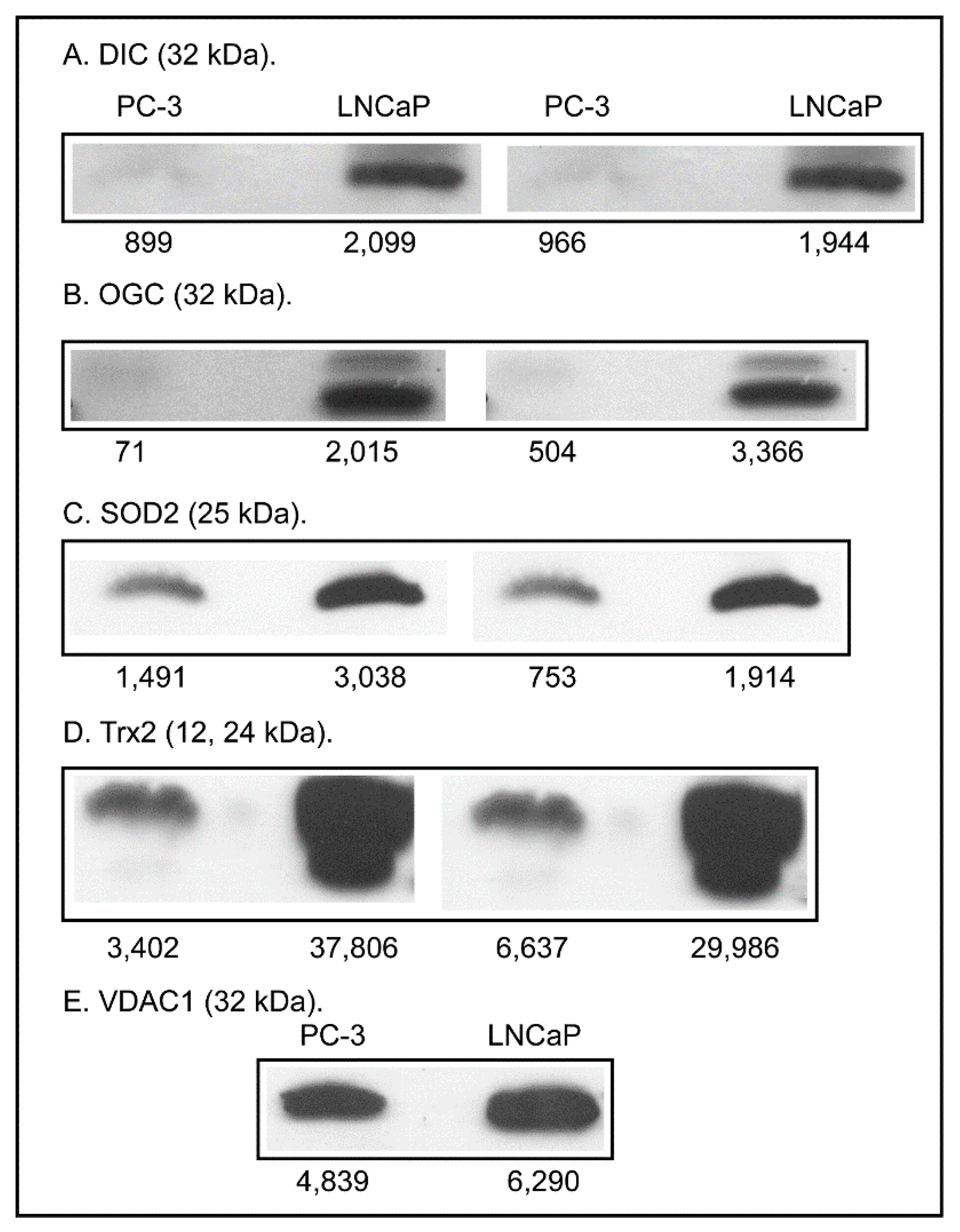
2.3. Expression of Apoptosis, Stress Response, and Mitochondrial Proteins in PC-3 and LNCaP Cells
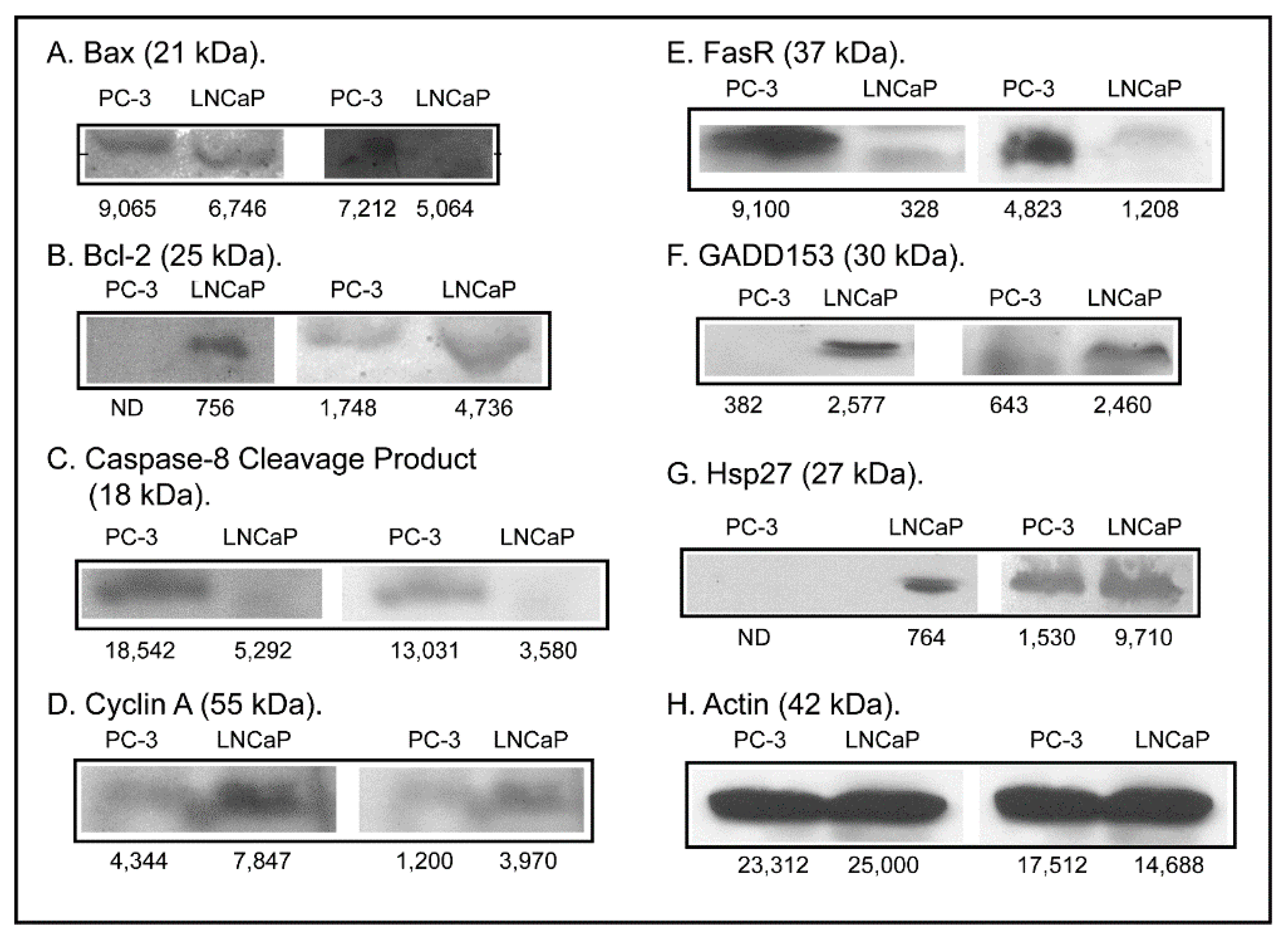
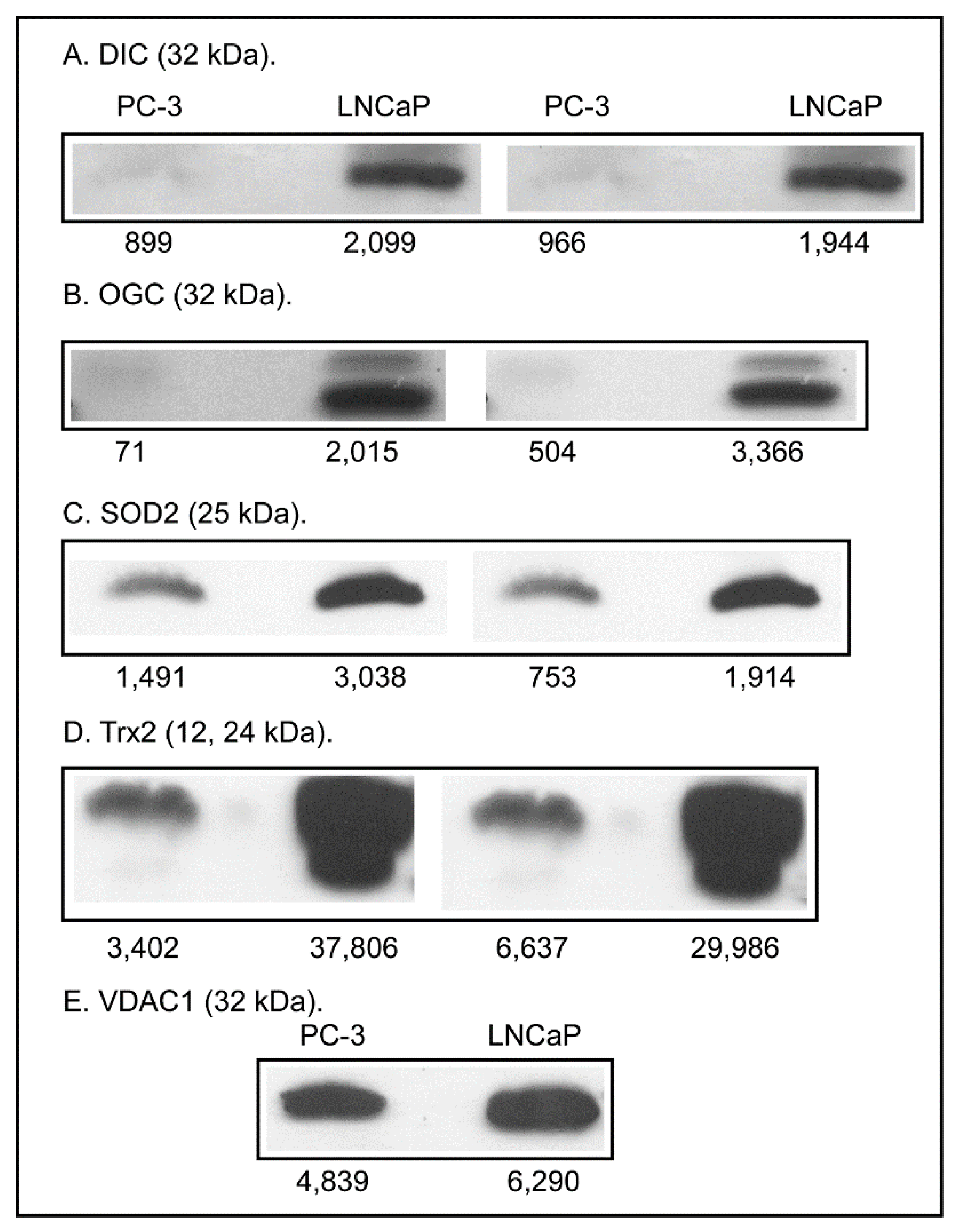
3. Discussion
4. Experimental Section
4.1. Cell Culture and Incubations
4.2. GSH and GSSG Measurements
4.3. LDH Release Cell Viability Assay
4.4. TUNEL Assay
4.5. Determination of Protein Expression by Western Blot Analyses
4.5.1. Basic Sample Preparation and General Assay Information
4.5.2. Antibody Information
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M. Cancer Statistics 2008. CA Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Abdalla, M.Y.; Aykin-Burns, N.; Simons, A.L.; Oberley, L.W.; Domannc, F.E.; Spitz, D.R. 2-Deoxyglucose combined with wild-type p53 overexpression enhances cytotoxicity in human prostate cancer cells via oxidative stress. Free Radic. Biol. Med. 2008, 44, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Aydina, A.; Arsova-Sarafinovskab, Z.; Sayala, A.; Ekena, A.; Erdema, O.; Ertenc, K.; Özgökc, Y.; Dimovski, A. Oxidative stress and antioxidant status in non-metastatic prostate cancer and benign prostatic hyperplasia. Clin. Biochem. 2006, 39, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; Bourdeau-Heller, J.M.; Zhong, W.; Oberley, T.D. Characterization of redox state of two human prostate carcinoma cell lines with different degrees of aggressiveness. Free Radic. Biol. Med. 2007, 43, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.K.R.; Gemin, A.; Singh, G. High activity of mitochondrial glycerophosphate dehydrogenase and glycerophosphate-dependent ROS production in prostate cancer cell lines. Biochem. Biophys. Res. Commun. 2005, 333, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Seidel, B.; Rudolph, B.; Lein, M.; Cronauer, M.V.; Henke, W.; Hampel, G.; Schnorr, D.; Loening, S.A. Antioxidant enzymes in malignant prostate cell lines and in primary cultured prostatic cells. Free Radic. Biol. Med. 1997, 23, 127–133. [Google Scholar] [CrossRef]
- Li, G.-X.; Hu, H.; Jiang, C.; Schuster, T.; Lu, J.X. Differential involvement of reactive oxygen species in apoptosis induced by two classes of selenium compounds in human prostate cancer cells. Int. J. Cancer 2007, 120, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Hong, S.; Jin, W.; Lim, S.; Kim, S.J.; Kang, H.J.; Park, E.H.; Ahn, K.; Lim, C.J. Up-regulation of defense enzymes is responsible for low reactive oxygen species in malignant prostate cancer cells. Exp. Biol. Med. 2005, 37, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Katiyara, S.; Katiyar, S.K. Berberine-induced apoptosis in human prostate cancer cells is initiated by reactive oxygen species generation. Toxicol. Appl. Pharmacol. 2008, 229, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Saglam, K.; Sonmez, A.; Gok, D.E.; Basal, S.; Kilic, S.; Akay, C.; Kocar, I.H. Antioxidant system activation in prostate cancer. Biol. Trace Elem. Res. 2004, 98, 13–19. [Google Scholar] [CrossRef]
- Zhong, W.; Oberley, T.D. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. 2001, 61, 7071–7078. [Google Scholar] [PubMed]
- Costello, L.C.; Franklin, R.B. Bioenergetic theory of prostate malignancy. Prostate 1994, 25, 62–66. [Google Scholar] [CrossRef]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002. [Google Scholar] [CrossRef] [Green Version]
- Coffey, R.N.T.; Watson, W.G.; Hegarty, N.J.; O’Neill, A.; Gibbons, N.; Brady, H.R.; Fitzpatrick, J.M. Thiol-mediated apoptosis in prostate carcinoma cells. Cancer 2000, 88, 2092–2104. [Google Scholar] [CrossRef]
- Curtin, J.F.; Cotter, T.G. Anisomycin activates JNK and sensitises DU145 prostate carcinoma cells to Fas-mediated apoptosis. Br. J. Cancer 2002, 87, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Guseva, N.V.; Taghiyev, A.F.; Rokhlin, O.W.; Cohen, M.B. Contribution of death receptor and mitochondrial pathways to Fas-mediated apoptosis in the prostatic carcinoma cell line PC3. Prostate 2002, 51, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, C.; O’Neill, A.; Spengler, B.; Christoffel, V.; Fitzpatrick, J.M.; Watson, R.W.G. Apigenin drives the production of reactive oxygen species and initiates a mitochondrial mediated cell death pathway in prostate epithelial cells. Prostate 2005, 63, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Nachshon-Kedmi, M.; Yannai, S.; Fares, F.A. Induction of apoptosis in human prostate cancer cell line, PC3, by 3,3′-diindolylmethane through the mitochondrial pathway. Br. J. Cancer 2004, 91, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ngo, L.; Perez, G.; Dokmanovic, M.; Marks, P.A. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc. Natl. Acad. Sci. USA 2006, 103, 15540–15545. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Jankovich, A. Glutathione levels and susceptibility to chemically induced injury in human prostate cancer cell lines. In Proceedings of the 53rd Annual Meeting and ToxExpo, Phoenix, AZ, USA, 23–27 March 2014; p. 77.
- Lash, L.H.; Putt, D.A.; Jankovich, A. Glutathione levels and susceptibility to chemically induced injury in human prostate cancer cell lines. FASEB J. 2014, 28, 663.11. [Google Scholar]
- McKernan, T.B.; Woods, E.B.; Lash, L.H. Uptake of glutathione by renal cortical mitochondria. Arch. Biochem. Biophys. 1991, 288, 653–663. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem.-Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Mitochondrial glutathione in toxicology and disease of the kidneys. Toxicol. Res. 2012, 1, 39–46. [Google Scholar] [CrossRef]
- Schnelldorfer, T.; Gansauge, S.; Gansauge, F.; Schlosser, S.; Beger, H.G.; Nussler, A.K. Glutathione depletion causes cell growth inhibition and enhanced apoptosis in pancreatic cancer cells. Cancer 2000, 89, 1440–1447. [Google Scholar] [CrossRef]
- Jayakumar, S.; Kunwar, A.; Sandur, S.K.; Pandey, B.N.; Chaubey, R.C. Differential response of DU145 and PC3 prostate cancer cells to ionizing radiation: Role of reactive oxygen species, GSH and Nrf2 in radiosensitivity. Biochim. Biophys. Acta 2014, 1840, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Blask, D.E.; Wilson, S.T.; Zalatan, F. Physiological melatonin inhibition of human breast cancer cell growth in vitro: Evidence for a glutathione-mediated pathway. Cancer Res. 1997, 15, 1909–1914. [Google Scholar]
- Han, Y.H.; Park, W.H. The effects of N-acetyl cysteine, buthionine sulfoximine, diethyldithiocarbamate or 3-amino-1,2,4-triazole on antimycin A-treated Calu-6 lung cells in relation to cell growth, reactive oxygen species and glutathione. Oncol. Rep. 2009, 22, 385–391. [Google Scholar] [PubMed]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in cancer cell death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta 2012, 1823, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Gumulec, J.; Balvan, J.; Sztalmachova, M.; Raudenska, M.; Dvorakova, V.; Knopfova, L.; Polanska, H.; Hudcova, K.; Ruttkay-Nedecky, B.; Babula, P.; et al. Cisplatin-resistant prostate cancer model: Differences in antioxidant system, apoptosis and cell cycle. Int. J. Oncol. 2014, 44, 923–933. [Google Scholar] [PubMed]
- Skjoth, I.H.E.; Issinger, O.G. Profiling of signaling molecules in four different human prostate carcinoma cell lines before and after induction of apoptosis. Int. J. Oncol. 2006, 28, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Terlecky, S.R.; Lash, L.H. Diabetes increases susceptibility of primary cultures of rat proximal tubular cells to chemically induced injury. Toxicol. Appl. Pharmacol. 2009, 241, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Putt, D.A.; Zhong, Q.; Lash, L.H. Adaptive changes in renal mitochondrial redox status in diabetic nephropathy. Toxicol. Appl. Pharmacol. 2012, 258, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Benipal, B.; Lash, L.H. Influence of renal compensatory hypertrophy on mitochondrial energetics and redox status. Biochem. Pharmacol. 2011, 81, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Benipal, B.; Lash, L.H. Modulation of mitochondrial glutathione status and cellular energetics in primary cultures of proximal tubular cells from remnant kidney of uninephrectomized rats. Biochem. Pharmacol. 2013, 85, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Griffith, O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980, 106, 207–212. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lash, L.H.; Putt, D.A.; Jankovich, A.D. Glutathione Levels and Susceptibility to Chemically Induced Injury in Two Human Prostate Cancer Cell Lines. Molecules 2015, 20, 10399-10414. https://doi.org/10.3390/molecules200610399
Lash LH, Putt DA, Jankovich AD. Glutathione Levels and Susceptibility to Chemically Induced Injury in Two Human Prostate Cancer Cell Lines. Molecules. 2015; 20(6):10399-10414. https://doi.org/10.3390/molecules200610399
Chicago/Turabian StyleLash, Lawrence H., David A. Putt, and Adam D. Jankovich. 2015. "Glutathione Levels and Susceptibility to Chemically Induced Injury in Two Human Prostate Cancer Cell Lines" Molecules 20, no. 6: 10399-10414. https://doi.org/10.3390/molecules200610399