
Green Polymer Chemistry: Enzyme Catalysis for Polymer Functionalization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
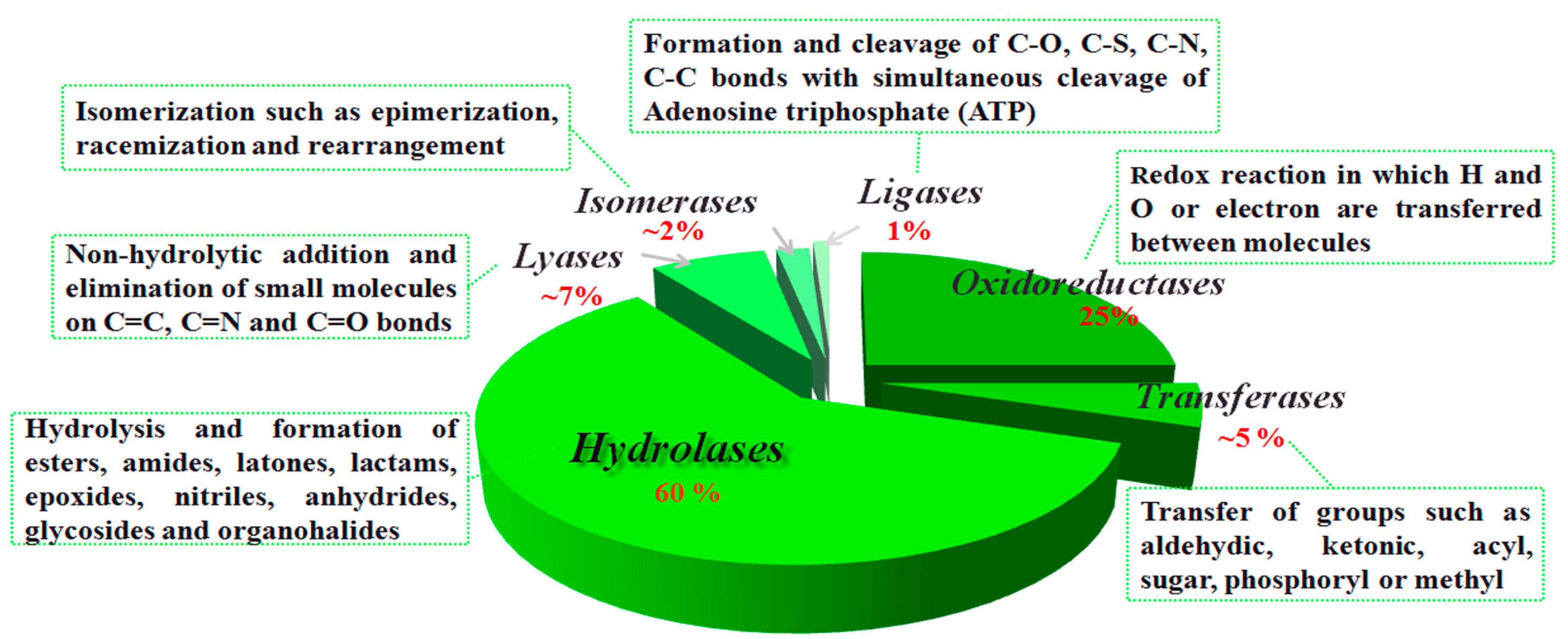
:1. Introduction
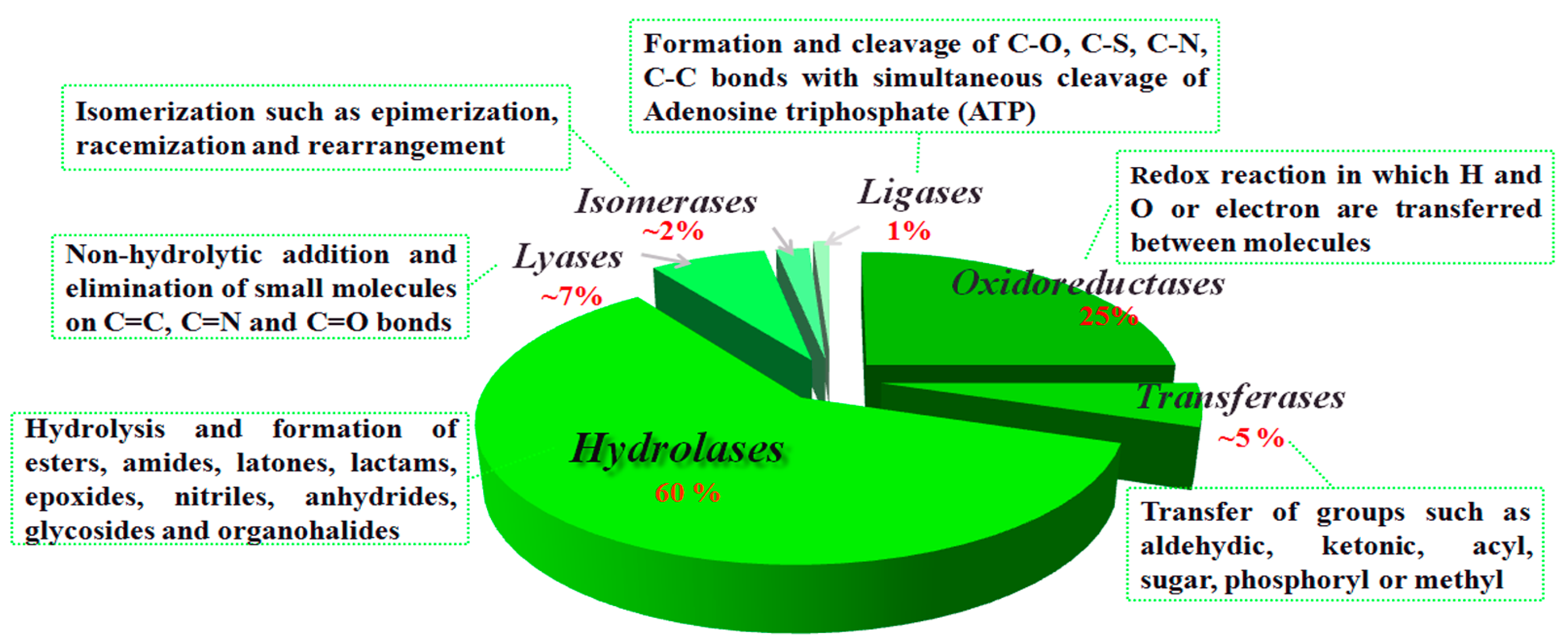
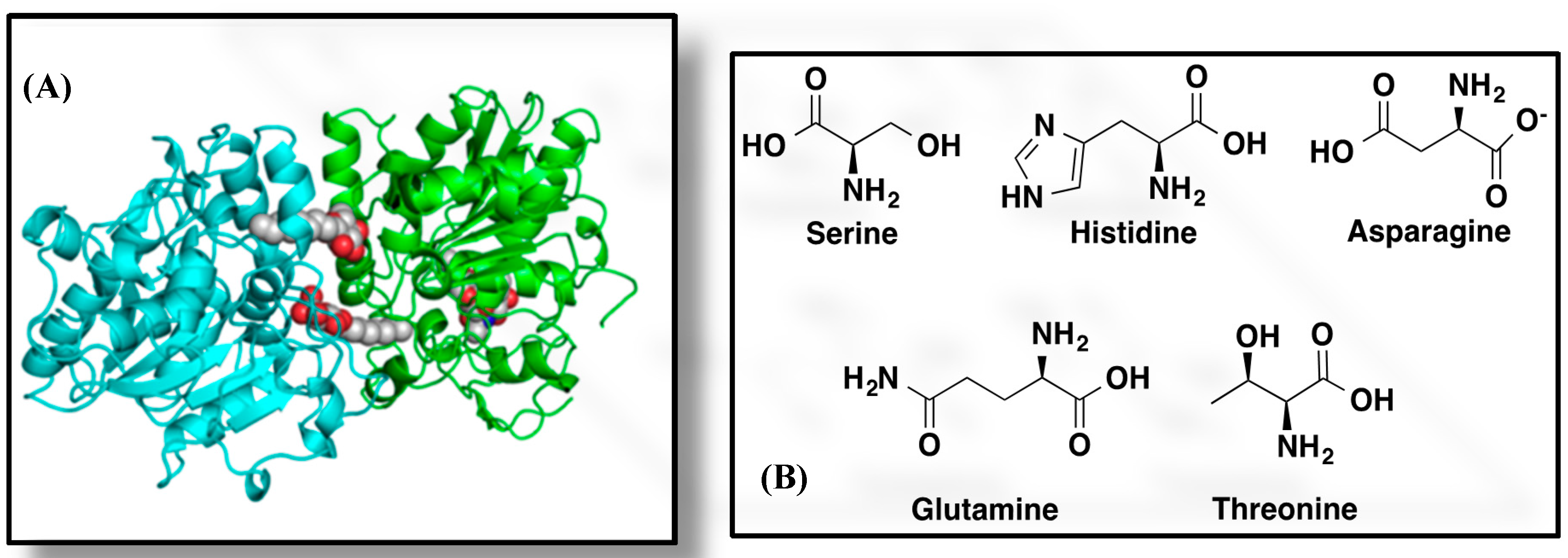
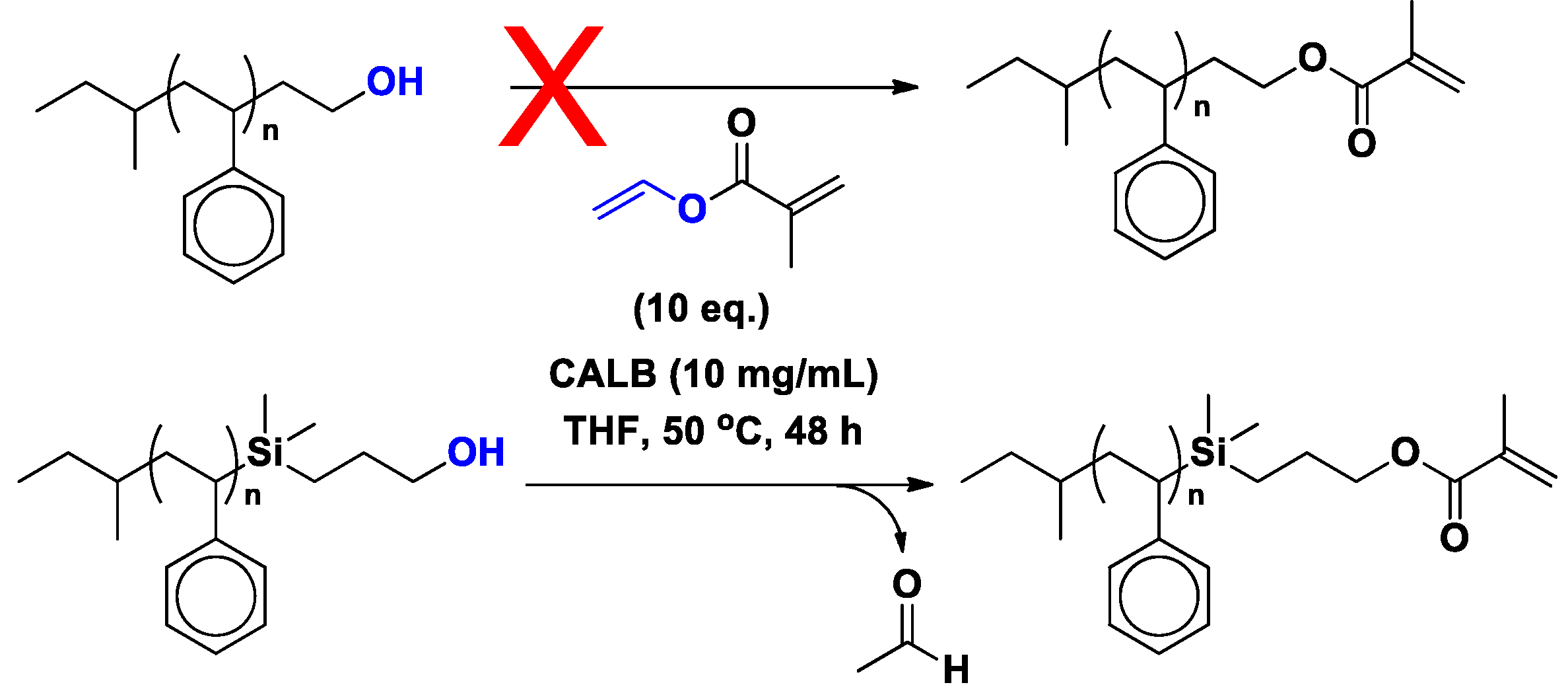
2. Fundamental Aspects of CALB-Catalyzed Transesterification, Michael Addition and Epoxidation
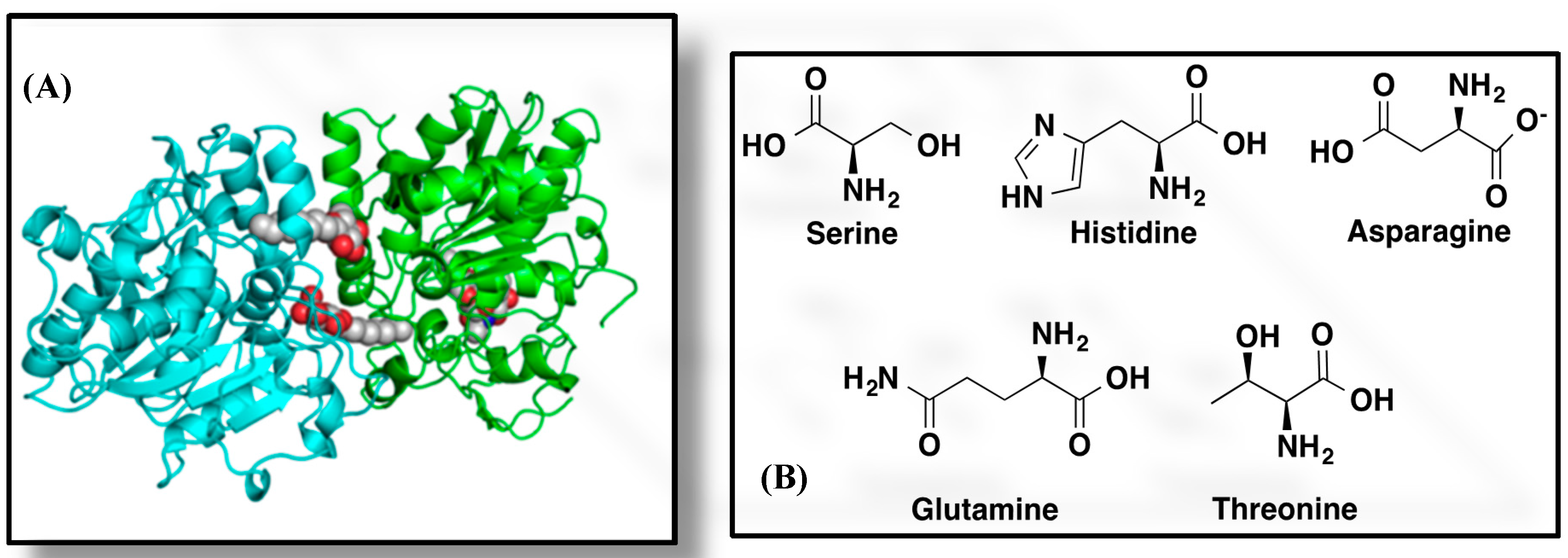
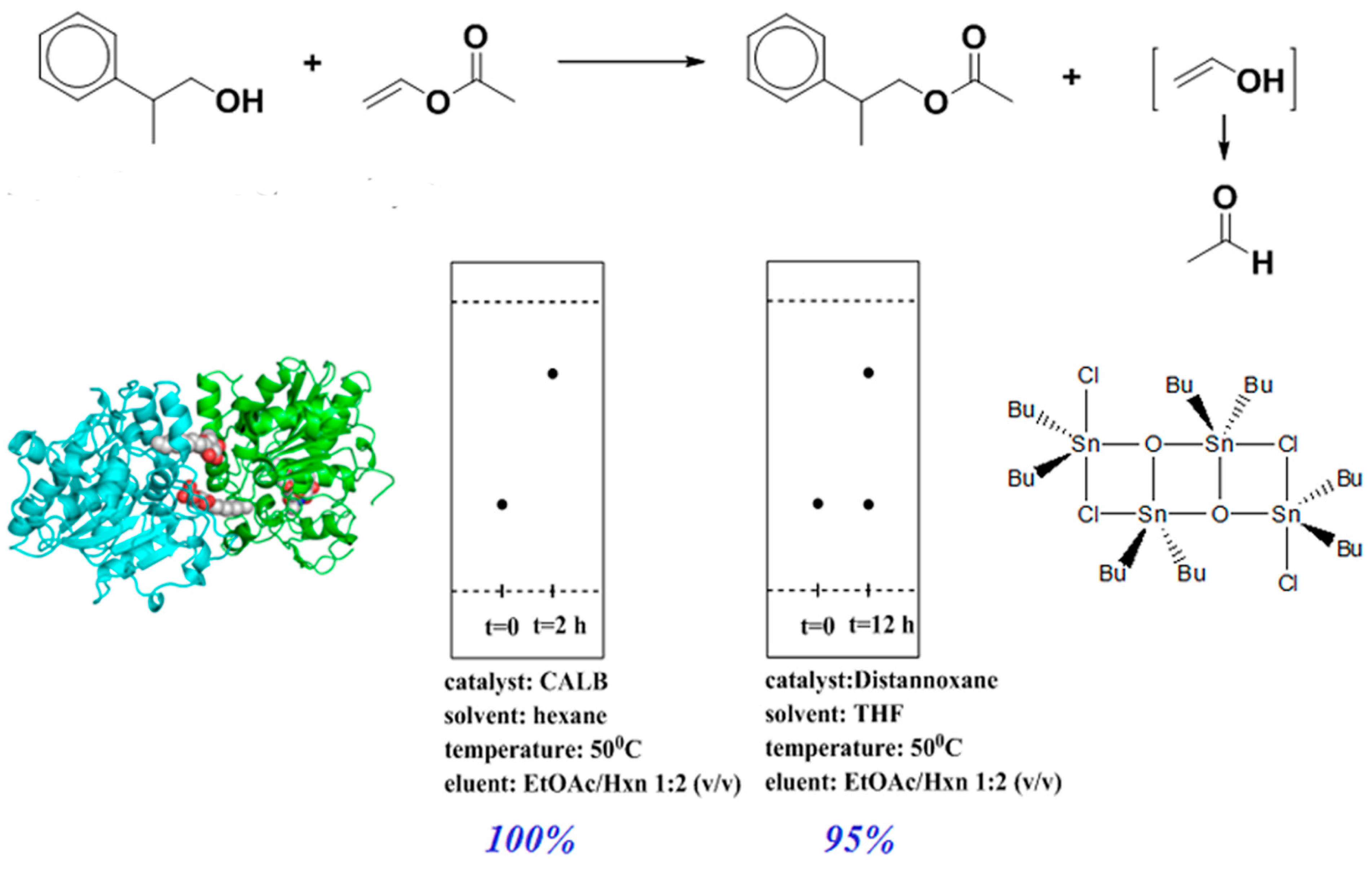
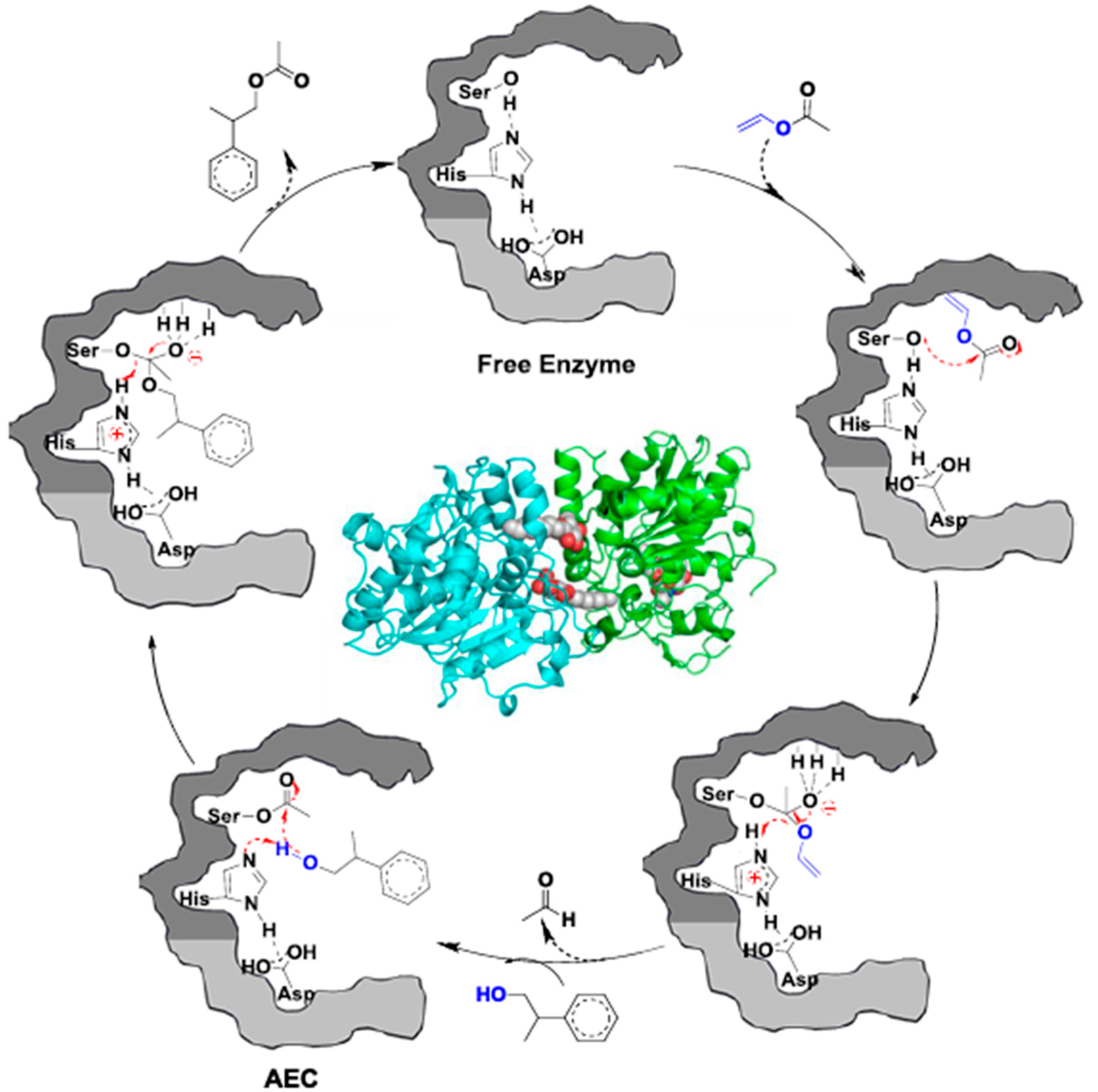
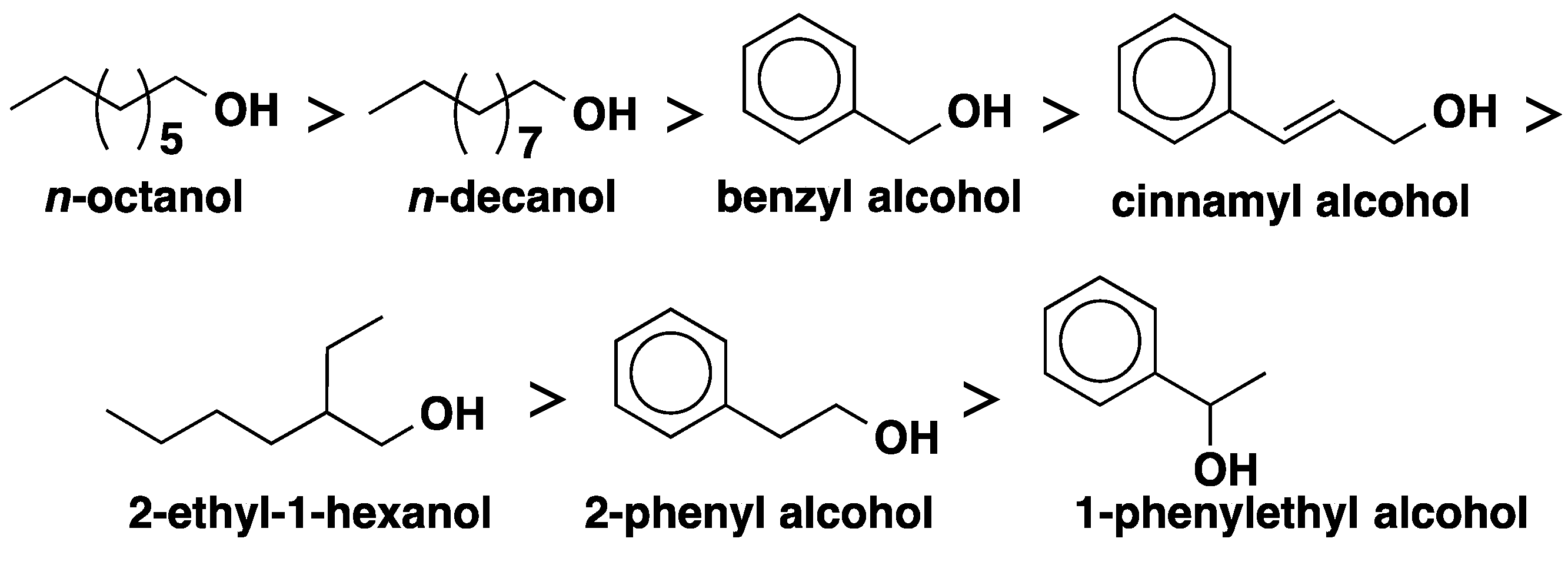
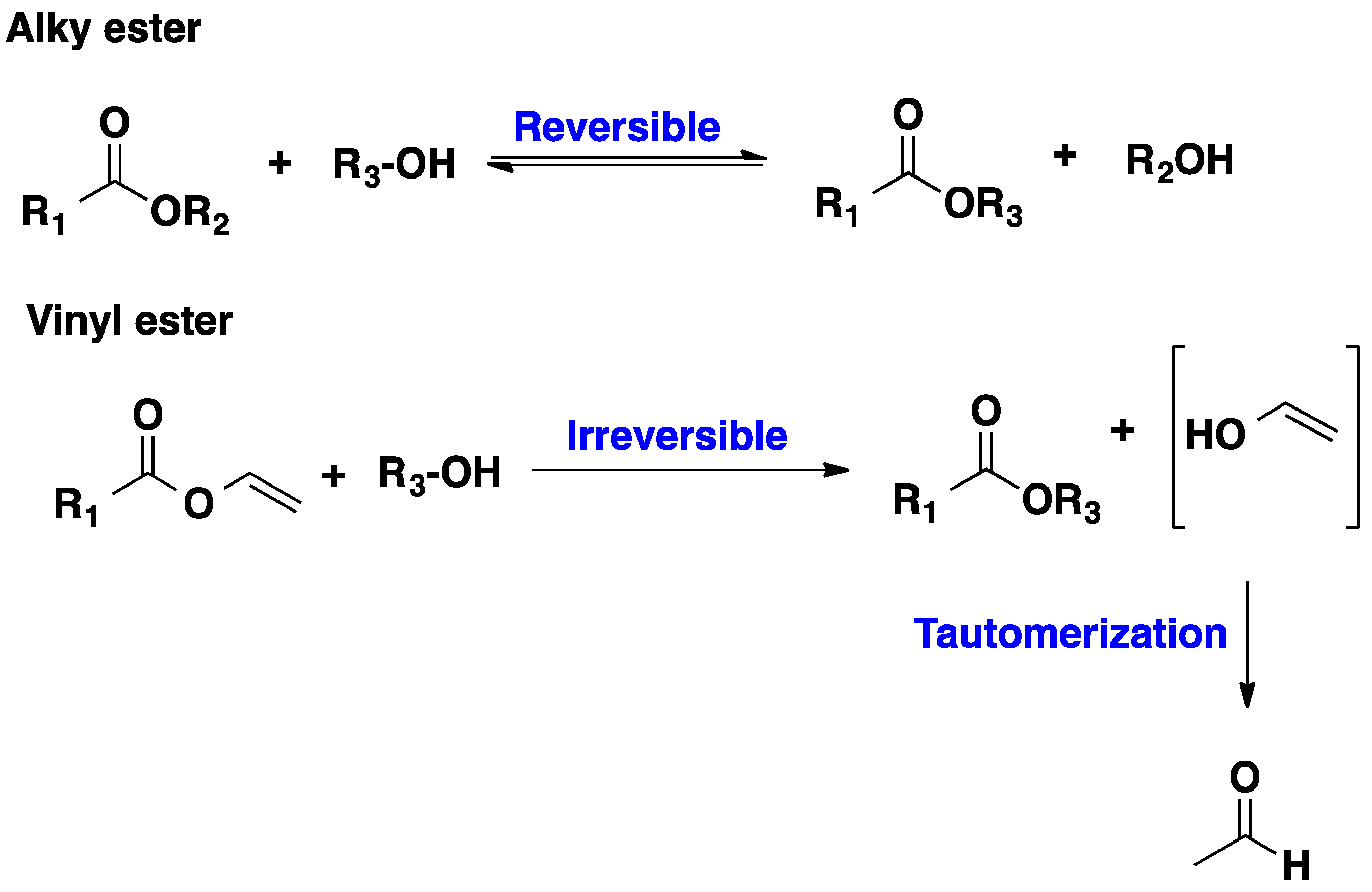
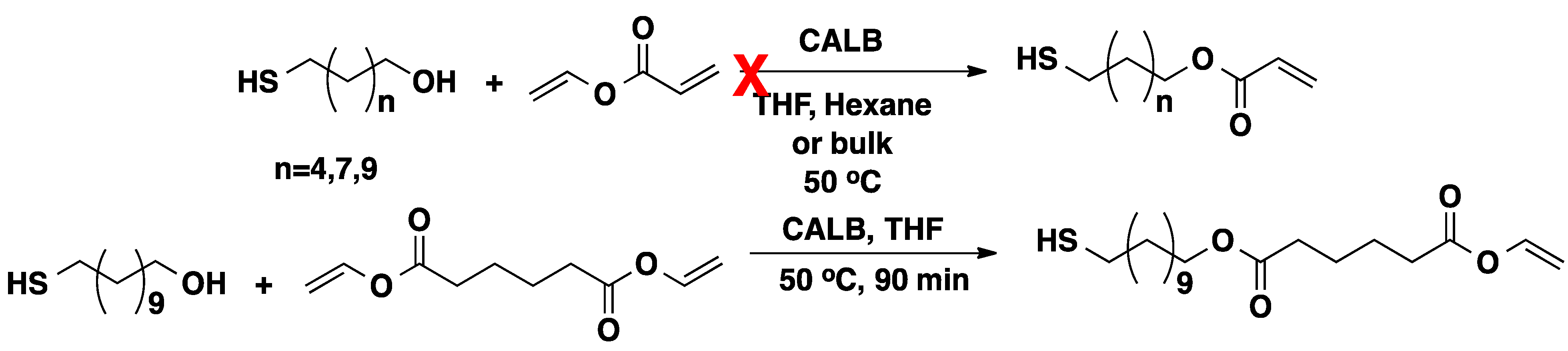
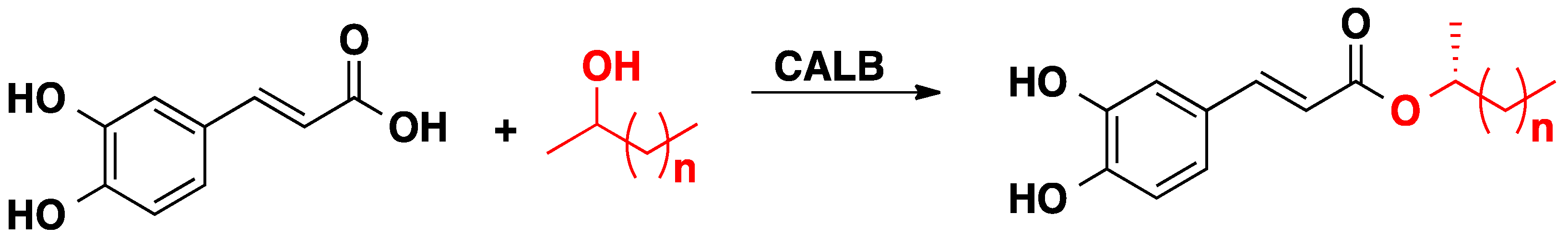
2.1. CALB-Catalyzed Transesterification
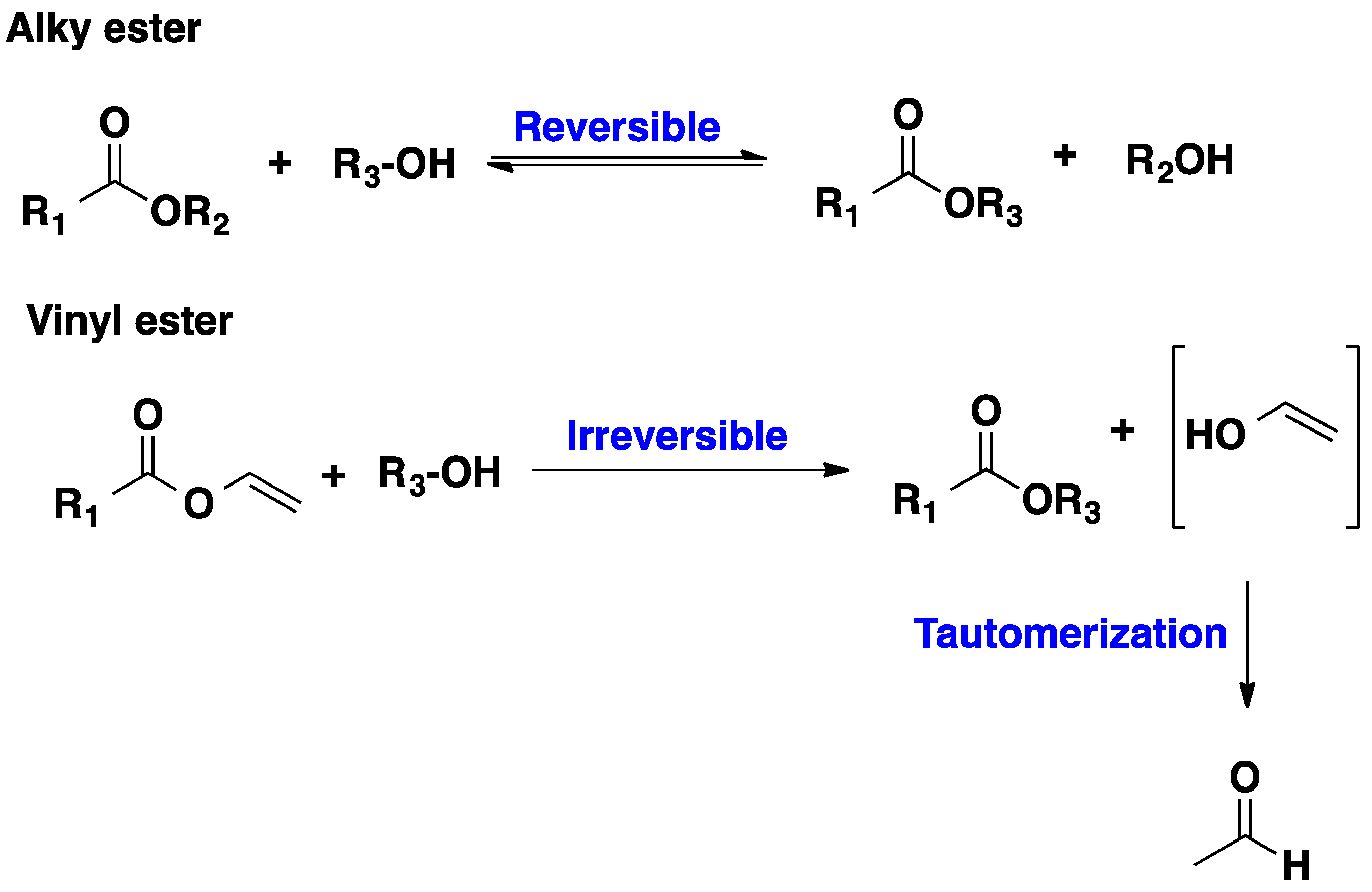
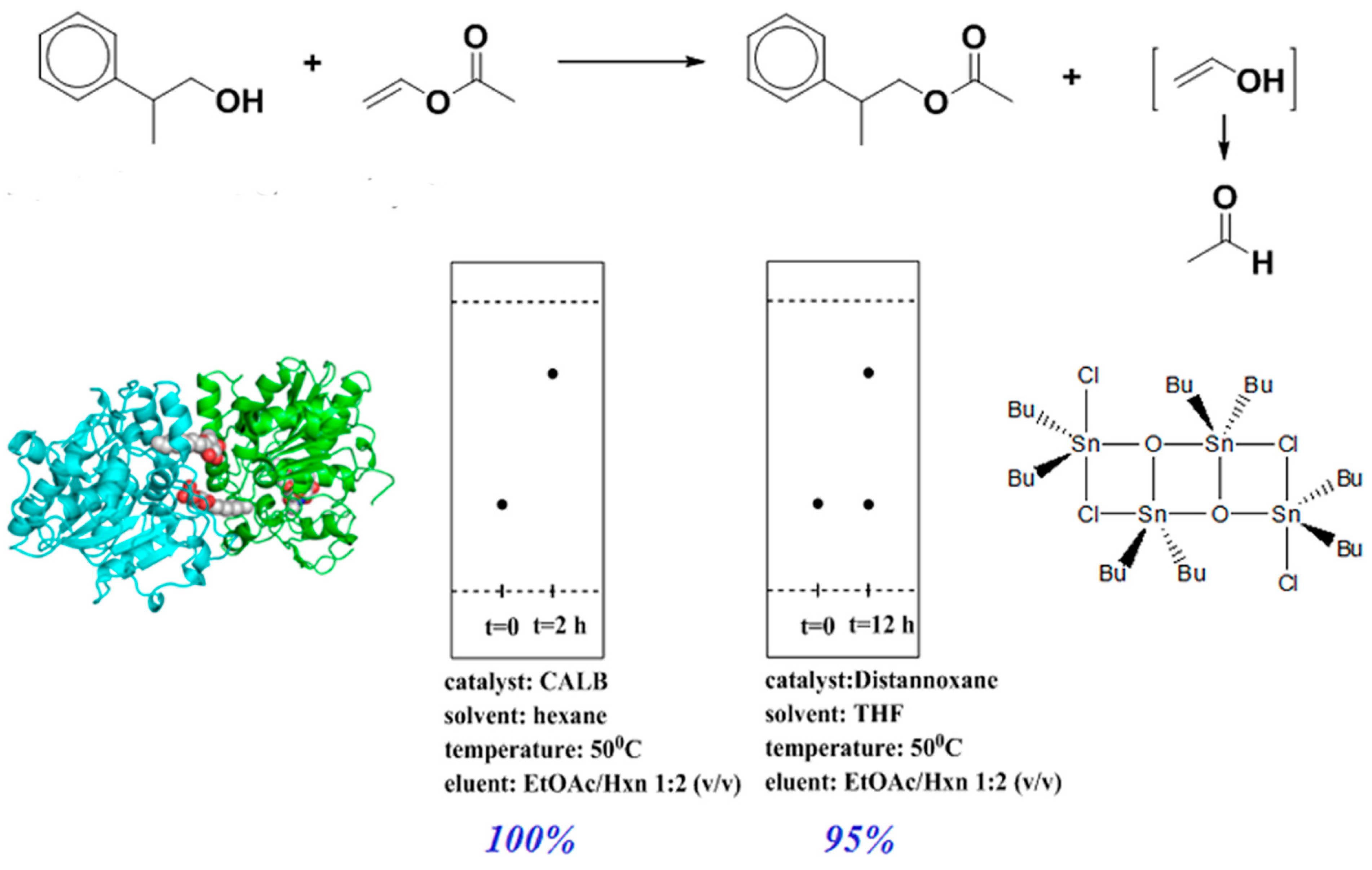
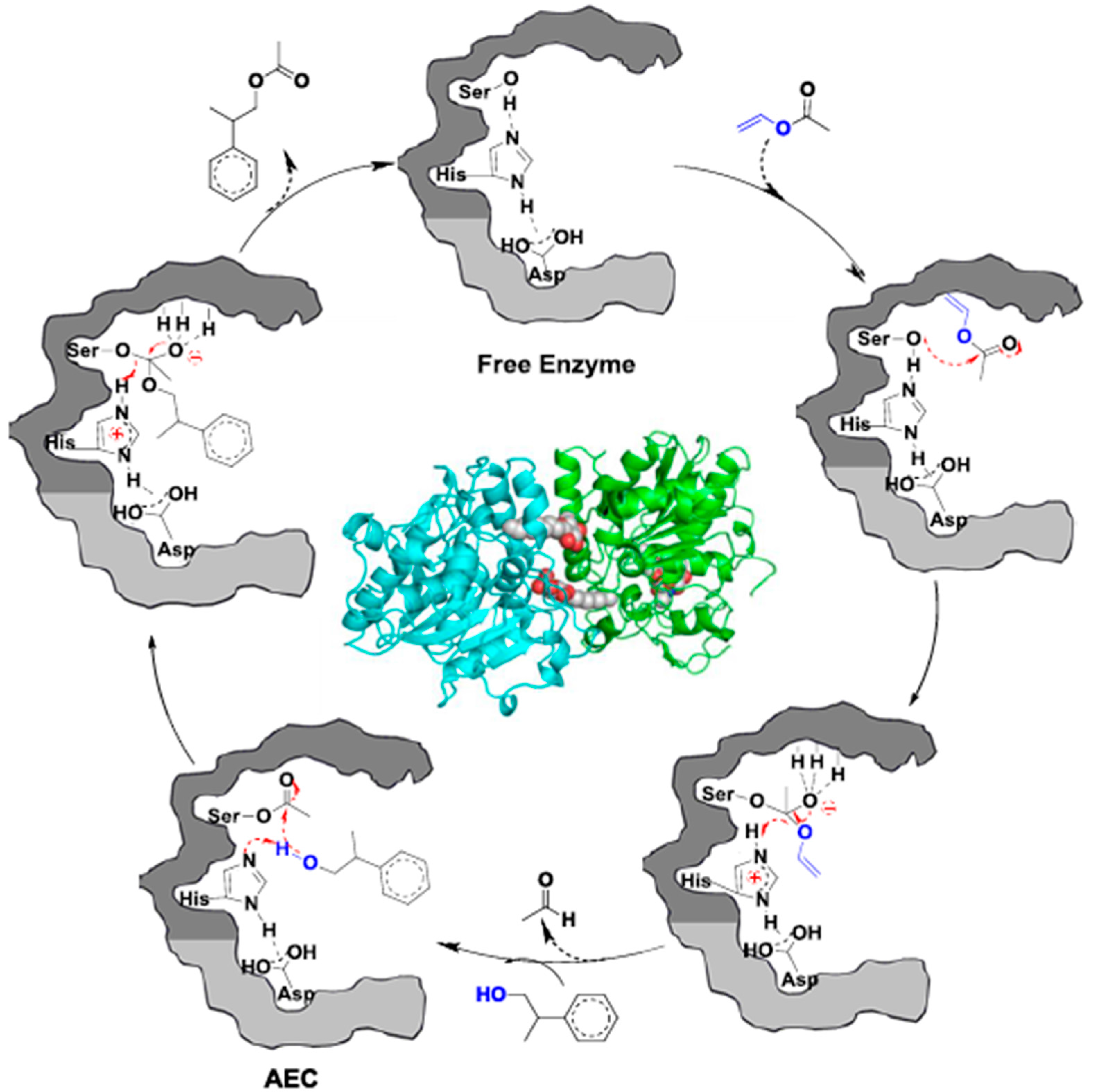
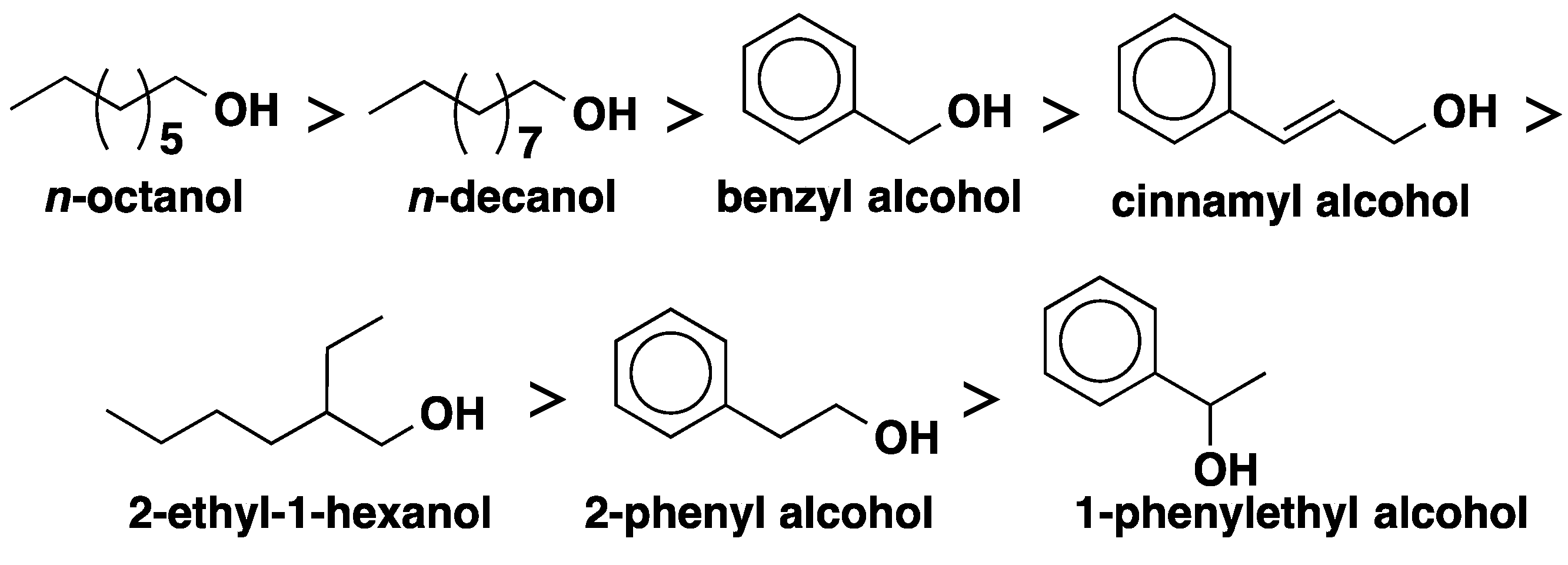
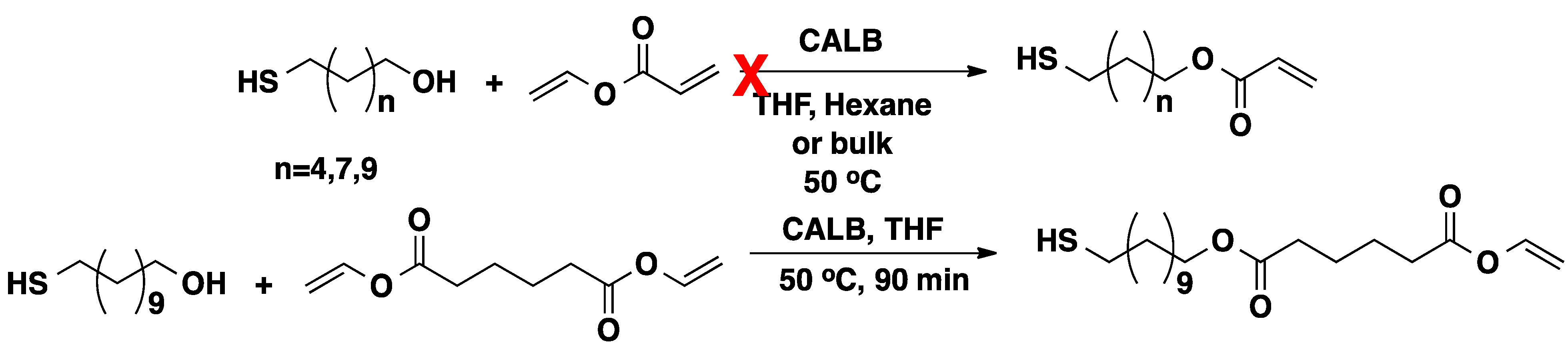

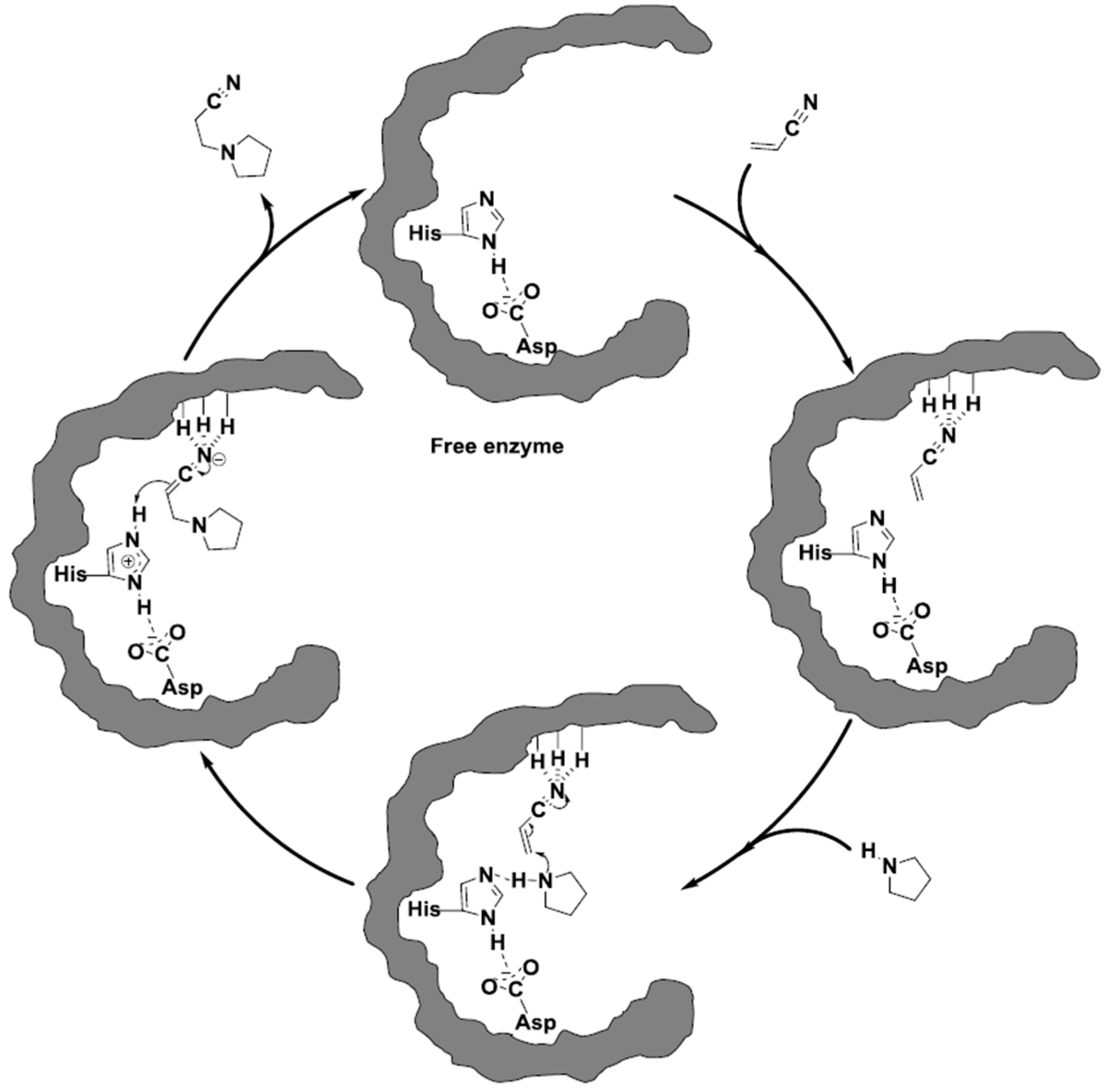
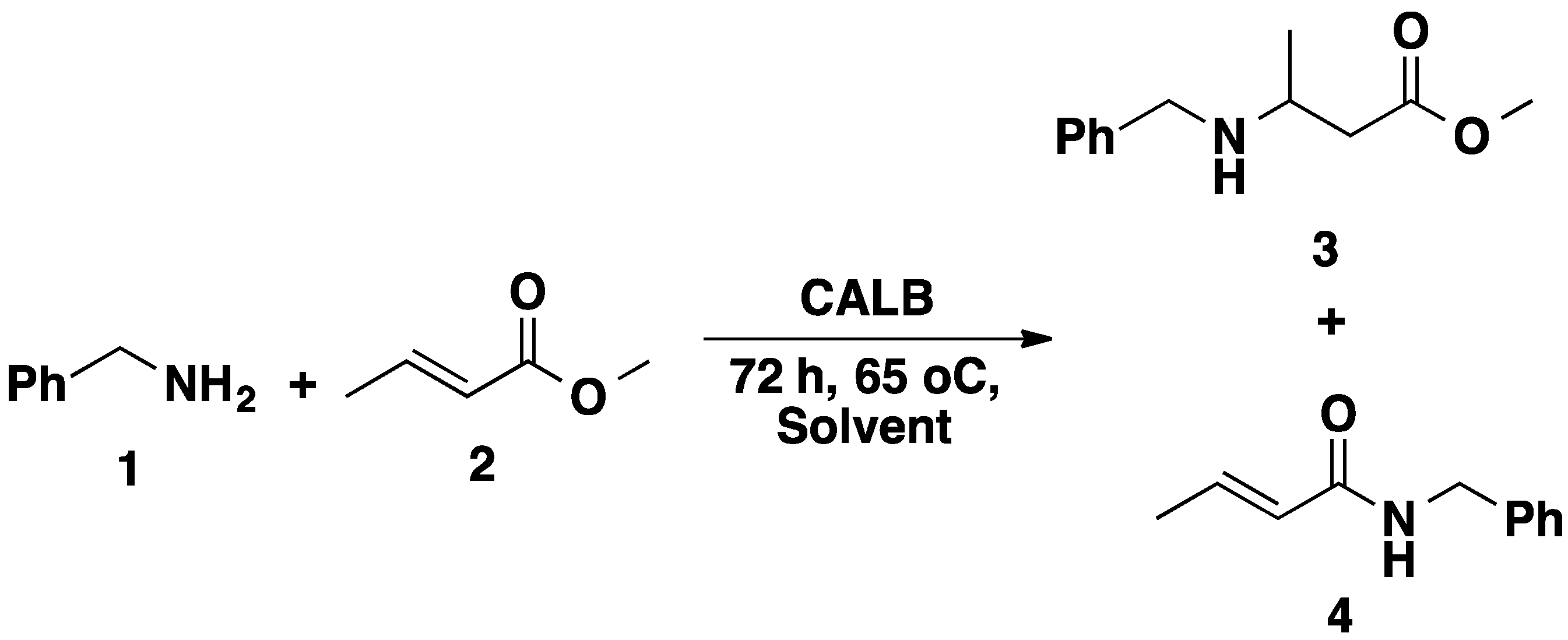
2.2. CALB-Catalyzed Michael Additions



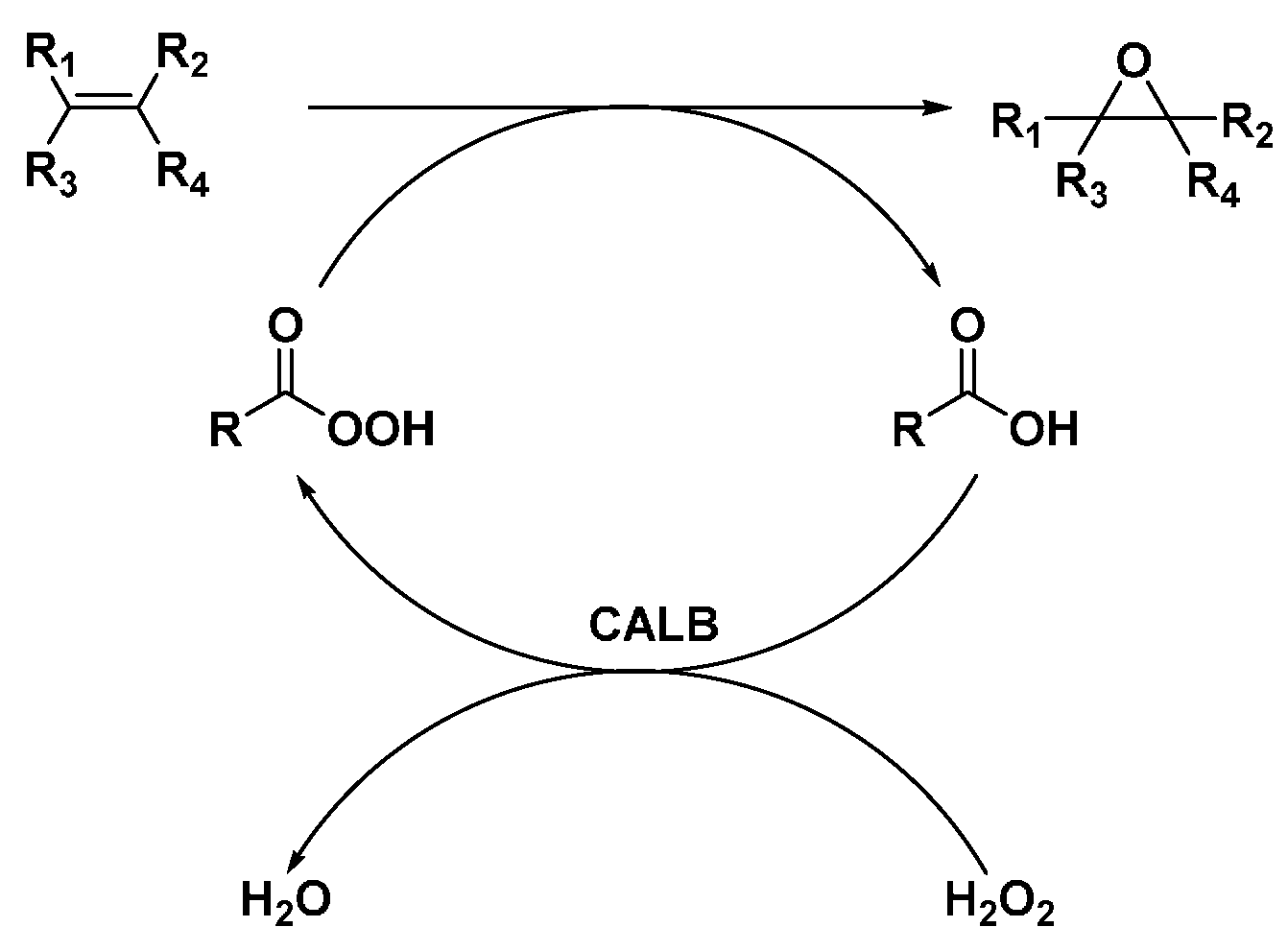
2.3. CALB-Catalyzed Epoxidation

3. Enzyme Catalysis in Polymer End-Functionalization
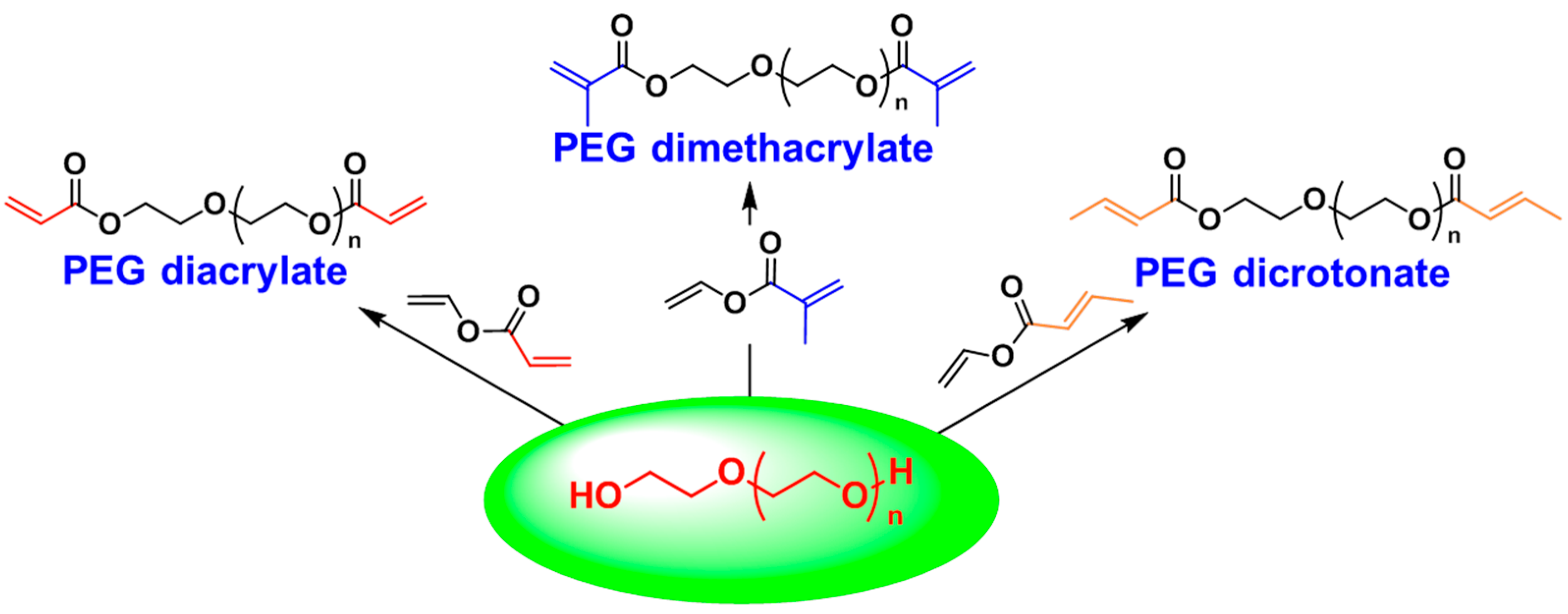
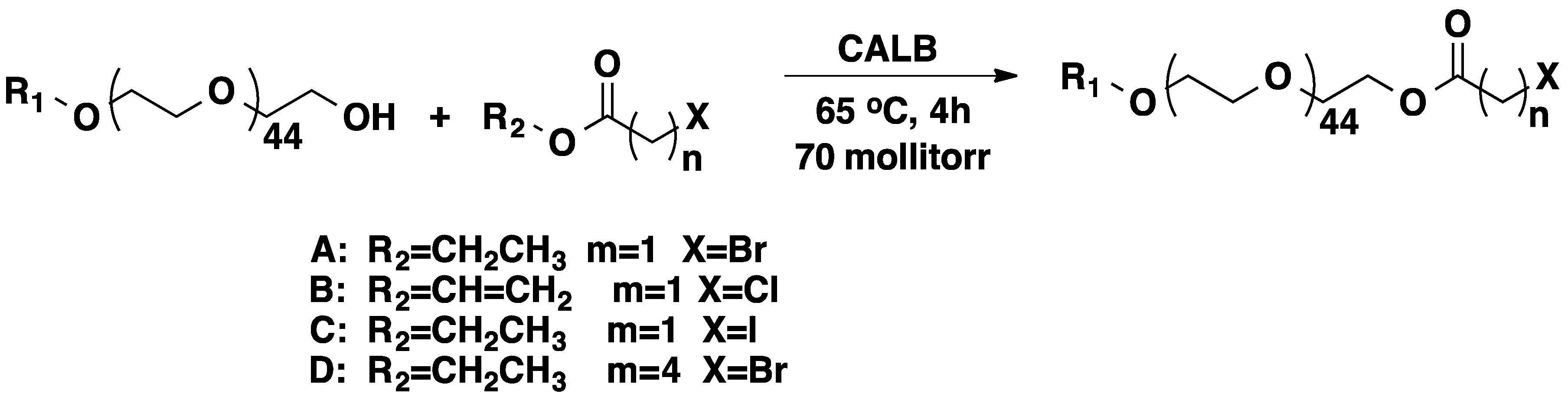
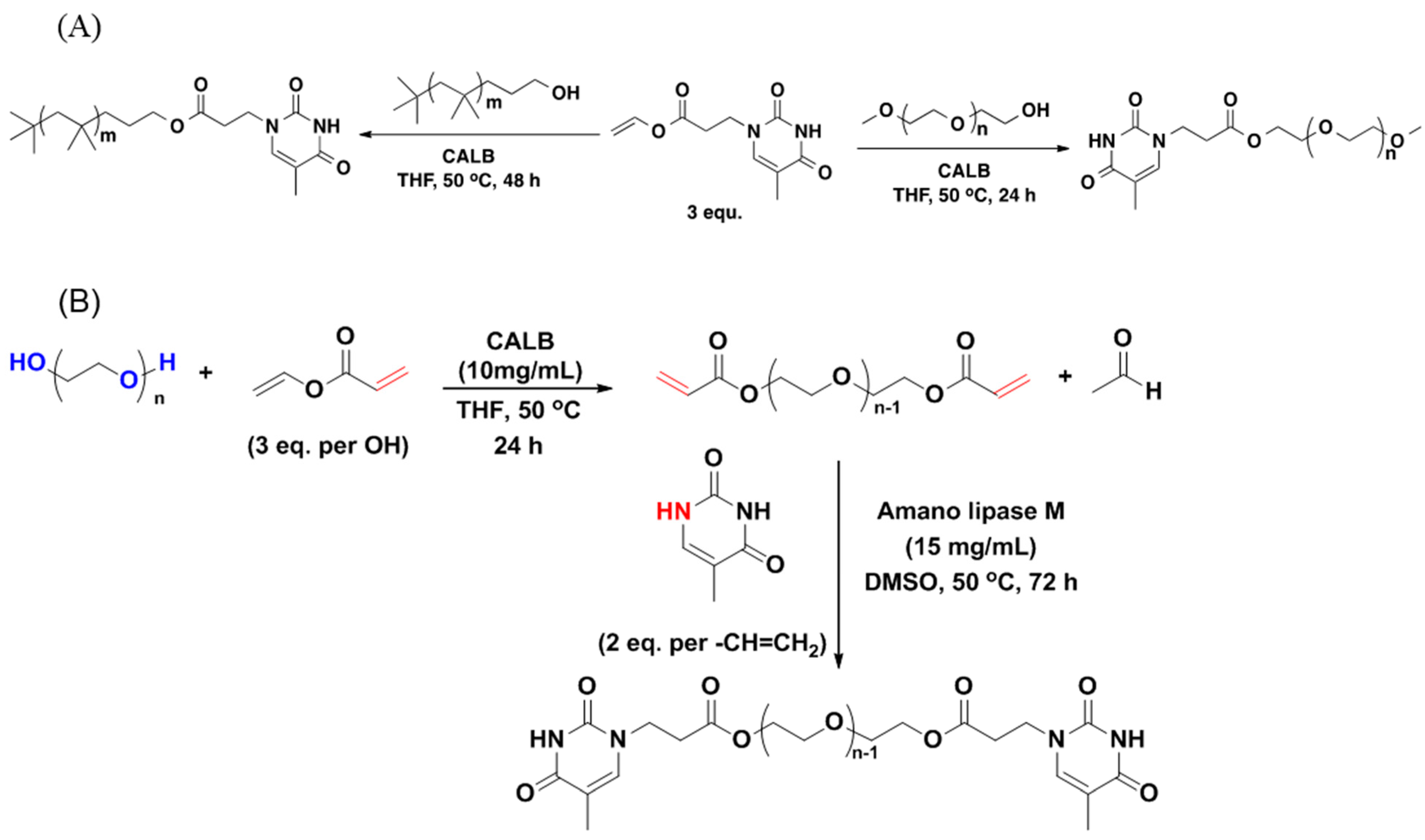
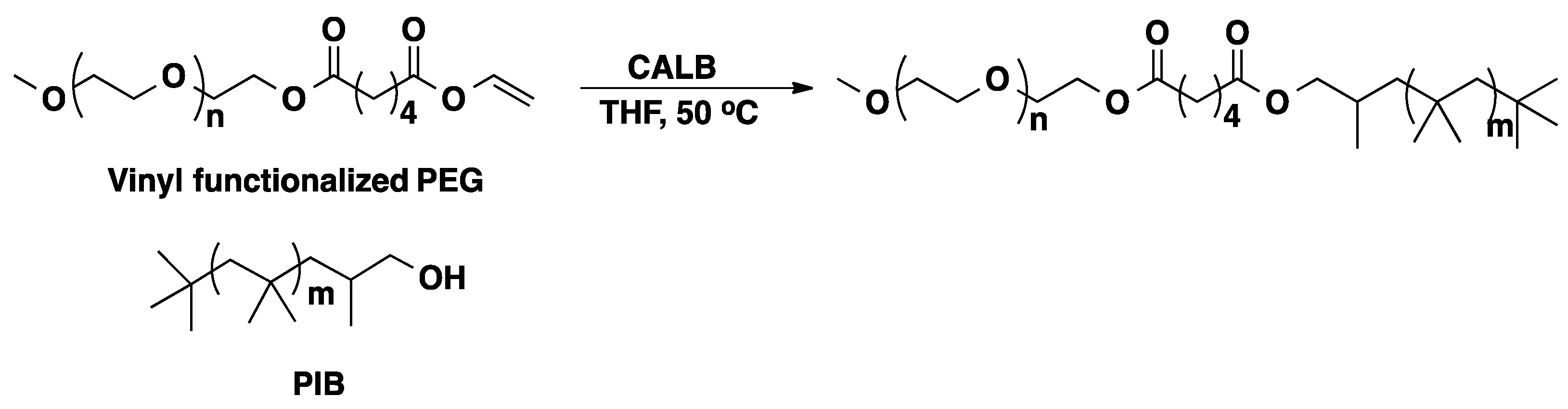
3.1. Poly(ethylene glycol) (PEG)
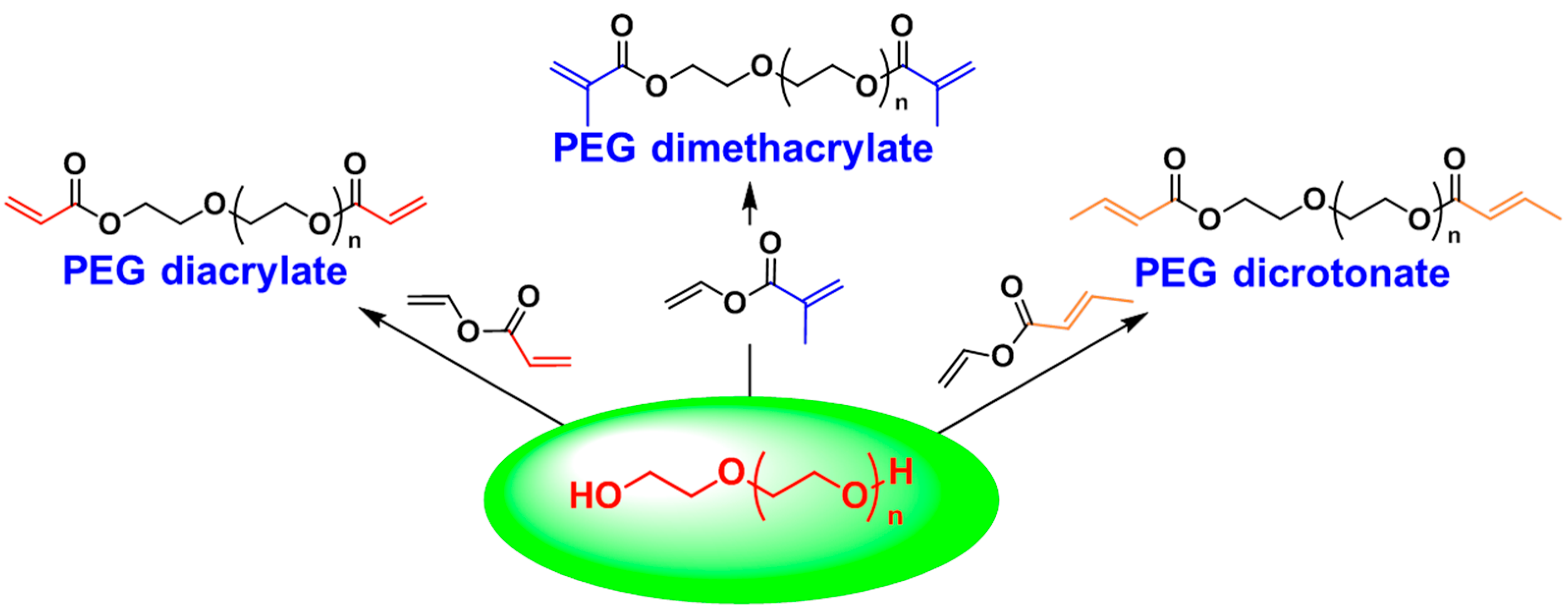
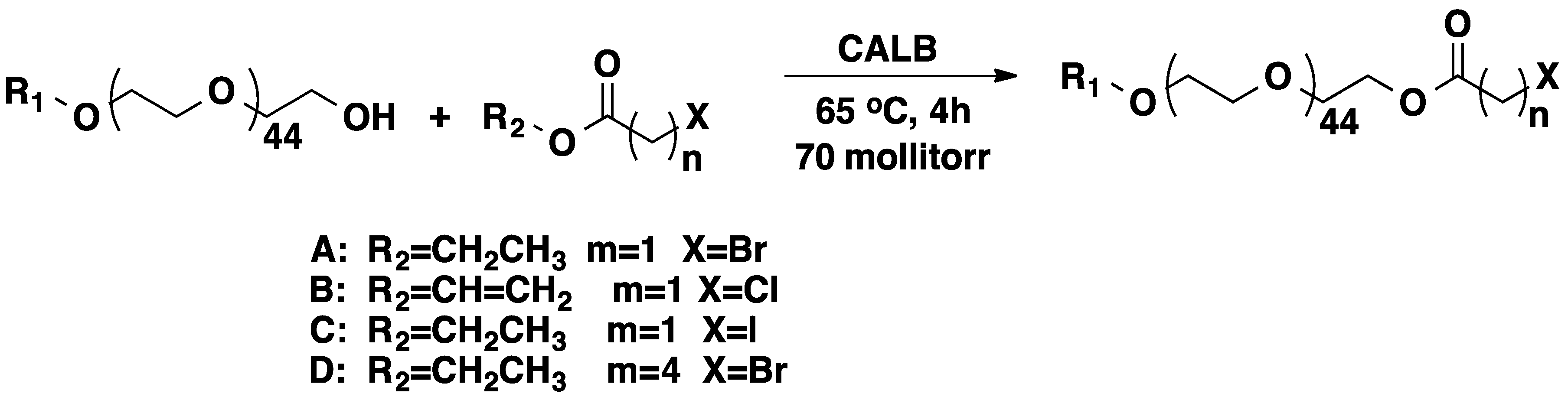

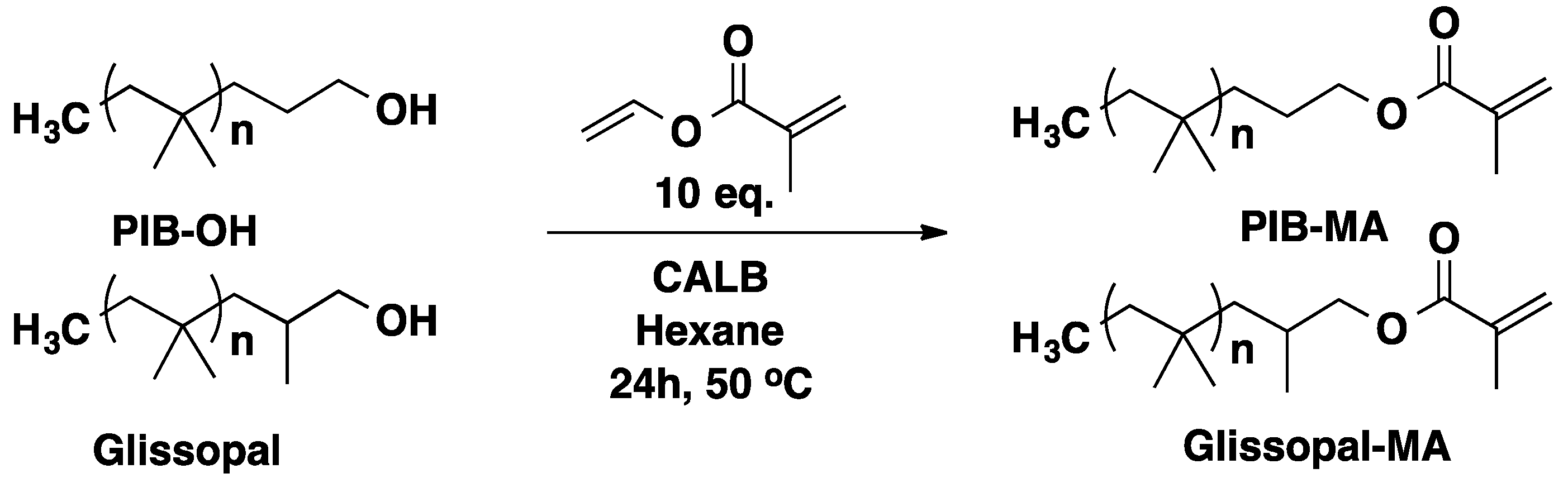
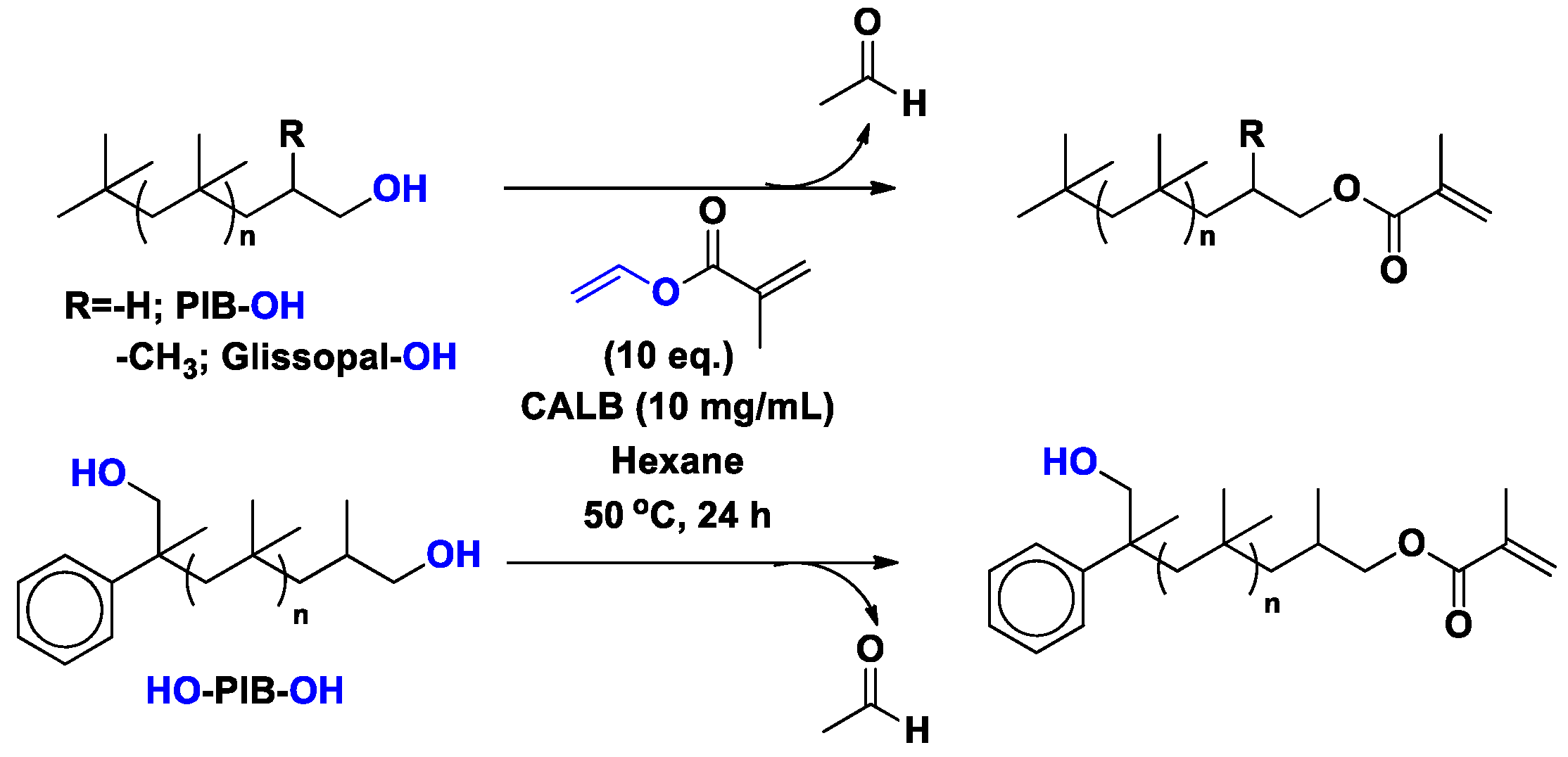
3.2. Polyisobutylene (PIB)
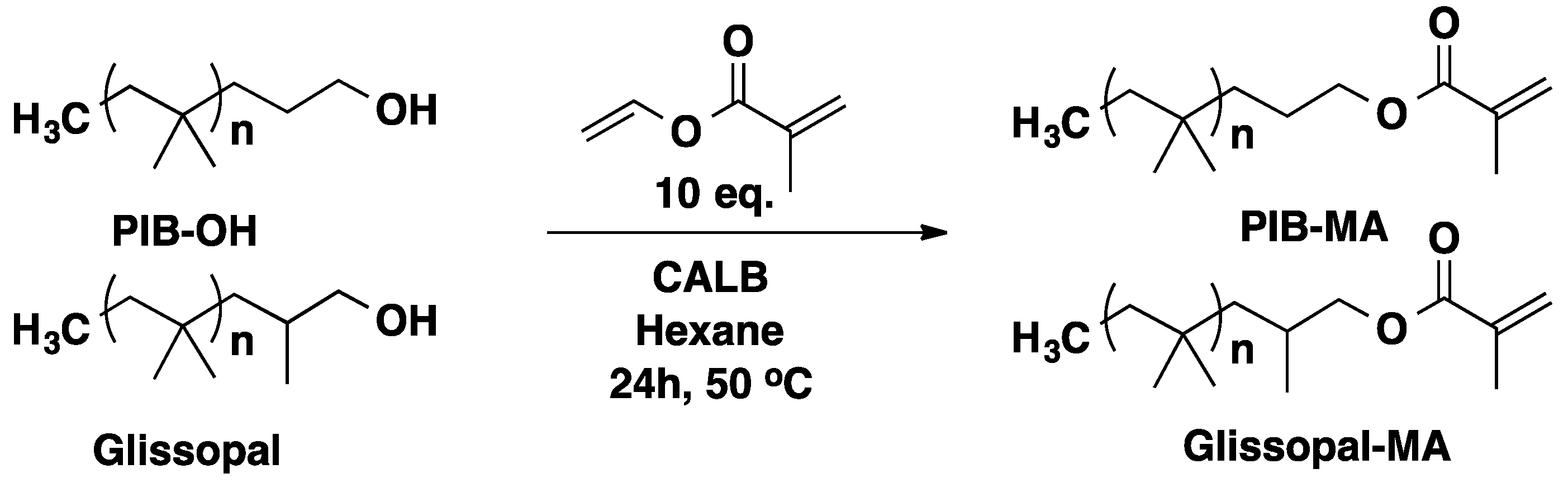
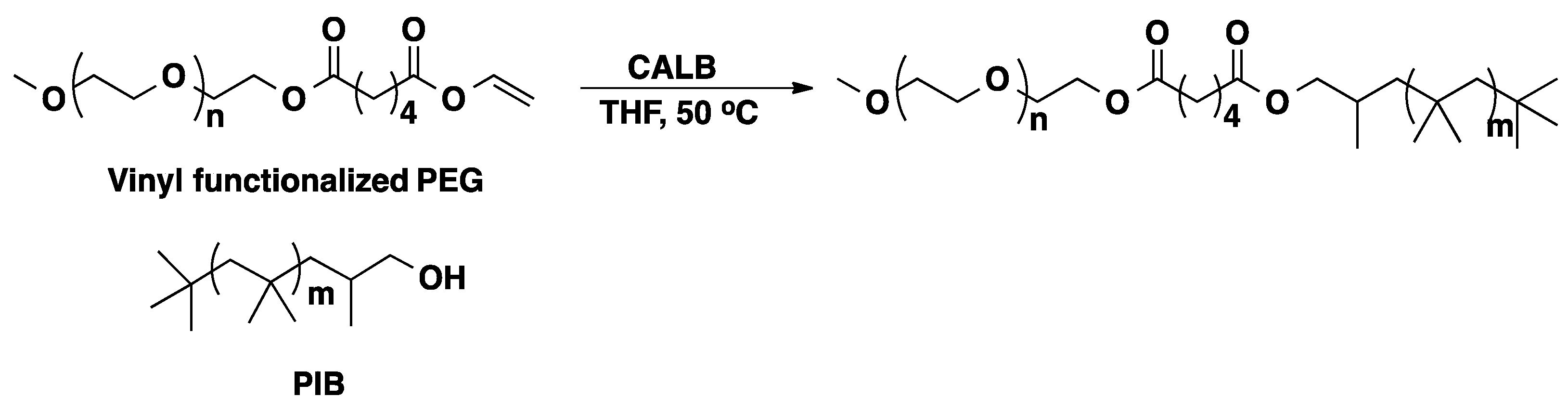

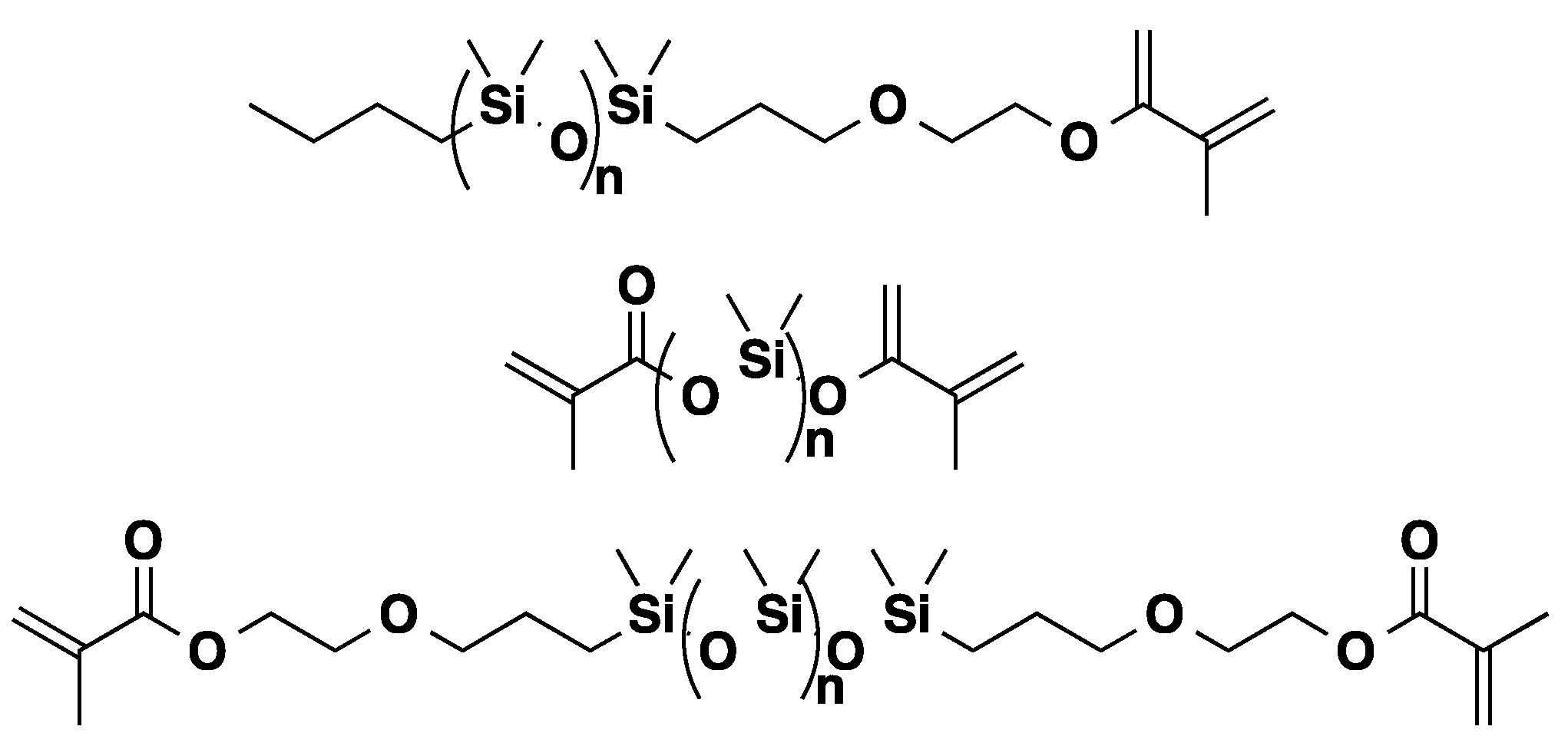
3.3. Polysiloxanes

3.4. Polystyrene
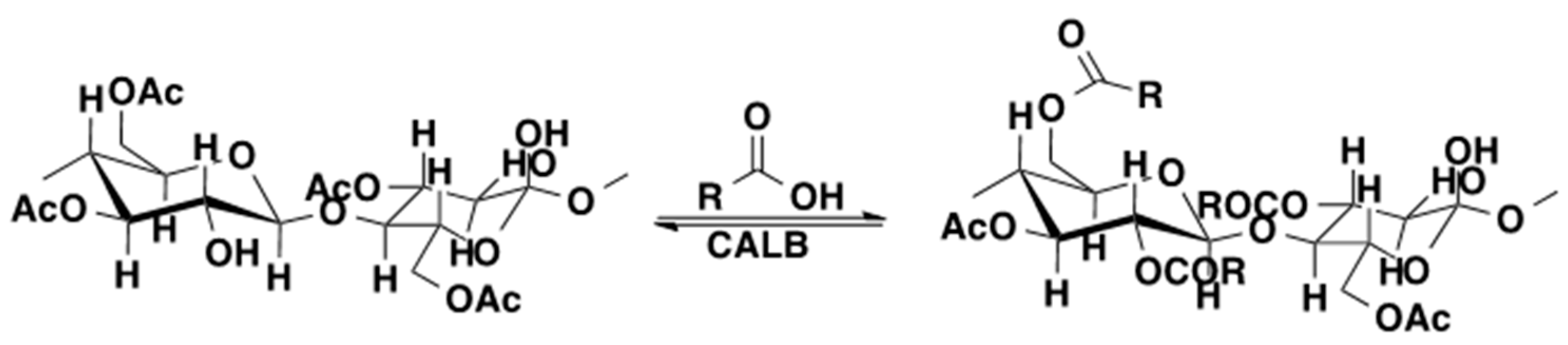
3.5. Oligoesters
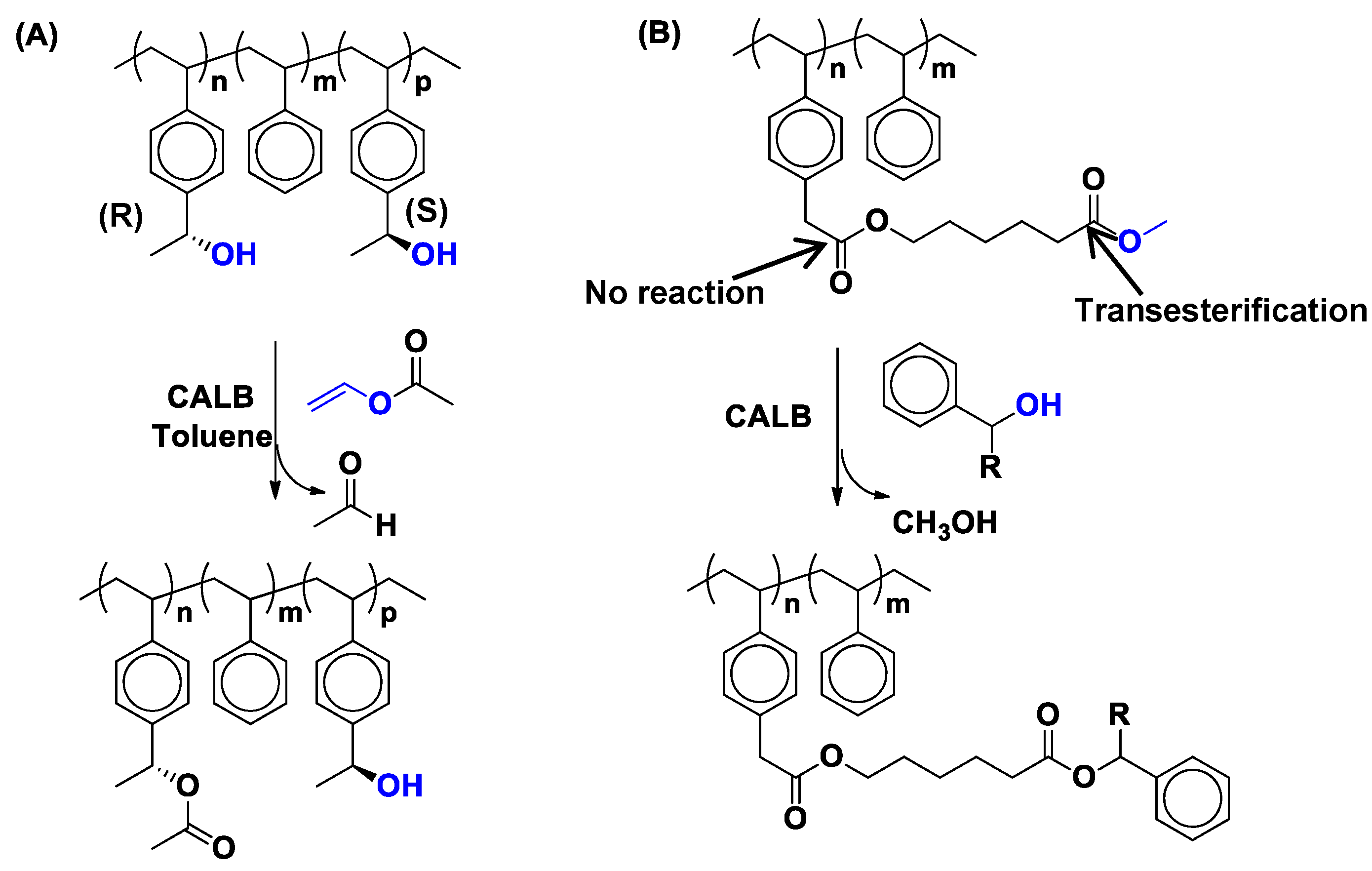
4. Regio- and Chemoselectivity in Enzyme-Catalyzed Polymer Functionalization
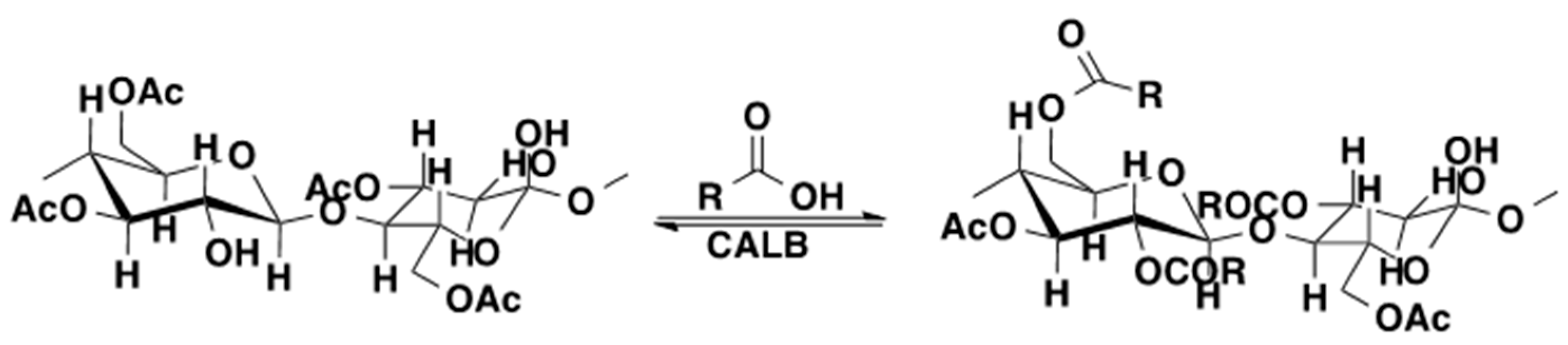
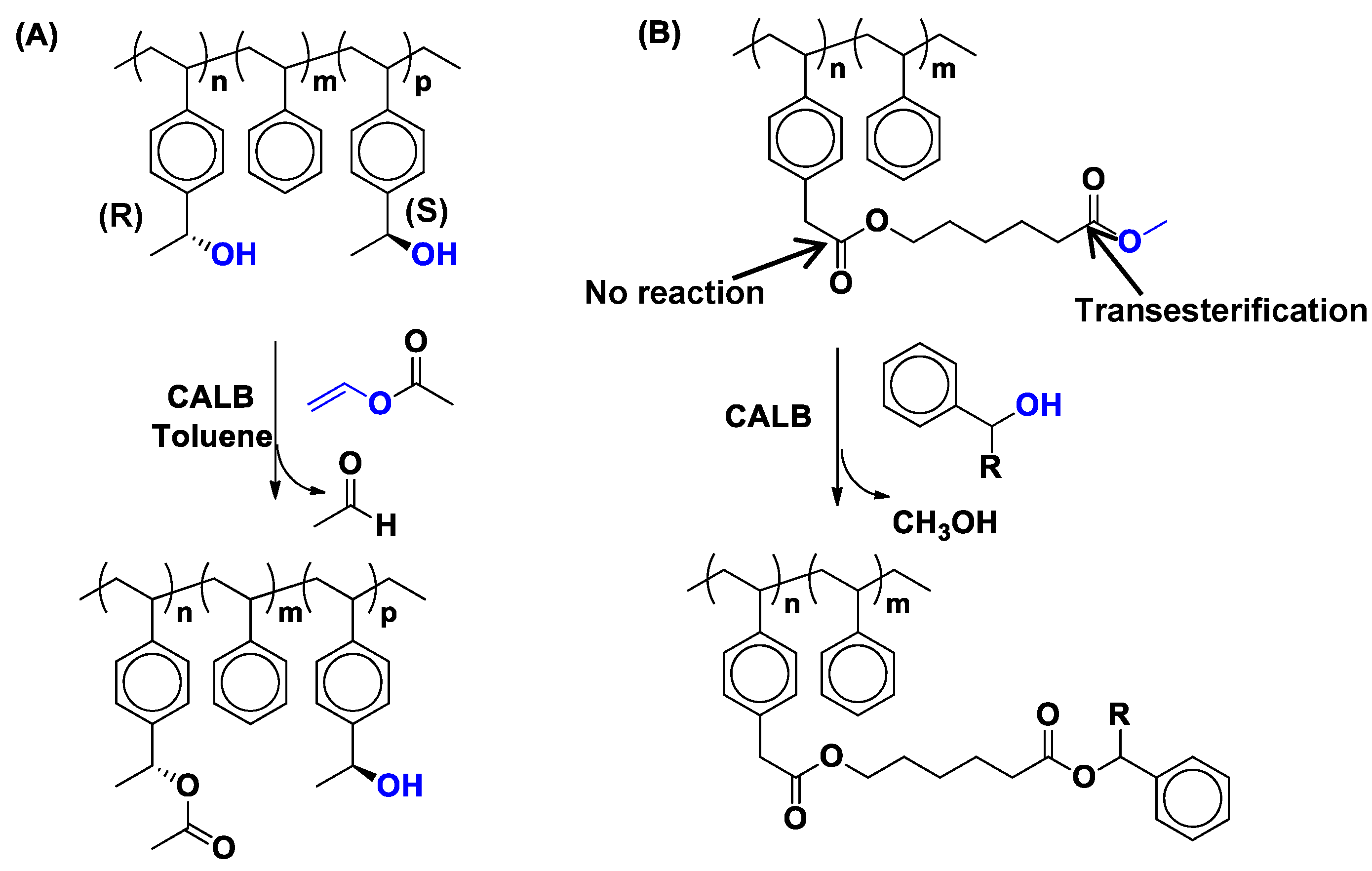

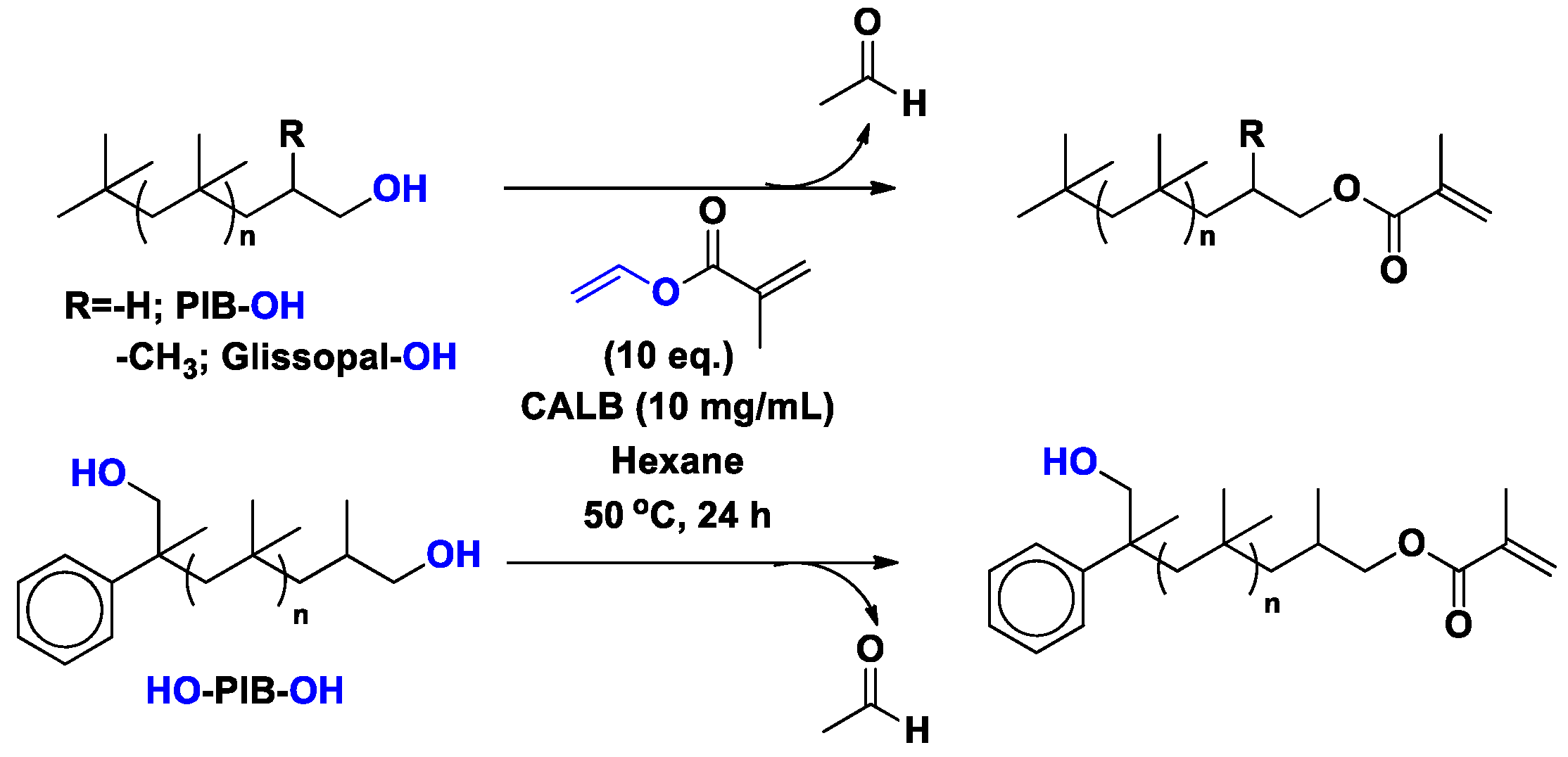
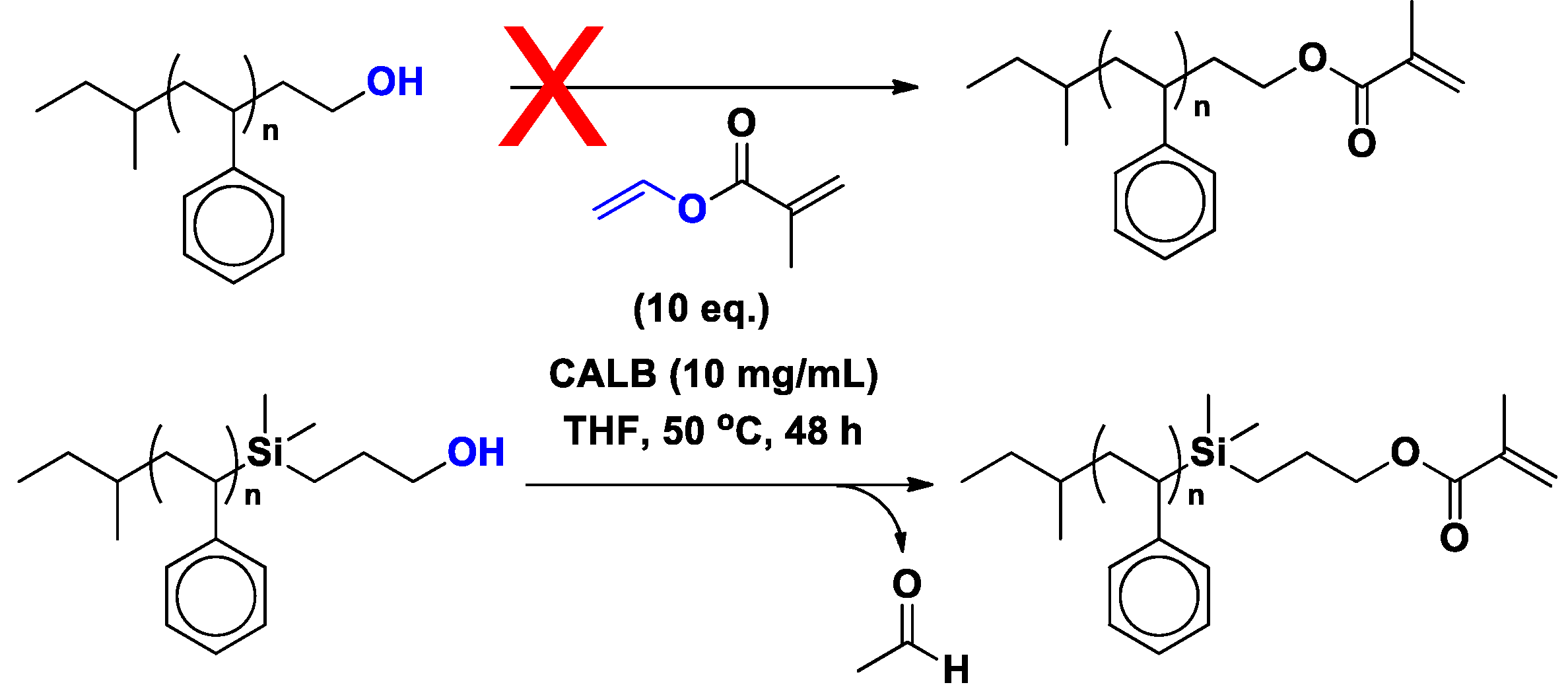
5. Limitations
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, A.S.; Mathias, G.; Inglis, A.J.; Barner-Kowollik, C. Post-Functionalization of Polymers via Orthogonal Ligation Chemistry. Macromol. Rapid Commun. 2013, 34, 810–849. [Google Scholar] [CrossRef] [PubMed]
- Hawker, C.J.; Wooley, K.L. The Convergence of Synthetic Organic and Polymer Chemistries. Science 2005, 309, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Hawker, C.J.; Fokin, V.V.; Finn, M.G.; Sharpless, K.B. Bringing Efficiency to Materials Synthesis: The Philosophy of Click Chemistry. Aust. J. Chem. 2007, 60, 381–383. [Google Scholar] [CrossRef]
- Iha, R.K.; Wooley, K.L.; Nystrom, A.M.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of Orthogonal “Click” Chemistries in the Synthesis of Functional Soft Materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar]
- Puskas, J.E.; Seo, K.S.; Sen, M.Y. Green Polymer Chemistry: Precision Synthesis of Novel Multifunctional oly(ethylene glycol)s Using Enzymatic Catalysis. Eur. Polym. J. 2011, 47, 524–534. [Google Scholar] [CrossRef]
- Gross, R.A.; Kumar, A.; Kalra, B. Polymer Synthesis by in Vitro Enzyme Catalysis. Chem. Rev. 2001, 101, 2097–2124. [Google Scholar] [CrossRef] [PubMed]
- Verger, R. ‘Interfacial Activation’ of Lipases: Facts and Artifacts. Trends Biotechnol. 1997, 15, 32–38. [Google Scholar] [CrossRef]
- Sarda, L.; Desnuelle, P. Action of Pancreatic Lipase on Emulsified Esters. Biochim. Biophys. Acta 1958, 30, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B. Grease Pit Chemistry Exposed. Nat. Struct. Biol. 1994, 1, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Ransac, S.; Carriere, F.; Rogalska, E.; Verger, R.; Marguet, F.; Buono, G.; Melo, E.P.; Cabral, J.M.S.; Egloff, M.E.; Tubeurgh, H.; et al. The kinetics, specifities and structural features of lipases. In Molecular Dynamics of Biomembranes; Kamp, A.F.O.D., Ed.; Springer: Heidelberg, Germany, 1996; pp. 265–304. [Google Scholar]
- Bornscheuer, U.T.; Kazlauskas, R.J. Hydrolases in Organic Synthesis: Regio- and Stereoselective Biotransformations, 2nd ed.; WILEY-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2006. [Google Scholar]
- Uppenberg, J.; Ohmer, N.; Norin, M.; Hult, K.; Kleywegt, G.J.; Patkar, S.; Waagen, V.; Anthomen, T.; Jones, T.A. Crystallographic and Molecular-modeling Studies of Lipase B from Candida Antarctica Reveal a Stereospecificity Pocket for Secondary Alcohols. Biochemistry 1995, 34, 16838–16851. [Google Scholar] [CrossRef] [PubMed]
- Faber, K. Biotransformations in Organic Chemistry, 5th ed.; Springer-Verlag: New York, NY, USA, 2004. [Google Scholar]
- Gotor-Fernandez, V.; Busto, E.; Gotor, V. Candida Antarctica Lipase B: An Ideal Biocatalyst for the Preparation of Nitrogenated Organic Compounds. Adv. Synth.Catal. 2006, 348, 797–812. [Google Scholar] [CrossRef]
- Uppenberg, J.; Hansen, M.T.; Patkar, S.; Jones, T.A. Sequence, Crystal-Structure Determination and Refinement of 2 Crystal Forms of Lipase-B from Candida-Antarctica. Structure 1994, 2, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Uppenberg, J.; Patkar, S.; Bergfors, T.; Jones, T.A. Crystalization and Preliminary-X-ray Studies of Lipase-B from Candida Antarctica. J. Mol. Biol. 1994, 235, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Miletic, N.; Nastasovic, A.; Loos, K. Immobilization of Biocatalysts for Enzymatic Polymerizations: Possibilities, Advantages, Applications. Bioresour. Technol. 2012, 115, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. Recent Developments in Lipase-Catalyzed Synthesis of Polyesters. Macromol. Rapid Commun. 2009, 30, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mishra, M.K.; Kennedy, J.P. Living Carbcationic Polymerization 13. Telechelic Polyisobutylenes by the Linear Bifunctional Aliphatic Initiator System-CH3OC(CH3)2CH2C(CH3)2CH2C(CH3)2OCH3/BCl3. Polym. Bull. 1987, 17, 213–219. [Google Scholar]
- Gil, Y.M.C.; Marcela, Y. Green Polymer Chemistry: The Role of Candida Antarctica Lipase B in Polymer Functionalization. Doctoral Dissertation, The University of Akron, Akron, OH, USA, 2014. [Google Scholar]
- Kirchner, G.; Scollar, M.P.; Klibanov, A.M. Resolution of Racemic Mixtures via Lipase Catalyst in Organic-solvents. J. Am. Chem. Soc. 1985, 107, 7072–7076. [Google Scholar] [CrossRef]
- Wang, Y.F.; Lalonde, J.J.; Momongan, M.; Bergbreiter, D.E.; Wong, C.-H. Lipase-catalyzed Irreversible Transesterifications Using Enol Esters as Acylating Reagents—Preparative Enantioselective and Regioselective Syntheses of Alcohols, Glycerol Derivatives, Sugars, and Organometallics. J. Am. Chem. Soc. 1988, 110, 7200–7205. [Google Scholar] [CrossRef]
- Yadav, G.D.; Trivedi, A.H. Kinetic Modeling of Immobilized-lipase Catalyzed Transesterification of n-octanol with Vinyl Acetate in Non-aqueous Media. Enzym. Microb. Technol. 2003, 32, 783–789. [Google Scholar] [CrossRef]
- Weber, H.K.; Stecher, H.; Faber, K. Sensitivity of Microbial Lipase to Acetaldehyde Formed by Acyl-Transfer Reactions from Vinyl Ester. Biotechnol. Lett. 1995, 17, 803–808. [Google Scholar] [CrossRef]
- Castano, M.; Seo, K.S.; Becker, M.L.; Wesdemiotis, C.; Puskas, J.E. Green polymer chemistry: Synthesis of symmetric and asymmetric telechelic ethylene glycol oligomers. Polym. Chem. 2015, 6, 1137–1142. [Google Scholar] [CrossRef]
- Yadav, G.D.; Lathi, P.S. Lipase Catalyzed Transesterification of Methyl Acetoacetate with n-butanol. J. Mol. Catal. B: Enzym. 2005, 32, 107–113. [Google Scholar] [CrossRef]
- Rizzi, M.; Stylos, P.; Riek, A.; Reuss, M. A Kinetic-study of Immobilized Lipase Catalyzing the Synthesis of Isoamyl Acetate by Transesterification in Normal-hexane. Enzym. Microb. Technol. 1992, 14, 709–714. [Google Scholar] [CrossRef]
- Martinelle, M.; Hult, K. KInetics of Acyl Transfer-Reactions in Organic Media Catalyzed by Candida-Antarctica Lipase-B Biochim. Biophys. Acta 1995, 1251, 191–197. [Google Scholar]
- Lutolf, M.P.; Tirelli, N.; Cerritelli, S.; Cavalli, L.; Hubbell, J.A. Systematic Modulation of Michael-type Reactivity of Thiols Through the Use of Charged Amino Acids. Bioconjugate Chem. 2001, 12, 1051–1056. [Google Scholar] [CrossRef]
- Jadzinsky, P.D.; Calero, G.; Ackerson, C.J.; Bushnell, D.A.; Kornberg, R.D. Structure of a Thiol Monolayer-protected Gold Nanoparticle at 1.1 Angstrom Resolution. Science 2007, 318, 430–433. [Google Scholar]
- Phillips, T.L.; Wasserman, T.H. Promise of Radiosensitizers and Radioprotectors in the Treatment of Human Cancer. Cancer Treat. Rep. 1984, 68, 291–302. [Google Scholar] [PubMed]
- Maheswara, M.; Kim, M.; Yun, S.-J.; Ju, J.J.; Do, J.Y. A new Strategy for Chemoselective O-acylation of Beta-mercapto Alcohols Via Alkylsilyl and Stannyl Protection. Tetrahedron Lett. 2009, 50, 480–483. [Google Scholar] [CrossRef]
- Seo, K.S. Design and Synthesis of Multifunctional Poly(ethylene glycol) Using Enzymatic Catalysis for Multivalent Cancer Drug Delivery. Doctoral Dissertation, The University of Akron, Akron, OH, USA, 2012. [Google Scholar]
- Xiao, P.; Zhang, A.; Zheng, L.; Song, Y. Straightforward Enzyme-Catalyzed Asymmetric Synthesis of Caffeic Acid Esters in Enantioenriched Form. Ind. Eng. Chem. Res. 2014, 53, 11638–11645. [Google Scholar] [CrossRef]
- Dhake, K.P.; Tambade, P.J.; Singhal, R.S.; Bhanage, B.M. Promiscuous Candida Antarctica Lipase B-catalyzed Synthesis of Beta-amino Esters via Aza-Michael Addition of Amines to Acrylates. Tetrahedron Lett. 2010, 51, 4455–4458. [Google Scholar] [CrossRef]
- Torre, O.; Alfonso, I.; Gotor, V. Lipase Catalysed Michael Addition of Secondary Amines to Acrylonitrile. Chem. Commun. 2004, 1724, 1724–1725. [Google Scholar] [CrossRef]
- Svedendahl, M.; Hult, K.; Berglund, P. Fast Carbon-carbon Bond Formation by a Promiscuous Lipase. J. Am. Chem. Soc. 2005, 127, 17988–17989. [Google Scholar] [CrossRef] [PubMed]
- Carlqvist, P.; Svedendahl, M.; Branneby, C.; Hult, K.; Brinck, T.; Berglund, P. Exploring the Active-site of a Rationally Redesigned Lipase for Catalysis of Michael-type Additions. ChemBioChem 2005, 6, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Priego, J.; Ortiz-Nava, C.; Carrillo-Morales, M.; Lopez-Munguıa, A.; Escalante, J.; Castillo, E. Solvent Engineering: An Effective Tool to Direct Chemoselectivity in a Lipase-catalyzed Michael Addition. Tetrahedron 2009, 65, 536–539. [Google Scholar] [CrossRef]
- Kukula, H.; Schlaad, H.; Antonietti, M.; Forster, S. The Formation of Polymer Vesicles or “Peptosomes” by Polybutadiene-block-poly(l-glutamate)s in Dilute Aqueous Solution. J. Am. Chem. Soc. 2002, 124, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.Y.; Karwa, A.; Kostelc, J.G.; Lee, N.S.; Dorshow, R.B.; Wooley, K.L. Paclitaxel-Loaded SCK Nanoparticles: An Investigation of Loading Capacity and Cell Killing Abilities in Vitro. Mol. Pharm. 2012, 9, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Shen, Y.; Pollack, K.A.; Taylor, J.-S.A.; Wooley, K.L. Dual Peptide Nucleic Acid- and Peptide-Functionalized Shell Cross-Linked Nanoparticles Designed to Target mRNA toward the Diagnosis and Treatment of Acute Lung Injury. Bioconjugate Chem. 2012, 23, 574–585. [Google Scholar] [CrossRef]
- Sun, X.K.; Rossin, R.; Turner, J.L.; Becker, M.L.; Joralemon, M.J.; Welch, M.J.; Wooley, K.L. An Assessment of the Effects of Shell Cross-linked Nanoparticle Size, Core Composition, and Surface PEGylation on in Vivo Biodistribution. Biomacromolecules 2005, 6, 2541–2554. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Vicent, M.J. Polymer Therapeutics-prospects for 21st century: The End of the Beginning. Adv. Drug Deliv. Rev. 2013, 65, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Jevsevar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of Therapeutic Proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.K.; Leena, S.; Kumar, K.S. Peptide-polymer Biotherapeutic Synthesis on Novel Cross-linked Beads with “Spatially Tunable” and “Isolated” Functional Sites. Biopolymers 2008, 90, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Puskas, J.E.; Sen, M.Y.; Kasper, J.R. Green Polymer Chemistry: Telechelic Poly(ethylene glycol)s via Enzymatic Catalysis. J. Polym. Sci. Pol. Chem. 2008, 46, 3024–3028. [Google Scholar] [CrossRef]
- Sen, M.Y.; Puskas, J.E.; Ummadisetty, S.; Kennedy, J. P. Green Polymer Chemistry: II Enzymatic Synthesis of Methacrylate-Terminated Polyisobutylenes. Macromol. Rapid Commun. 2008, 29, 1598–1602. [Google Scholar] [CrossRef]
- Seo, K.S.; Puskas, J.E.; Ummadisetty, S.; Kennedy, J.P. Enzyme-catalyzed Quantitative Chain-end Functionalization of Poly(ethylene glycol)s Under Solventless Conditions. RSC Adv. 2014, 4, 1683–1688. [Google Scholar] [CrossRef]
- Jankova, K.; Kops, J. 1H-NMR Investigation of Quantitative Functionallizatin of Poly(ethylene glycol)s. J. Appl. Polym. Sci. 1994, 54, 1027–1032. [Google Scholar]
- Dau, J.; Lagaly, G. Surface Modification of Bentonites. II. Modification of Montmorillonite with Cationic Poly(ethylene oxides). Croatica Chemica Acta 1998, 71, 983–1004. [Google Scholar]
- Castano, M.; Seo, K.S.; Kim, E.H.; Becker, M.L.; Puskas, J.E. Green Polymer Chemistry VIII: Synthesis of Halo-ester-Functionalized Poly(ethylene glycol)s via Enzymatic Catalysis. Macromol. Rapid Commun. 2013, 34, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Puskas, J.E.; Sen, M.Y.; Seo, K.S. Green polymer chemistry using nature’s catalysts, enzymes. J. Polym. Sci. Pol. Chem. 2009, 47, 2959–2976. [Google Scholar] [CrossRef]
- Puskas, J.E.; Chen, Y.; Dahman, Y.; Padavan, D. Polyisobutylene-based Biomaterials. J. Polym. Sci. Pol. Chem. 2004, 42, 3091–3109. [Google Scholar] [CrossRef]
- Pinchuk, L.; Wilson, G.J.; Barry, J.J.; Schoephoerster, R.T.; Parel, J.-M.; Kennedy, J.P. Medical Applications of Poly(styrene-block-isobutylene-block-styrene) (“SIBS”). Biomaterials 2008, 29, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Puskas, J.E.; Munoz-Robledo, L.G.; Hoerr, R.A.; Foley, J.; Schmidt, S.P.; Evancho-Chapman, M.; Dong, J.; Frethem, C.; Haugstad, G. Drug-eluting Stent Coatings. WIRES Nanomed. Nanobiotechnol. 2009, 1, 451–462. [Google Scholar] [CrossRef]
- Puskas, J.E.; Sen, M.Y. Green Polymer Chemistry: Enzymetic Functionalization of Liquid Polymers in Bulk. Polym. Prepr. 2009, 50, 34–35. [Google Scholar]
- Sen, M.Y. Functionalization of Polymers Using Enzymatic Catalysis. Doctoral Dissertation, The University of Akron, Akron, OH, USA, 2009. [Google Scholar]
- Holmberg, K. Applications of Block Copolymers. In Amphiphilic Block Copolymers: Self-Assembly and Applications; Alexandridis, P., Lindman, B., Eds.; Elsevier: Oxford, UK, 2000; pp. 305–318. [Google Scholar]
- Sereti, V.; Stamatis, H.; Koukios, E.; Kolisis, F.N. Enzymatic Acylation of Cellulose Acetate in Organic Media. J. Biotechnol. 1998, 66, 219–223. [Google Scholar] [CrossRef]
- Sereti, V.; Stamatis, H.; Pappas, C.; Polissiou, M.; Kolisis, F.N. Enzymatic Acylation of Hydroxypropyl Cellulose in Organic Media and Determination of Ester Formation by Diffuse Reflectance Infrared Fourier Transform (DRIFT) Spectroscopy. Biotechnol. Bioeng. 2001, 72, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Sahoo, B.; Teraoka, I.; Miller, L.M.; Gross, R.A. Enzyme-catalyzed Regioselective Modification of Starch Nanoparticles. Macromolecules 2005, 38, 61–68. [Google Scholar] [CrossRef]
- Chen, Z.-G.; Zong, M.-H.; Li, G.-J. Lipase-catalyzed Modification of Konjac Glucomannan. J. Appl. Polym. Sci. 2006, 102, 1335–1340. [Google Scholar] [CrossRef]
- Duxbury, C.J.; Hilker, I.; de Wildeman, S.M.A.; Heise, A. Enzyme-responsive Materials: Chirality to Program Polymer Reactivity. Angew. Chem. Int. Ed. 2007, 46, 8452–8454. [Google Scholar] [CrossRef]
- Padovani, M.; Hilker, I.; Duxbury, C.J.; Heise, A. Functionalization of Polymers with High Precision by Dual Regio-and Stereoselective Enzymatic Reactions. Macromolecules 2008, 41, 2439–2444. [Google Scholar] [CrossRef]
- Pavel, K.; Ritter, H. Enzymes in polymer chemistry, 6. Lipase-catalyzed acylation of comb-like methacrylic polymers containing OH groups in the side chains. Die Makromolekulare Chemie 1992, 193, 323–328. [Google Scholar]
- Shao, L.; Kumar, G.; Lenhart, J.L.; Smith, P.J.; Payne, G.F. Enzymatic modification of the synthetic polymer polyhydroxystyrene. Enzym. Microb. Technol. 1999, 25, 660–668. [Google Scholar] [CrossRef]
- Jarvie, A.W.P.; Overton, N.; St Pourcain, C.B. Enzyme Catalysed Modification of Synthetic Polymers. J. Chem. Soc. Perkin Trans. 1999, 1, 2171–2176. [Google Scholar] [CrossRef]
- Bjorkling, F.; Godtfredsen, S.E.; Kirk, O. Lipase-mediated Formation of Peroxycarboxylic Acids used in Catalytic Epoxidation of Alkenes. J. Chem. Soc. Chem. Commun. 1990, 1301, 1301–1303. [Google Scholar] [CrossRef]
- Quirk, R.P.; Kim, H.; Polce, M.J.; Wesdemiotis, C. Anionic synthesis of primary amine functionalized polystyrenes via hydrosilation of allylamines with silyl hydride functionalized polystyrenes. Macromolecules 2005, 38, 7895–7906. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, S.; Puskas, J.E. Green Polymer Chemistry: Enzyme Catalysis for Polymer Functionalization. Molecules 2015, 20, 9358-9379. https://doi.org/10.3390/molecules20059358
Sen S, Puskas JE. Green Polymer Chemistry: Enzyme Catalysis for Polymer Functionalization. Molecules. 2015; 20(5):9358-9379. https://doi.org/10.3390/molecules20059358
Chicago/Turabian StyleSen, Sanghamitra, and Judit E. Puskas. 2015. "Green Polymer Chemistry: Enzyme Catalysis for Polymer Functionalization" Molecules 20, no. 5: 9358-9379. https://doi.org/10.3390/molecules20059358