
Absorption and Metabolism Characteristics of Triptolide as Determined by a Sensitive and Reliable LC-MS/MS Method

Abstract
:1. Introduction
2. Results and Discussion
2.1. Chromatography and Mass Spectrometry
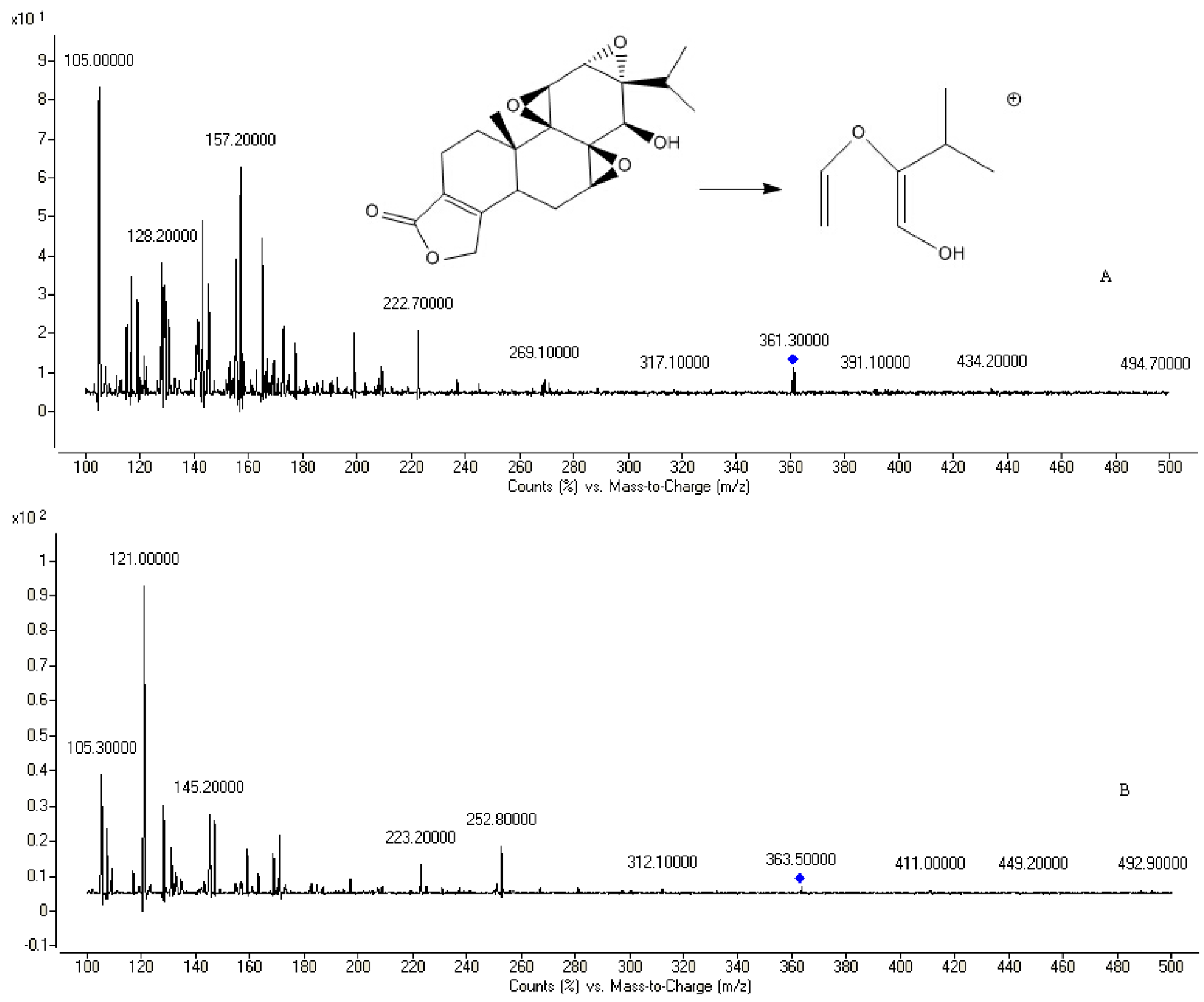
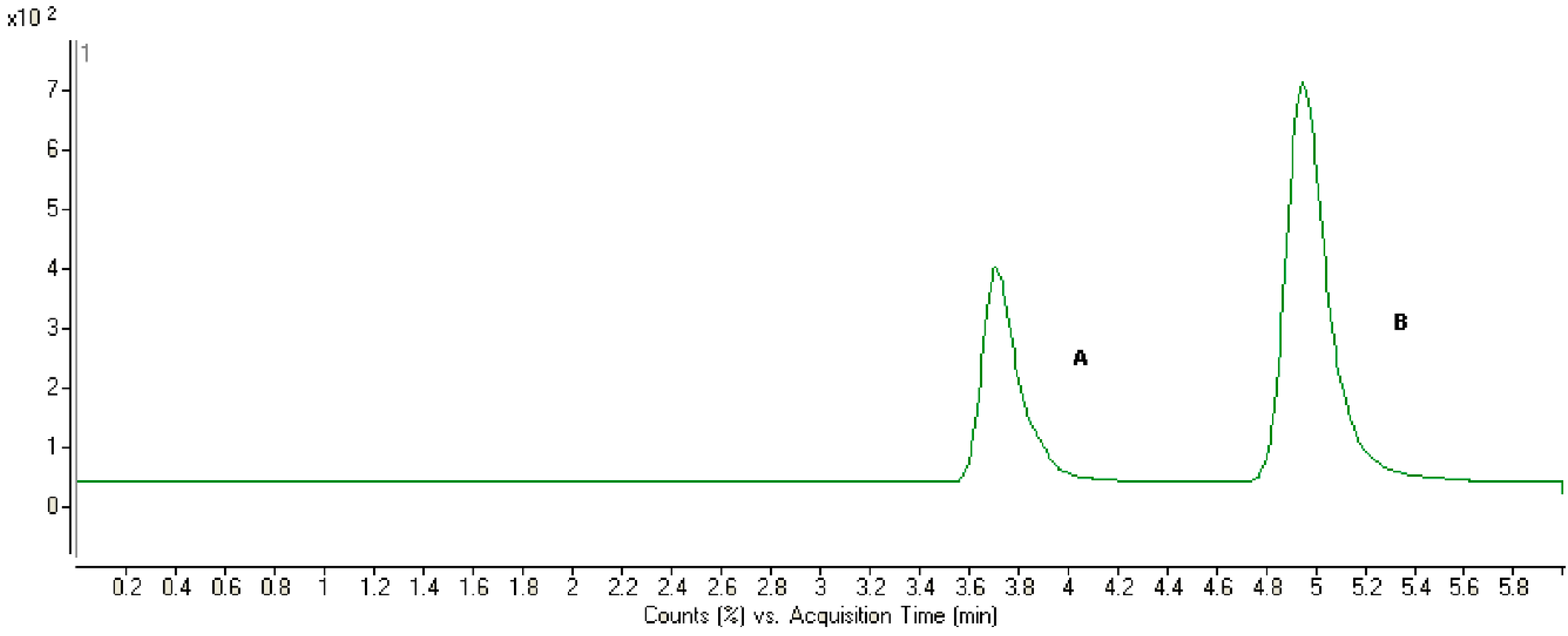
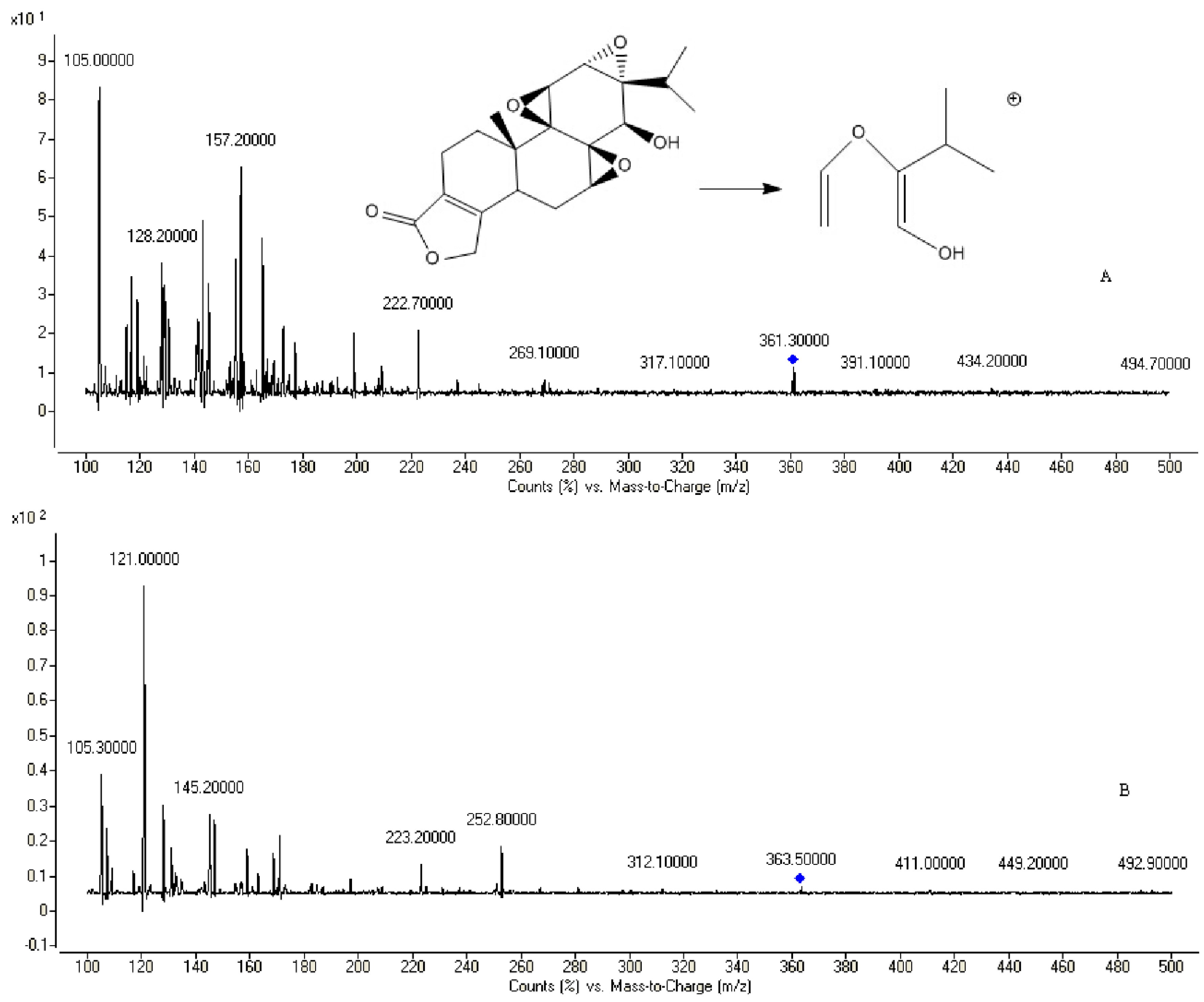
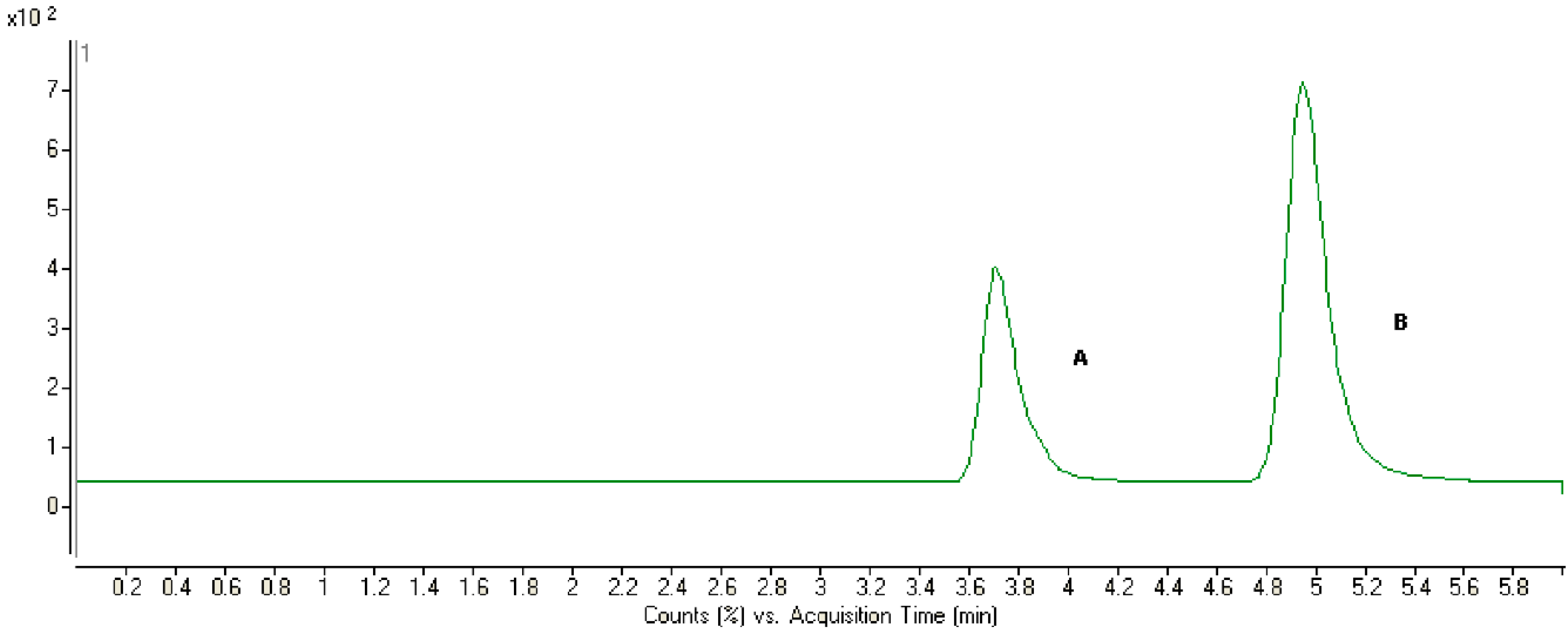
2.2. Method Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Plasma Samples (ng/mL) | Intra-Day | Inter-Day | ||||
|---|---|---|---|---|---|---|---|
| Concentration measured (ng/mL) | Precision (%, RSD) | Accuracy (%, RE) | Concentration measured (ng/mL) | Precision (%, RSD) | Accuracy (%, RE) | ||
| Triptolide | 10 | 10.58 | 5.4 | 5.80 | 9.23 | 8.2 | −7.70 |
| 50 | 46.35 | 4.8 | −7.30 | 54.35 | 7.5 | 8.70 | |
| 500 | 441.54 | 2.3 | −11.69 | 551.24 | 4.5 | 10.24 | |
| Analyte | Plasma Samples (ng/mL) | Stability (%, RE) | ||
|---|---|---|---|---|
| Short-Term (24 h at room temperature) | Long-Term (30 days at −40 °C) | Three Freeze-Thaw Cycles at −40 °C | ||
| Triptolide | 10 | 8.1 | 3.8 | −4.1 |
| 50 | −7.5 | 7.4 | 6.8 | |
| 500 | 9.98 | 8.5 | 9.54 | |
2.3. Pharmacokinetic Studies

| Parameter | Oral | Intravenous |
|---|---|---|
| Tmax (h) | 0.17 ± 0.03 | - |
| Cmax (μg·L−1) | 293.19 ± 24.43 | - |
| t1/2 (h) | 0.42 ± 0.23 | 0.19 ± 0.01 |
| AUC (0-t) (μg·h·L−1) | 151.16 ± 17.69 | 236.50 ± 26.40 |
| AUMC (0-t) (μg·h·L−1) | 112.99 ± 21.20 | 57.35 ± 9.76 |
| CL (L·h−1·kg−1) | 6.67 ± 0.78 | 4.26 ± 0.48 |
| MRT (h) | 0.74 ± 0.05 | 0.24 ± 0.02 |
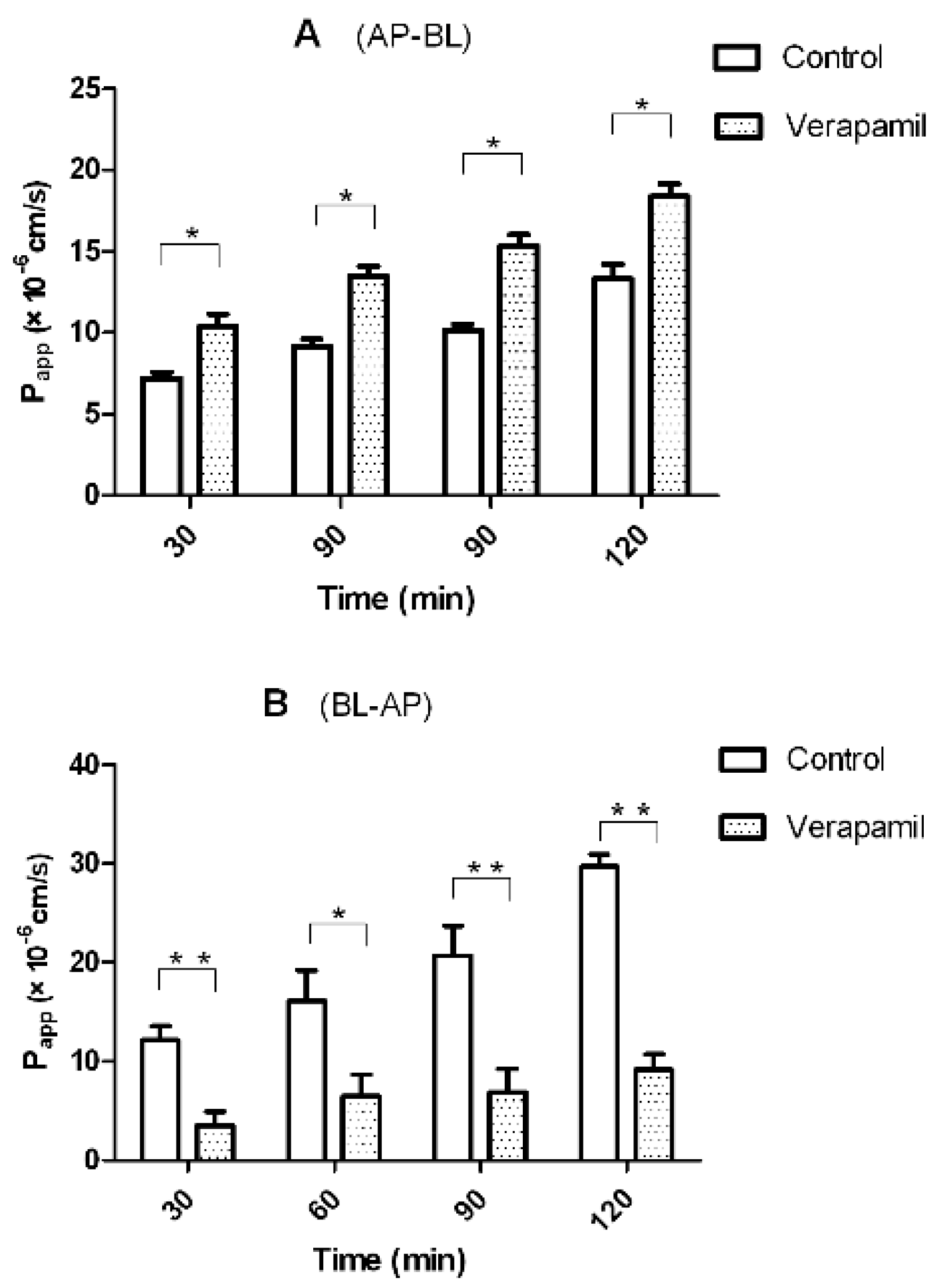
2.4. Transport of Triptolide across a Caco-2 Cell Monolayer Model

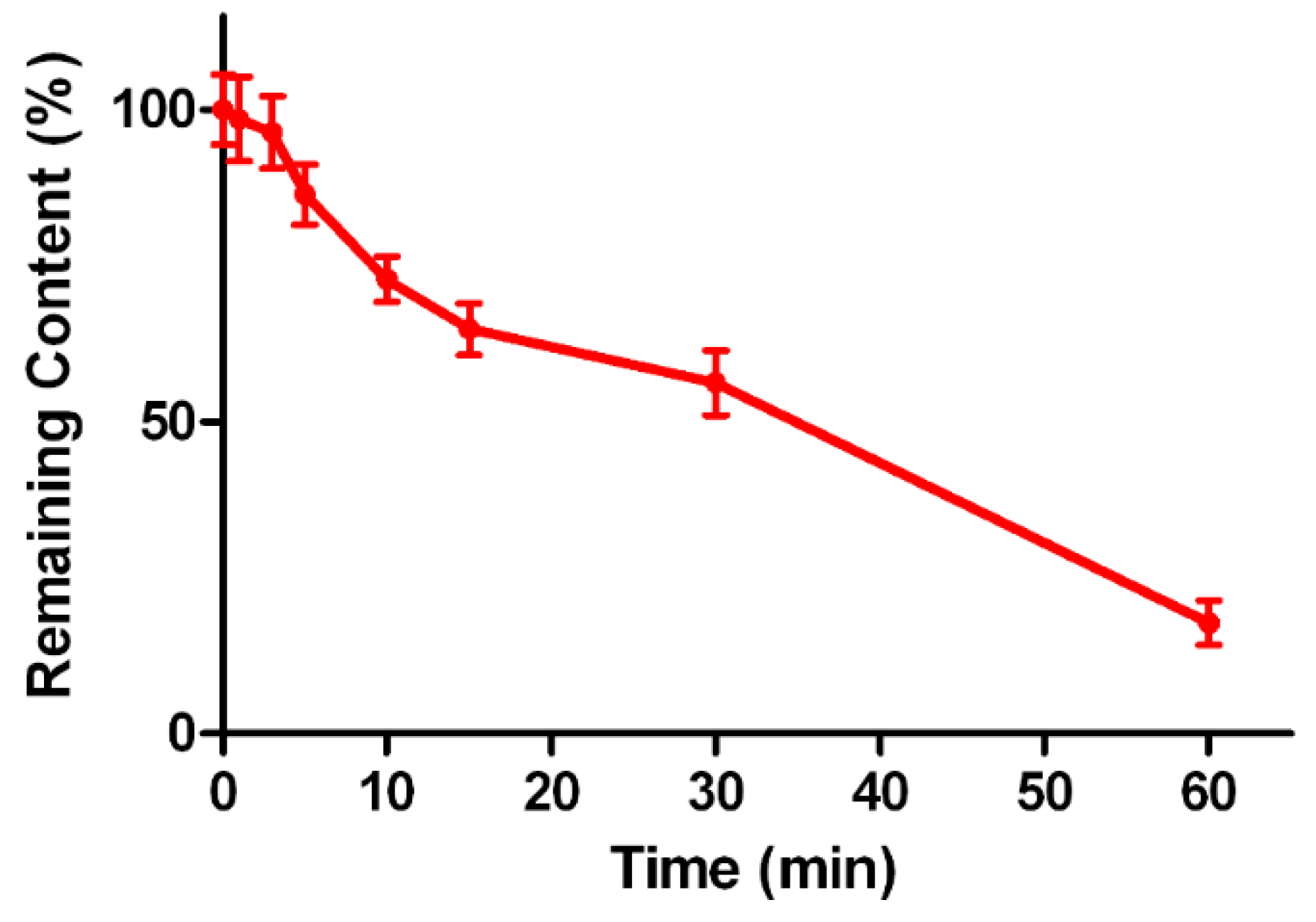
2.5. Metabolic Stability of Triptolide in Human Liver Microsomes
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Instrumentation and Conditions
3.3. Pharmacokinetic Study in Vivo
3.3.1. Animals
3.3.2. In Vivo Pharmacokinetic Study
3.3.3. Plasma Sample Preparation
3.3.4. Preparation of Standard and Quality Control Samples
3.3.5. Method Validation
3.3.6. Data Analysis
3.4. Caco-2 Cell Monolayer Model
3.4.1. Cell Culture
3.4.2. Transport Studies
3.4.3. Data Analysis in the Caco-2 Cell Monolayer Model
3.5. Metabolic Stability in Human Liver Microsomes
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brinker, A.M.; Raskin, I. Determination of triptolide in root extracts of Tripterygium wilfordii by solid-phase extraction and reverse-phase high-performance liquid chromatography. J. Chromatogr. A 2005, 1070, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Jiang, Z.Z.; Zhang, L.Y. Triptolide: Progress on research in pharmacodynamics and toxicology. J. Ethnopharmacol. 2014, 155, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; LeBlanc, K.J.; Li, L. Triptolide preserves cognitive function and reduces neuropathology in a mouse model of Alzheimer’s disease. PLoS ONE 2014, 9, e108845. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.; Xia, H.; Park, W.; Hackett, M.J.; Song, C.; Na, K.; Hui, K.M.; Hyeon, T. pH-Sensitive Nanoformulated Triptolide as a Targeted Therapeutic Strategy for Hepatocellular Carcinoma. ACS Nano 2014, 8, 8027–8039. [Google Scholar] [CrossRef] [PubMed]
- Park, B. Triptolide, a diterpene, inhibits osteoclastogenesis, induced by RANKL signaling and human cancer cells. Biochimie 2014, 105, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, L.; Zhou, G.B. The main anticancer bullets of the Chinese medicinal herb, thunder god vine. Molecules 2011, 16, 5283–5297. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.; Challana, S. Reproductive toxicity of triptolide in male house rat, Rattus rattus. Sci. World J. 2014, 2014, 879405. [Google Scholar]
- Li, W.; Liu, Y.; He, Y.Q.; Zhang, J.W.; Gao, Y.; Ge, G.B.; Liu, H.X.; Huo, H.; Liu, H.T.; Wang, L.M.; et al. Characterization of triptolide hydroxylation by cytochrome P450 in human and rat liver microsomes. Xenobiotica 2008, 38, 1551–1565. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L.; Zhou, X.; Chen, X.; Huang, H.; Zhao, S.; Li, X.; Zhong, D. Metabolite profiling and identification of triptolide in rats. J. Chromatogr. B 2013, 939, 51–58. [Google Scholar] [CrossRef]
- Shao, F.; Wang, G.; Xie, H.; Zhu, X.; Sun, J.; A, J. Pharmacokinetic study of triptolide, a constituent of immunosuppressive chinese herb medicine, in rats. Biol. Pharm. Bull. 2007, 30, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.; Huang, X.; Su, Y.; Ji, J.; Jiang, Z.; Zhang, L. Glycyrrhizin accelerates the metabolism of triptolide through induction of CYP3A in rats. J. Ethnopharmacol. 2014, 152, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.L.; Shao, Q.; Qu, H.B.; Cheng, Y.Y. Simple method for determination of five terpenoids from different parts of Tripterygium wilfordii and its preparations by HPLC coupled with evaporative light scattering detection. J. Sep. Sci. 2007, 30, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Schmidt, B.M.; Poulev, A.; Raskin, I. Determination of tripdiolide in root extracts of Tripterygium wilfordii by solid-phase extraction and reversed-phase high-performance liquid chromatography. Phytochem. Anal. 2008, 19, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhang, L.; Zhao, X.; Hu, L.; Jiang, Z. Simultaneous determination of triptolide, wilforlide A and triptonide in human plasma by high-performance liquid chromatography-electrospray ionization mass spectrometry. Biol. Pharm. Bull. 2006, 29, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.M.; Liu, P.X.; Zhang, Y.J.; Li, C.K.; Li, Y.; Wang, J.; Zhou, L.; Zhang, Z.Q. Simultaneous determination of triptolide and its prodrug MC002 in dog blood by LC-MS/MS and its application in pharmacokinetic studies. J. Ethnopharmacol. 2013, 150, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Su, M.X.; Song, M.; Yang, D.S.; Shi, J.F.; Di, B.; Hang, T.J. Simultaneous LC-MS/MS determination of five tripterygium pyridine alkaloids in dog plasma and its application to their pharmacokinetic study after oral administration of tripterygium glycosides tablets. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 990, 31–38. [Google Scholar] [CrossRef]
- Su, P.; Cheng, Q.; Wang, X.; Cheng, X.; Zhang, M.; Tong, Y.; Li, F.; Gao, W.; Huang, L. Characterization of eight terpenoids from tissue cultures of the Chinese herbal plant, Tripterygium wilfordii, by high-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Biomed. Chromatogr. 2014, 28, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, Q.; Li, W.; Sun, S.; Zhang, W.; Zhu, Z.; Zhang, G.; Chai, Y. Absorption and metabolism of three monoester-diterpenoid alkaloids in Aconitum carmichaeli after oral administration to rats by HPLC-MS. J. Ethnopharmacol. 2014, 154, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Sun, F.; Zhao, L.; Zhong, Y.; Chai, Y.; Zhang, G. Intestinal transport of sophocarpine across the Caco-2 cell monolayer model and quantification by LC/MS. Biomed. Chromatogr. 2014, 28, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.Y.; Liang, S.C.; Ge, G.B.; Liu, Y.; Dong, P.P.; Zhang, J.W.; Wang, A.X.; Hou, J.; Zhu, L.L.; Yang, L.; et al. Inhibitory effects of sanguinarine on human liver cytochrome P450 enzymes. Food Chem. Toxicol. 2013, 56, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, X.; Chen, Y.; Wu, Y. Absorption and Metabolism Characteristics of Triptolide as Determined by a Sensitive and Reliable LC-MS/MS Method. Molecules 2015, 20, 8928-8940. https://doi.org/10.3390/molecules20058928
Gong X, Chen Y, Wu Y. Absorption and Metabolism Characteristics of Triptolide as Determined by a Sensitive and Reliable LC-MS/MS Method. Molecules. 2015; 20(5):8928-8940. https://doi.org/10.3390/molecules20058928
Chicago/Turabian StyleGong, Xiaomei, Yan Chen, and Yi Wu. 2015. "Absorption and Metabolism Characteristics of Triptolide as Determined by a Sensitive and Reliable LC-MS/MS Method" Molecules 20, no. 5: 8928-8940. https://doi.org/10.3390/molecules20058928