
A Structural Hierarchy Matching Approach for Molecular Similarity/Substructure Searching

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Substructures and Fuzzy Fingerprints
2.2. Similarity Searching

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Hits | Rank | Hits | Rank | Hits | Rank | Hits | Rank | Hits | Rank | Hits |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10,583 | 8 | 55 | 15 | 9 | 22 | 3 | 29 | 1 | 36 | 1 |
| 2 | 1773 | 9 | 40 | 16 | 8 | 23 | 2 | 30 | 1 | 37 | 1 |
| 3 | 622 | 10 | 30 | 17 | 8 | 24 | 2 | 31 | 1 | 38 | 1 |
| 4 | 312 | 11 | 22 | 18 | 7 | 25 | 4 | 32 | 1 | 39 | 1 |
| 5 | 165 | 12 | 17 | 19 | 6 | 26 | 2 | 33 | 1 | 40 | 1 |
| 6 | 111 | 13 | 13 | 20 | 7 | 27 | 3 | 34 | 1 | 41 | 1 |
| 7 | 73 | 14 | 11 | 21 | 5 | 28 | 4 | 35 | 1 | 42 | 1 |
2.3. Substructure Searching
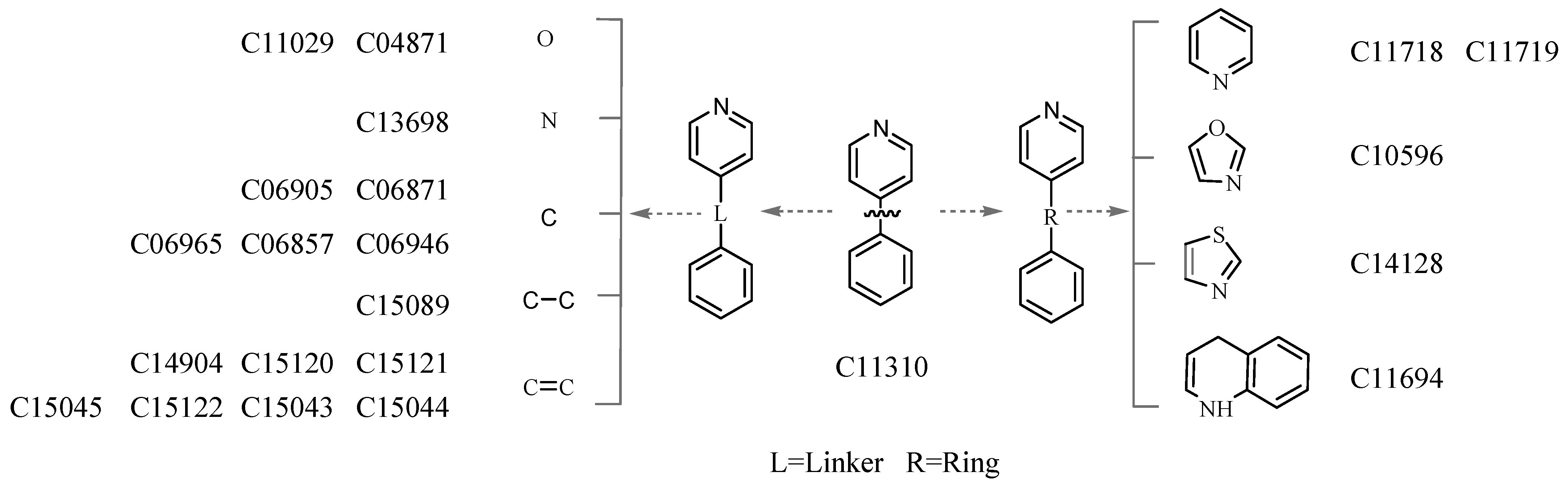
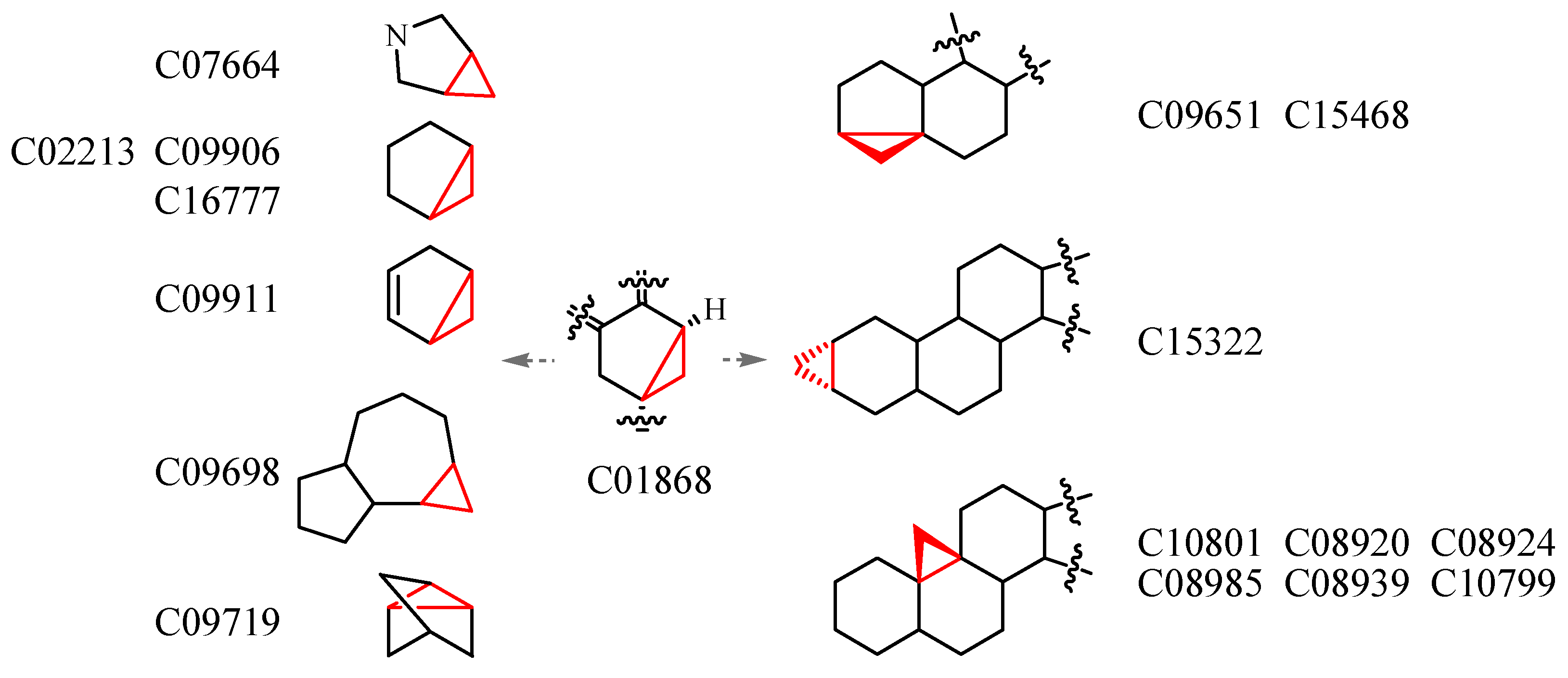
3. Experimental Section
3.1. Chemical Dataset
3.2. Converting Molecular ConneZction Tables to Canonical SMILES
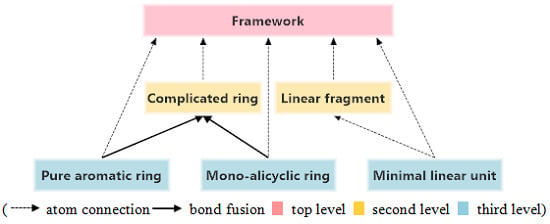
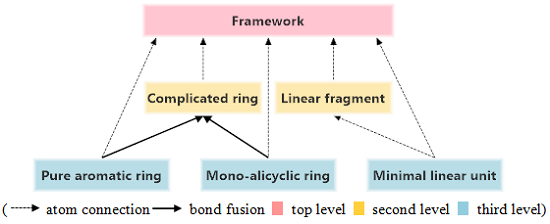
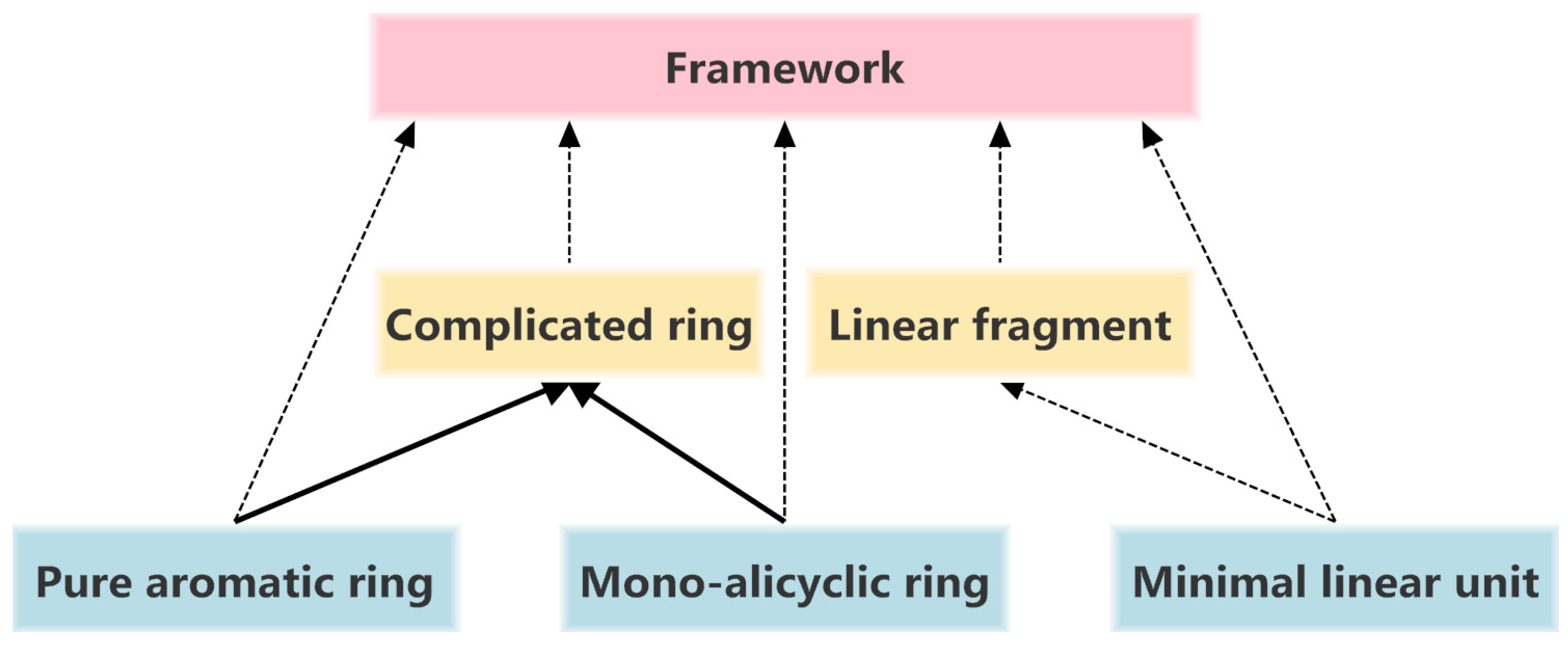
3.3. Structure Hierarchies
 top level,
top level,  second level,
second level,  third level).
top level, second level, third level).
third level).
top level, second level, third level).
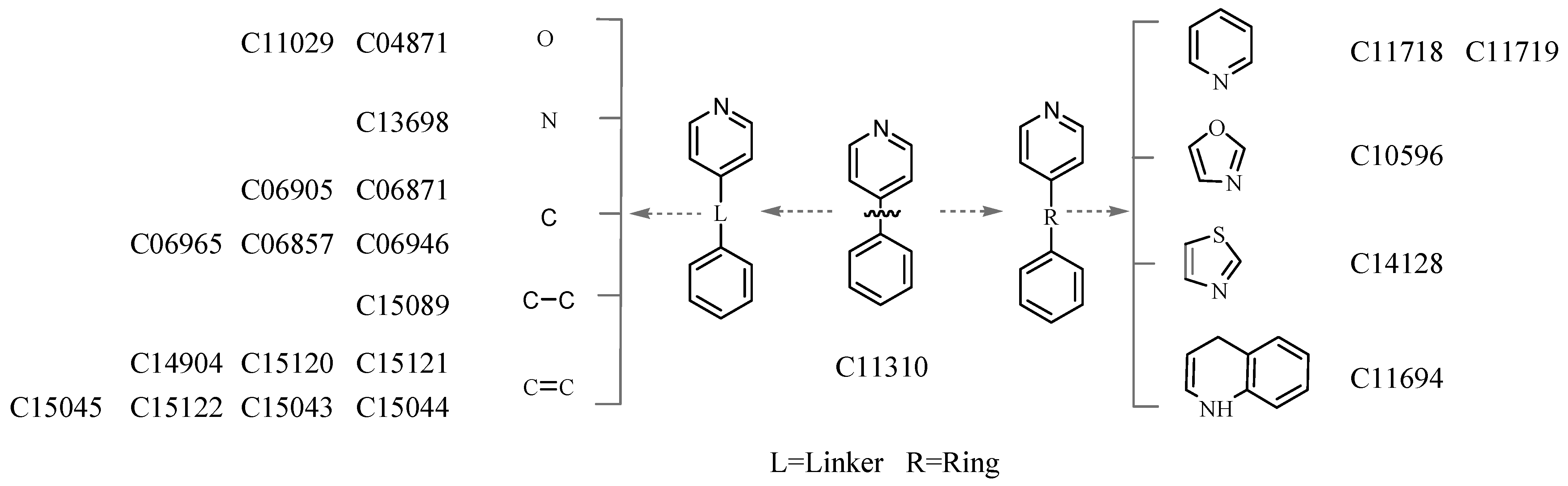
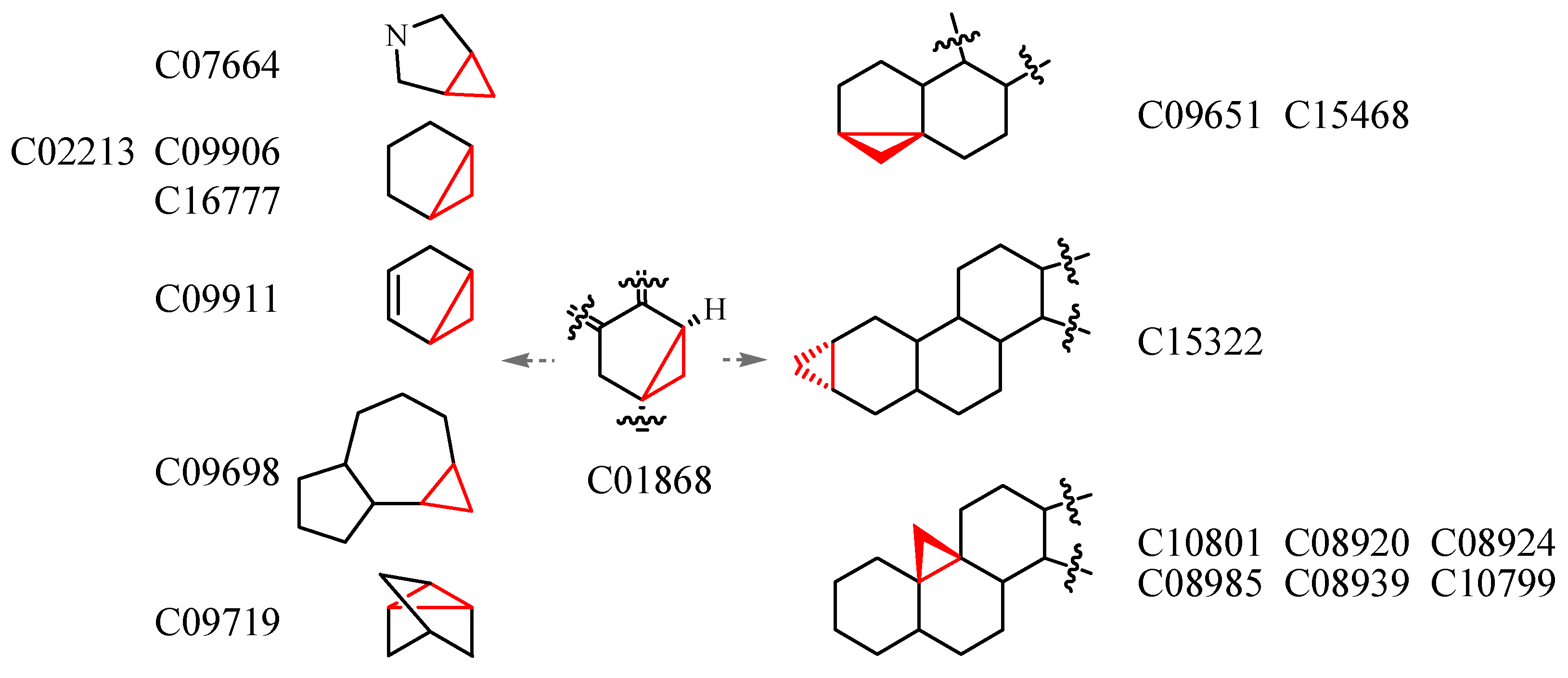
3.4. Deriving the Substructures
SET temp_list substructure_list
READ dataset
FOR each_molecule IN dataset
DETERMINE molecule_hierarchy
IF Framework THEN
PUSH ring linker side_chain TO temp_list
ELSE IF Complicated_ring THEN
PUSH ring side_chain TO temp_list
ELSE IF Unit_ring THEN
PUSH ring TO substructure_list
PUSH side_chain TO temp_list
ELSE
PUSH Linear_fragment TO temp_list
ENDIF
ENDFOR
FOR each_ring IN temp_list
DETERMINE Unit_ring
IF TRUE THEN
PUSH TO substructure_list
ELSE
GET Unit_ring
PUSH Unit_ring TO substructure_list
ENDIF
ENDFOR
FOR each_non-ring IN temp_list
DETERMINE Unit_line
IF TRUE THEN
PUSH TO substructure_list
ELSE
REPEAT
GET longer_linear_fragment
UNTIL Unit_line
PUSH Line TO substructure_list
ENDIF
ENDFOR
3.5. Substructure and Its Chemical Environment
3.6. Fuzzy Fingerprints
3.7. Ranking of the Retrieved Molecules
READ query_molecule_hierarchy
IF Complicated_ring THEN
RANK Complicated_ring AS first_level
RANK side_chain AS second_level_or_third_level
ELSE
RANK Framework AS first_level
RANK side_chain AS second_level_or_third_level
ENDIF
3.8. Testing the Matching Precision in Similarity Searching
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1988, 38, 983–996. [Google Scholar] [CrossRef]
- Johnson, A.M.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: New York, NY, USA, 1990; pp. 384–393. [Google Scholar]
- Hattori, M.; Okuno, Y.; Goto, S.; Kanehisa, M. Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc. 2003, 125, 11853–11865. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Baskin, I.; Varnek, A. Fragment descriptors in SAR/QSAR/QSPR studies, molecular similarity analysis and in virtual screening. In Chemoinformatics Approaches to Virtual Screening; Varnek, A., Tropsha, A., Eds.; Royal Society of Chemistry: Cambridge, UK, 2008; Chapter 1; pp. 539–540. [Google Scholar]
- Yan, X.; Gu, Q.; Lu, F.; Li, J.B.; Xu, J. GSA: A GPU-accelerated structure similarity algorithm and its application in progressive virtual screening. Mol. Divers. 2012, 16, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Jilek, R.J.; Guessregen, S.; Clark, S.J.; Wendt, B.; Clark, R.D. Lead-Hopping. Validation of topomer similarity as a superior predictor of similar biological activities. J. Med. Chem. 2004, 47, 6777–6791. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3. [Google Scholar] [CrossRef]
- The ChinMedNetworks. Available online: http://chinmednetworks.org/chem (accessed on 1 March 2015).
- Goto, S.; Okuno, Y.; Hattori, M.; Nishioka, T.; Kanehisa, M. LIGAND: Database of chemical compounds and reactions in biological pathways. Nucleic Acids Res. 2002, 30, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Nishioka, T.; Kanehisa, M. LIGAND: Chemical database for enzyme reactions. Bioinformatics 1998, 14, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system.1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES.2. Algorithm for generation of unique SMILES notation. J. Am. Chem. Soc. 1989, 29, 97–101. [Google Scholar]
- Guha, R.; Howard, M.T.; Hutchison, G.R.; Murray-Rust, P.; Rzepa, H.; Steinbeck, C.; Wegner, J.; Willighagen, E.L. The Blue Obelisk-interoperability in chemical informatics. J. Chem. Inf. Model. 2006, 46, 991–998. [Google Scholar] [CrossRef] [PubMed]
- The Open Babel Package. Available online: http://openbabel.org (accessed on 1 October 2011).
- Siegel, M.G.; Vieth, M. Drugs in other drugs: A new look at drugs as fragments. Drug Discov. Today 2007, 12, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Bemis, G.W.; Murcko, M.A. Properties of known drugs. 2. Side chains. J. Med. Chem. 1999, 42, 5095–5099. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gu, Q.; Liu, H.; Zhou, J.; Bu, X.; Huang, Z.; Lu, G.; Li, D.; Wei, D.; Wang, L.; et al. Chemomics and drug innovation. Sci. China Chem. 2013, 56, 71–85. [Google Scholar] [CrossRef]
- Yan, X.; Ding, P.; Liu, Z.; Wang, L.; Liao, C.; Gu, Q.; Xu, J. Big data in drug design. Chin. Sci. Bull. 2015, 60, 558–565. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, S.-S.; Dong, H.-J.; Zhou, X.-X.; Liu, Y.-M.; Zhang, F.-X.; Wang, Q.; Huang, X.-A. A Structural Hierarchy Matching Approach for Molecular Similarity/Substructure Searching. Molecules 2015, 20, 8791-8799. https://doi.org/10.3390/molecules20058791
Ji S-S, Dong H-J, Zhou X-X, Liu Y-M, Zhang F-X, Wang Q, Huang X-A. A Structural Hierarchy Matching Approach for Molecular Similarity/Substructure Searching. Molecules. 2015; 20(5):8791-8799. https://doi.org/10.3390/molecules20058791
Chicago/Turabian StyleJi, Shu-Shen, Hong-Ju Dong, Xin-Xin Zhou, Ya-Min Liu, Feng-Xue Zhang, Qi Wang, and Xin-An Huang. 2015. "A Structural Hierarchy Matching Approach for Molecular Similarity/Substructure Searching" Molecules 20, no. 5: 8791-8799. https://doi.org/10.3390/molecules20058791