
Antitrypanosomal Acetylene Fatty Acid Derivatives from the Seeds of Porcelia macrocarpa (Annonaceae)

Abstract
:1. Introduction
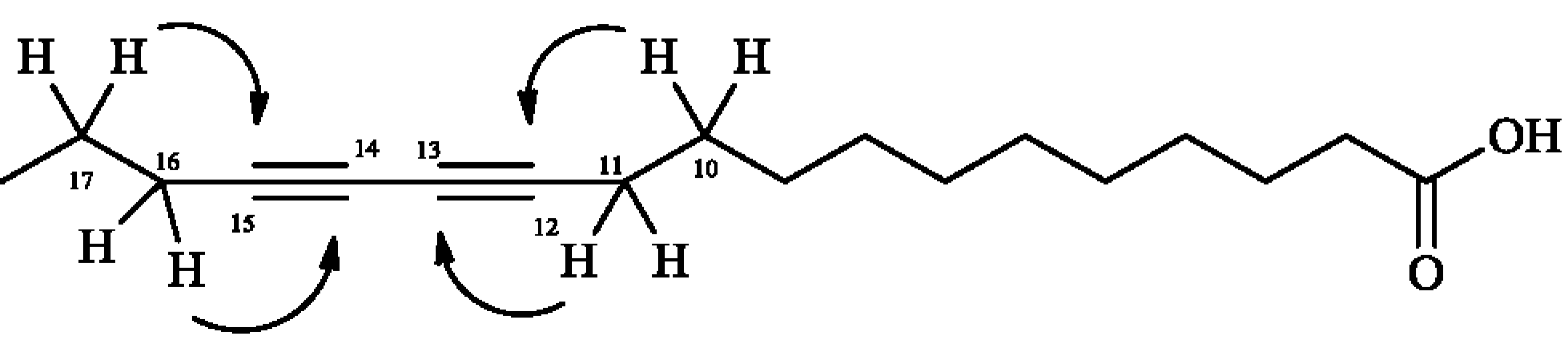
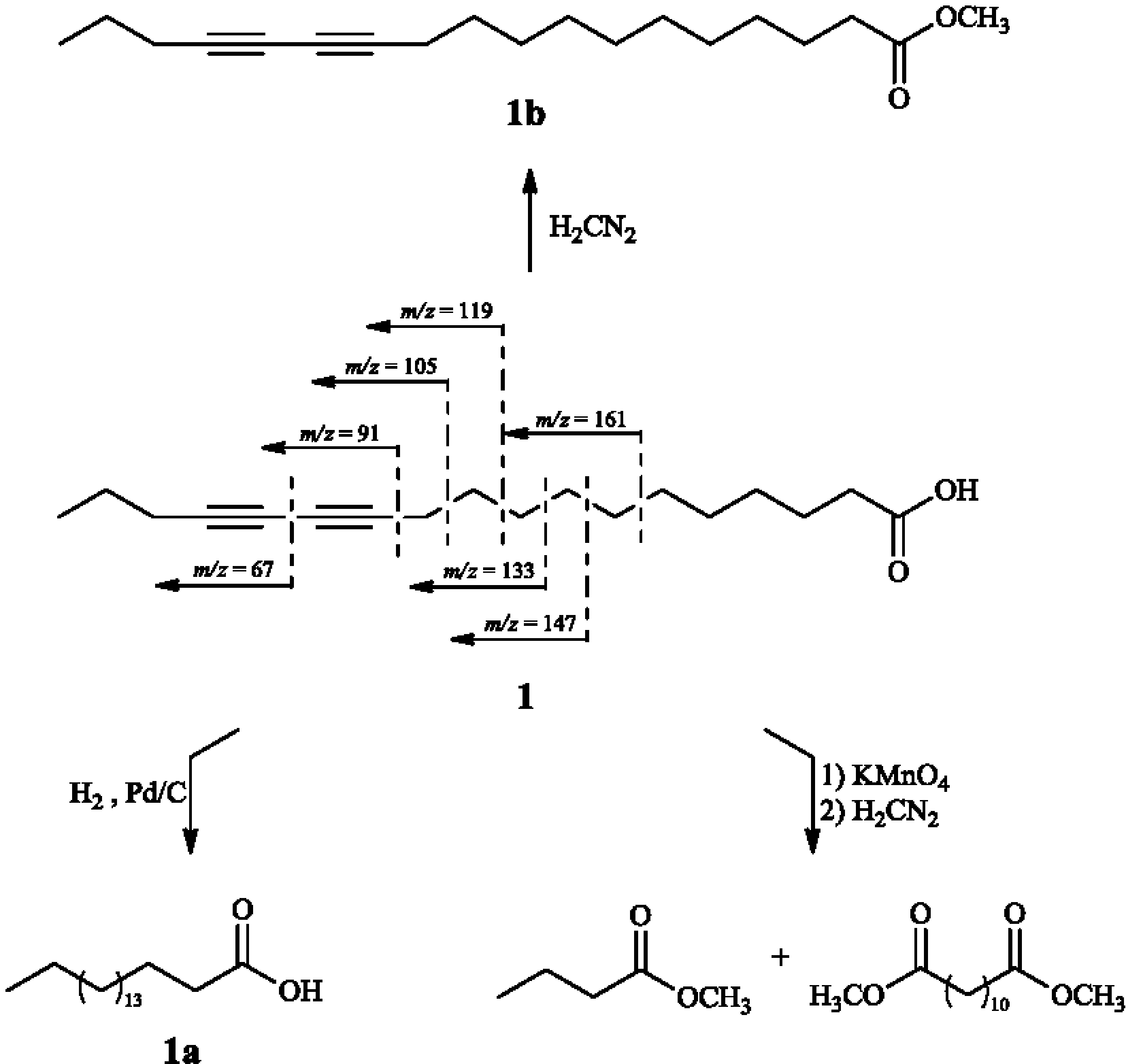
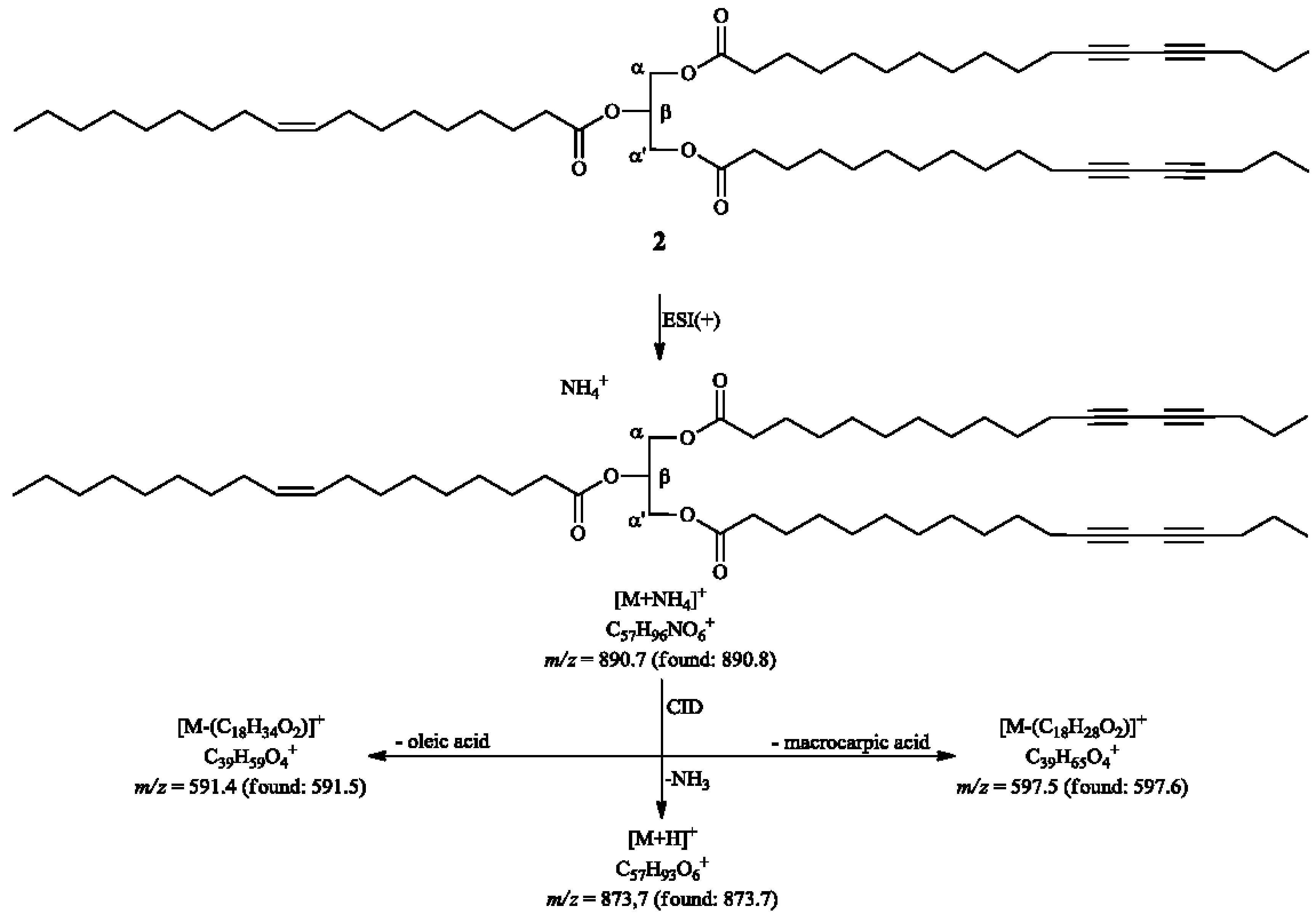
2. Results and Discussion
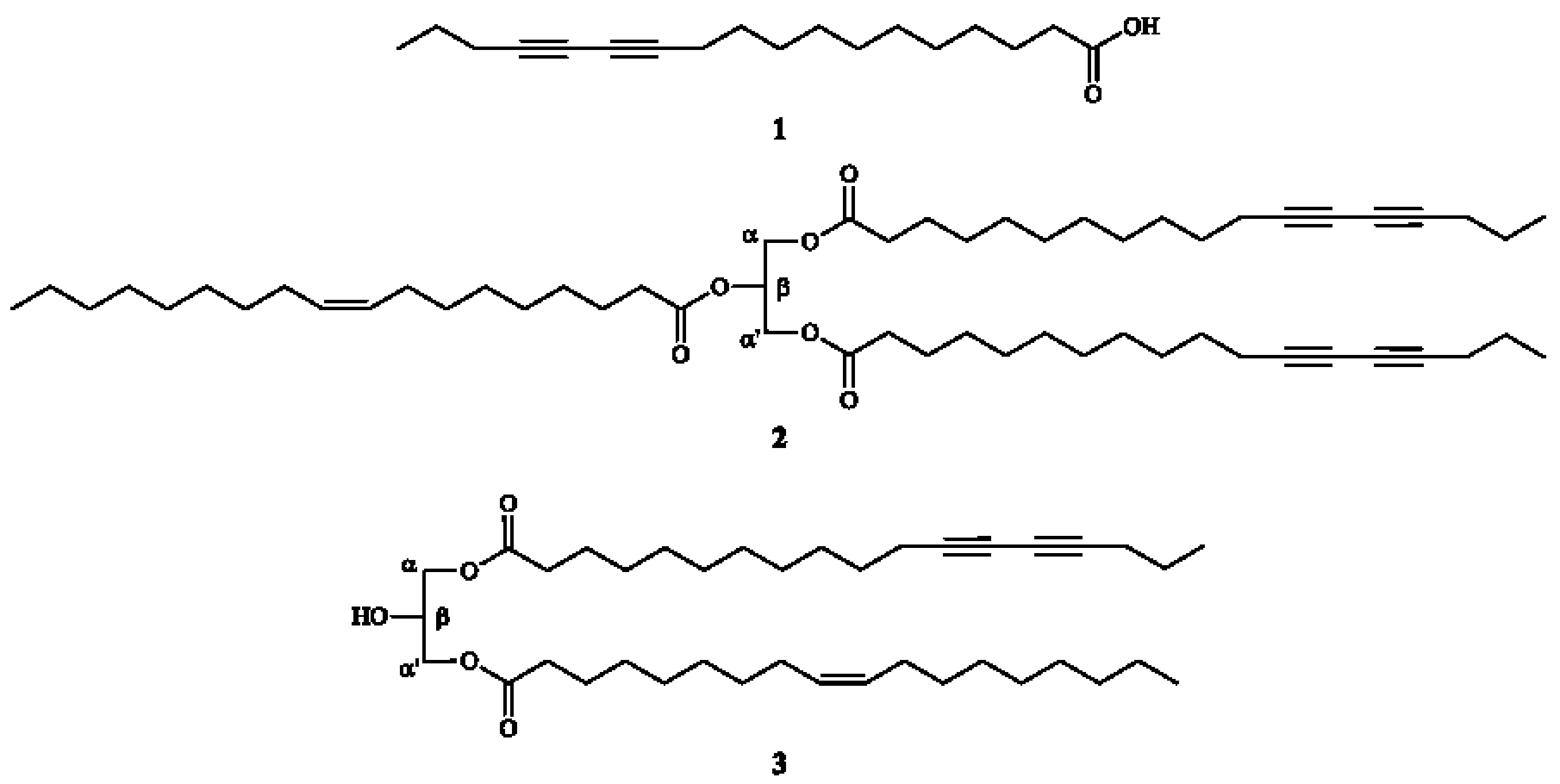
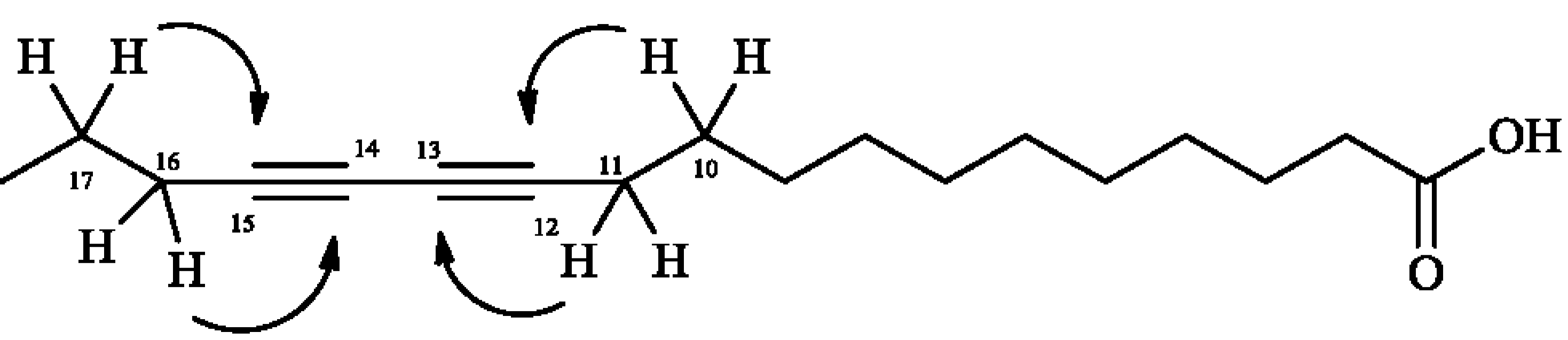
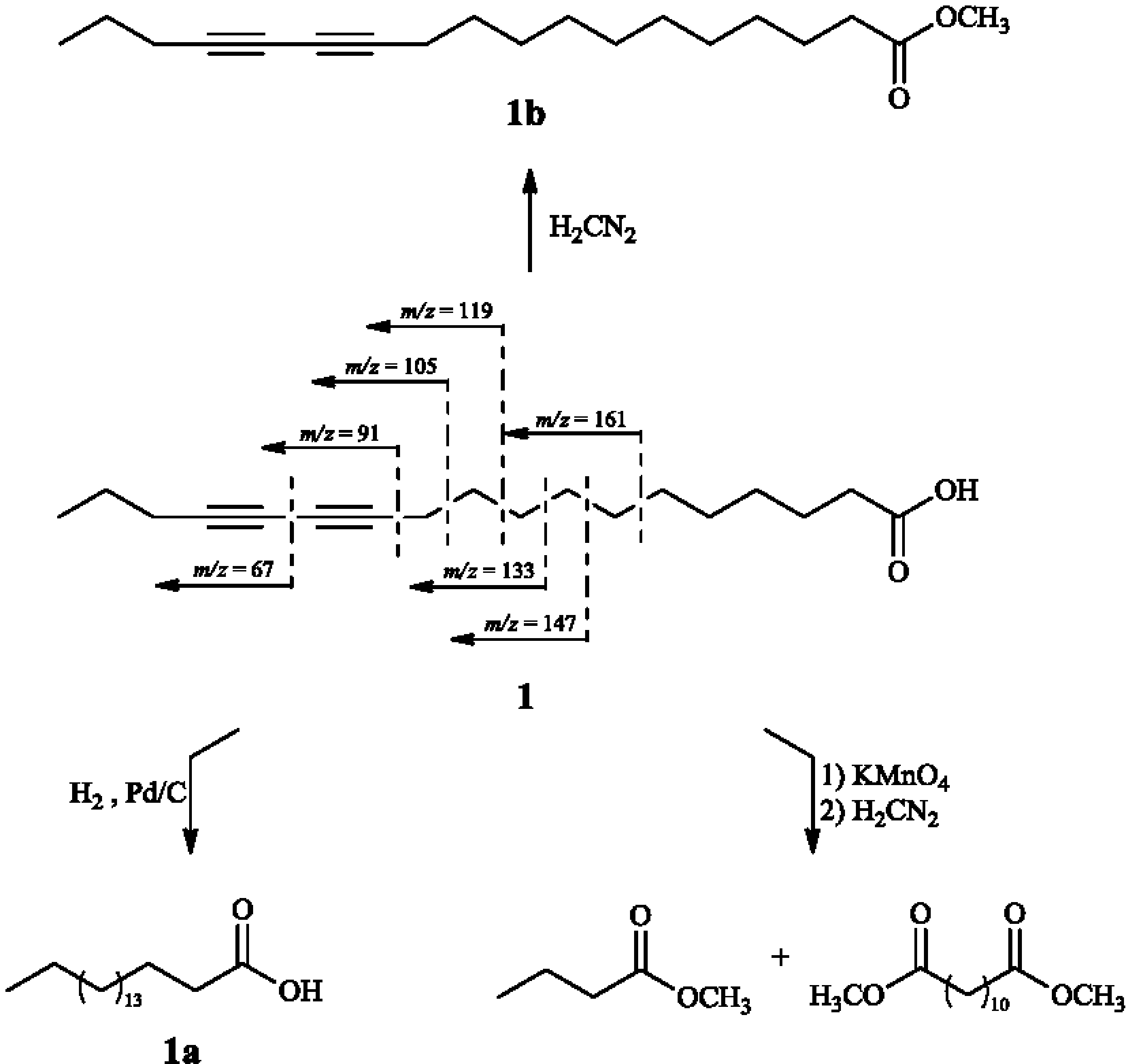
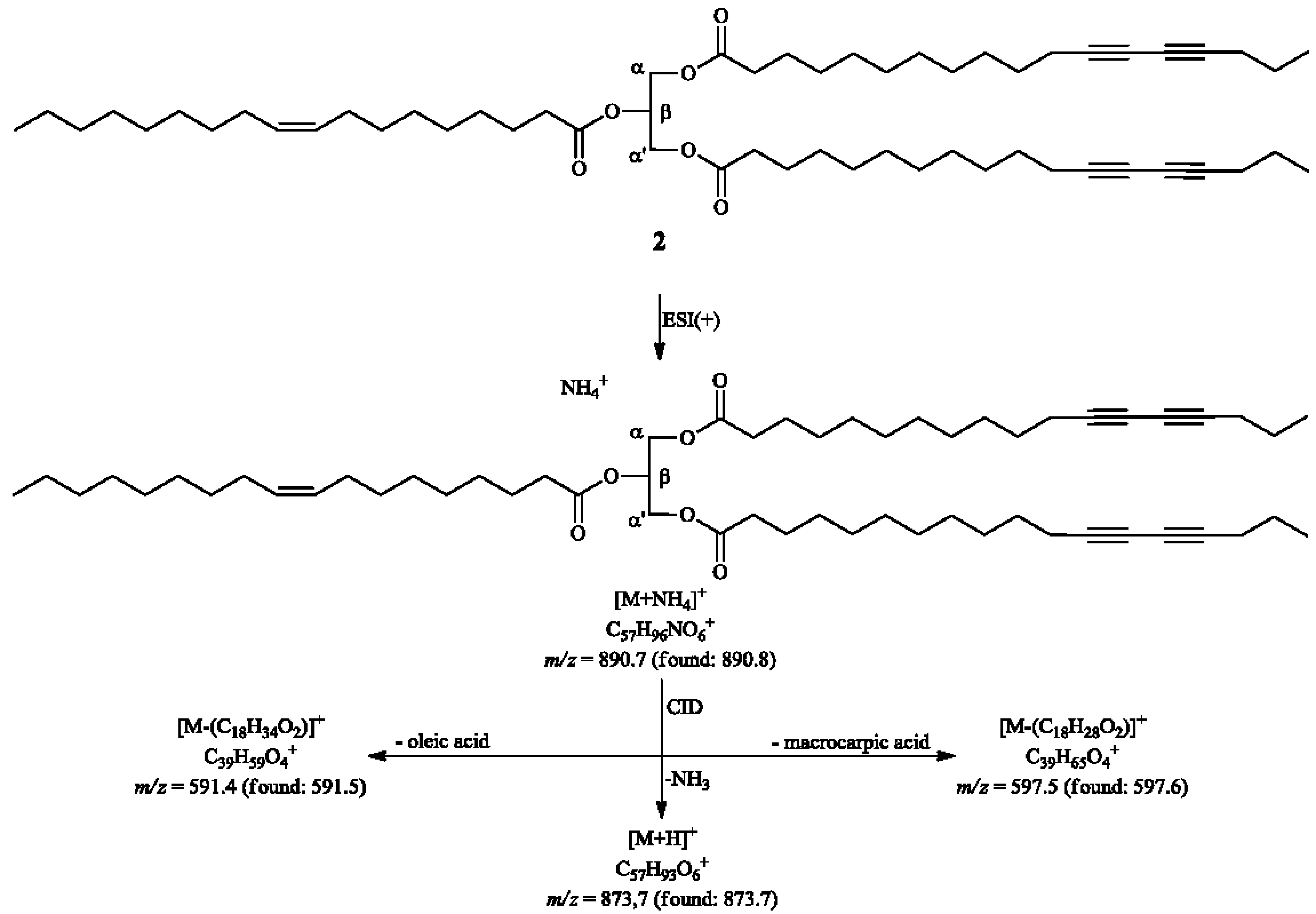

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract/Fraction/ compound | IC50 (µg/mL) a CI95% | CC50 (µg/mL) b CI95% | SI |
|---|---|---|---|
| T. cruzi trypomastigote | NCTC | ||
| Hexane extract | 65.44 (55.34–70.23) | >100 | >1.5 |
| Fraction I | NA | ND | - |
| Fraction II | NA | ND | - |
| Fraction III | 5.32 (4.24–6.68) | >100 | >18.8 |
| 1 | 10.70 (4.34–26.39) 38.77 µM | 44.27 (31.03–63.14) 160.40 µM | 4.1 |
| 1a | NA | ND | - |
| 2 | NA | ND | - |
| 3 | NA | ND | - |
| Benznidazole | 139.00 (118.80–162.74) 534.2 µM | 125.90 (102.87–142.32) 483.7 µM | 0.9 |
3. Experimental
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Product Characterization
3.4.1. 12,14-Octadecadyinoic acid (Macrocarpic acid, 1)
3.4.2. α,α'-Dimacrocarpoyl-β-oleylglycerol (2)
3.4.3. α-Macrocarpoyl-α'-oleylglycerol (3)
3.5. Preparation of Semi-Synthetic Derivatives of 1–3
3.5.1. Catalytic Hydrogenation of 1
3.5.2. Methylation of 1
3.5.3. Oxidative Cleavage of 1
3.5.4. Transesterification of 2 or 3
3.6. Parasite Maintenance
3.7. Determination of the Activity against T. cruzi—Trypomastigotes
3.8. Mammalian Cells
3.9. Determination of the Cytotoxicity against Mammalian Cells
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barrett, M.P.; Croft, S.L. Management of trypanosomiasis and leishmaniasis. Br. Med. Bull. 2012, 104, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Ketter, H.; Marjanovic, S. Engaging biotechnology companies in the development of innovative solutions for diseases of poverty. Nature Rev. Drug Discov. 2004, 3, 171–176. [Google Scholar] [CrossRef]
- Engels, D.; Savioli, L. Reconsidering the underestimated burden caused by neglected tropical diseases. Trends Parasitol. 2006, 22, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.; Biavatti, M.W.; Brun, R.; da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—part I. Curr. Med. Chem. 2012, 19, 2128–2175. [Google Scholar] [PubMed]
- Murray, N.A. Revision of Cimbopetalum and Porcelia (Annonaceae). Syst. Bot. Monogr. 1993, 40, 8–121. [Google Scholar] [CrossRef]
- Chaves, M.H.; Roque, N.F. Amides and liganamides from Porcelia macrocarpa. Phytochemistry 1997, 46, 879–881. [Google Scholar] [CrossRef]
- Chaves, M.H.; Santos, L.À.; Lago, J.H.G.; Roque, N.F. Alkaloids from Porcelia macrocarpa. J. Nat. Prod. 2001, 64, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Lago, J.H.G.; Chaves, M.H.; Ayres, M.C.C.; Agripino, D.G.; Young, M.C.M. Evaluation of antifungal and DNA-damaging activities of alkaloids from branches of Porcelia macrocarpa. Planta Med. 2007, 73, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Chaves, M.H.; Roque, N.F.; Ayres, M.C.C. Steroids and flavonoids of Porcelia macrocarpa. J. Braz. Chem. Soc. 2004, 15, 608–613. [Google Scholar] [CrossRef]
- Chaves, M.H.; Lago, J.H.G.; Roque, N.F. Macrocarpane, a new sesquiterpene skeleton from the leaves of Porcelia macrocarpa. J. Braz. Chem. Soc. 2003, 14, 16–19. [Google Scholar]
- Chaves, M.H.; Freitas, A.; Roque, N.F.; Cavalheiro, A.J. Separação e identificação de constituintes químicos polares de Porcelia macrocarpa. Quím. Nova 2000, 23, 307–309. [Google Scholar] [CrossRef]
- Chaves, M.H.; Roque, N.F. Acetogenins from Porcelia macrocarpa: Stereochemical determination of 2-alkyl-3-hidroxy-4-methyl-γ lactones by 13C-NMR spectroscopy. Phytochemistry 1997, 44, 523–527. [Google Scholar] [CrossRef]
- Grecco, S.S.; Felix, M.J.P.; Lago, J.H.G.; Pinto, E.G.; Tempone, A.G.; Romoff, P.; Ferreira, M.J.P.; Sartorelli, P. Anti-trypanosomal phenolic derivatives from Baccharis uncinella C. DC. (Asteraceae). Nat. Prod. Commun. 2014, 9, 171–173. [Google Scholar] [PubMed]
- Morais, T.R.; Costa-Silva, T.; Tempone, A.G.; Borborema, S.E.T.; Scotti, M.T.; Souza, R.M.F.; Araujo, A.; Oliveira, A.; Morais, S.; Sartorelli, P.; et al. Antiparasitic activity of natural and semi-synthetic tirucallane triterpenoids from Schinus terebinthifolius (Anacardiaceae). Molecules 2014, 19, 5761–5776. [Google Scholar] [CrossRef] [PubMed]
- Picolo, C.D.; Palmeira, M.; Souza, K.; Passero, L.F.D.; Laurenti, M.D.; Martins, E.G.A.; Sartorelli, P.; Lago, J.H.G. Antileishmanial activity evaluation of adunchalcone, a new prenylated dihydrochalcone from Piper aduncum L. Fitoterapia 2014, 97, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Rea, A.; Tempone, A.G.; Pinto, E.G.; Mesquista, J.T.; Silva, L.G.; Rodrigues, E.; Sartorelli, P.; Lago, J.H.G. Soulamarin isolated from Calophyllum brasiliense (Clusiaceae) induces plasma membrane permeabilization of Trypanosoma cruzi and mytochondrial dysfunction. PLoS Negl. Trop. Dis. 2013, 7, e2556–e2563. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Wang, H.; Kirisawa, M.; Satoh, M.; Takeuchi, N. Acetylenes from Panax quinquefolium. Phytochemistry 1992, 31, 3499–3506. [Google Scholar] [CrossRef]
- Setzer, W.N.; Green, T.J.; Whitakerm, K.W.; Moriaritym, D.M.; Yanceym, C.A.; Lawtonm, R.O.; Bates, R.B. A cytotoxic diacetylene from Dendropanax arboreus. Planta Med. 1995, 61, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Knothe, G.; Jie, M.S.F.L.; Lam, C.C.; Bagby, M.O. Evaluation of the 13C-NMR signals of unsaturated carbons of triacylglycerols. Chem. Phys. Lipids 1995, 77, 187–191. [Google Scholar] [CrossRef]
- Jie, M.S.F.L.; Lam, C.C. 1H-Nuclear magnetic resonance spectroscopic studies of saturated, acetylenic and ethylenic triacylglycerols. Chem. Phys. Lipids 1995, 77, 155–171. [Google Scholar] [CrossRef]
- Jie, M.S.F.L.; Lam, C.C. 13C-NMR studies of polyunsaturated triacylglycerols of type AAA and mixed triacylglycerols containing saturated, acetylenic and ethylenic acyl groups. Chem. Phys. Lipids 1995, 78, 1–13. [Google Scholar] [CrossRef]
- Henderson, J.M.; Petersheim, M.; Templeman, G.J.; Softly, B.J. Quantitation and structure elucidation of the positional isomers in a triacylglycerol mixture using proton and carbon one- and two-dimensional NMR. J. Agric. Food Chem. 1994, 42, 435–441. [Google Scholar] [CrossRef]
- Buchanan, M.S.; Toshihiro, H.; Asakawa, Y. Acylglycerols from the slime mould Lycogala epidendrum. Phytochemistry 1996, 41, 791–794. [Google Scholar] [CrossRef]
- Grecco, S.S.; Reimão, J.Q.; Tempone, A.G.; Sartorelli, P.; Romoff, P.; Ferreira, M.J.P.; Fávero, O.A.; Lago, J.H.G. Isolation of an antileishmanial and antitrypanosomal flavanone from the leaves of Baccharis retusa DC. (Asteraceae). Parasitol. Res. 2010, 106, 111–113. [Google Scholar]
- Muller, P.Y.; Milton, M.K. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. 2012, 11, 751–761. [Google Scholar]
- Kraus, C.M.; Neszmelyi, A.; Holly, S.; Wiedemann, B.; Nenninger, A.; Torsell, K.B.; Bohlin, L.; Wagner, H. New acetylenes isolated from the bark of Heisteria acuminata. J. Nat. Prod. 1998, 61, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Yamazoe, S.; Hasegawa, K.; Shigemori, H. Structure-activity relationship of acetylenes from galls of Hedera rhombea as plant growth inhibitors. Z. Naturforsch. 2006, 61c, 536–540. [Google Scholar]
- Marles, R.J.; Farnswort, N.R. Isolation of a novel cytotoxic polyacetylene from a traditional anthelmintic medicinal plant, Minquartia guianensis. J. Nat. Prod. 1989, 52, 261–266. [Google Scholar] [CrossRef]
- Kuklev, D.V.; Domb, A.J.; Dembitsky, V.M. Bioactive acetylenic metabolites. Phytomedicine 2013, 20, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Brandão, M.G.; Krettli, A.U.; Soares, L.S.; Nery, C.G.; Marinuzzi, H.C. Antimalarial activity of extracts and fractions from Bidens pilosa and other Bidens species (Asteraceae) correlated with the presence of acetylene and flavonoid compounds. J. Ethnopharmacol. 1997, 57, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Senn, M.; Gunzenhauser, S.; Brun, R.; Séquin, U. Antiprotozoal polyacetylenes from the Tanzanian medicinal plant Cussonia zimmermannii. J. Nat. Prod. 2007, 70, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Carballeira, N.M.; Cartagena, M.; Sanabria, D.; Tasdemir, D.; Prada, C.F.; Reguera, R.M.; Balaña-Fouce, R. 2-Alkynoic fatty acids inhibit topoisomerase IB from Leishmania donovani. Bioorg. Med. Chem. Lett. 2012, 22, 6185–6189. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Grecco Sdos, S.; Reimão, J.Q.; Tempone, A.G.; Sartorelli, P.; Cunha, R.L.; Romoff, P.; Ferreira, M.J.; Fávero, O.A.; Lago, J.H. In vitro antileishmanial and antitrypanosomal activities of flavanones from Baccharis retusa DC. (Asteraceae). Exp. Parasitol. 2012, 130, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, D.S.; Tempone, A.G.; Reimão, J.Q.; Taniwaki, N.N.; Romoff, P.; Fávero, O.A.; Sartorelli, P.; Mecchi, M.C.; Lago, J.H.G. Anti-leishmanial and anti-trypanosomal potential of polygodial isolated from stem barks of Drimys brasiliensis Miers (Winteraceae). Parasitol. Res. 2011, 109, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.; Mesquita, J.T.; Tempone, A.G.; Lago, J.H.G.; Guimarães, E.F.; Kato, M.J. Leishmanicidal activity of an alkenylphenol from Piper malacophyllum is related to plasma membrane disruption. Exp. Parasitol. 2012, 132, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1, 1a, 2 and 3 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Á. Santos, L.; Cavalheiro, A.J.; Tempone, A.G.; Correa, D.S.; Alexandre, T.R.; Quintiliano, N.F.; Rodrigues-Oliveira, A.F.; Oliveira-Silva, D.; Martins, R.C.C.; Lago, J.H.G. Antitrypanosomal Acetylene Fatty Acid Derivatives from the Seeds of Porcelia macrocarpa (Annonaceae). Molecules 2015, 20, 8168-8180. https://doi.org/10.3390/molecules20058168
De Á. Santos L, Cavalheiro AJ, Tempone AG, Correa DS, Alexandre TR, Quintiliano NF, Rodrigues-Oliveira AF, Oliveira-Silva D, Martins RCC, Lago JHG. Antitrypanosomal Acetylene Fatty Acid Derivatives from the Seeds of Porcelia macrocarpa (Annonaceae). Molecules. 2015; 20(5):8168-8180. https://doi.org/10.3390/molecules20058168
Chicago/Turabian StyleDe Á. Santos, Luciana, Alberto J. Cavalheiro, Andre G. Tempone, Daniela S. Correa, Tatiana R. Alexandre, Natalia F. Quintiliano, André F. Rodrigues-Oliveira, Diogo Oliveira-Silva, Roberto Carlos C. Martins, and João Henrique G. Lago. 2015. "Antitrypanosomal Acetylene Fatty Acid Derivatives from the Seeds of Porcelia macrocarpa (Annonaceae)" Molecules 20, no. 5: 8168-8180. https://doi.org/10.3390/molecules20058168