
Investigation of Carbohydrate Recognition via Computer Simulation

{kind=link}
{kind=link}
Abstract
:1. Introduction
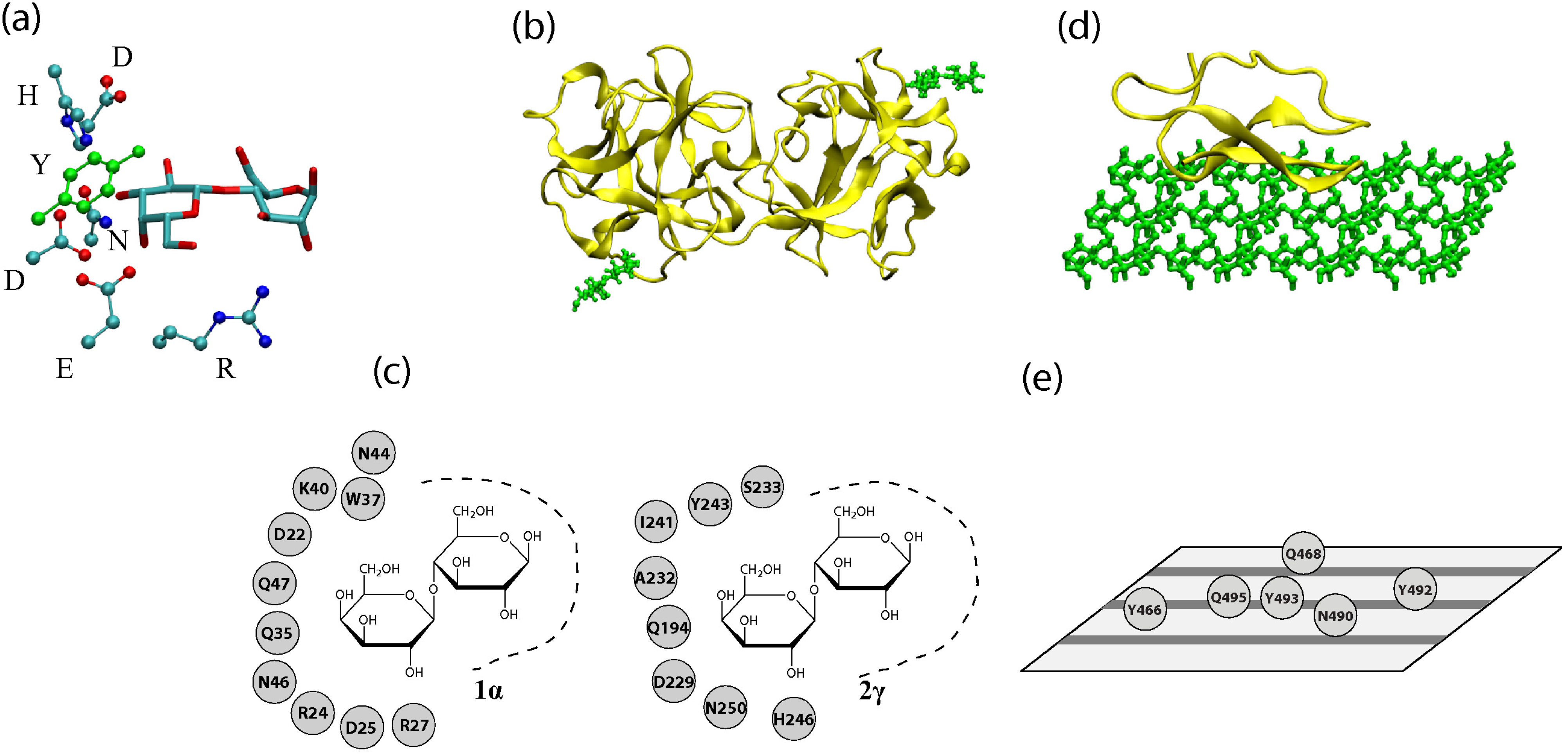
2. Structural Features of Carbohydrate-Recognizing Proteins
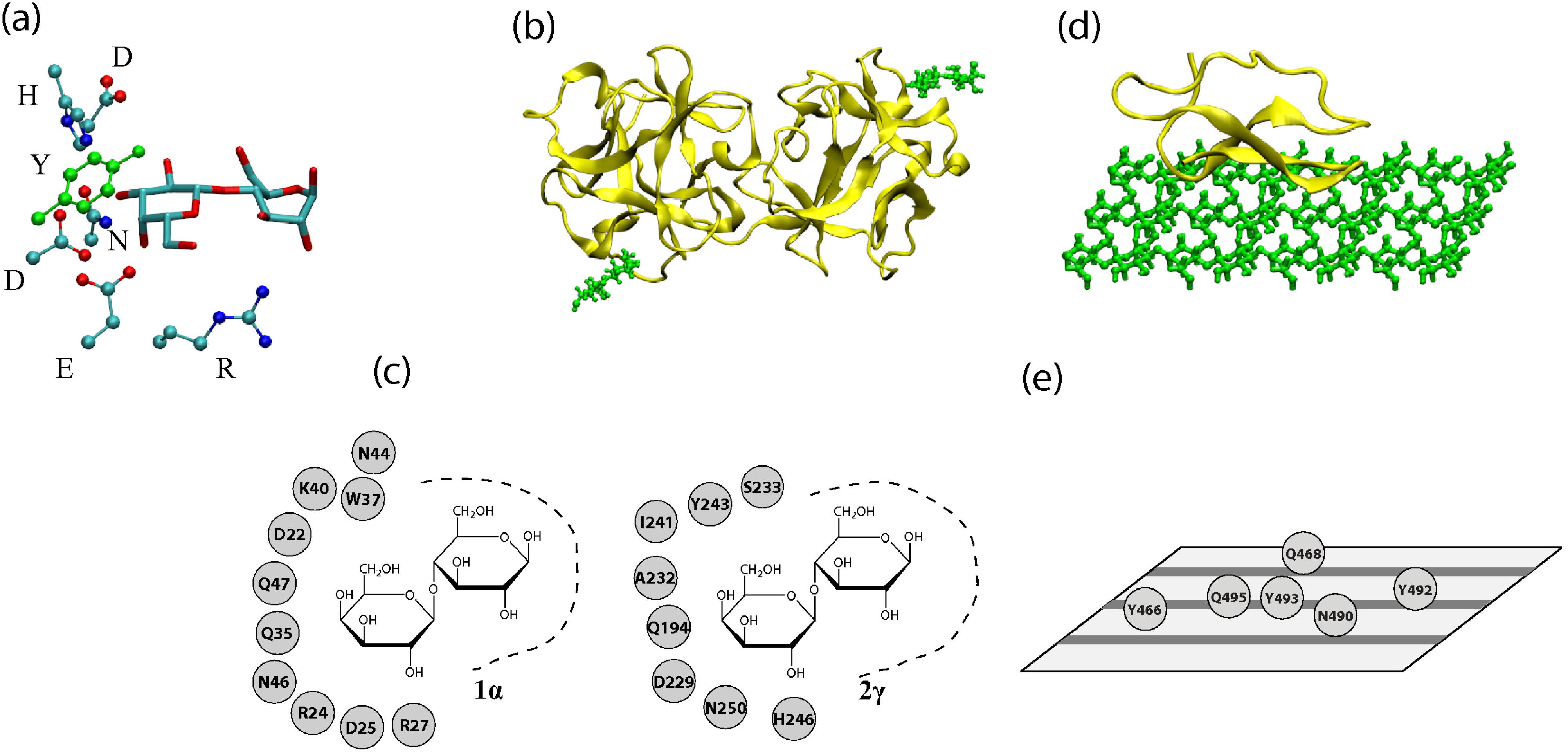
3. Physical Interactions at the Recognition Sites
4. Computer Simulation of Protein-Carbohydrate Complexes
5. Free Energy Calculation of Protein-Carbohydrate Interactions
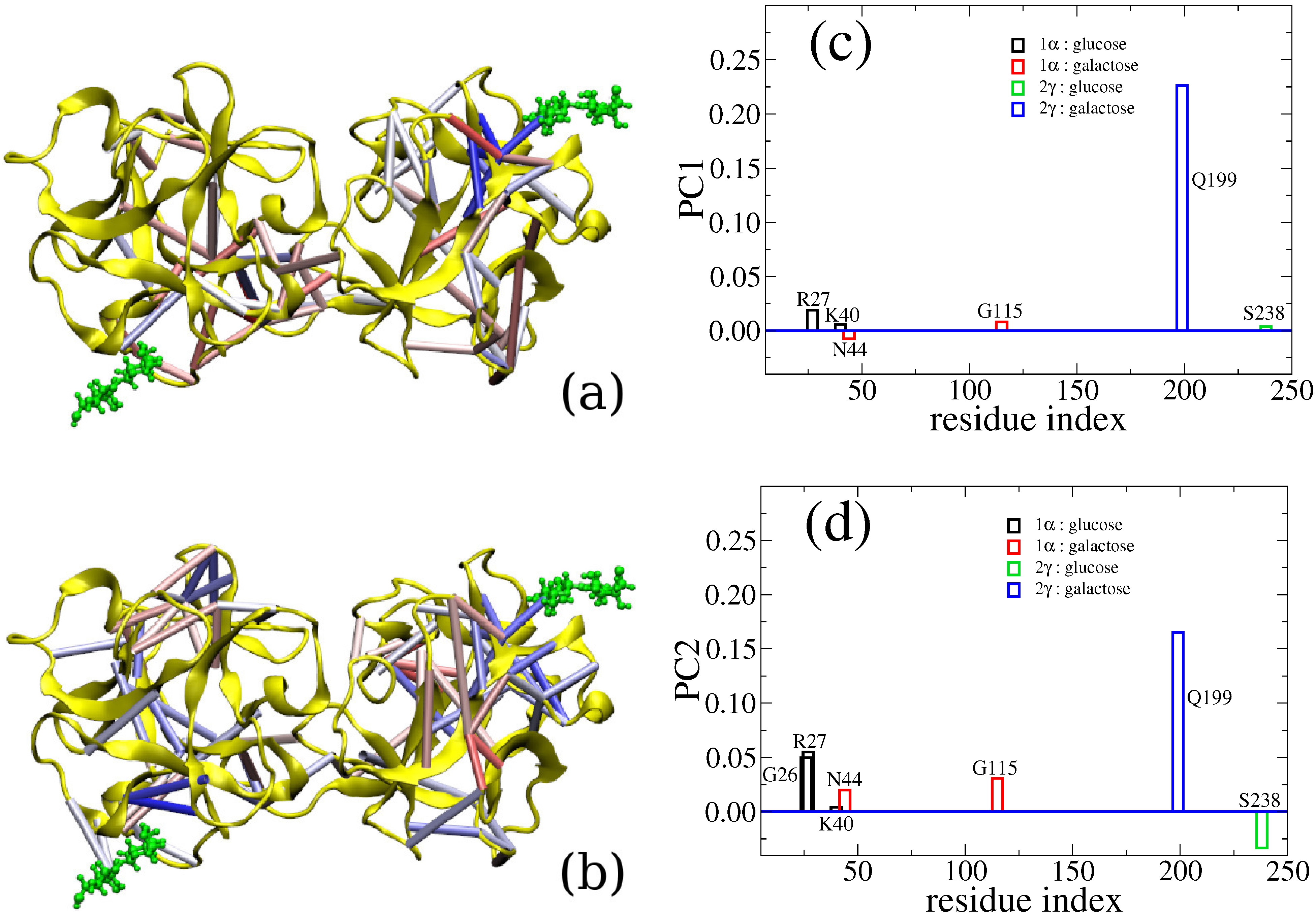
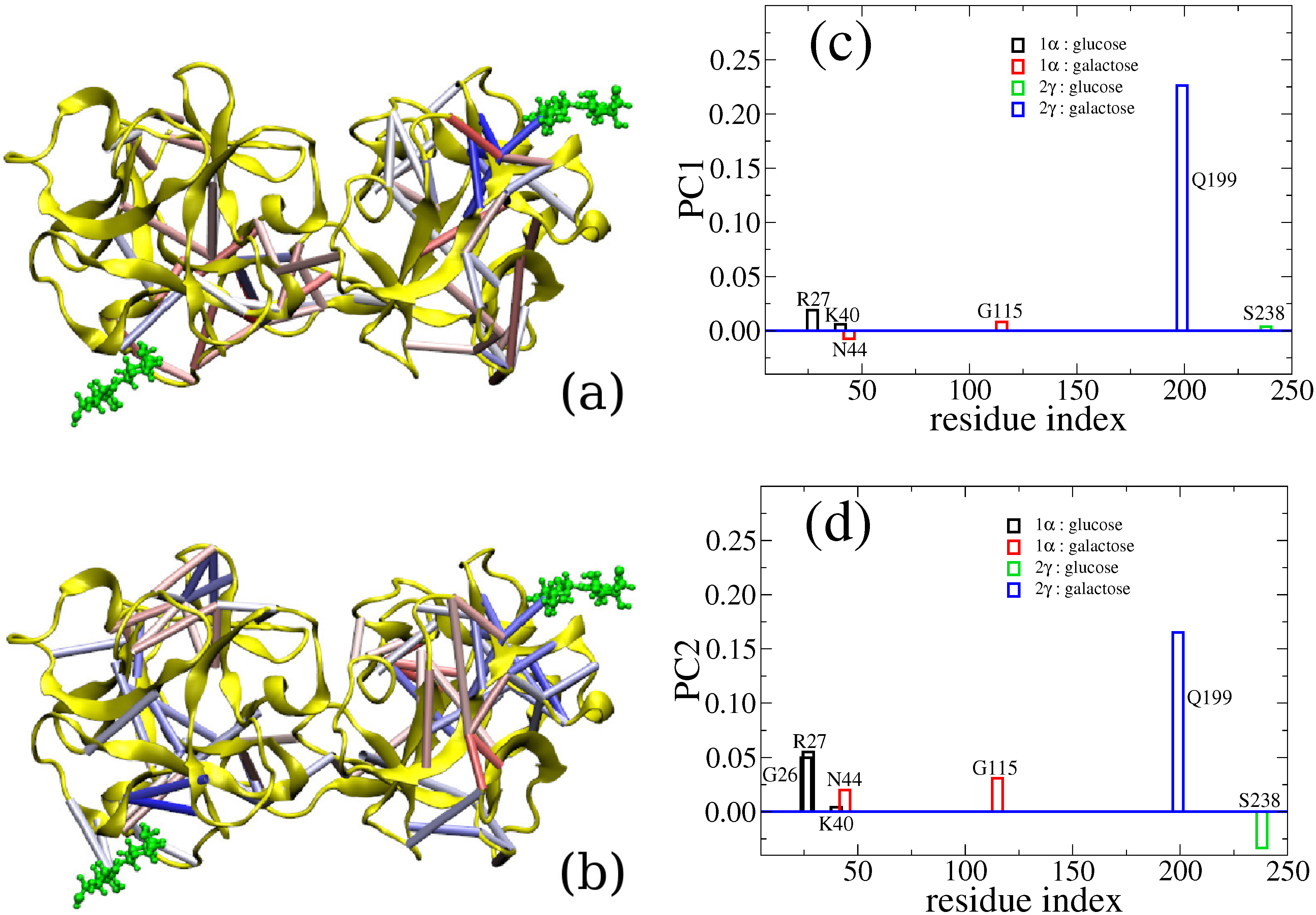
6. Cross-Talk Between Recognition Sites
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Taylor, M.E.; Drickamer, K. Introduction to Glycobiology, 3rd ed.; Oxford University Press: New York, NY, USA, 2011. [Google Scholar]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Sphyris, N.; Lord, J.M.; Wales, R.; Roberts, L.M. Mutational analysis of the Ricinus lectin B-chains galactose-binding ability of the 2γ subdomain of Ricinus communis agglutinin B-chain. J. Biol. Chem. 1995, 270, 20292–20297. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Doyle, M.L.; Casper, J.D.; Cicala, C.; Leavitt, S.A.; Majeed, S.; Steenbeke, T.D.; Venturi, M.; Chaiken, I.; Fung, M.; et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002, 420, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; Gonzalez-outeirino, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2007, 29, 622–655. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, M.L.; Woods, R.J. Structural Glycobiology: A Game of Snakes and Ladders. Glycobiology 2008, 18, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Pathiaseril, A.; Woods, R.J. Relative Energies of Binding for Antibody-Carbohydrate-Antigen Complexes Computed from Free-Energy Simulations. J. Am. Chem. Soc. 2000, 122, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q. Molecular Dynamics Simulaitons of Biomimetic Carbohydrate Materials. Ph.D. Thesis, Royal Institute of Technology, Stockholm, Sweden, 2011. [Google Scholar]
- Moscona, A. Neuraminidase Inhibitors for Influenza. N. Engl. J. Med. 2005, 353, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Akkouh, O.; Ng, T.B.; Singh, S.S.; Yin, C.; Dan, X.; Chan, Y.S.; Pan, W.; Cheung, R.C.F. Lectins with Anti-HIV Activity: A Review. Molecules 2015, 20, 648–668. [Google Scholar] [CrossRef] [PubMed]
- Francois, B.; Russell, R.J.M.; Murray, J.B.; Aboul-ela, F.; Masquida, B.; Vicens, Q.; Westhof, E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Mayer, R.; Roche, A. Sugar-lectin interactions: Sugar clusters, lectin multivalency and avidity. Carbohydr. Lett. 2000, 4, 35–52. [Google Scholar]
- Drickamer, K.; Taylor, M.E. Biology of Animal Lectins. Annu. Rev. Cell Biol. 1993, 9, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Lee, Y.C. Affinity enhancement by multivalent lectin-carbohydrate interaction. Glycoconj. J. 2000, 17, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Rini, J.M. Lectin Structure. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 551–577. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Drickamer, K. Structural Basis of Lectin-Carbohydrate Recognition. Annu. Rev. Biochem. 1996, 65, 441–473. [Google Scholar] [CrossRef] [PubMed]
- Sharon, N.; Lis, H. History of lectins: From hemagglutinins to biological recognition molecules. Glycobiology 2004, 14, 53R–62R. [Google Scholar] [CrossRef] [PubMed]
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed]
- Drickamer, K. C-type lectin-like domains. Curr. Opin. Struct. Biol. 1999, 9, 585–590. [Google Scholar] [CrossRef]
- Loris, R. Principles of structures of animal and plant lectins. Biochim. Biophys. Acta 2002, 1572, 198–208. [Google Scholar] [CrossRef]
- Boraston, A.B.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. [Google Scholar] [PubMed]
- Shoseyov, O.; Shani, Z.; Levy, I. Carbohydrate Binding Modules: Biochemical Properties and Novel Applications. Microbiol. Mol. Biol. Rev. 2006, 70, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Z. Structure and Function of Carbohydrate-Binding Module Families 13 and 42 of Glycoside Hydrolases, Comprising a β-Trefoil Fold. Biosci. Biotechnol. Biochem. 2013, 77, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Lutteke, T.; Bohne-Lang, A.; Loss, A.; Goetz, T.; Frank, M.; von der Lieth, C.W. GLYCOSCIENCES.de: An Internet portal to support glycomics and glycobiology research. Glycobiology 2006, 16, 71R–81R. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Sarkar, A.; Breton, C.; Drouillard, S.; Rivet, A.; Imberty, A. Glyco3D: A Portal for Structural Glycoscience; 2013. Methods Mol. Biol. 2015, 1273, 241–258. [Google Scholar] [PubMed]
- Lutteke, T.; Frank, M.; von der Lieth, C. Carbohydrate Structure Suite (CSS): Analysis of carbohydrate 3D structures derived from the PDB. Nucleic Acids Res. 2005, 33, D242–D246. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Im, W. Glycan fragment database: A database of PDB-based glycan 3D structures. Nucleic Acids Res. 2013, 41, D470–D474. [Google Scholar] [CrossRef] [PubMed]
- Kuttel, M.; Mao, Y.; Widmalm, G.; Lundborg, M. CarbBuilder: An Adjustable Tool for Building 3D Molecular Structures of Carbohydrates for Molecular Simulation. In Proceedings of the 2011 IEEE 7th International Conference on E-Science (e-Science), Stockholm, Sweden, 5–8 December 2011; pp. 395–402.
- Quiocho, F.A. Carbohydrate-Binding Proteins: Tertiary Structures and Protein-Sugar Interactions. Annu. Rev. Biochem. 1986, 55, 287–315. [Google Scholar] [CrossRef] [PubMed]
- Marechal, Y. The Hydrogen Bond and the Water Molecule: The Physics and Chemistry of Water, Aqueous and Bio-Media; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Sheu, S.Y.; Yang, D.Y.; Selzle, H.L.; Schlag, E.W. Energetics of hydrogen bonds in peptides. Proc. Natl. Acad. Sci. USA 2003, 100, 12683–12687. [Google Scholar] [CrossRef] [PubMed]
- Asensio, J.L.; Arda, A.; Canada, F.J.; Jimenez-Barbero, J. Carbohydrate-Aromatic Interactions. Acc. Chem. Res. 2013, 46, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Laughrey, Z.R.; Kiehna, S.E.; Riemen, A.J.; Waters, M.L. Carbohydrate-pi interactions: What are they worth? J. Am. Chem. Soc. 2008, 130, 14625–14633. [Google Scholar] [CrossRef] [PubMed]
- Faller, C.E.; Guvench, O. Terminal sialic acids on CD44 N-glycans can block hyaluronan binding by forming competing intramolecular contacts with arginine side chains. Proteins Struct. Funct. Bioinform. 2014, 82, 3079–3089. [Google Scholar] [CrossRef] [PubMed]
- Angata, T.; Hayakawa, T.; Yamanaka, M.; Varki, A.; Nakamura, M. Discovery of Siglec-14, a novel sialic acid receptor undergoing concerted evolution with Siglec-5 in primates. FASEB J. 2006, 20, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 2007, 446, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Stencel-Baerenwald, J.E.; Reiss, K.; Reiter, D.M.; Stehle, T.; Dermody, T.S. The sweet spot: Defining virus-sialic acid interactions. Natl. Rev. Microbiol. 2014, 12, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Vinson, M.; van der Merwe, P.A.; Kelm, S.; May, A.; Jones, E.Y.; Crocker, P.R. Characterization of the Sialic Acid-binding Site in Sialoadhesin by Site-directed Mutagenesis. J. Biol. Chem. 1996, 271, 9267–9272. [Google Scholar] [PubMed]
- Jamal-Talabani, S.; Boraston, A.B.; Turkenburg, J.P.; Tarbouriech, N.; Ducros, V.M.A.; Davies, G.J. Ab Initio Structure Determination and Functional Characterization Of CBM36: A New Family of Calcium-Dependent Carbohydrate Binding Modules. Structure 2004, 12, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Elcock, A.H.; Sept, D.; McCammon, J.A. Computer Simulation of Protein-Protein Interactions. J. Phys. Chem. B 2001, 105, 1504–1518. [Google Scholar] [CrossRef]
- Lehtio, J.; Sugiyama, J.; Gustavsson, M.; Fransson, L.; Linder, M.; Teeri, T.T. The binding specificity and affinity determinants of family 1 and family 3 cellulose binding modules. Proc. Natl. Acad. Sci. USA 2003, 100, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Beckham, G.T.; Matthews, J.F.; Bomble, Y.J.; Bu, L.; Adney, W.S.; Himmel, M.E.; Nimlos, M.R.; Crowley, M.F. Identification of Amino Acids Responsible for Processivity in a Family 1 Carbohydrate-Binding Module from a Fungal Cellulase. J. Phys. Chem. B 2010, 114, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Nimlos, M.R.; Beckham, G.T.; Matthews, J.F.; Bu, L.; Himmel, M.E.; Crowley, M.F. Binding Preferences, Surface Attachment, Diffusivity, and Orientation of a Family 1 Carbohydrate-binding Module on Cellulose. J. Biol. Chem. 2012, 287, 20603–20612. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Petsko, G.A. Molecular-dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Petridis, L.; Smith, J.C. Biomolecular Structure and Dynamics with Neutrons: The View from Simulation. Isr. J. Chem. 2014, 54, 1264–1273. [Google Scholar] [CrossRef]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W.H. Freeman: New York, NY, USA, 1999. [Google Scholar]
- Tinoco, I.; Sauer, K.; Wang, J.C.; Puglisi, J.D.; Harbison, G.; Rovnyak, D. Physical Chemistry: Principles and Applications in Biological Sciences; Prentice Hall: Upper Saddle River, NJ, USA, 2013. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Knott, B.; Crowley, M.; Himmel, M.; Stahlberg, J.; Beckham, G. Carbohydrate-Protein Interactions That Drive Processive Polysaccharide Translocation in Enzymes Revealed from a Computational Study of Cellobiohydrolase Processivity. J. Am. Chem. Soc. 2014, 136, 8810–8819. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Huang, J.; MacKerell, A. Enhanced Conformational Sampling of Carbohydrates using Biasing Potential and Solute Tempering Replica Exchange: Application to the N-glycan on the HIV gp120 Envelope Protein. Biophys. J. 2015, 108, 157a. [Google Scholar] [CrossRef]
- McCammon, J.A.; Harvey, S.C. Dynamics of Proteins and Nucleic Acids; Press Syndicate of the University of Cambridge: New York, NY, USA, 1987. [Google Scholar]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Greene, S.N.; Kamath, G.; Brady, J.W.; Venable, R.M.; Pastor, R.W.; Mackerell, A.D., Jr. Additive Empirical Force Field for Hexopyranose Monosaccharides. J. Comput. Chem. 2008, 29, 2543–2564. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Hatcher, E.; Venable, R.M.; Pastor, R.W.; Mackerell, A.D., Jr. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W.; MacKerell, A.D. CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate-Protein Modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.N.; Lins, R.D.; Maass, A.; Soares, T.A. A Glycam-Based Force Field for Simulations of Lipopolysaccharide Membranes: Parametrization and Validation. J. Chem. Theory Comput. 2012, 8, 4719–4731. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hunenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Kruger, P.; van Gunsteren, W.F. The GROMOS Biomolecular Simulation Program Package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Xiong, X.; Chen, Z.; Cossins, B.; Shi, J. Force fields and scoring functions for carbohydrate simulation. Carbohydr. Res. 2015, 401, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Foley, B.L.; Tessier, M.B.; Woods, R.J. Carbohydrate force fields. WIRES Comput. Mol. Sci. 2012, 2, 652–697. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; Rzepiela, A.J.; de Vries, A.H.; Dijkhuizen, L.; Hunenberger, P.H.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to Carbohydrates. J. Chem. Theory Comput. 2009, 5, 3195–3210. [Google Scholar] [CrossRef]
- Lopez, C.A.; Bellesia, G.; Redondo, A.; Langan, P.; Chundawat, S.P.S.; Dale, B.E.; Marrink, S.J.; Gnanakaran, S. MARTINI Coarse-Grained Model for Crystalline Cellulose Microfibers. J. Phys. Chem. B 2015, 119, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Kara, M.; Zacharias, M.; Koca, J. Enhanced conformational sampling of carbohydrates by Hamiltonian replica-exchange simulation. Glycobiology 2014, 24, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Langan, P.; French, A.; Johnson, G.P.; Gnanakaran, S. Conformational Flexibility of Soluble Cellulose Oligomers: Chain Length and Temperature Dependence. J. Am. Chem. Soc. 2009, 131, 14786–14794. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.R.; Guvench, O.; MacKerell, A.D. CHARMM Additive All-Atom Force Field for Acyclic Polyalcohols, Acyclic Carbohydrates, and Inositol. J. Chem. Theory Comput. 2009, 5, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Nivedha, A.K.; Makeneni, S.; Foley, B.L.; Tessier, M.B.; Woods, R.J. Importance of ligand conformational energies in carbohydrate docking: Sorting the wheat from the chaff. J. Comput. Chem. 2014, 35, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.C.; Anderson, K.W.; Merz, K.M. Force field design for metalloproteins. J. Am. Chem. Soc. 1991, 113, 8262–8270. [Google Scholar] [CrossRef]
- Xantheas, S. Cooperativity and hydrogen bonding network in water clusters. Chem. Phys. 2000, 258, 225–231. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta 2015, 1850, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, G.; Roux, B. Modeling induced polarization with classical Drude oscillators: Theory and molecular dynamics simulation algorithm. J. Chem. Phys. 2003, 119, 3025–3039. [Google Scholar] [CrossRef]
- Patel, D.; He, X.; MacKerell, A.J. Polarizable Empirical Force Field for Hexopyranose Monosaccharides Based on the Classical Drude Oscillator. J. Phys. Chem. B 2015, 119, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowly, M.; Walker, R.C.; Zhang, W.; et al. AMBER Molecular Dymnamics Package; AMBER 10 University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Q.R.; Lindsay, R.J.; Raval, S.R.; Dobbs, J.S.; Nellas, R.B.; Shen, T. Effects of Branched O-Glycosylation on a Semiflexible Peptide Linker. J. Phys. Chem. B 2014, 118, 2050–2057. [Google Scholar] [CrossRef] [PubMed]
- Meynier, C.; Guerlesquin, F.; Roche, P. Computational Studies of Human Galectin-1: Role of Conserved Tryptophan Residue in Stacking Interaction with Carbohydrate Ligands. J. Biomol. Struct. Dyn. 2009, 27, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.G.; Weimar, T.; Kohli, T.; Woods, R.J. Molecular dynamics simulations of galectin-1-oligosaccharide complexes reveal the molecular basis for ligand diversity. Proteins Struct. Funct. Bioinf. 2003, 53, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Butenhof, K.J.; Gerken, T.A. Structure and dynamics of mucin-like glycopeptides. Examination of peptide chain expansion and peptide-carbohydrate interactions by stochastic dynamics simulations. Biochemistry 1993, 32, 2650–2663. [Google Scholar] [CrossRef] [PubMed]
- Mandal, T.K.; Mukhopadhyay, C. Effect of glycosylation on structure and dynamics of MHC class I glycoprotein: A molecular dynamics study. Biopolymers 2001, 59, 11–23. [Google Scholar] [CrossRef]
- Mark, P.; Zhang, Q.; Czjzek, M.; Brumer, H.; Agren, H. Molecular dynamics simulations of a branched tetradecasaccharide substrate in the active site of a xyloglucan endo-transglycosylase. Mol. Simul. 2011, 37, 1001–1013. [Google Scholar] [CrossRef]
- Favreau, A.; Faller, C.E.; Guvench, O. CD44 Receptor Unfolding Enhances Binding by Freeing Basic Amino Acids to Contact Carbohydrate Ligand. Biophys. J. 2013, 105, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Plazinski, W.; Knys-Dzieciuch, A. The ‘order-to-disorder’ conformational transition in CD44 protein: An umbrella sampling analysis. J. Mol. Graph. Model. 2013, 45, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Jamison, F.W., II; Foster, T.J.; Barker, J.A.; Hills, R.D., Jr.; Guvench, O. Mechanism of Binding Site Conformational Switching in the CD44-Hyaluronan Protein-Carbohydrate Binding Interaction. J. Mol. Biol. 2011, 406, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.E.; Gilson, M.K. Free Energy, Entropy, and Induced Fit in Host-Guest Recognition: Calculations with the Second-Generation Mining Minima Algorithm. J. Am. Chem. Soc. 2004, 126, 13156–13164. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Bandyopadhyay, S. Conformational flexibility of a protein-carbohydrate complex and the structure and ordering of surrounding water. Phys. Chem. Chem. Phys. 2012, 14, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Shenkin, P.S.; Hollinger, F.P.; Still, W.C. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J. Phys. Chem. A 1997, 101, 3005–3014. [Google Scholar] [CrossRef]
- David, L.; Luo, R.; Gilson, M. Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2000, 21, 295–309. [Google Scholar] [CrossRef]
- Shen, T.; Wong, C.F.; McCammon, J.A. Brownian dynamics simulation of helix-capping motif. Biopolymers 2003, 70, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Given, J.A.; Bush, B.L.; McCammon, J.A. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997, 72, 1047–1069. [Google Scholar] [CrossRef]
- Hamelberg, D.; McCammon, A. Standard Free Energy of Releasing a Localized Water Molecule from the Binding Pockets of Proteins: Double-Decoupling Method. J. Am. Chem. Soc. 2004, 126, 7683–7689. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A.; Lybrand, T.P.; Allison, S.A.; Northrup, S.H. Ligand binding: New theoretical approaches to molecular recognition. In Biomolecular Stereodynamics; Adenine Press: New York, NY, USA, 1986; Volume 3. [Google Scholar]
- Koppisetty, C.; Frank, M.; Lyubartsev, A.; Nyholm, P. Binding energy calculations for hevein-carbohydrate interactions using. J. Comput. Aided Mol. Des. 2015, 29, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Bryce, R.; Hillier, I.; Naismith, J. Carbohydrate-Protein Recognition: Molecular Dynamics Simulations and Free Energy Analysis of Oligosaccharide Binding to Concanavalin A. Biophys. J. 2001, 81, 1373–1388. [Google Scholar] [CrossRef]
- Liang, G.; Schmidt, R.; Yu, H.; Cumming, D.; Brady, J. Free Energy Simulation Studies of the Binding Specificity of Mannose-Binding Protein. J. Phys. Chem. 1996, 100, 2528–2534. [Google Scholar] [CrossRef]
- Masukawa, K.; Kollman, P.; Kuntz, I. Investigation of Neuraminidase-Substrate Recognition Using Molecular Dynamics and Free Energy Calculations. J. Med. Chem. 2003, 46, 5628–5637. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The Oxygen Equilibrium of Hemoglobin and Its Structural Interpretation. Proc. Natl. Acad. Sci. USA 1935, 21, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.L. Cooperativity Theory in Biochemistry: Steady-State and Equilibrium Systems, 1st ed.; Springer: New York, NY, USA, 2011. [Google Scholar]
- Changeux, J.P.; Edelstein, S.J. Allosteric Mechanisms of Signal Transduction. Science 2005, 308, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Karplus, M. Allostery and cooperativity revisited. Protein Sci. 2008, 17, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.; Chervenak, M.C.; Toone, E.J. Energetics of lectin-carbohydrate binding. A microcalorimetric investigation of concanavalin A-oligomannoside complexation. J. Biol. Chem. 1992, 267, 22907–22911. [Google Scholar] [PubMed]
- Popovych, N.; Sun, S.; Ebright, R.H.; Kalodimos, C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006, 13, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Zentz, C.; Frenoy, J.P.; Bourrillon, R. Binding of galactose and lactose to ricin. Biochem. Biophys. Acta 1978, 536, 18–26. [Google Scholar] [PubMed]
- Adair, W.L.; Kornfeld, S. Isolation of the Receptors for Wheat Germ Agglutinin and the Ricinus communis Lectins from Human Erythrocytes Using Affinity Chromatography. J. Biol. Chem. 1974, 249, 4696–4704. [Google Scholar]
- Yao, J.; Nellas, R.B.; Glover, M.M.; Shen, T. Stability and Sugar Recognition Ability of Ricin-like Carbohydrate Binding Domains. Biochemistry 2011, 50, 4097–4104. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Q.R.; Lindsay, R.J.; Nellas, R.B.; Fernandez, E.J.; Shen, T. Mapping Allostery through Computational Glycine Scanning and Correlation Analysis of Residue-Residue Contacts. Biochemistry 2015, 54, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, Q.R.; Lindsay, R.J.; Petridis, L.; Shen, T. Investigation of Carbohydrate Recognition via Computer Simulation. Molecules 2015, 20, 7700-7718. https://doi.org/10.3390/molecules20057700
Johnson QR, Lindsay RJ, Petridis L, Shen T. Investigation of Carbohydrate Recognition via Computer Simulation. Molecules. 2015; 20(5):7700-7718. https://doi.org/10.3390/molecules20057700
Chicago/Turabian StyleJohnson, Quentin R., Richard J. Lindsay, Loukas Petridis, and Tongye Shen. 2015. "Investigation of Carbohydrate Recognition via Computer Simulation" Molecules 20, no. 5: 7700-7718. https://doi.org/10.3390/molecules20057700