
Synthesis, Antiproliferative Activity and Molecular Properties Predictions of Galloyl Derivatives

Abstract
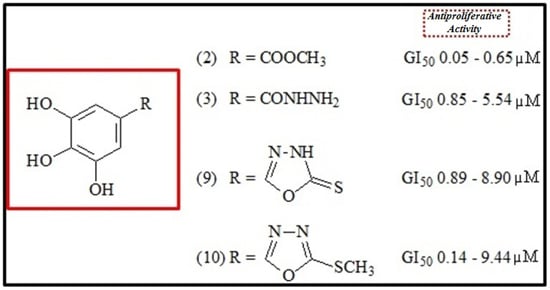
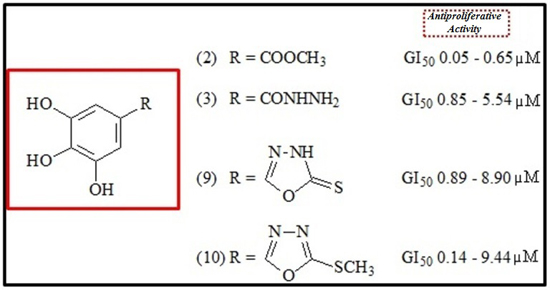
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
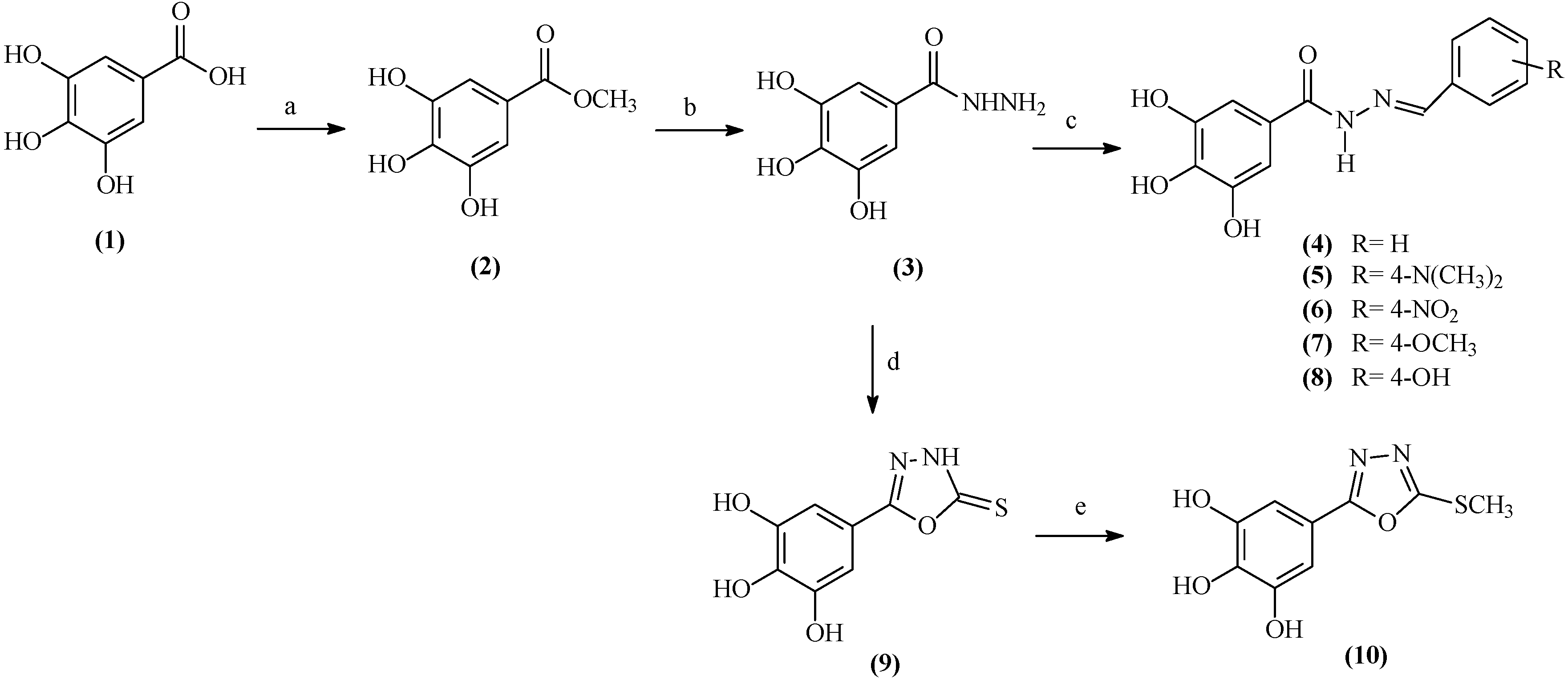
2.2. Antiproliferative Activity

{kind=link}
{kind=link}
| Cancer Cell Lines | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| U251 | MCF-7 | NCI/ADR-RES | 786-0 | NCI-H460 | PC-3 | OVCAR-3 | HT-29 | K-562 | HaCaT | |
| 2 | 0.16 | 0.16 | 0.12 | 0.65 | 0.13 | 0.57 | 0.05 | 0.49 | 0.31 | 0.26 |
| 3 | 1.11 | 0.85 | 0.91 | 1.56 | 5.54 | 3.53 | 0.88 | 1.21 | 1.17 | 2.50 |
| 4 | 7.80 | 5.55 | 7.16 | 9.06 | 11.20 | 5.07 | 1.24 | 7.53 | 9.96 | 8.29 |
| 5 | 24.10 | 8.61 | 4.68 | 8.93 | 21.64 | - | 2.93 | 16.24 | 2.05 | 4.05 |
| 6 | 5.12 | - | 8.46 | 7.60 | - | 13.99 | 5.91 | - | - | 31.55 |
| 7 | 9.14 | 15.66 | 11.34 | 18.85 | 41.05 | 19.64 | 5.98 | 25.12 | 8.93 | 18.08 |
| 8 | 6.60 | - | - | 26.80 | 9.38 | - | 1.35 | - | 9.17 | 8.72 |
| 9 | 4.20 | 1.08 | 2.83 | 8.90 | 8.77 | 6.05 | 0.89 | 2.61 | 1.72 | 2.25 |
| 10 | 5.20 | 0.34 | 0.17 | 9.20 | 9.44 | 7.43 | 0.14 | 2.77 | 0.60 | 2.50 |
| Dox | 0.02 | 0.01 | 0.20 | 0.04 | 0.01 | 0.18 | 0.02 | 0.09 | 0.03 | 0.01 |
| Cancer Cell Lines | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| U251 | MCF-7 | NCI/ADR-RES | 786-0 | NCI-H460 | PC-3 | OVCAR-3 | HT-29 | K-562 | HaCaT | |
| 2 | 4.10 | 5.12 | 3.62 | 8.62 | 7.97 | 13.10 | 1.59 | 8.52 | 5.67 | 16.74 |
| (29.19) | (-) | (30.26) | (31.36) | (34.92) | (-) | (33.69) | (30.60) | (-) | (-) | |
| 3 | 6.13 | 2.36 | 2.56 | 17.98 | 17.35 | 18.31 | 9.77 | 3.27 | - | 29.02 |
| (31.36) | (-) | (-) | (-) | (-) | (-) | (-) | (26.21) | - | >100 | |
| 4 | - | 23.54 | - | - | - | - | 29.61 | - | - | - |
| (-) | (-) | |||||||||
| 5 | - | 20.26 | 83.07 | - | - | - | 41.52 | 48.50 | 8.98 | 18.41 |
| (47.93) | (-) | (-) | (-) | (-) | (55.32) | |||||
| 6 | - | - | - | - | - | 84.67 | - | - | - | - |
| (-) | ||||||||||
| 7 | 64.22 | - | 66.56 | - | - | - | - | - | 41.76 | - |
| (-) | (-) | (-) | ||||||||
| 8 | 26.07 | - | - | - | - | - | - | - | - | - |
| (-) | ||||||||||
| 9 | 14.58 | 5.06 | 15.86 | 24.68 | 19.43 | 18.09 | 13.62 | 13.74 | 11.35 | 16.53 |
| (41.26) | (-) | (-) | (-) | (42.67) | (56.99) | (58.99) | (58.99) | (58.99) | (-) | |
| 10 | 16.40 | 3.20 | 2.33 | 29.40 | 29.52 | 28.45 | 2.38 | 17.66 | 3.70 | 18.00 |
| (30.43) | (20.24) | (19.65) | (-) | (65.78) | (69.30) | (17.34) | (60.32) | (20.20) | (-) | |
| Dox | 2.68 | 2.52 | 22.42 | 0.51 | 3.73 | 1.03 | 1.47 | 26.03 | 2.60 | 0.77 |
| (-) | (-) | (-) | (17.82) | (-) | (7.95) | (-) | (-) | (20.09) | (-) | |
2.3. Lipinski’s Rule of Five
| Lipinski’s Parameters | ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | %ABS | TPSA a (Å2) | nHBA a (nON) | nHBD a (nOHNH) | Log P a | MW a | n violations a | Log S b |
| 2 | 78.98 | 86.99 | 5 | 3 | 0.85 | 184.15 | 0 | −0.87 |
| 3 | 69.04 | 115.80 | 6 | 6 | −0.94 | 184.15 | 1 | −1.09 |
| 4 | 73.75 | 102.15 | 6 | 4 | 1.84 | 272.26 | 0 | −2.83 |
| 5 | 72.64 | 105.38 | 7 | 4 | 1.94 | 315.33 | 0 | −2.87 |
| 6 | 57.95 | 147.97 | 9 | 4 | 1.80 | 317.26 | 0 | −3.47 |
| 7 | 70.57 | 111.38 | 7 | 4 | 1.89 | 302.29 | 0 | −2.85 |
| 8 | 66.78 | 122.37 | 7 | 5 | 1.36 | 288.26 | 0 | −2.54 |
| 9 | 73.63 | 102.51 | 6 | 4 | 0.34 | 226.21 | 0 | −2.10 |
| 10 | 74.63 | 99.61 | 6 | 3 | 1.27 | 240.24 | 0 | −3.07 |
3. Experimental Section
3.1. Chemistry
3.1.1. General
3.1.2. Galloyl hydrazide (3)
3.1.3. General Procedure for the Preparation of N'-(Substituted)-arylidene galloyl hydrazides 4–8
3.1.4. Galloyl-2-thioxo-1,3,4-oxadiazole (9)
3.1.5. Galloyl-2-methylthio-1,3,4-oxadiazole (10)
3.2. Anticancer Assay in Vitro
3.3. In Silico Study
- (I)
- hydrogen bond donors ≤ 5 (OH and NH groups);
- (II)
- hydrogen bond acceptors ≤ 10 (N and O atoms);
- (III)
- molecular weight < 500;
- (IV)
- calculated logP < 5.
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Golumbic, C.; Mattill, H.A. The antioxidant properties of gallic acid and allied compounds. J. Am. Chem. Soc. 1942, 19, 144–145. [Google Scholar]
- Kim, D.O.; Lee, K.W.; Lee, H.J.; Lee, C.Y. Vitamin C equivalent antioxidant capacity of phenolic phytochemicals. J. Agric. Food Chem. 2002, 50, 3713–3717. [Google Scholar] [CrossRef] [PubMed]
- Kroes, B.H.; van den Berg, A.J.J.; Quarles van Ufford, H.C.; van Dijk, H.; Labadie, R.P. Anti-inflammatory activity of gallic acid. Planta Med. 1992, 58, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Gichner, T. Two types of antimutagenic effects of gallic and tannic acids towards N-nitroso-compounds-induced mutagenicity in the Ames Salmonella assay. Folia Microbiol. 1987, 32, 55–62. [Google Scholar] [CrossRef]
- Mirvish, S.S.; Cardesa, A.; Wallcave, L.; Shubik, P. Induction of mouse lung adenomas by amines or ureas plus nitrite and by N-nitroso compounds: Effect of ascorbate, gallic acid, thiocyanate and caffeine. J. Natl. Cancer Inst. 1975, 55, 633–666. [Google Scholar] [PubMed]
- Inoue, M.; Suzuki, R.; Sakaguchi, N.; Li, Z.; Takeda, T.; Ogihara, Y.; Jiang, B.; Chen, Y. Role of reactive oxygen species in gallic acid-induced apoptosis. Biol. Pharm. Bull. 1995, 18, 1526–1530. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Kataoka, T.; Hayashi, T.; Hasegawa, M.; Ishi, Y.; Hibasami, H. Induction of apoptosis by gallic acid in human stomach cancer KATO III and colon adenocarcinoma COLO205 cell lines. Oncol. Rep. 2000, 6, 1221–1223. [Google Scholar]
- Hsu, C.; Lo, W.; Yen, G. Gallic acid induces apoptosis in 3T3-L1 pre-adipocytes via a Fas- and mitochondrial-mediated pathway. J. Agric. Food Chem. 2007, 55, 7359–7365. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Velmurugan, B.; Rajamanickam, S.; Agarwal, R.; Agarwal, C. Gallic acid, an active constituent of grape seed extract, exhibits antiproliferative, pro apoptotic and anti-tumorigenic effects against prostate carcinoma xenograft growth in nude mice. Pharm. Res. 2009, 26, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. Gallic acid induced lung cancer cell death is related to glutathione depletion as well as reactive oxygen species increase. Toxicol. In Vitro 2010, 24, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Moon, H.J.; Han, Y.H.; Park, W.H. Gallic acid inhibits the growth of HeLa cervical cancer cells via apoptosis and/or necrosis. Food Chem. Toxicol. 2010, 48, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, F.; Jiang, H.; Wu, K.; Zheng, X.; Cai, Y.; Katakowski, M.; Chopp, M.; To, S.T. Gallic acid suppresses cell viability, proliferation, invasion and angiogenesis in human glioma cells. Eur. J. Pharmacol. 2010, 641, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Madlener, S.; Illmer, C.; Horvath, Z.; Saiko, P.; Losert, A.; Herbacek, I.; Grusch, M.; Elford, H.L.; Krupitza, G.; Bernhaus, A.; et al. Gallic acid inhibits ribonucleotide reductase and cyclooxygenases in human HL-60 promyelocytic leukemia cells. Cancer Lett. 2007, 245, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Bernhaus, A.; Fritzer-Szekeres, M.; Grusch, M.; Saiko, P.; Krupitza, G.; Venkateswarlu, G.; Trimurtulu, G.; Jaeger, W.; Szekeres, T. Digalloylresveratrol, a new phenolic acid derivative induces apoptosis and cell cycle arrest in human HT-29 colon cancer cells. Cancer Lett. 2009, 274, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Chia, Y.C.; Rajbanshi, R.; Calhoun, C.; Chiu, R.H. Anti-neoplastic effects of gallic acid, a major component of Toona sinensis leaf extract, on oral squamous carcinoma cells. Molecules 2010, 15, 8377–8389. [Google Scholar] [CrossRef] [PubMed]
- Horvath, Z.; Saiko, P.; Illmer, C.; Madlener, S.; Hoechtl, T.; Bauer, W.; Erker, T.; Jaeger, W.; Fritzer-Szekeres, M.; Szekeres, T. Synergistic action of resveratrol, an ingredient of wine, with Ara-C and tiazofurin in HL-60 human promyelocytic leukemia cells. Exp. Hematol. 2005, 33, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Tsai, S.; Chen, L.; Lin-Shiau, L.; Lin, J. Resveratrol-induced G2 arrest through the inhibition of CDK7 and p34CDC2 kinases in colon carcinoma HT29 cells. Biochem. Pharmacol. 2003, 65, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y. Development of Gallic Acid-Conjugated Linoleic Acid Ester as Novel Functional Materials and Its Biological Activity. Ph.D. Thesis, Chonbuk National University, Jeonju, Korea, 2007. [Google Scholar]
- Lee, H.; Kwon, Y.; Lee, J.H.; Kim, J.; Shin, M.K.; Kim, S.H.; Bae, H. Methyl gallate exhibits potent antitumor activities by inhibiting tumor infiltration of CD4+CD25+ regulatory T cells. J. Immunol. 2010, 185, 6698–6705. [Google Scholar] [CrossRef] [PubMed]
- Wacher, V.J. Use of Gallic Acid Esters to Increase Bioavailability of Orally Administered Pharmaceutical Compounds. Ph.D. Thesis, University of California, San Francisco, CA, USA, 2001. [Google Scholar]
- Spickenreither, M.; Braun, S.; Bernhardt, G.; Dove, S.; Buschauer, A. Novel 6-O-acylated vitamin C derivatives as hyaluronidase inhibitors with selectivity for bacterial lyases. Bioorg. Med. Chem. Lett. 2006, 16, 5313–5316. [Google Scholar] [CrossRef] [PubMed]
- Isoyama, T.; Thwaites, D.; Selzer, M.G.; Carey, R.I.; Barbucci, R.; Lokeshwar, V.B. Differential selectivity of hyaluronidase inhibitors toward acidic and basic hyaluronidases. Glycobiology 2006, 16, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reaction, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, M.A.; Shaharyar, M. Synthesis, Characterization, and In Vitro Anticancer Evaluation of Novel 2,5-Disubstituted 1,3,4-Oxadiazole Analogue. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Kothayer, H.; Elshanawani, A.A.; Abu Kull, M.E.; El-Sabbagh, O.I.; Shekhar, M.P.; Brancale, A.; Jones, A.T.; Westwell, A.D. Design, synthesis and in vitro anticancer evaluation of 4,6-diamino-1,3,5-triazine-2-carbohydrazides and -carboxamides. Bioorg. Med. Chem. Lett. 2013, 23, 6886–6889. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.R.; Charris, J.; Camacho, J.; Barazarte, A.; Gamboa, N.; Antunes, F. Cytotoxic effects of N'-formyl-2-(5-nitrothiophen-2-yl)benzothiazole-6-carbohydrazide in human breast tumor cells by induction of oxidative stress. Anticancer Res. 2012, 32, 2721–2726. [Google Scholar] [PubMed]
- Ali, M.A.; Shaharyar, M. Oxadiazole mannich bases: Synthesis and antimycobacterial activity. Bioorg. Med. Chem. Lett. 2007, 17, 3314–3316. [Google Scholar] [CrossRef] [PubMed]
- Formagio, A.S.N.; Tonin, L.T.D.; Foglio, M.A.; Madjarof, C.; de Carvalho, J.E.; da Costa, W.F.; Cardoso, F.P.; Sarragiotto, M.H. Synthesis and antitumoral activity of novel 3-(2-substituted-1,3,4-oxadiazol-5-yl) and 3-(5-substituted-1,2,4-triazol-3-yl) β-carboline derivatives. Bioorg. Med. Chem. Lett. 2008, 16, 9660–9667. [Google Scholar] [CrossRef]
- Savariz, F.C.; Formagio, A.S.N.; Barbosa, V.A.; Foglio, M.A.; de Carvalho, J.E.; Duarte, M.C.T.; Filho, B.P.D.; Sarragiotto, M.H. Synthesis, Antitumor and Antimicrobial Activity of Novel 1-Substituted Phenyl-3-[3-alkylamino(methyl)-2-thioxo-1,3,4-oxadiazol-5-yl] β-Carboline Derivatives. J. Braz. Chem. Soc. 2010, 21, 288–298. [Google Scholar] [CrossRef]
- Barbosa, V.A.; Formagio, A.S.N.; Savariz, F.C.; Foglio, M.A.; Spindola, H.M.; Carvalho, J.E.; Meyer, E.; Sarragiotto, M.H. Synthesis and antitumor activity of β-carboline 3-(substituted-carbohydrazide) derivatives. Bioorg. Med. Chem. 2011, 19, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.I. A Textbook of Practical Organic Chemistry, 3rd ed.; Wiley: New York, NY, USA, 1957; pp. 538–539. [Google Scholar]
- Ilango, K.; Arunkumar, S. Synthesis, antimicrobial and antitubercular activities of some novel trihydroxy benzamido azetidin-2-one derivatives. Trop. J. Pharm. Res. 2011, 10, 219–229. [Google Scholar] [CrossRef]
- Hsieh, T.J.; Liu, T.Z.; Chi, Y.C.; Chern, C.L.; Lu, F.J.; Chuang, M.C.; Mau, S.Y.; Chen, S.H.; Syu, Y.H.; Chen, C.H. Protective effect of methyl gallate from Toona sinensis (Meliaceae) against hydrogen peroxide-induced oxidative stress and DNA damage in MDCK cells. Food Chem. Toxicol. 2004, 42, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Kang, O.H.; Lee, Y.S.; Oh, Y.C.; Chae, H.S.; Jang, H.J.; Shin, D.W.; Kwon, D.Y. Antibacterial activity of methyl gallate isolated from Galla Rhois or carvacrol combined with nalidixic acid against nalidixic acid resistant bacteria. Molecules 2009, 14, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Jang, H.S.; Oh, J.S.; Yang, K.H.; Choi, N.K.; Lim, H.S.; Kim, S.M. Effects of methyl gallate and gallic acid on the production of inflammatory mediators interleukin-6 and interleukin-8 by oral epithelial cells stimulated with Fusobacterium nucleatum. J. Microbiol. 2009, 47, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Oh, J.S.; Kang, I.C.; Hong, S.J.; Choi, C.H. Inhibitory effect of methyl gallate and gallic acid on oral bacteria. J. Microbiol. 2008, 46, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, J.K.; Kim, D.W.; Hwang, H.S.; Eum, W.S.; Park, J.; Han, K.H.; Oh, J.S.; Choi, S.Y. Antitumor activity of methyl gallate by inhibition of focal adhesion formation and Akt phosphorylation in glioma cells. Biochim. Biophys. Acta 2013, 1830, 4017–4029. [Google Scholar] [CrossRef] [PubMed]
- Tsuruo, T.; Naito, M.; Tomida, A.; Fujita, N.; Mashima, T.; Sakamoto, H.; Haga, N. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003, 94, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.C.; Francis, R.E.; Guest, S.K.; Costa, J.R.; Gomes, A.R.; Myatt, S.S.; Brosens, J.J.; Lam, E.W.F. Doxorubicinactivates FOXO3a to induce the expression of multidrug resistance gene ABCB1(MDR1) in K562 leukemic cells. Mol. Cancer Ther. 2008, 7, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Harisi, R.; Dudas, J.; Nagy-Olah, J.; Timar, F.; Szendroi, M.; Jeney, A. Extracellular matrixinduces doxorubicin-resistance in human osteosarcoma cells by suppression of p53 function. Cancer Biol. Ther. 2007, 6, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.J.; Duarte, N.; Gyemant, N.; Radics, R.; Cherepnev, G.; Varga, A.; Molnár, J. Inter-action between doxorubicin and the resistance modifier stilbene on multidrugresistant mouse lymphoma and human breast cancer cells. Anticancer Res. 2006, 26, 3541–3546. [Google Scholar] [PubMed]
- Song, X.; Liu, X.; Chi, W.; Liu, Y.; Wei, L.; Wang, X.; Jinming, Y. Hypoxia-induced resistance tocisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencingof HIF-1alpha gene. Cancer Chemother. Pharmacol. 2006, 58, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Biing, J.T.; Yang, Y.F.; Liu, H.S.; Ye, C.T.; Chao, C.F. The induction of multidrug resistancein human cervical carcinoma cell lines by estrogenic hormones. Proc. Natl. Sci. Counc. Repub. China Part B: Life Sci. 1994, 18, 64–70. [Google Scholar]
- Turner, J.G.; Sullivan, D.M. CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr. Med. Chem. 2008, 15, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; Pedretti, A.; Testa, B. Assessing drug-likeness e what are we missing? Drug Discov. Today 2008, 13, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P. Calculation of Molecular Properties and Bioactivity Score. Available online: http://www.molinspiration.com (accessed on 2 November 2014).
- Sander, T. Molecular Property Explorer. Available online: http://www.organic-chemistry.org/prog/peo (accessed on 2 November 2014).
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langlet, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Bi, L.; Wang, W.; Wang, C.; Baudy-Floc’h, M.; Ju, J.; Peng, S. Synthesis and cytotoxic activities of β-carboline amino acid ester conjugates. Bioorg. Med. Chem. 2006, 14, 6998–7010. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 2–10 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maximo da Silva, M.; Comin, M.; Santos Duarte, T.; Foglio, M.A.; De Carvalho, J.E.; Do Carmo Vieira, M.; Nazari Formagio, A.S. Synthesis, Antiproliferative Activity and Molecular Properties Predictions of Galloyl Derivatives. Molecules 2015, 20, 5360-5373. https://doi.org/10.3390/molecules20045360
Maximo da Silva M, Comin M, Santos Duarte T, Foglio MA, De Carvalho JE, Do Carmo Vieira M, Nazari Formagio AS. Synthesis, Antiproliferative Activity and Molecular Properties Predictions of Galloyl Derivatives. Molecules. 2015; 20(4):5360-5373. https://doi.org/10.3390/molecules20045360
Chicago/Turabian StyleMaximo da Silva, Marciane, Marina Comin, Thiago Santos Duarte, Mary Ann Foglio, João Ernesto De Carvalho, Maria Do Carmo Vieira, and Anelise Samara Nazari Formagio. 2015. "Synthesis, Antiproliferative Activity and Molecular Properties Predictions of Galloyl Derivatives" Molecules 20, no. 4: 5360-5373. https://doi.org/10.3390/molecules20045360