2. Results and Discussion
Photophysical properties associated with highly enhanced NLO absorption cross sections in the NIR region were achieved by the attachment of one or multiple 9,9'-dialkyl-2-diphenylaminofluorene moieties on a spherical C
60 molecule using a periconjugated keto-bridging unit [
30]. It also indicated the improvement of light harvesting efficiency by using hindered and branched 3',5',5'-trimethylhexyl (C
9) arms to maintain the space-separation of fluorophores intramolecularly within the nanostructure and intermolecularly during possible cluster formation or aggregation in solution. These conclusions became the basis of our new molecular design of tris(DPAF-C
9) analogous structures
5–
7. In this case, the central benzene ring was shared by two and three phenylaminofluorene antenna unit in the molecular structure of
6 and
7, respectively, as a fused dendron. Since tris(DPAF-C
9)
7 is highly fluorescence emissive, it can be utilized as a charge-injecting host fluorophore for the study of PLED devices.
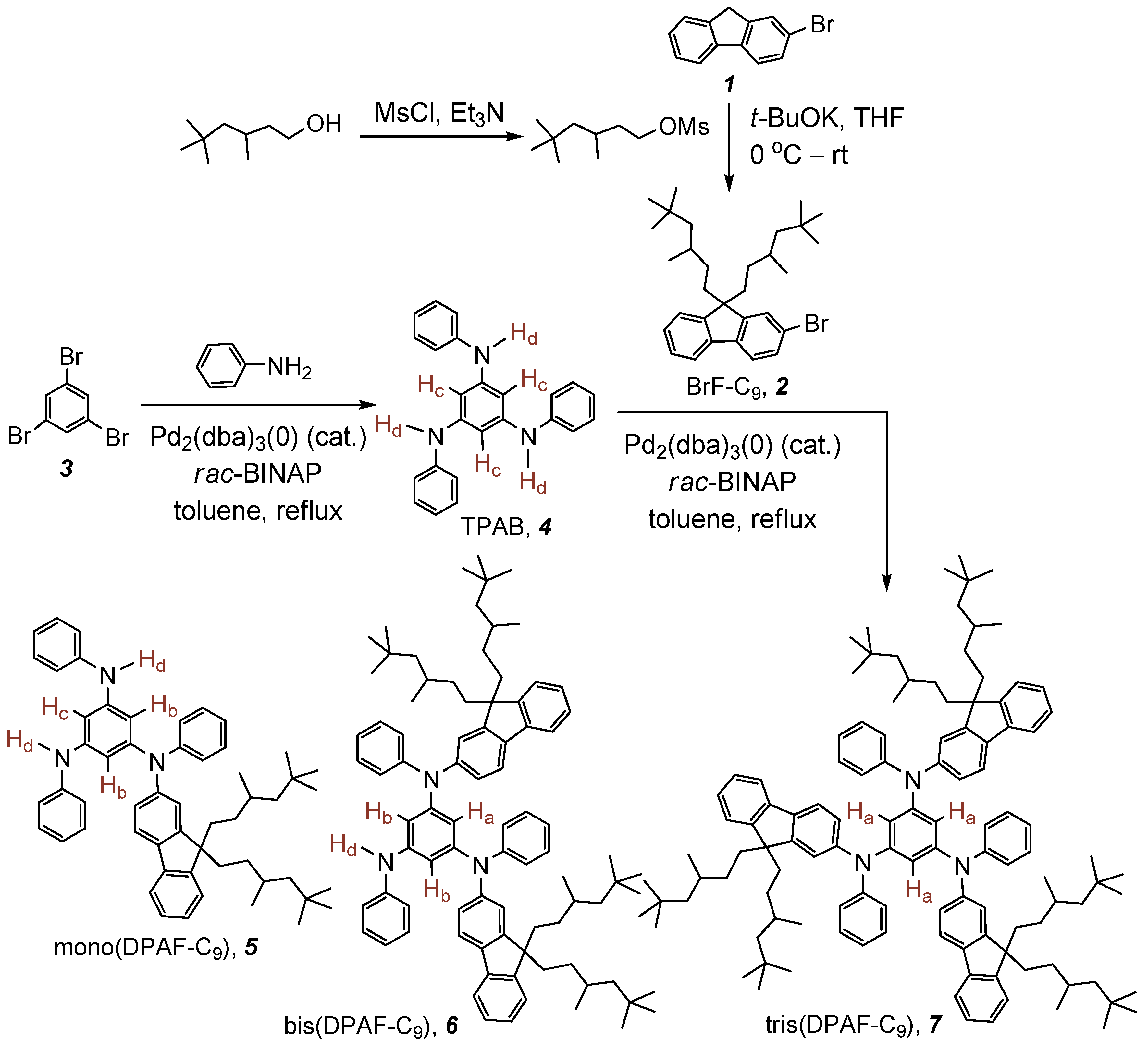
The synthetic route for the preparation of asymmetrical chromophore mono(DPAF-C
9)
5 and bis(DPAF-C
9)
6, and
C3-symmetrical chromophore tris(DPAF-C
9)
7, having both dialkylated fluorenyl and phenyl groups on each nitrogen atom of a central 1,3,5-triaminobenzene core was performed, as shown in
Scheme 1. Alkylation of 2-bromofluorene
1 with mesylated 3,5,5-trimethylhexanol giving 2-bromo-9,9-bis(3',5',5'-trimethylhexyl)fluorene, BrF-C
9 2, was carried out using potassium
t-butoxide as a base. The reaction proceeded in a high yield of 90%. The synthesis of 1,3,5-tris(phenylamino)benzene, TPAB
4, was reported previously [
31,
32]. However, in order to improve the yield of product, we employed an modified procedure of the palladium-catalyzed amination reaction with tris(dibenzylideneacetone)dipalladium(0) [Pd
2(dba)
3(0)] as the catalyst. The reaction was carried out by using five equivalents (excess) of aniline in respect to the quantity of 1,3,5-tribromobenzene
3 in the presence of
rac-2,2'-bis(diphenylphosphino)-1,1'-binaphthyl (
rac-BINAP) that resulted in a good tendency for the tris-product formation. Accordingly, a higher yield of
4 in 75% was obtained. A similar palladium-catalyzed amination was also applied in the next step synthesis. For instance, the reaction of TPAB with six equivalents of BrF-C
9 in dry toluene at refluxing temperature for a period of 72 h was conducted to afford the final product tris(DPAF-C
9)
7. The crude product of
7 was then purified subsequently by column chromatography (silica gel) using only hexane and followed by hexane–ethyl acetate (9:1) as the eluent to give light yellow solids or clear thick sticky gel-like paste, while residual solvents are present, in a yield of 88%.
For the synthesis of asymmetrical fluorophores, a different equivalent ratio of starting materials TPAB and BrF-C9 in 1:2 was applied for the preparation of bis(DPAF-C9) 6 and in 1:1 for the synthesis of mono(DPAF-C9) 5. In the case of the compound 6, similar reaction conditions as those for 7 in dry toluene were performed to give the product bis(DPAF-C9). The crude thick liquid of 6 was purified by column chromatography (SiO2) using hexane–ethyl acetate (9:1) as the eluent to yield white to light yellow glassy solids in 52%. For the synthesis of mono(DPAF-C9) 5, a shorter solvent refluxing period of 48 h was used for the reaction of TPAB with one equivalent of 2 in dry toluene, giving the product of mono(DPAF-C9) 5 as white to light yellow glassy solids in a yield of 68% after column chromatography (SiO2) purification using hexane–ethyl acetate (9:1) initially and then 7:3 as the eluent. All products 5, 6, and 7 were found to be photosensitive in the presence of electrophilic solvents and stable as a dry solid. Therefore, dark conditions were necessary during the workup and purification procedures.
Structural characterization of 5, 6, and 7 was made using various spectroscopic techniques. By comparison between the IR spectra of tris(DPAF-C9) 7 and TPAB, the introduction of dialkylated fluorenyl moiety to 4 was clearly confirmed by the appearance of strong aliphatic C–H stretching vibration bands centered at 2953 cm−1 (supporting information) that was absent for TPAB. It also accompanied with several characteristic bands at 3063 and 1583 cm−1 assigned, for aromatic C-H stretching and C=C absorption, respectively. Anti-symmetric deformations of CH3 groups and scissor vibrations of CH2 groups appeared as medium intensity bands centered around 1493 (s) and 1450 cm−1, while symmetric deformations of CH3 groups exhibited the absorption around 1363 (m) cm−1. Two other bands at 1294 (m) and 1249 (m) were assigned to the asymmetric stretching vibrations of C-N-C moieties. Close resemblance of these IR absorption bands was observed for both mono(DPAF-C9) 5 and bis(DPAF-C9) 6 indicating high similarity of functional groups consisting of DPAF-C9 and TPAB moieties. All compounds 5–7 showed strong C–H out-of-plan deformation bands at 756 and 711 cm−1, for example, for 7. Furthermore, systematic decrease in intensity of the broad N–H absorption band centered at 3403 and 3375 cm−1 for TPAB to 3396 cm−1 for 5, 3402 cm−1 for 6, and complete disappearance for 7, indicated clearly the corresponding progressive amination of fluorenyl moieties (F-C9) from 5 to 7.
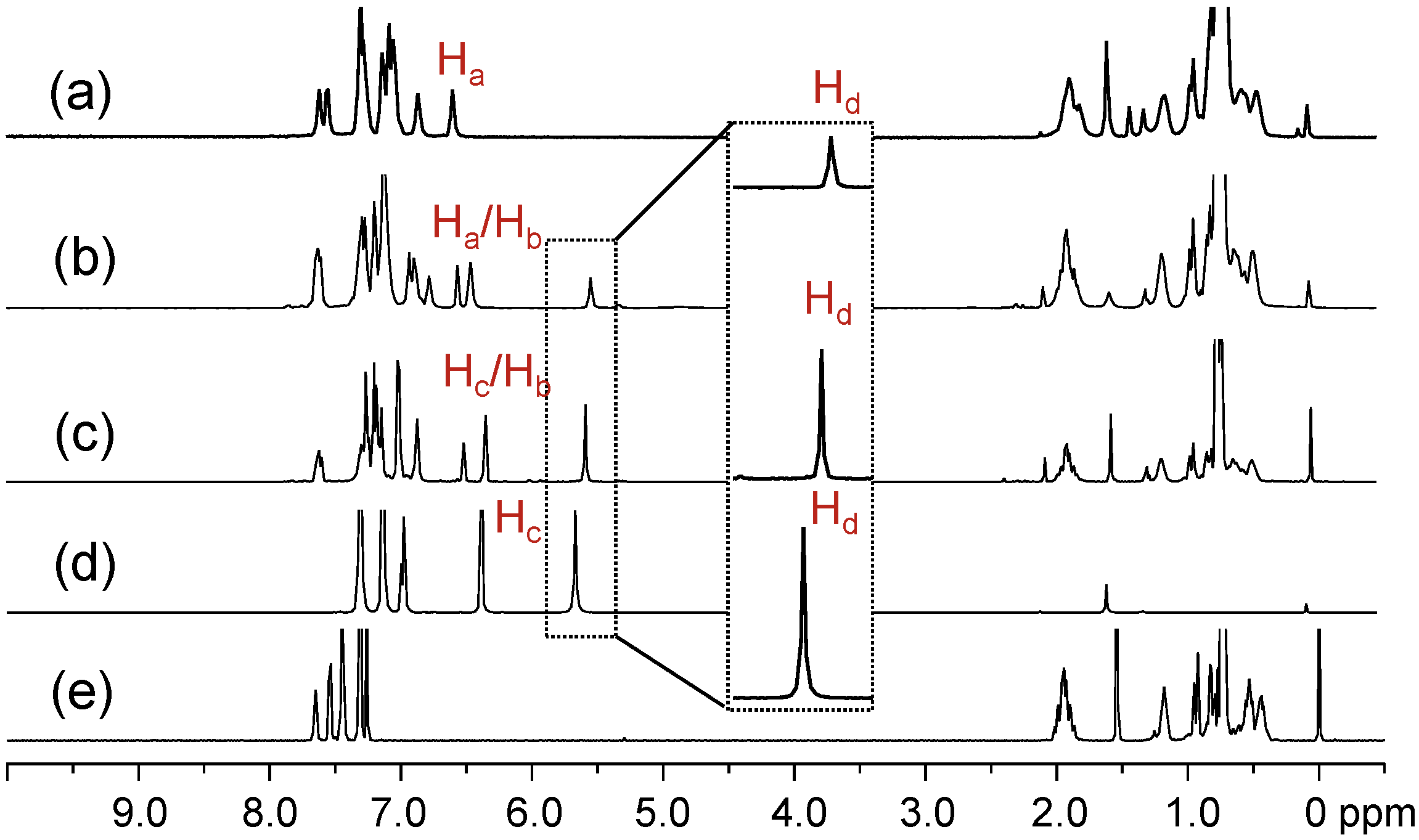
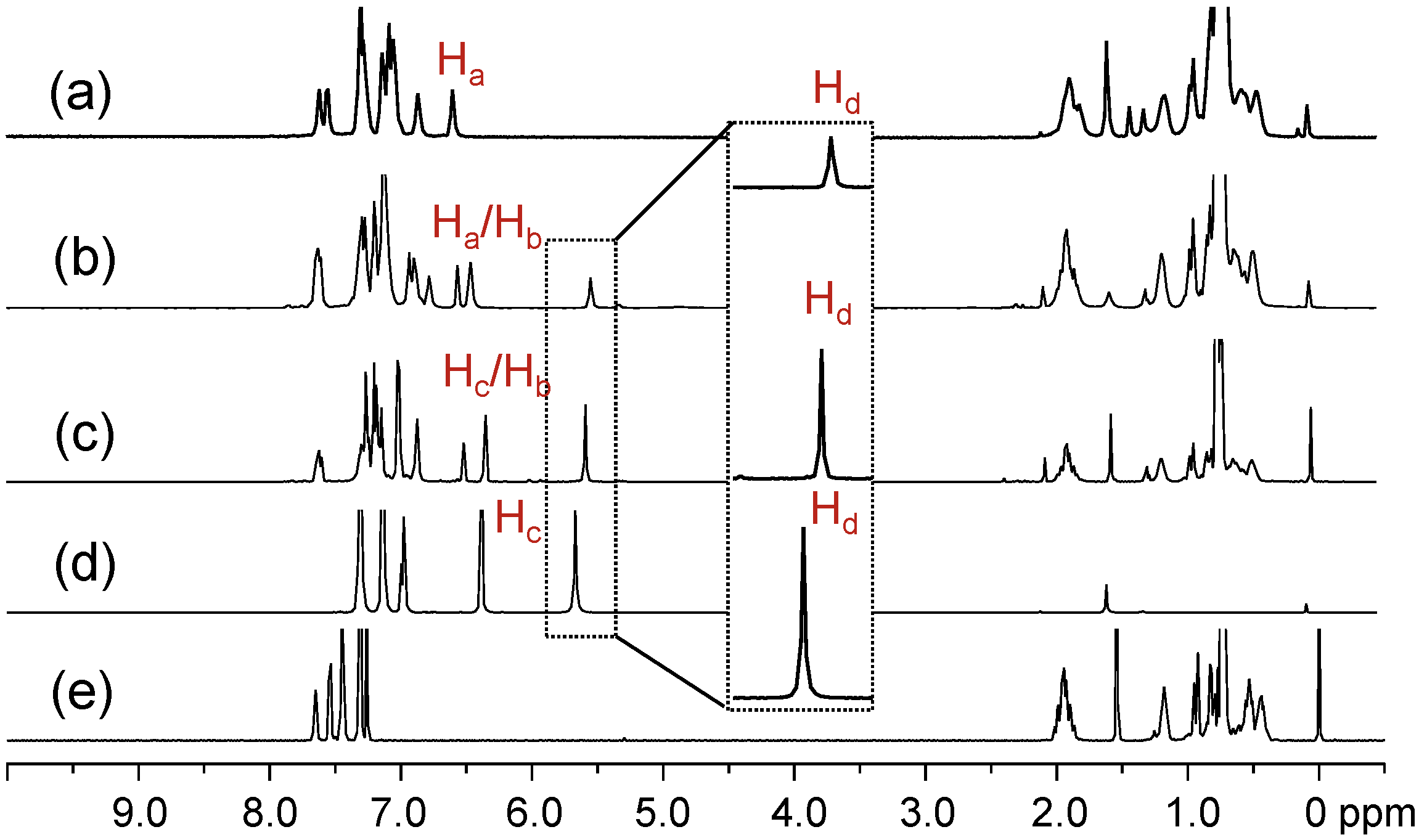
Figure 1.
1H-NMR spectra of (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, (c) mono(DPAF-C9) 5, (d) TPAB 4, and (e) BrF-C9 2.
Figure 1.
1H-NMR spectra of (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, (c) mono(DPAF-C9) 5, (d) TPAB 4, and (e) BrF-C9 2.
1H-NMR spectra of the precursor molecules TPAB and BrF-C
9 were used as the reference, as shown in
Figure 1d,e, for the comparison with those of mono-, bis-, and tris(DPAF-C
9). The most critical evidence of the sequential attachment of 9,9-bis(3',5',5'-trimethylhexyl)fluorene dendrons to
4 was given by the systematic decrease in the integrated intensity ratio of the singlet signal at δ 5.67, assigned for the chemical shift of N–H (H
d) protons of TPAB (
Figure 1d). The peak intensity of this peak matched well with the proton ratio of H
d as two, one, and zero for
5 (at δ 5.56,
Figure 1c),
6 (at δ 5.52,
Figure 1b), and
7 (
Figure 1a), respectively. The spectra also indicated the corresponding ratio increase of C
9-alkyl proton peaks at δ 0.4–2.1 in intensity going from
5 to
7 with roughly similar multiplet peak patterns as those of the parent BrF-C
9. Characteristics of the central benzene protons can be used for the analysis of the relative geometrically configuration of fluorenyl rings with respect to each other. In the case of mono(DPAF-C
9)
5 (
Figure 1c), two peaks centered at δ 6.33 and 6.50 in a peak integration ratio of 2:1 were clearly detected that allowed us to assign them as the chemical shifts of H
b and H
c, respectively, with the latter having a slight down-fielded shift of 0.18 ppm from that of the singlet H
c of
4 at δ 6.32. As the number of fluorene addend (F-C
9) increased to the structure of bis(DPAF-C
9)
6, the central benzene protons changed to H
a (fluorene rings at both sides, 1H) and H
b (one fluorene ring and one phenyl ring at either sides, 2H) types. Two peak groups located at δ 6.42 (broad singlet) and 6.53 (singlet) in an integration ratio of 2:1 (
Figure 1b) allowed a clear assignment to the chemical shift of H
b and H
a, respectively. The peak broadening and splitting at δ 6.42 revealed two non-equivalent H
bs, perhaps, as the result of a mixture of two possible geometrical conformational isomers of
6 with two fluorene rings either at a
cis-conformation (up-up) or
trans-conformation (up-down) since free rotation around the benzene–N bond is unlikely to occur. In addition, an increased complexity and number of fluorenyl proton peaks at δ 6.70–7.70 also supported the existence of two non-equivalent geometrical conformers. Interestingly, the only major product of tris(DPAF-C
9)
7 isolated showed full disappearance of N–H
d proton signal at δ 5.5–6.0 and a singlet H
a proton peak at δ 6.55 (
Figure 1a) in a similar chemical shift as that of H
a in
6. The former indicated the completion of palladium-catalyzed amination reactions with three equivalents of light-harvesting DPAF-antenna moieties. The latter revealed three equivalent H
as that led us to suggest a
cis-conformation (geometrically at the same side of the central benzene ring as up-up-up configuration for three fluorenyl rings) for the structure of
7 (more discussion below by structural simulation and calculation). Even though the peak and coupling pattern were more complex in the aromatic region of δ 7.0–8.0 in the spectrum as compared to those of the precursor TPAB, however, without a symmetrical environment for three fluorenyl rings in the structure of
7, the number of aromatic proton peaks each with coupling multiplet should lead to a much more complicated spectrum than that of
6 in the same chemical shift region. Therefore, the observation of only a limit number of fluorenyl aryl proton peak groups at δ 6.82–7.57 with an overall higher proton integration ratio to the peak of H
a supported the argument of a quasi-symmetrical
cis-conformation for
7.
Figure 2.
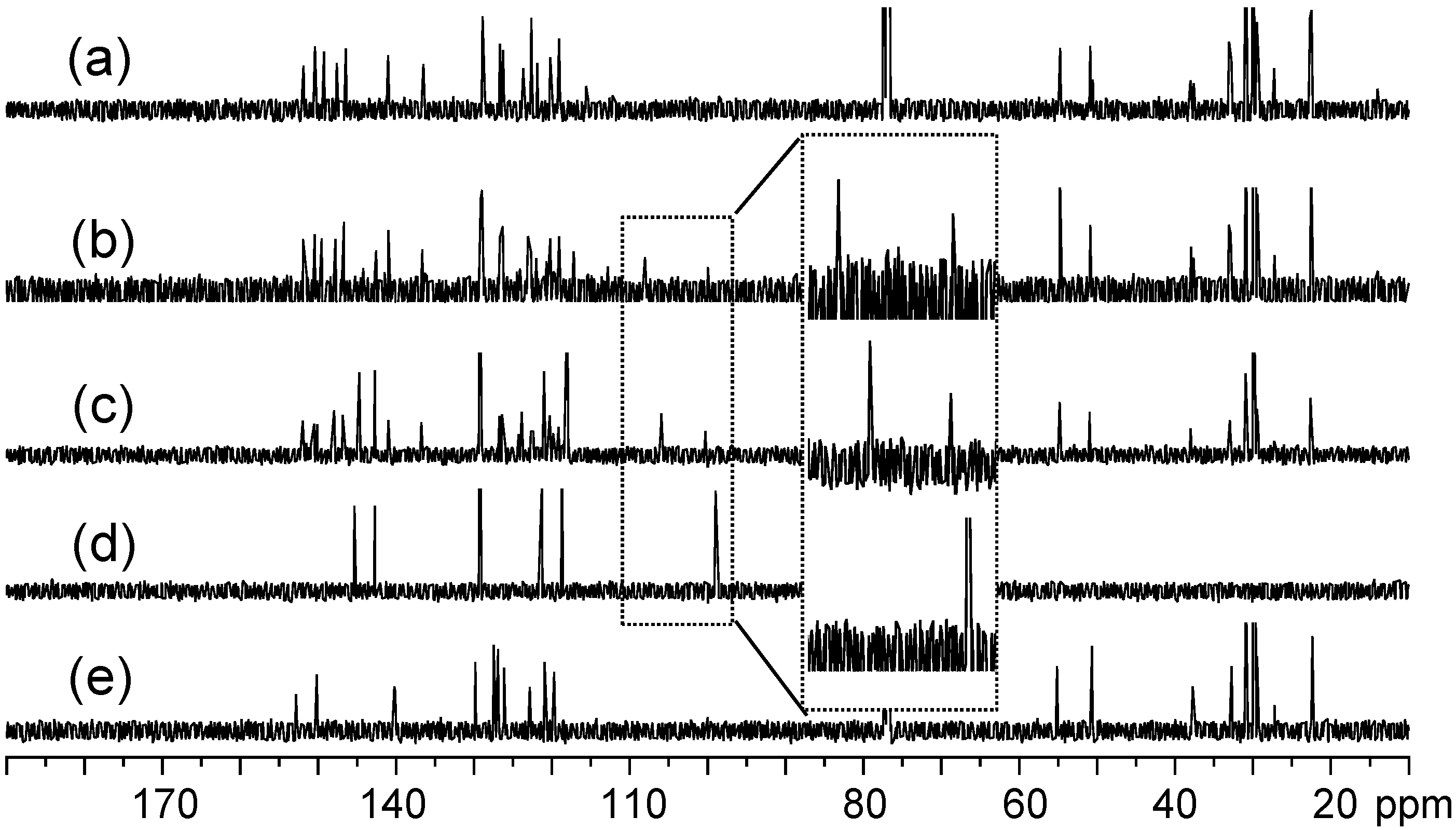
13C-NMR spectra of (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, (c) mono(DPAF-C9) 5, (d) TPAB 4, and (e) BrF-C9 2.
Figure 2.
13C-NMR spectra of (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, (c) mono(DPAF-C9) 5, (d) TPAB 4, and (e) BrF-C9 2.
13C-NMR spectra of the precursor molecules TPAB were also used as the reference for the comparison with those of mono-, bis-, and tris(DPAF-C
9). Attachment of one fluorenyl ring to TPAB led clearly to two sets of aromatic carbon (sp
2) peaks at δ 115–155 in the
13C-NMR spectrum of
5 (
Figure 2c). Chemical shifts of the group of five sp
2 carbon peaks having higher peak intensity were roughly identical to those of TPAB (
Figure 2d). This allowed us to assign the rest of multiple peaks in lower peak intensity to the fluorenyl carbons. The relative intensity of these two peak groups was found to increase progressively for the latter as the number of attached fluorenyl ring increased from
5,
6 (
Figure 2b), to
7 (
Figure 2a), consistent with their corresponding structural composition. Upon the attachment of one DPAF-C
9 to
4, the central phenyl carbon (C
c) peak of TPAB at δ 99.0 was found to split and shift down-field to δ 105.9 (C
b) and 100.3 (C
c), in an intensity ratio of roughly 2:1 consistent with the structure of
5, in
Figure 2c of mono(DPAF-C
9). Similar shifts to δ 108.0 (C
a,
Figure 2b) and 100.0 (C
b) for bis(DPAF-C
9)
6 and to δ 115.6 (H
a,
Figure 2a) for tris(DPAF-C
9)
7 were detected. These spectroscopic data provided an additional verification of the structures
5–
7. High stability of
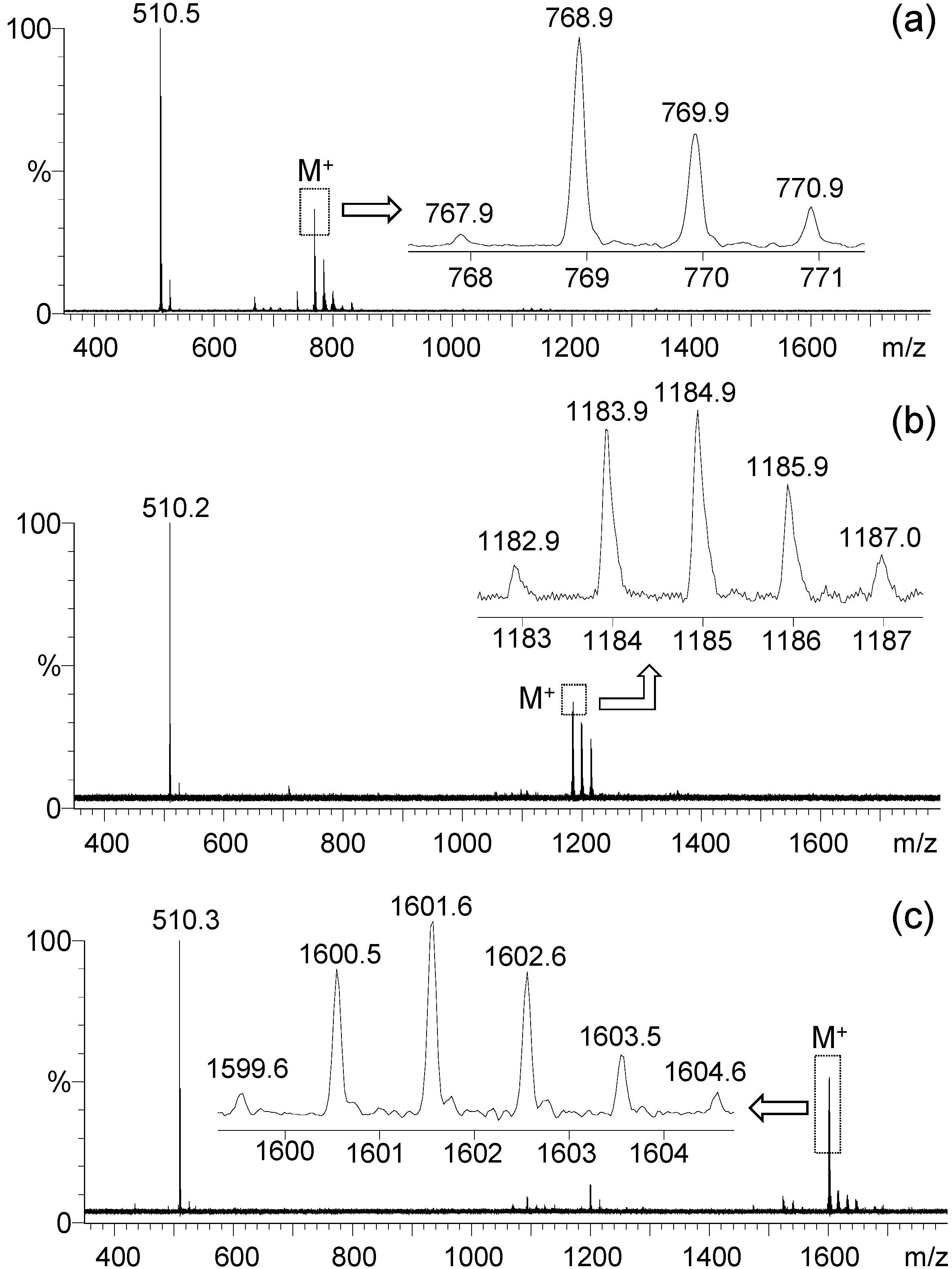
5–
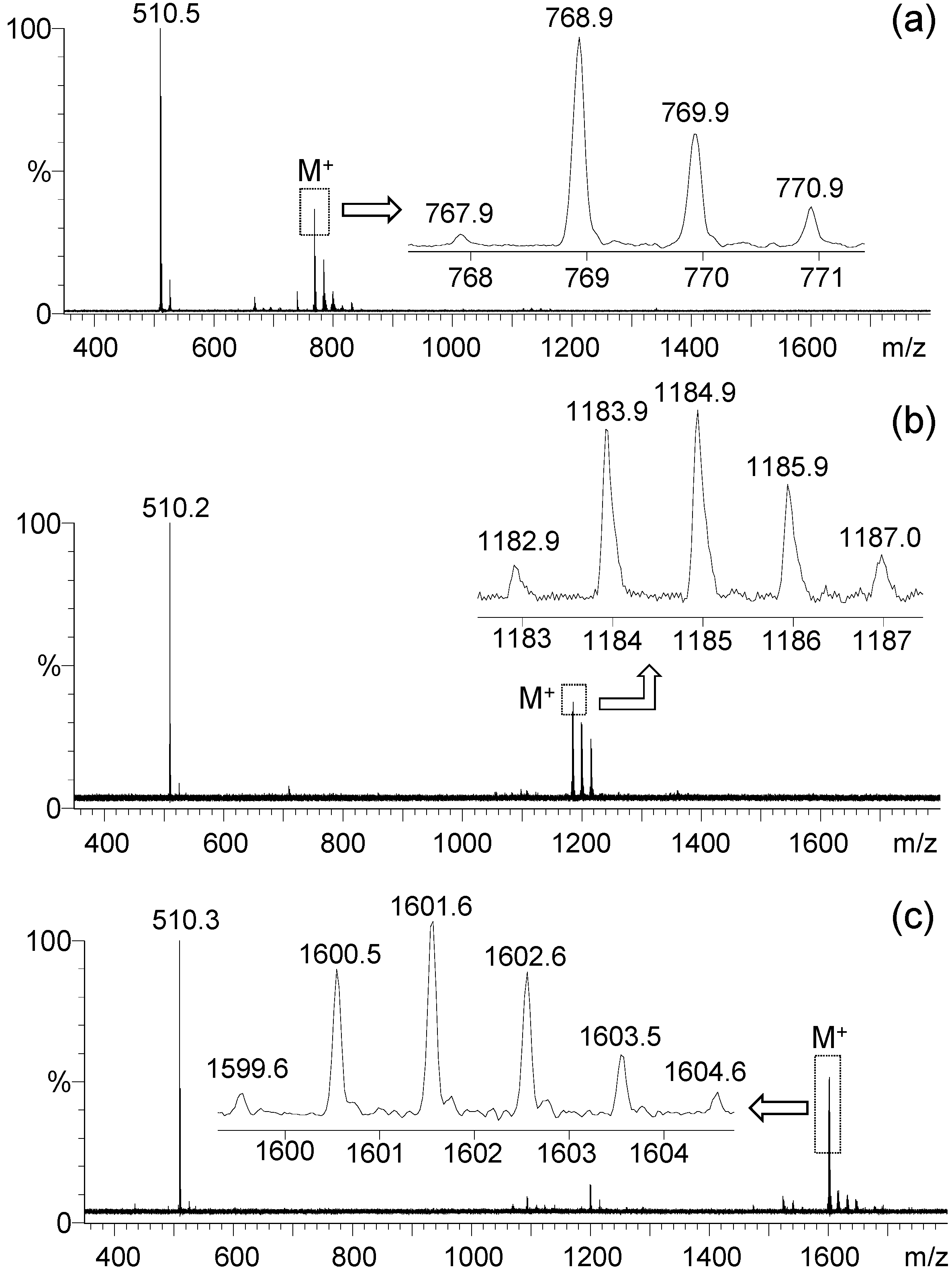
7 was observed during positive ion matrix-assisted laser desorption ionization (MALDI–TOF) mass spectroscopic measurements using 3,5-dimethoxy-4-hydroxycinnamic acid (sinapic acid) as the matrix, as shown in
Figure 3a–c, respectively. All spectra of
5,
6, and
7 displayed a group of molecular ion mass (MH
+) peaks at
m/z 768.9, 1184.9, and 1601.6 in good agreement with the calculated mass of M
+ as
m/z 767.52, 1183.86, and 1600.21, respectively. It also showed a clean fragmentation pattern consistent with a main fragmented ion mass peak at
m/z 510.2–510.5, corresponding to the mass of PhN
+H-fluorene-C
9. This revealed the central benzene-N bond as the weakest one to undergo dissociation. The data also provided unambiguous evidence of the successful synthesis of mono-, bis-, and tris(DPAF-C
9).
Figure 3.
MALDI–TOF mass spectra of (a) mono(DPAF-C9) 5, (b) bis(DPAF-C9) 6, and (c) tris(DPAF-C9) 7.
Figure 3.
MALDI–TOF mass spectra of (a) mono(DPAF-C9) 5, (b) bis(DPAF-C9) 6, and (c) tris(DPAF-C9) 7.
Effect of TPAB ratio with respect to BrF-C9 on the synthesis of tris(DPAF-C9) was also investigated. Our initial synthesis of tris(DPAF-C9) was carried out by using an exact three-equivalent amount of BrF-C9 in respect to the central TPAB core. It was found that this ratio of starting materials did not lead to a clean product, instead resulted in two different product fractions identified as bis- and tris(DPAF-C9) showing Ha and Hb proton peaks (supporting information) at δ 6.56 and 6.48, respectively, slightly down-field shifted in the mixture from those of the corresponding pure sample. By the integration ratio of these two peaks, the major product 7 and the minor product 6 were accounted for 69.0% and 31.0%, respectively. Accordingly, we undertook the TPAB ratio-dependent evaluation of the production formation with three additional ratio of 1:2, 1:4, and 1:6 (excess). As a result, the decrease of TPAB quantity applied to 1:2, the Hb peak intensity at δ 6.48 increased significantly indicating the presence of mono(DPAF-C9) in addition to bis(DPAF-C9) as a mixture. Contrarily, the Hb peak decreased sharply in intensity, corresponding to the yield of 6 as 17.3%, as the TPAB ratio being increased to 1:4. We found that a full reaction conversion to 7 became possible by using a TPAB ratio of 1:6, leading to the complete disappearance of Hb peak in the product spectrum.
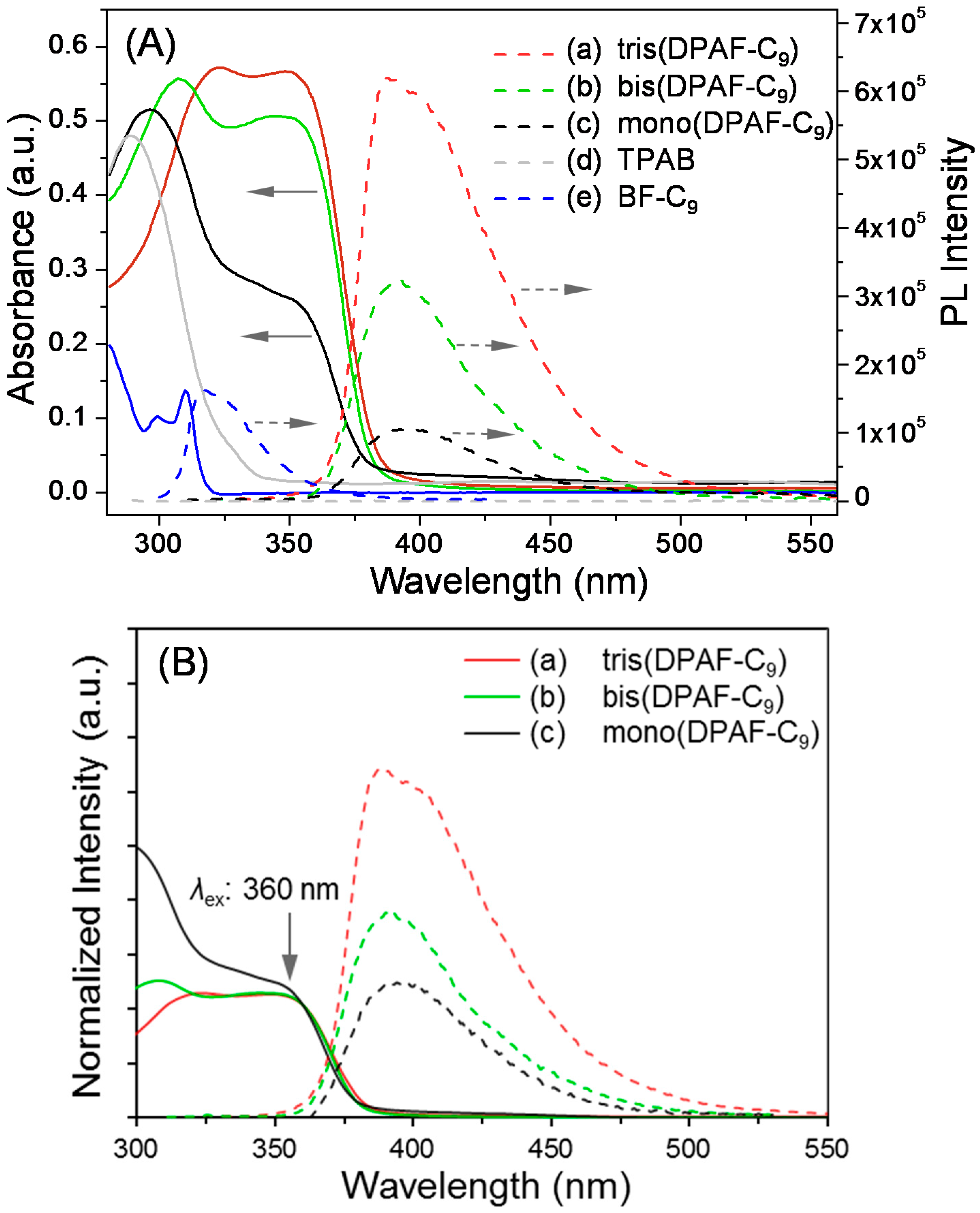
UV-vis spectra of TPAB
4, mono-
5, and bis-
6, and tris-
7 collected at a concentration of 1.0 × 10
‒5 M in CHCl
3 were shown in
Figure 4A (solid lines). Chemical modification of TPAB with one or more fluorenyl moieties converted the secondary to corresponding tertiary amines that led to a gradual bathochromic shift from the absorption of
4 centered at 289 nm (λ
max, ε = 4.80 × 10
4 L·mol
−1·cm
−1) to that of
5 at 297 nm (ε = 5.16 × 10
4), of
6 at 308 nm (ε = 5.56 × 10
4), and of
7 at 323 nm (ε = 5.71 × 10
4 L·mol
−1·cm
−1). It also accompanied with a n ew band centered at 349 nm (ε = 2.64 × 10
4) for
5, 348 nm (ε = 5.07 × 10
4) for
6, and 348 nm (ε = 5.66 × 10
4 L·mol
−1·cm
−1) for
7. Intensity of the latter band was found to increase progressively from
5 to
7 corresponding to the increasing number of fluorenyl moiety that allowed us to assign this peak to the characteristic absorption band of F-C
9 moiety. With a new electron-releasing 3,5-diaminophenylamino group being bonded at C
2 position of a F-C
9, these bands presented a large bathochromic shift from those of BrF-C
9 (λ
max 299 and 310 nm) with an electron-withdrawing bromo group instead at the same carbon position. The shift also corresponds to an extended fluorenyl ring system interconnected by a 1,3,5-triaminobenzene central core, consistent with the structure of
5–
7 synthesized.
All mono-
5, and bis-
6, and tris(DPAF-C
9) are highly photoluminescent (PL) chromophores each showing an emission band at λ
em,max 391.2, 390.8, and 390.1 nm, respectively, (
Figure 4A, dashed lines; λ
ex = 360 nm). Intensity of the band increased roughly in proportion to the number of F-C
9 rings substituted to TPAB. For the clarity, the absorption intensity and relative PL intensity of compounds
5–
7 were normalized at the excitation wavelength of 360 nm as shown in
Figure 4B. Partly owing to the molecular rigidity in the particular stereo-configuration with partial orbital overlap via unpaired electrons of amino groups at the center core of
7 as compared with those of
5 and
6, an obvious enhancement in the PL intensity (
Figure 4B-a) was detected. Similar enhancement in a lesser extent was also observed in the bis(DPAF-C
9)
6 (
Figure 4B-b) in relation to that of mono(DPAF-C
9)
5. This revealed emission-independency among F-C
9 units of
7 with no π-stacking related excimer or self-quenching effect, presumably, owing to the geometrical stereo-conformation of F-C
9 moieties with respect to each other that prevented the close in-phase π-π plane contact of fluorene rings and yielded high PL in significant intensity. Unlike tris(DPAF-C
9), PL arising from the benzene and phenyl moieties of TPAB was fully quenched (
Figure 4A-d at the baseline) providing evidence of strong intermolecular hydrogen bonding interactions between nitrogen and hydrogen among molecules, leading to a strong self-quenching effect. Accordingly, the S
0 → S
1 band gaps (0,0) of
5,
6, and
7 can be estimated from the crossover onset position of their absorption and emission edges in CHCl
3 and were found to be 3.24, 3.26 and 3.28 eV, respectively.
Figure 4.
(A) UV-vis spectra (solid lines) and photoluminescence (dotted lines) of (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, (c) mono(DPAF-C9) 5, (d) TPAB 4, and (e) BrF-C9 2 in CHCl3 at a concentration of 1.0 × 10−5 M and (B) normalized absorption and PL intensity of 5–7 at the excitation wavelength of 360 nm.
Figure 4.
(A) UV-vis spectra (solid lines) and photoluminescence (dotted lines) of (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, (c) mono(DPAF-C9) 5, (d) TPAB 4, and (e) BrF-C9 2 in CHCl3 at a concentration of 1.0 × 10−5 M and (B) normalized absorption and PL intensity of 5–7 at the excitation wavelength of 360 nm.
The fluorescent emission wavelength of
cis-tris(DPAF-C
9) was found in a close range to that of PVK with both HOMO (−4.81 eV) and LUMO energy level (−1.57 eV) in 1.0 and 0.7 eV, respectively, higher than those of PVK. The former is only slightly lower than that of Ir(ppy)
3 (−4.9 eV). Therefore, this donor chromophore may serve as the secondary blue hole-transporting material, which is capable of minimizing the aggregation-related self-quenching effect, in the modulation of PLED performance (
supporting information Figure S5).
Figure 5.
Cyclic voltammograms (CV) of (A–C) at different voltages vs Ag/AgCl with (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, and (c) mono(DPAF-C9) 5 solution in a concentration of 1.75 × 10‒3 M in CH3CN–CH2Cl2, containing (n-butyl)4N+-PF6‒ electrolyte (0.4 M), using Pt as working and counter electrodes and Ag/AgCl as the reference electrode at a scan rate of 10 mV/s.
Figure 5.
Cyclic voltammograms (CV) of (A–C) at different voltages vs Ag/AgCl with (a) tris(DPAF-C9) 7, (b) bis(DPAF-C9) 6, and (c) mono(DPAF-C9) 5 solution in a concentration of 1.75 × 10‒3 M in CH3CN–CH2Cl2, containing (n-butyl)4N+-PF6‒ electrolyte (0.4 M), using Pt as working and counter electrodes and Ag/AgCl as the reference electrode at a scan rate of 10 mV/s.
We also carried out the cyclic voltammetry (CV) measurement of mono-
5, bis-
6, and tris-
7 in a solvent mixture of CH
2Cl
2–CH
3CN in the presence of (
n-butyl)
4N
+-PF
6‒ electrolyte, using Pt as working and counter electrodes and Ag/AgCl as the reference electrode. The CV characteristics of compounds
5–
7 under variation of the cyclic oxidation voltage vs Ag/AgCl from 1.0 to 1.3 V were evaluated, as shown in
Figure 5A–C. As a result, only one consistent reversible redox wave was detectable in a similar current range (0.0–1.1 V) for all three compounds indicating that only one F-C
9 ring moiety was participating the oxidation–reduction cycle at the same concentration regardless its structure being mono-, bis-, or tris-adduct of TPAB. This revealed full electrochemical independency of all F-C
9 rings in the structure. By taking the most stable (symmetrical) and reversible first cyclic wave of
Figure 5C as an example, a nearly identical onset oxidation potential (
Eoxonset) was measured for
5,
6, and
7 as 0.83 V. Under the same CV conditions, the first half-wave redox potential of ferrocence reference sample was measured as 0.82 V. Therefore, by taking the oxidation potential value of ferrocence at −4.8 eV below the vacuum level as the reference, the HOMO levels of mono-
5, bis-
6, and tris-7 were calculated as a nearly identical value of −4.81 eV that was summarized in
Table 1. Furthermore, the optical energy gap (
Eopt, S
0 → S
1 band gap) can be obtained and calculated from the absorption onset of all compounds
5,
6, and
7, via
Figure 4, in a value of 3.28, 3.26, and 3.24 eV, respectively, as discussed above. Consequently, corresponding LUMO levels were then calculated as −1.53, −1.55, and −1.57 eV, respectively.
Table 1.
Electrochemical properties of mono-5, bis-6, and tris(DPAF-C9) 7.
Table 1.
Electrochemical properties of mono-5, bis-6, and tris(DPAF-C9) 7.
| Compound | Eoxonset (V) | HOMO (eV) a | Eopt (eV) b | LUMO (eV) |
|---|
| Tris(DPAF-C9) 7 | 0.83 | −4.81 | 3.24 | −1.57 |
| Bis(DPAF-C9) 6 | 0.83 | −4.81 | 3.26 | −1.55 |
| Mono(DPAF-C9) 5 | 0.83 | −4.81 | 3.28 | −1.53 |
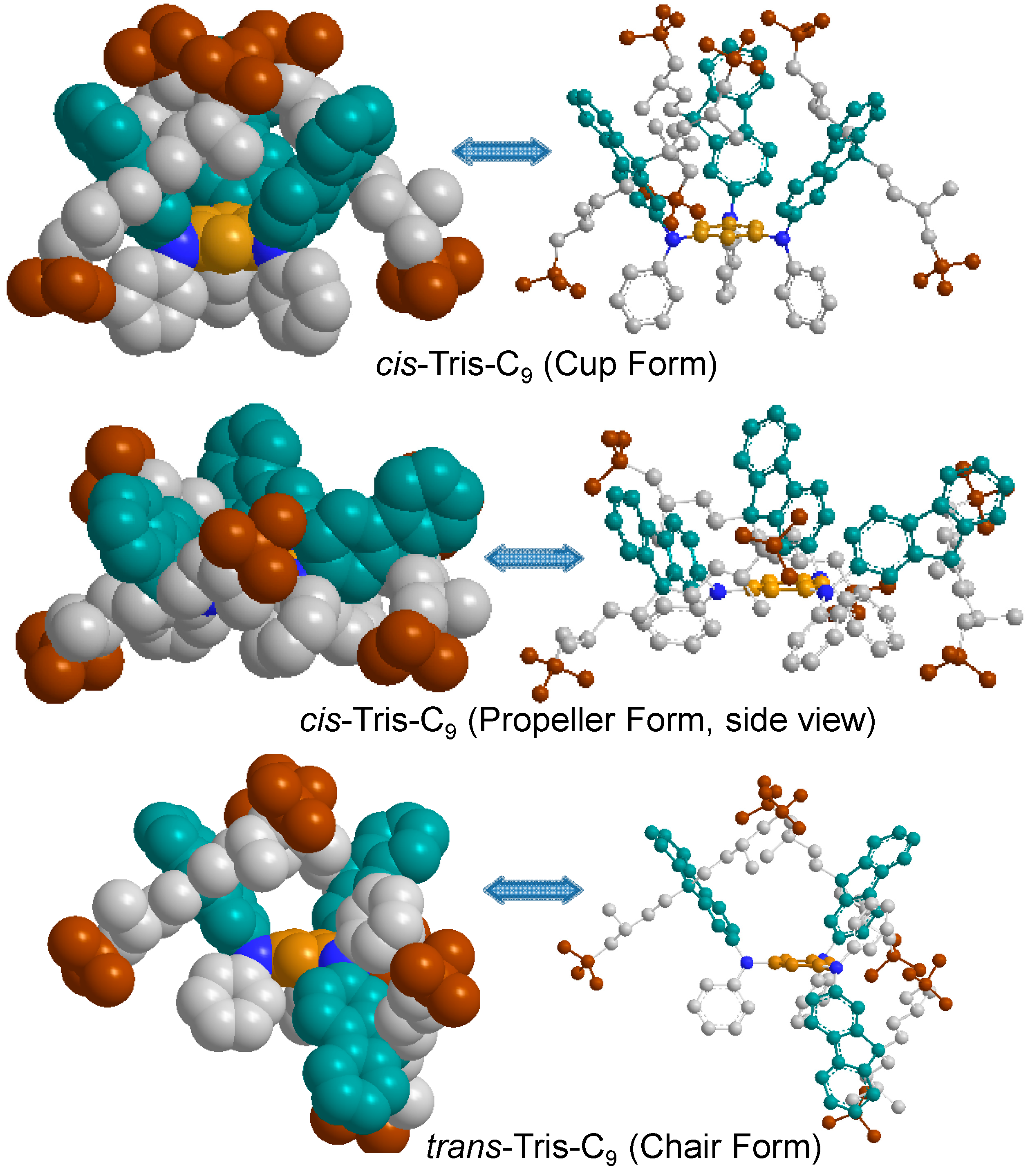
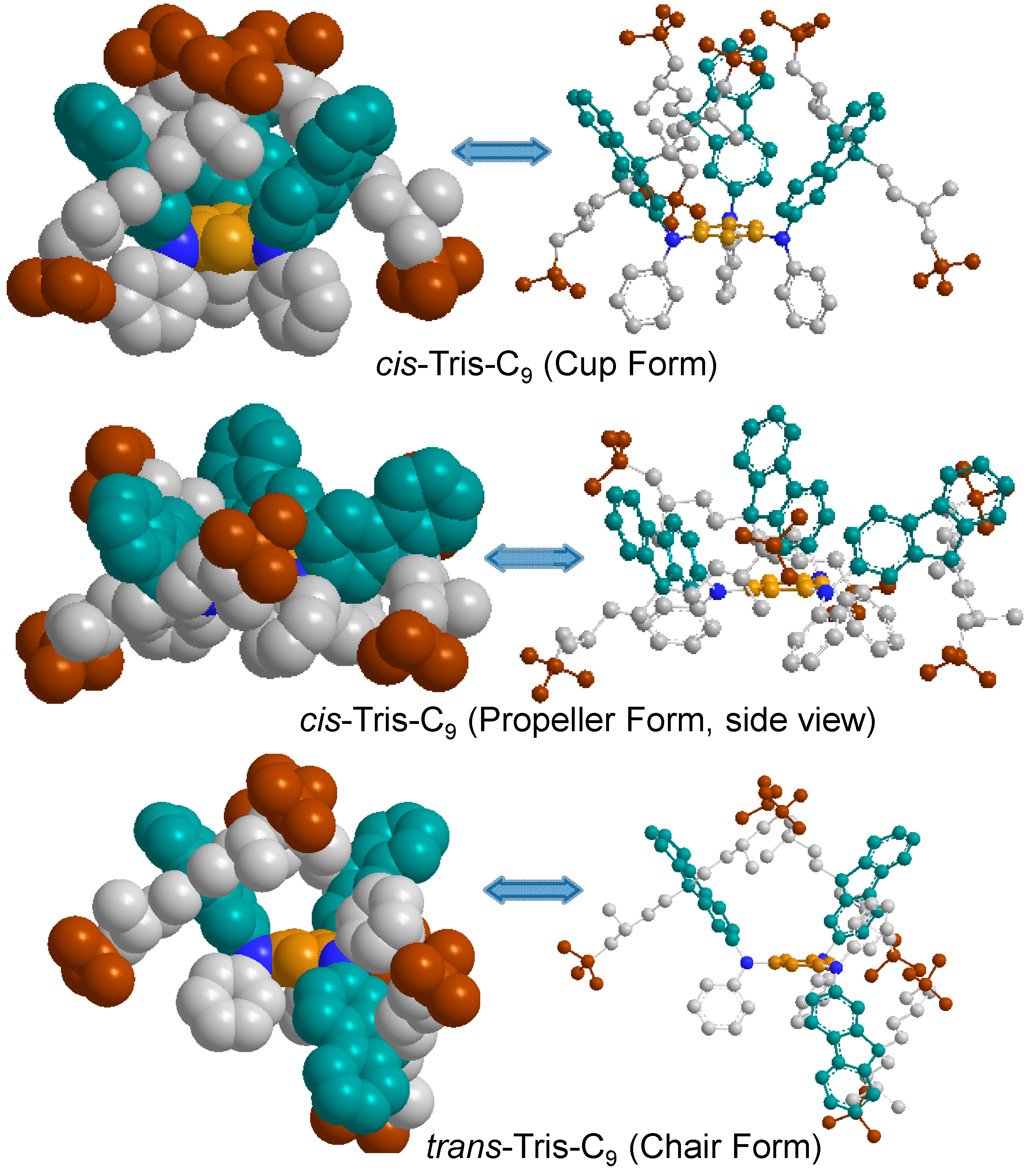
In the consideration of the structural configuration, it is reasonable to expect two possible nonplanar geometrical stereoisomers as, namely, tris-
trans- (with two fluorenyl rings at the same side of the central benzene ring and one fluorenyl rings located at the opposite side as an up-up-down configuration) and tris-
cis-conformation (geometrically at the same side of the central benzene ring as an up-up-up configuration for all three fluorenyl rings) analogues, for the structure of tris(DPAF-C
9)
7 in spite of only one chromatographic spot being detected on TLC (silica gel, toluene–hexane, 5:95). This observation clearly suggests that the 3D-structure of
7 may not be a planar configuration with the fluorenyl and/or the phenyl rings being possibly twisted relative to the center core position, forming a pair of
cis/trans stereo-conformers or isolable isomers, as discussed in
Figure 6. In order to estimate the C-C bond rotational barrier energy of tris(DPAF-C
9) between possible stereo-conformers, we conducted the variable temperature NMR measurement in the range of 300–335 K in CDCl
3. However, we did not observe any clear difference except small peak broadening. This result suggested that the rotational barrier energy is high enough to restrict the
cis–
trans isomerization and allowed the isolation of a single isomer at least in room temperature.
As we described above, uncongested
1H-NMR spectrum of tris(DPAF-C
9)
7 revealed a
C3-symmetrical
cis-conformation for its structure. Our puzzle existed regarding to the exclusive formation of, supposedly, sterically unfavorable
cis-isomer. Accordingly, we undertook the comparison of formation energy by calculation on the 3D-structure of three plausible isomers of tris(DPAF-C
9)
7, as shown in
Figure 6. Two
cis-forms, especially a
cis-(
cup-)form, seem to be more crowded than
trans-(
chair-)form. Surprisingly, heat of formation (Δ
Hf) calculated by the semi-empirical PM3 method revealed unexpectedly high stability of the
cup-form with other forms in an order of
chair ≈
cup >
propeller for C
9 substituents (−9.19, −9.13, and −4.40 kcal/mol, respectively) and the highest stability of
cup-forms (
cup >
chair >
propeller) for the slightly less crowded
n-C
6, C
7, and C
8 substituents. Such
cup-form formation tends to be more preferable as the alkyl chains at 9-fluorenyl position become longer as can be seen in the differential heat of formation ΔΔ
Hf for both
cup–
propeller and
cup–
chair energy gaps (
Table 2). These results imply that hydrophobic interaction between three alkyl chains as well as CH/π interaction between the alkyl group and the fluorenyl ring plays an important role for the preferential formation of the
cup-form. The DFT calculation at B3LYP/6-31G* level of theory for some alkyl groups resulted in the similar tendency, although the values suggested the slightly more stable
chair-form even for
n-C
6 group as compared with the
cup-form, probably due to the lack of consideration on the solvent polarity which affects to hydrophobic interactions (
Table 3).
Figure 6.
Optimized geometrical structures of tris(DPAF-C9) 7, by calculation, showing a different degree of interaction force among terminal t-butyl groups (brown).
Figure 6.
Optimized geometrical structures of tris(DPAF-C9) 7, by calculation, showing a different degree of interaction force among terminal t-butyl groups (brown).
Table 2.
Calculated heat of formation for tris(DPAF-Cn) a.
Table 2.
Calculated heat of formation for tris(DPAF-Cn) a.
| R b | ΔHf (kcal/mol) | ΔΔHf (kcal/mol) |
|---|
| (Cn) | cis | trans | cup–propeller | cup–chair |
|---|
| | cup | propeller | chair | | (cis–trans) |
|---|
| Me | 258.38 | 253.67 | 247.85 | 4.71 | 10.53 |
| Et | 231.59 | 223.91 | 233.48 | 7.68 | 1.89 |
| n-C6 | 87.01 | 95.12 | 93.67 | −8.11 | −6.66 |
| C7 | 59.59 | 64.00 | 60.07 | −4.41 | −0.48 |
| C8 | 22.23 | 28.10 | 22.52 | −5.87 | −0.29 |
| C9 | −9.13 | −4.40 | −9.19 | −4.73 | 0.06 |
Table 3.
DFT calculation of heat of formation for tris(DPAF-Cn) a.
Table 3.
DFT calculation of heat of formation for tris(DPAF-Cn) a.
| R b | ΔHf (kcal/mol) | ΔΔHf (kcal/mol) |
|---|
| (Cn) | cis (cup) | trans (chair) | cup–chair |
|---|
| Et | −1922585.7 | −1922609.1 | 23.4 |
| MeOC2H4 | −2353750.4 | −2353762.0 | 11.6 |
| n-C6 | −2514673.7 | −2514677.3 | 3.6 |
| C9 | −2958697.2 | −2958703.8 | 6.6 |
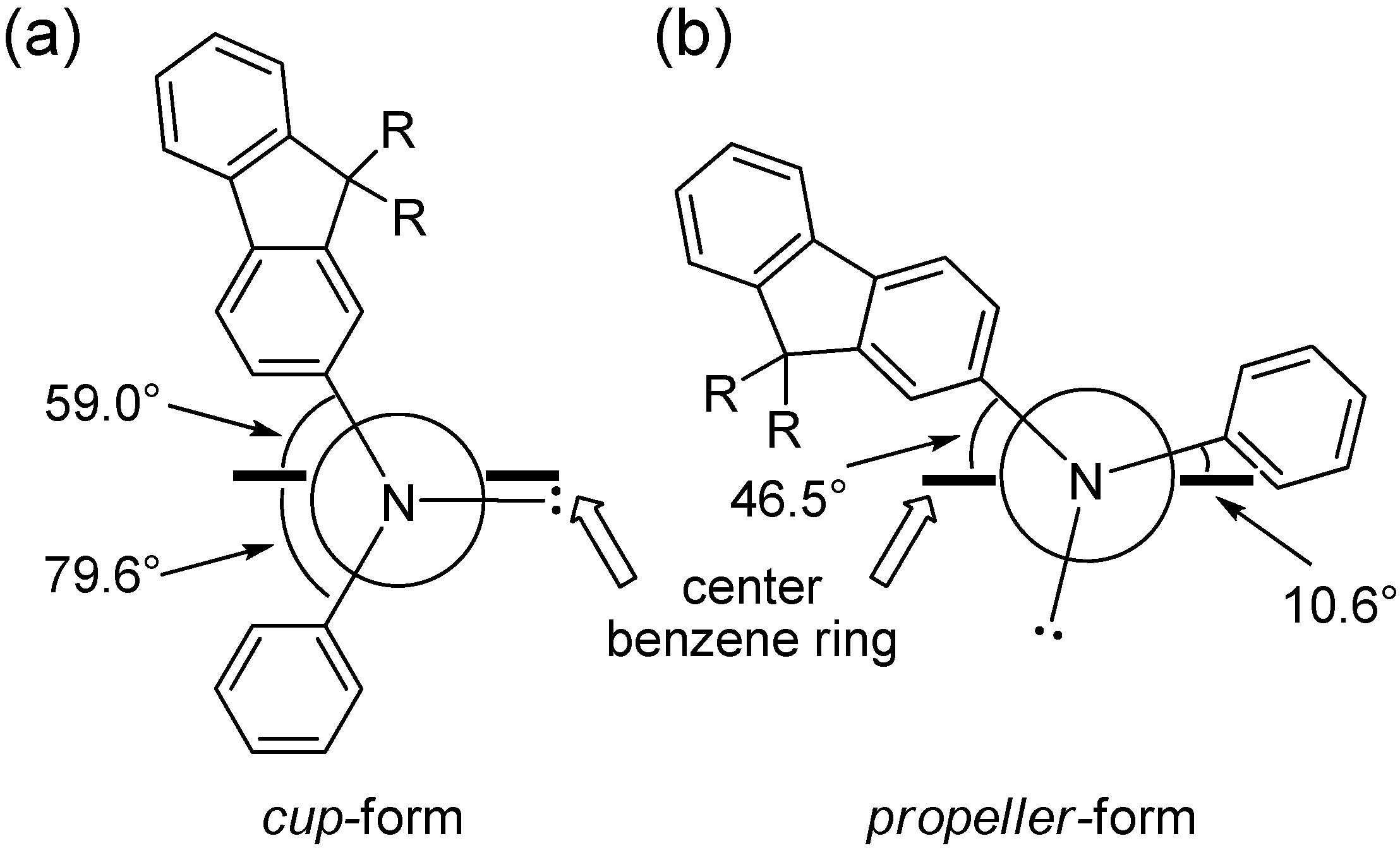
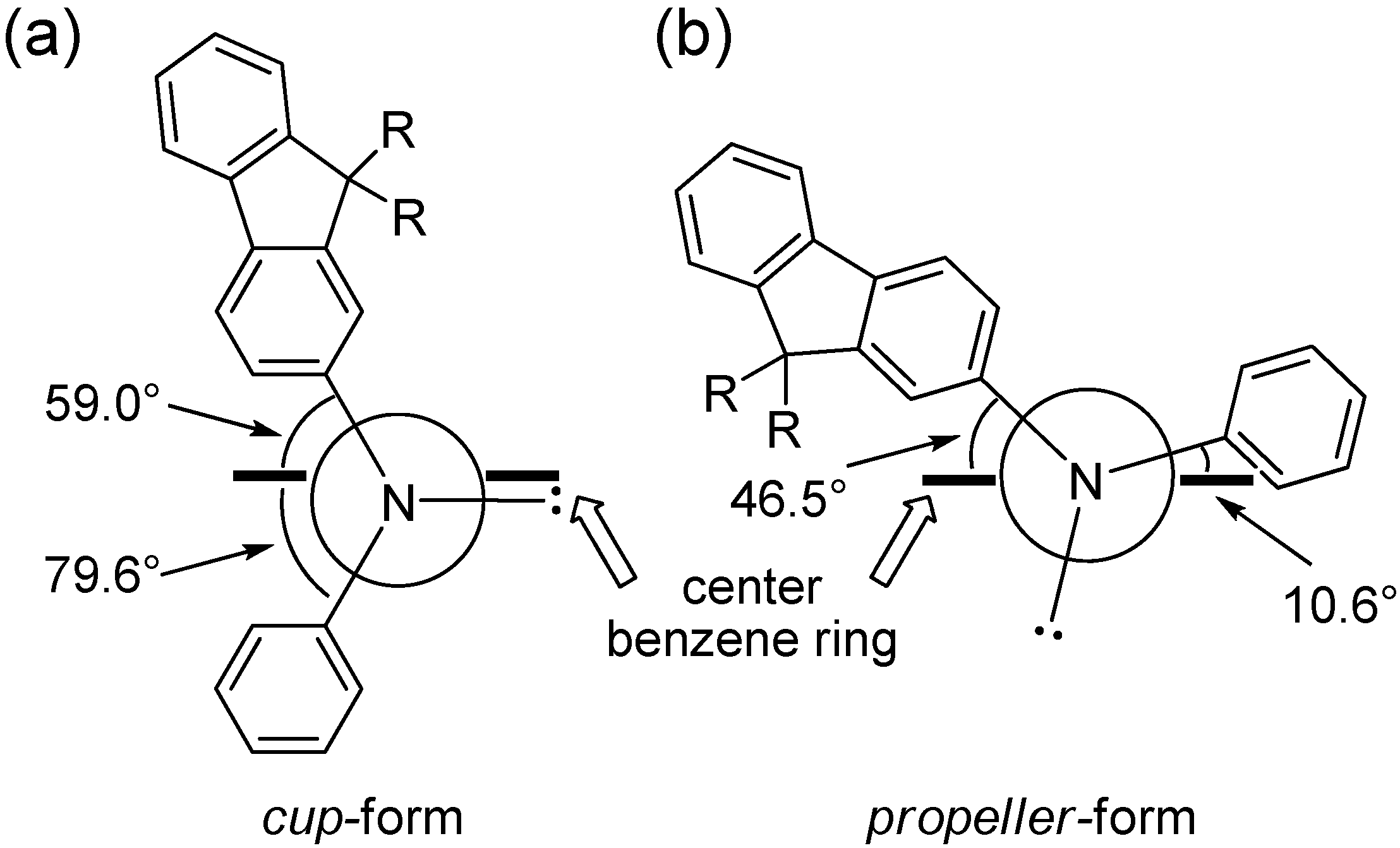
The unexpected stability of
cup-form may be explained by the torsional angle of the center benzene ring defined by the position of aryl groups on nitrogen atoms. The calculated torsional (dihedral) angles between the aryl–nitrogen C–N bond and the center benzene ring C–C bond for two
cis-conformers of tris(DPAF-C
9) (
cup-form and
propeller-form) obtained from the optimized 3D-structure are illustrated in
Figure 7. The angle strain of the
cup-form was found to be much less than that of the
propeller-form. The angle strain of
trans-conformers is somewhat higher than that of the symmetrical
cup-form due to unavoidable unsymmetrical conformation of the
chair-form. Therefore, the highly symmetrical
cis-
cup-isomer of tris(DPAF-C
9) can be formed exclusively.
Figure 7.
Torsional (dihedral) angles between aryl–nitrogen bond and center benzene ring for (a) cup-form and (b) propeller-form of tris(DPAF-C9) 7.
Figure 7.
Torsional (dihedral) angles between aryl–nitrogen bond and center benzene ring for (a) cup-form and (b) propeller-form of tris(DPAF-C9) 7.
In summary, above explanations for the preferential cis-cup-formation are supported by results of the calculated heat of formation and dihedral angle, which are based on the thermodynamically controlled mechanism. However, the restricted bond rotation may not give the thermodynamically stable isomer under the present condition. Therefore, an alternative explanation for the preferential cis-cup-formation of tris(DPAF-C9) is also considered based on the kinetically controlled mechanism in the critical amination reaction. Fluorenyl substitution at three phenylamino groups on the central benzene ring of TPAB occurs one by one to form mono(DPAF-C9), bis(DPAF-C9), and tris(DPAF-C9) sequentially. Owing to the unrestricted rotation of phenylamino group, two phenyl groups on nitrogen atoms can be escaped from a larger fluorenyl substituent group via the steric hindrance. Consequently, the tendency of subsequent substitution of a second fluorenyl group should favor the formation of cis-bis(DPAF-C9). This preferred configuration may hinder its isomerization to the trans-form due to the restricted bond rotation. Similarly, the third substitution of the last fluorenyl group should lead to the selective product of cis-tris(DPAF-C9) (supporting information).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}