
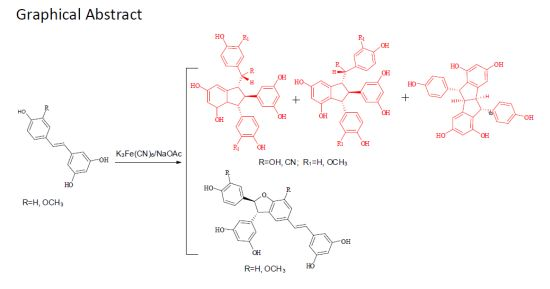
Potassium Hexacyanoferrate (III)-Catalyzed Dimerization of Hydroxystilbene: Biomimetic Synthesis of Indane Stilbene Dimers

 ,
,
Abstract
:
1. Introduction
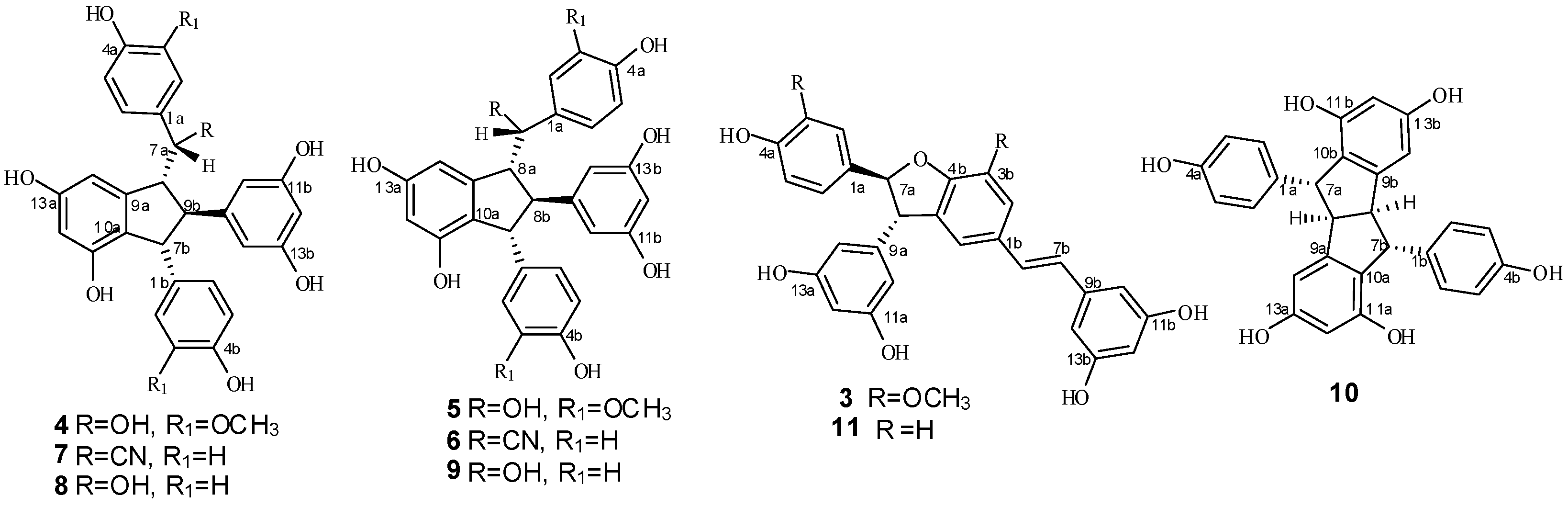
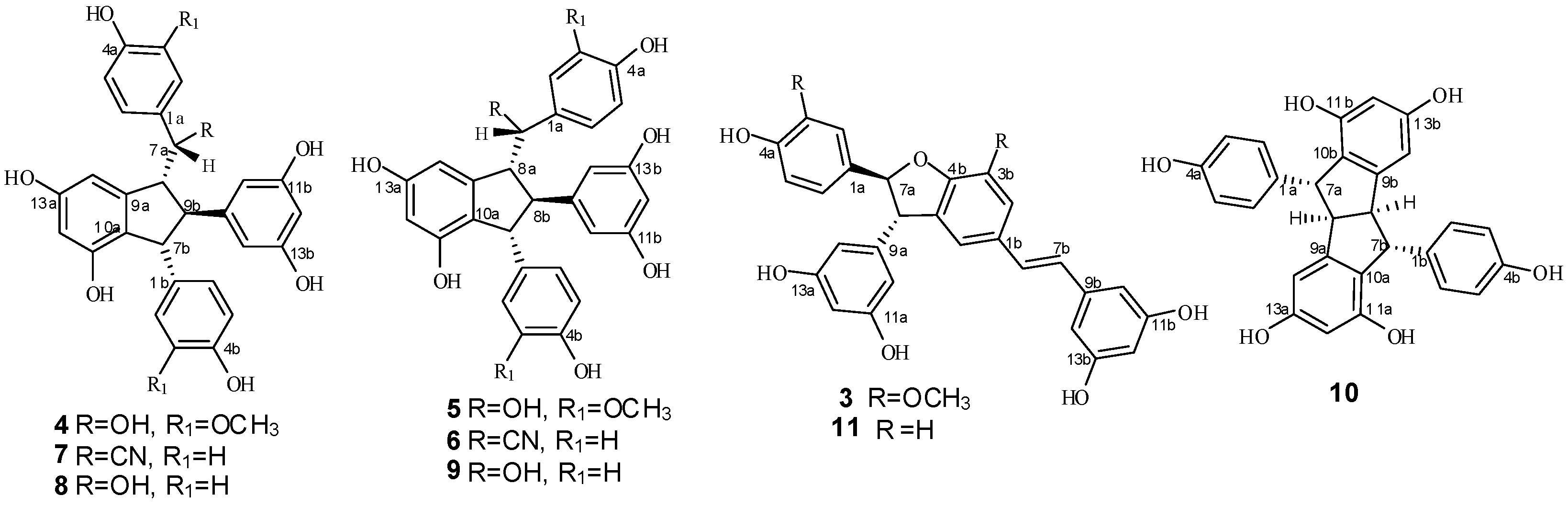
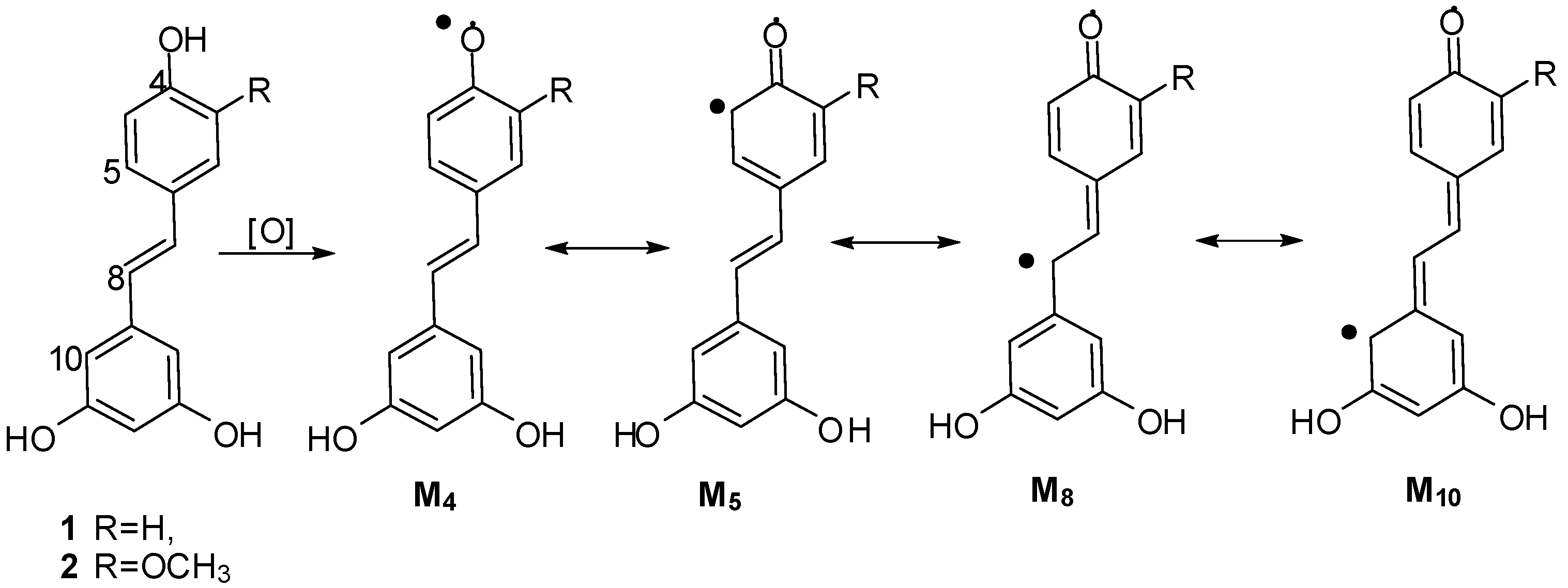
2. Results and Discussion
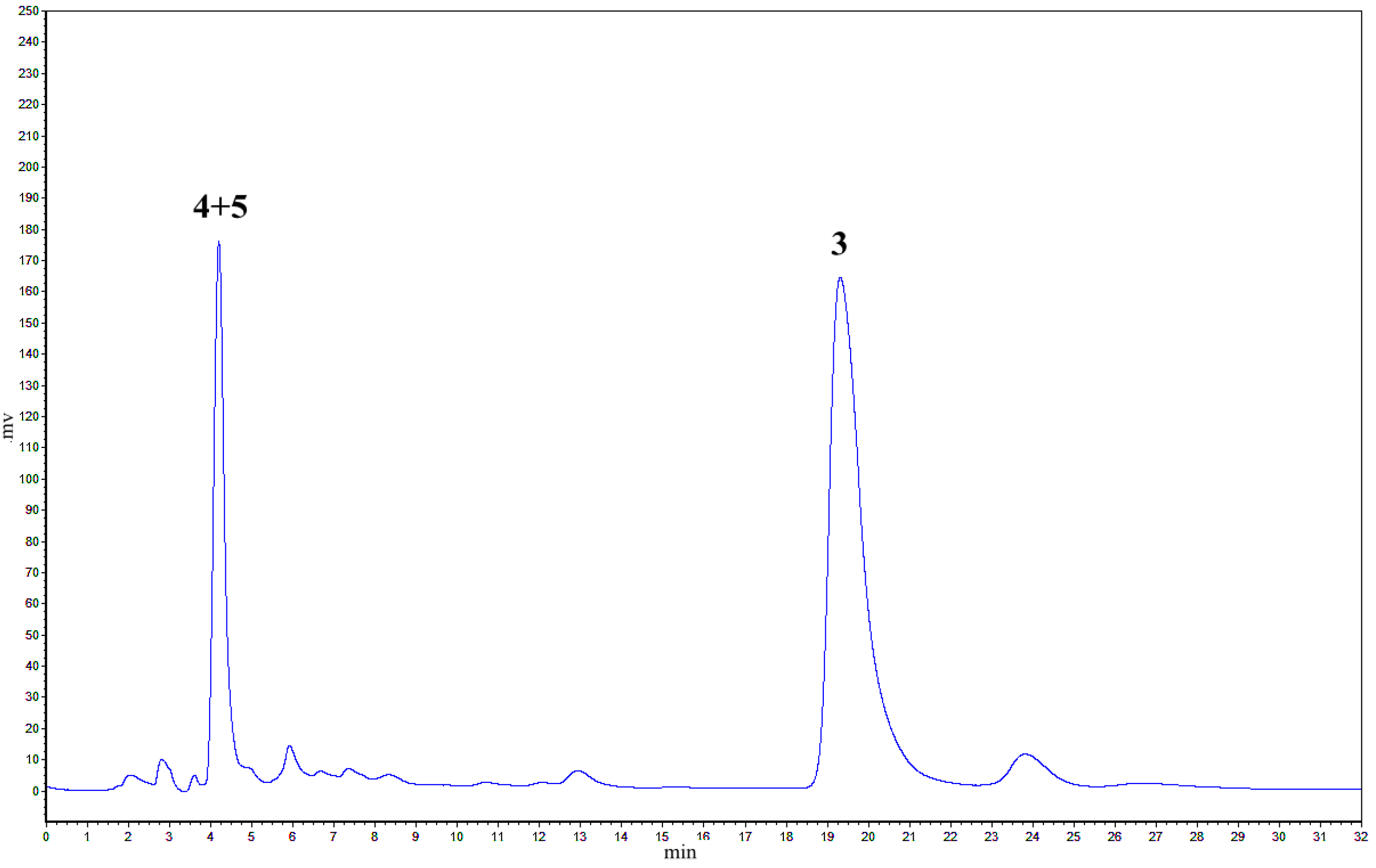
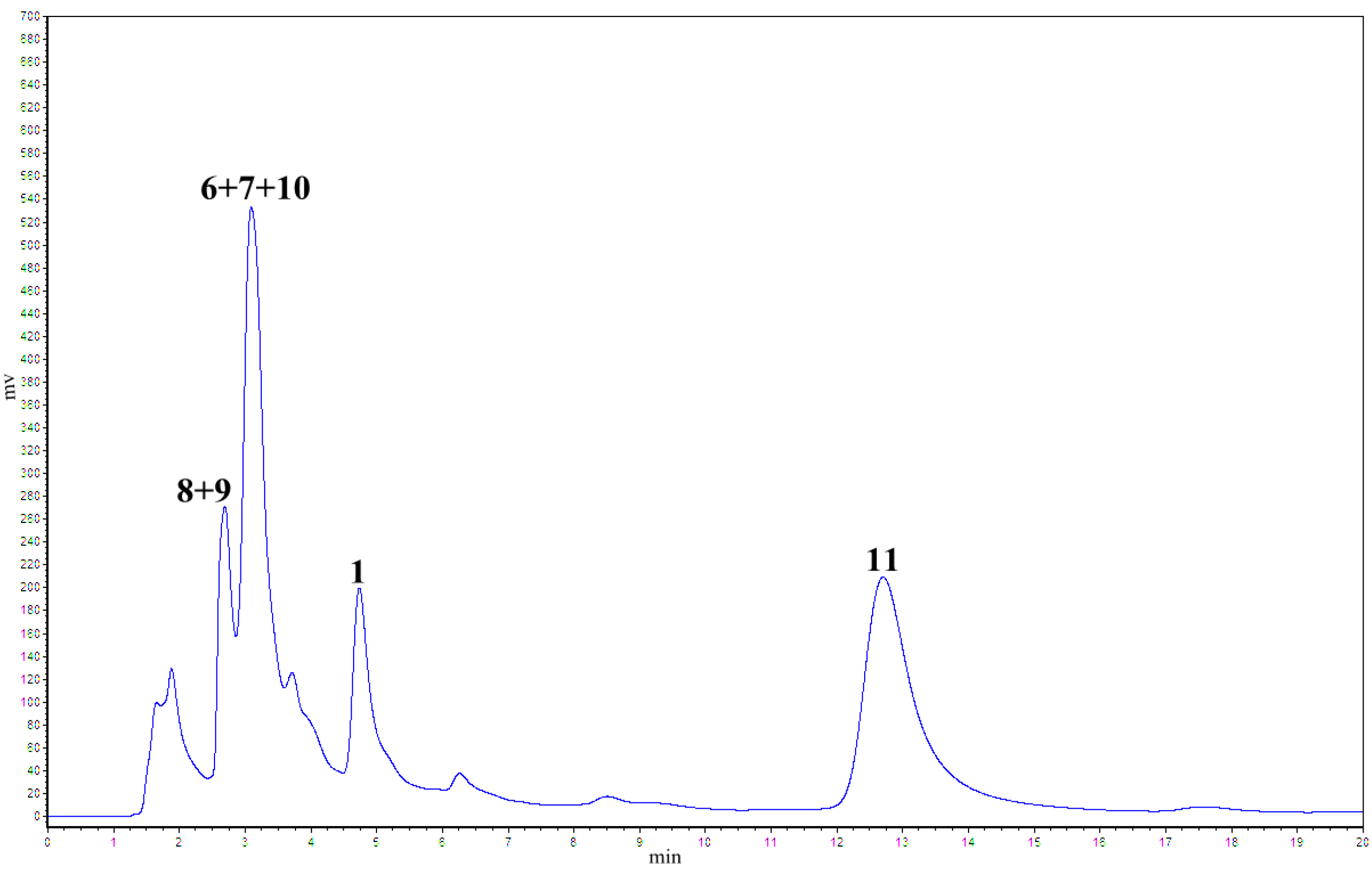
2.1. Treatment of 2 with Potassium Hexacyanoferrate (III)/Sodium Acetate
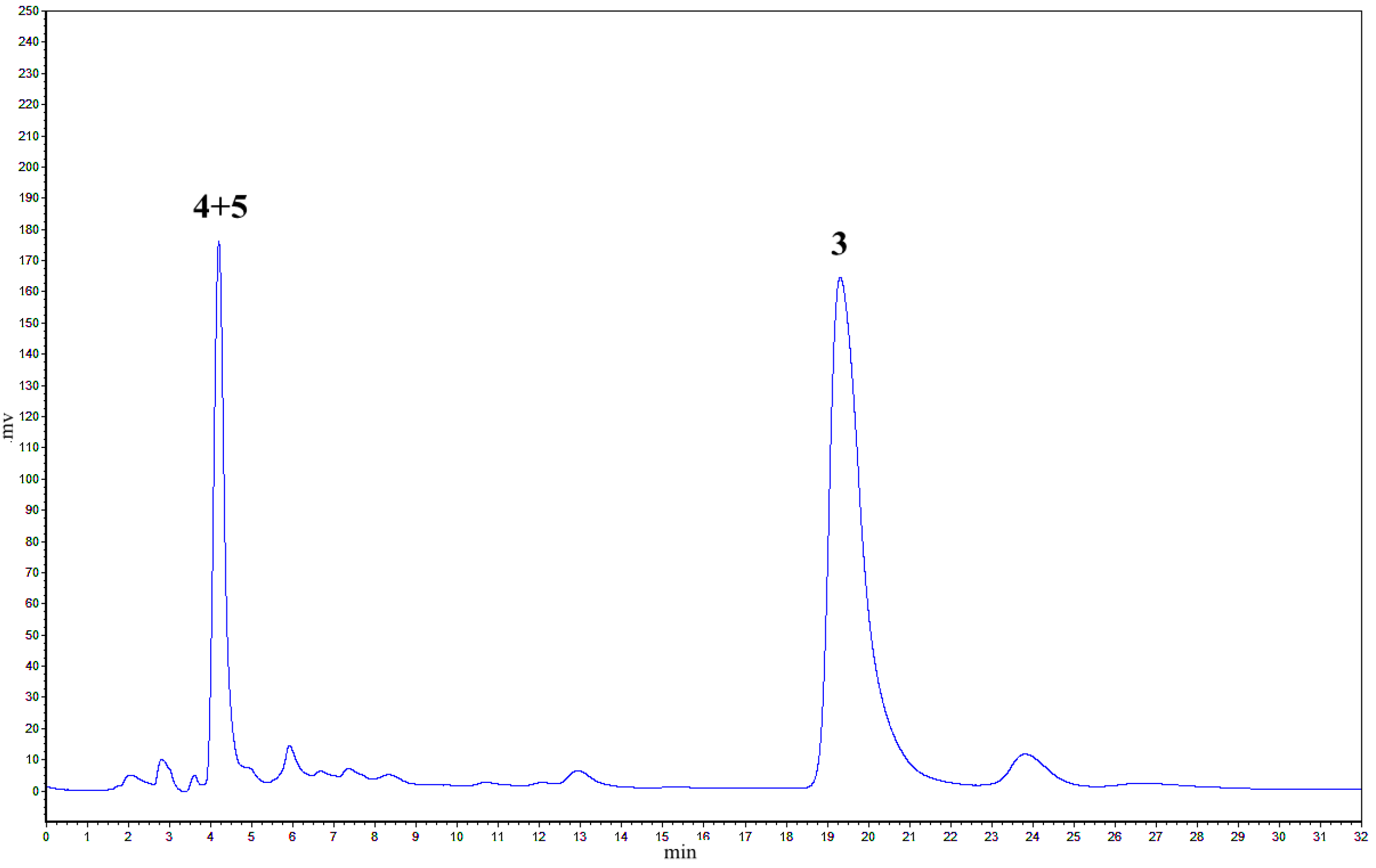
2.2. Treatment of 1 with Potassium Hexacyanoferrate (III)/Sodium Acetate

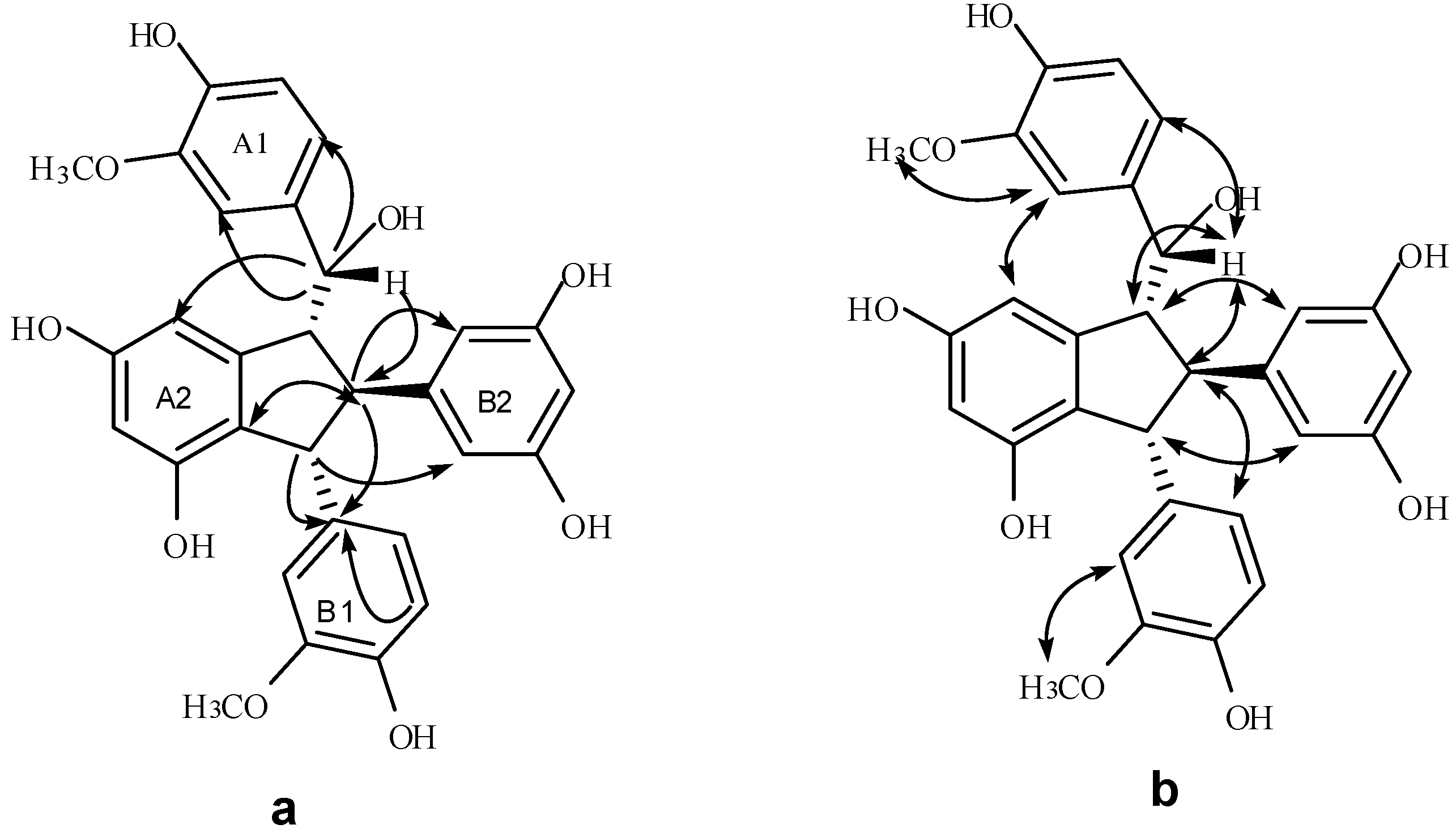
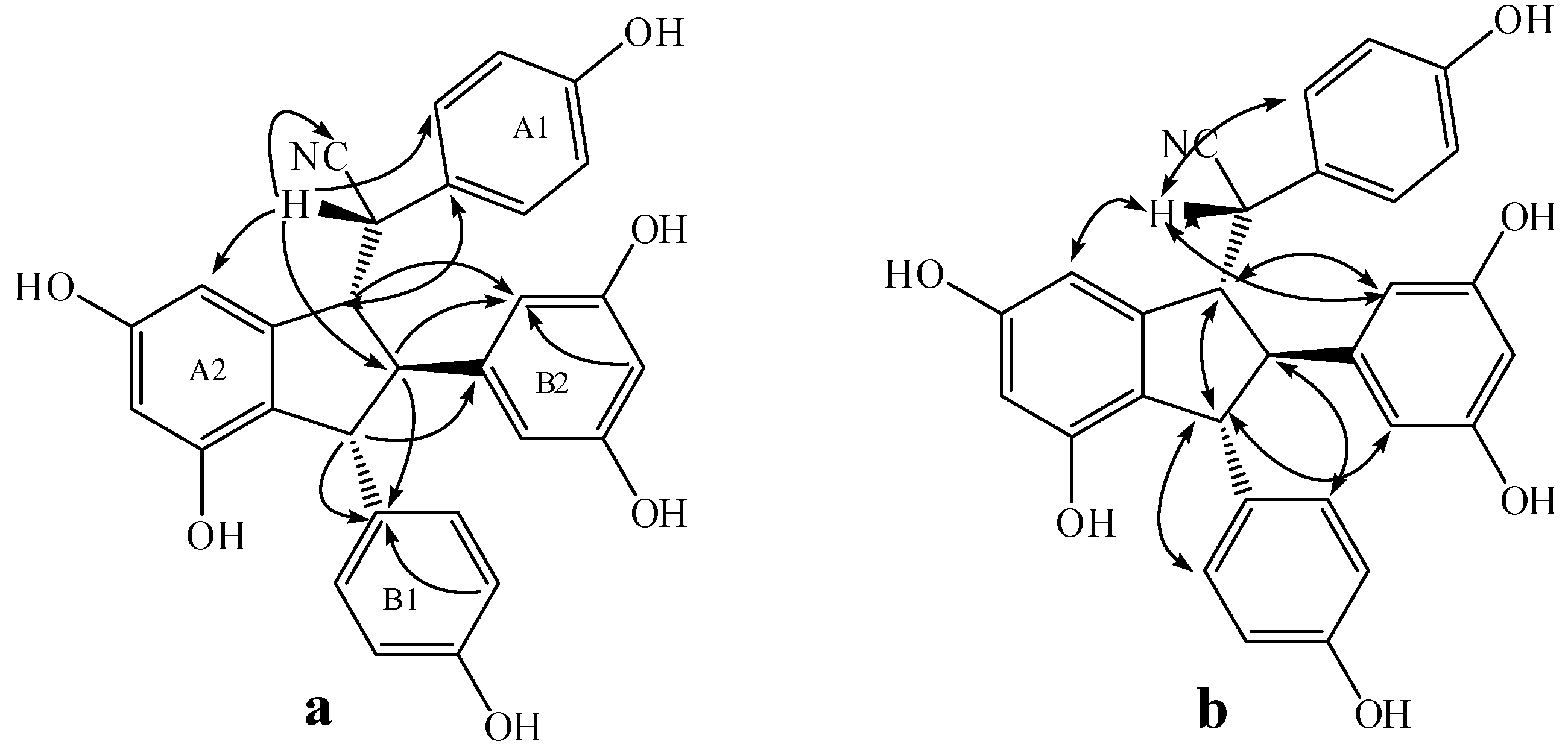
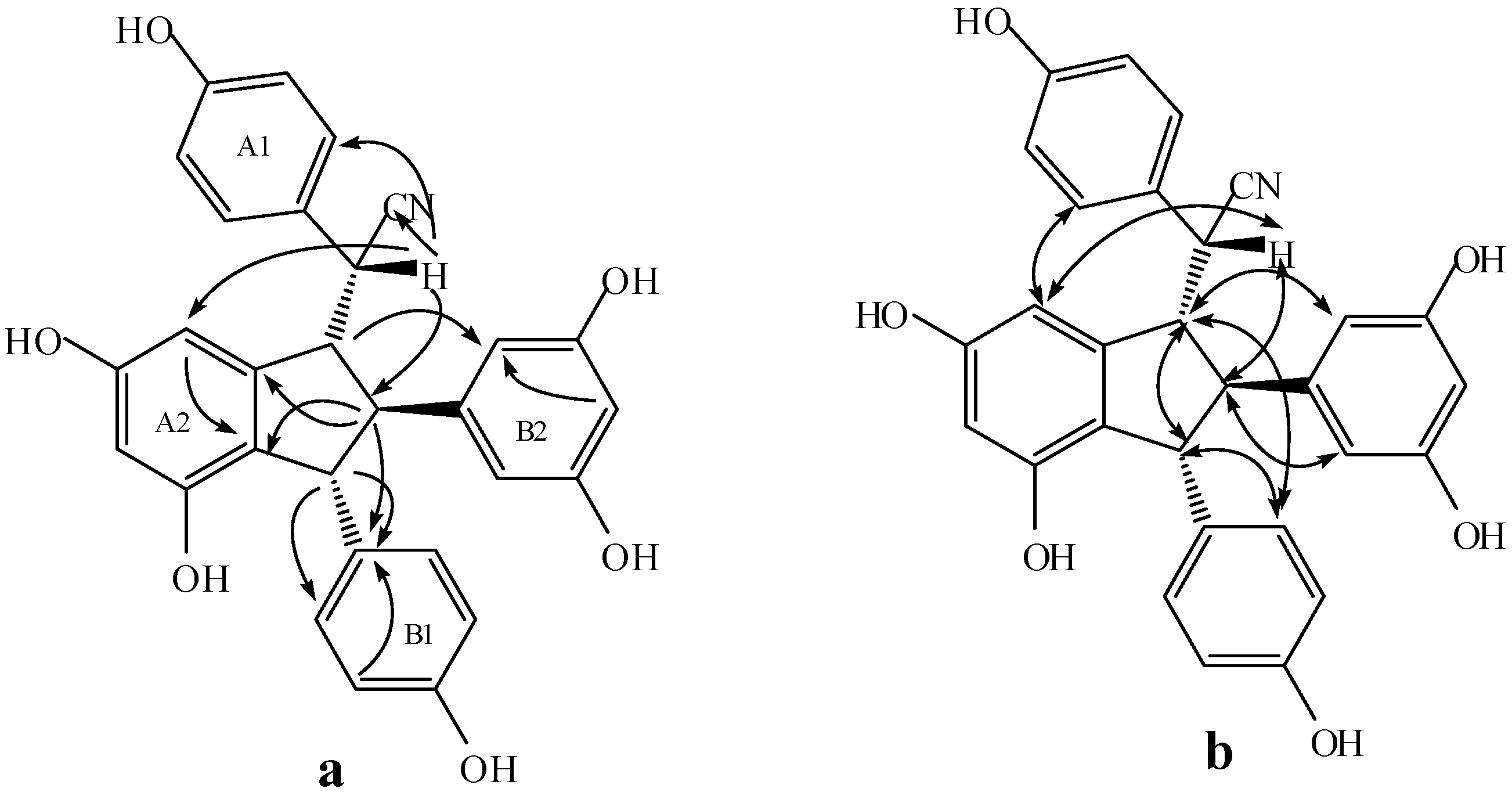
2.3. Structural Identification of New Dimers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 4 | 5 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 1a | 135.5s | 136.5s | ||
| 2a | 112.4d | 6.82 (d, 1.8) | 112.4d | 6.37 (d, 1.8) |
| 3a | 148.5s | 148.7d | ||
| 4a | 146.9s | 146.7s | ||
| 5a | 115.3d | 6.61 (d, 7.8) | 115.74d | 6.59 (d, 7.8), |
| 6a | 121.1d | 6.57 (dd, 7.8, 1.8) | 120.94d | 6.39 (dd, 7.8, 1.8) |
| 7a | 78.0d | 4.48 (d, 7.2) | 78.7d | 4.35 (d, 9.0) |
| 8a | 61.8d | 3.49 (t, 6.0) | 62.0d | 3.29 (overlap) |
| 9a | 148.7s | 149.5s | ||
| 10a | 123.9s | 123.0s | ||
| 11a | 155.4s | 155.1s | ||
| 12a | 102.6 | 6.08 (d, 1.8) | 102.6d | 6.19 (d, 1.8) |
| 13a | 158.8s | 159.2s | ||
| 14a | 106.0d | 6.07 (br s) | 106.30d | 6.58 (d, 1.8) |
| 1b | 139.0s | 138.8s | ||
| 2b | 112.0d | 6.37 (d, 1.8) | 111.9d | 6.49 (d, 1.8) |
| 3b | 147.4s | 148.5s | ||
| 4b | 145.5s | 145.6s | ||
| 5b | 115.6d | 6.55 (d, 8.4) | 115.69d | 6.62 (d, 7.8) |
| 6b | 120.0d | 6.30 (dd, 8.4, 1.8) | 120.93d | 6.35 (dd, 7.8, 1.8) |
| 7b | 57.4d | 4.08 (d, 4.8) | 56.4d | 4.14 (d, 3.0) |
| 8b | 60.0d | 3.17 (t, 5.4) | 60.2d | 2.74 (t, 3.0) |
| 9b | 150.7s | 151.3s | ||
| 10(14)b | 106.9d | 6.03 (d, 2.4) | 106.33d | 5.75 (d, 2.4) |
| 11(13)b | 159.4s | 159.3s | ||
| 12b | 101.3d | 6.02 (t, 2.4) | 101.2d | 5.96 (t, 2.4) |
| 3a-OCH3 | 56.3q | 3.61 (s) | 56.3q | 3.58 (s) |
| 3b-OCH3 | 56.2q | 3.65 (s) | 56.1q | 3.67 (s) |

| Position | 6 | 7 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 1a | 125.89s | 125.85s | ||
| 2(6)a | 129.31d | 6.89 (2H, d, 8.5) | 130.72d | 7.06 (2H, d, 8.5) |
| 3(5)a | 115.55d | 6.79 (2H, d, 8.5) | 116.09d | 6.74 (2H, d, 8.5) |
| 4a | 157.12s | 158.03s | ||
| 7a | 41.16d | 3.76 (1H, d, 8.5) | 42.09d | 3.96 (1H, d, 8.1) |
| 8a | 59.35d | 3.01 (1H, t, 4.0) | 61.31d | 3.26 (1H, t, 4.5) |
| 9a | 147.44s | 148.66s | ||
| 10a | 121.28s | 122.54s | ||
| 11a | 154.59s | 155.23s | ||
| 12a | 102.39d | 6.38 (1H, d, 2.0) | 103.18d | 6.30 (1H, d, 1.6) |
| 13a | 158.49s | 159.02s | ||
| 14a | 103.90d | 6.57 (1H, d, 2.0) | 104.72d | 5.95 (1H, brs) |
| 1b | 135.50s | 136.82s | ||
| 2(6)b | 128.40d | 6. 87 (2H, d, 8.5) | 129.21d | 6.84 (2H, d, 8.5) |
| 3(5)b | 114.97d | 6.76 (2H, d, 8.5) | 115.81d | 6.71 (2H, d, 8.5) |
| 4b | 155.72s | 156.51s | ||
| 7b | 53.84d | 4.28 (1H, d, 4.0) | 55.89d | 4.27 (1H, d, 4.5) |
| 8b | 56.70d | 3.52 (1H, dd, 8.5, 4.0) | 58.07d | 3.58 (1H, dd, 8.1, 4.5) |
| 9b | 145.39s | 145.53s | ||
| 10(14)b | 105.13d | 5.96 (2H, d, 2.0) | 106.10d | 6.07 (2H, d, 2.0) |
| 11(13)b | 158.49s | 159.31s | ||
| 12b | 100.75d | 6.17 (1H, t, 2.0) | 101.42d | 6.15 (1H, t, 2.0) |
| CN | 120.33s | 121.73s | ||

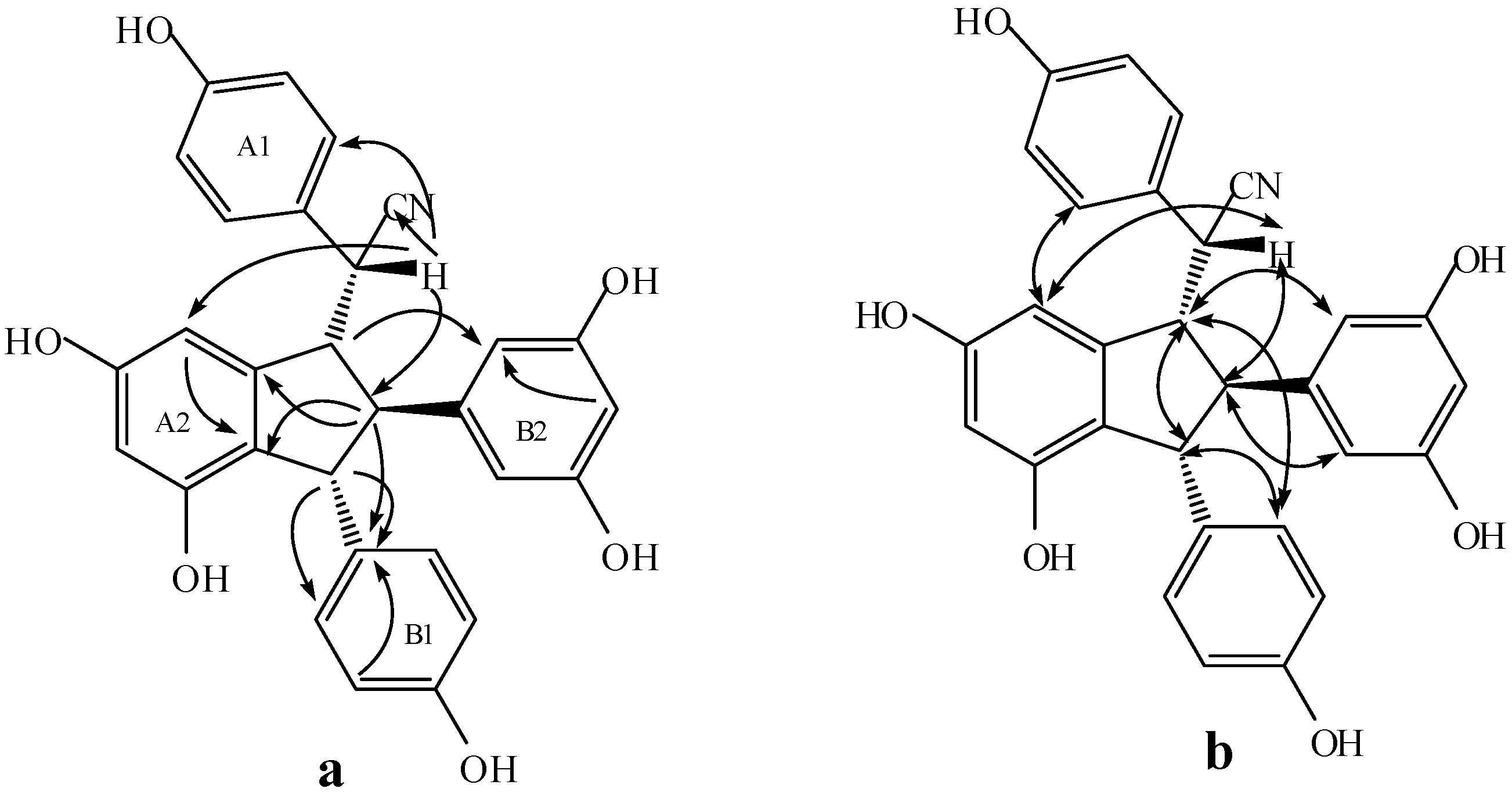
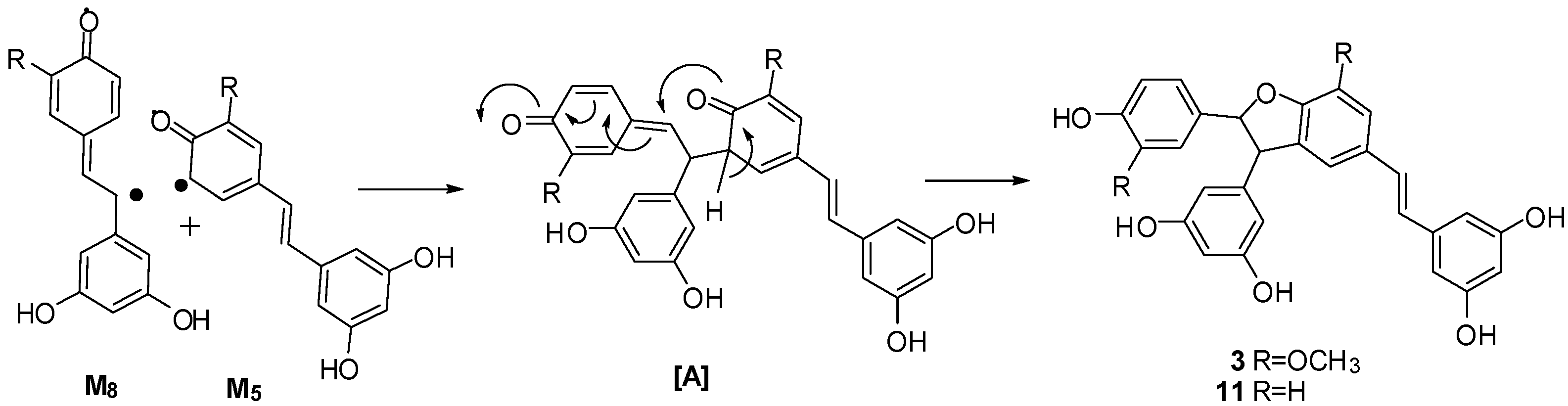
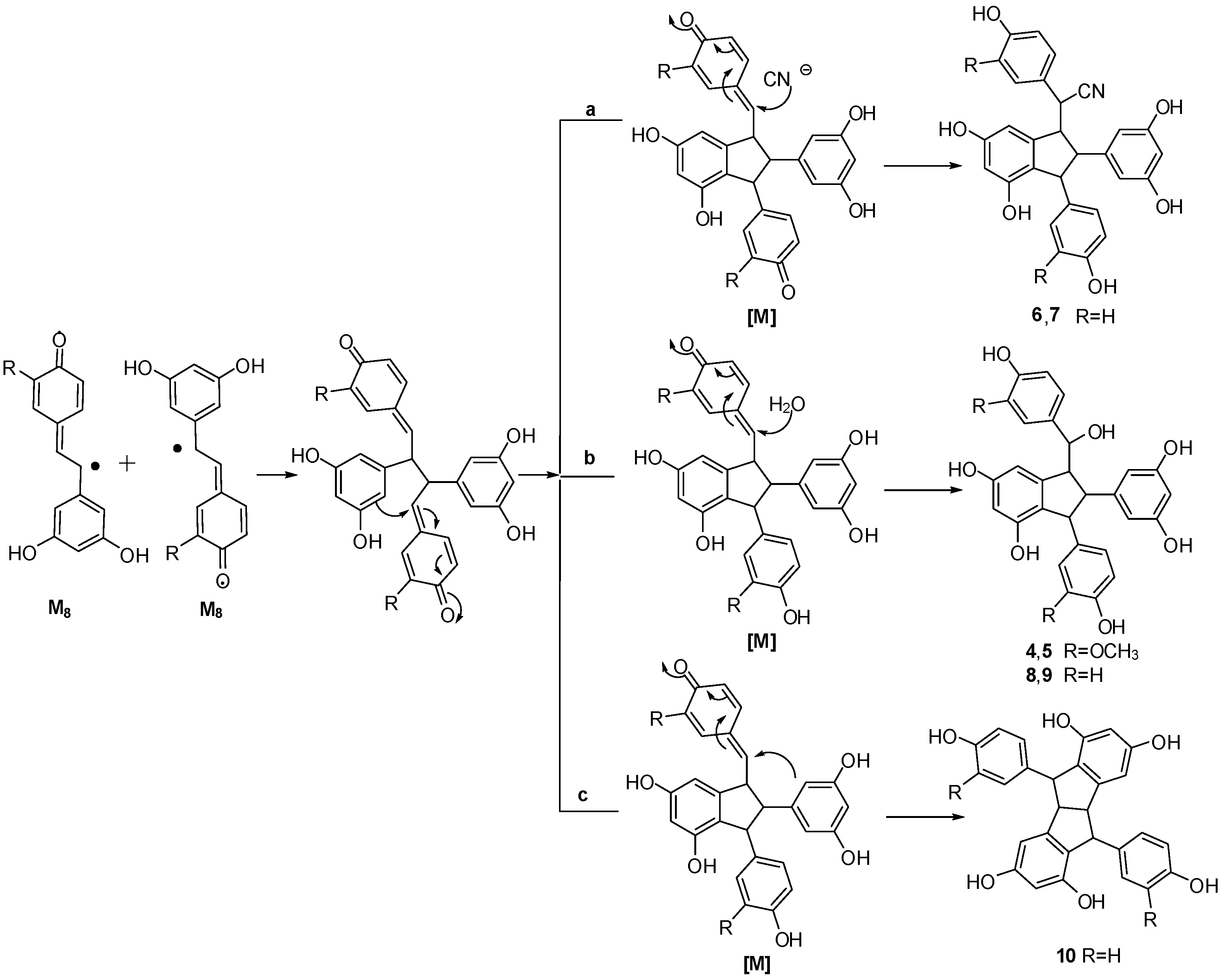
2.4. Discussion of the Probable Coupling Reaction Mechanism

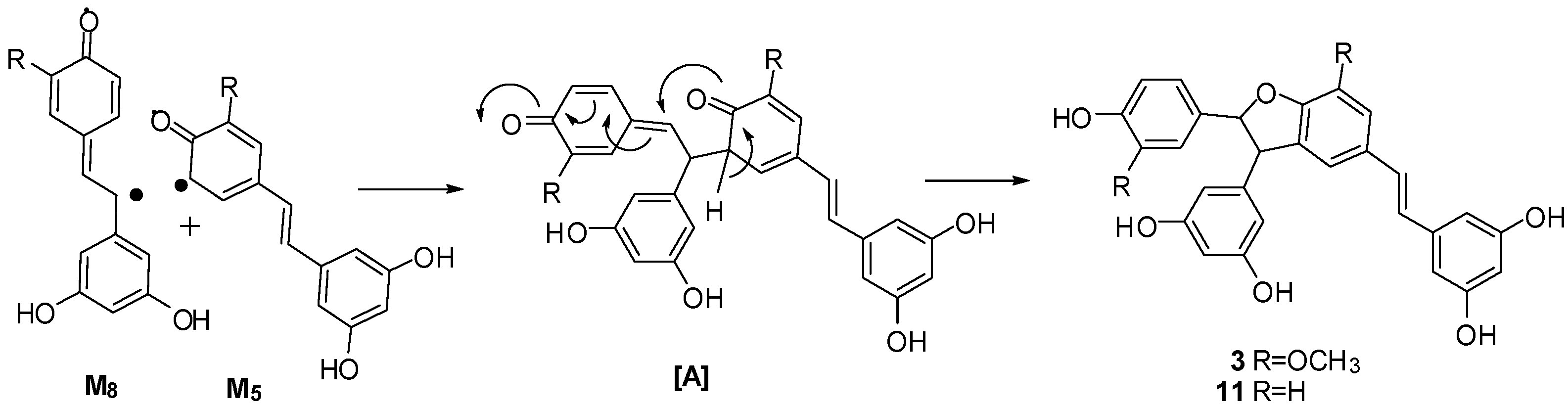
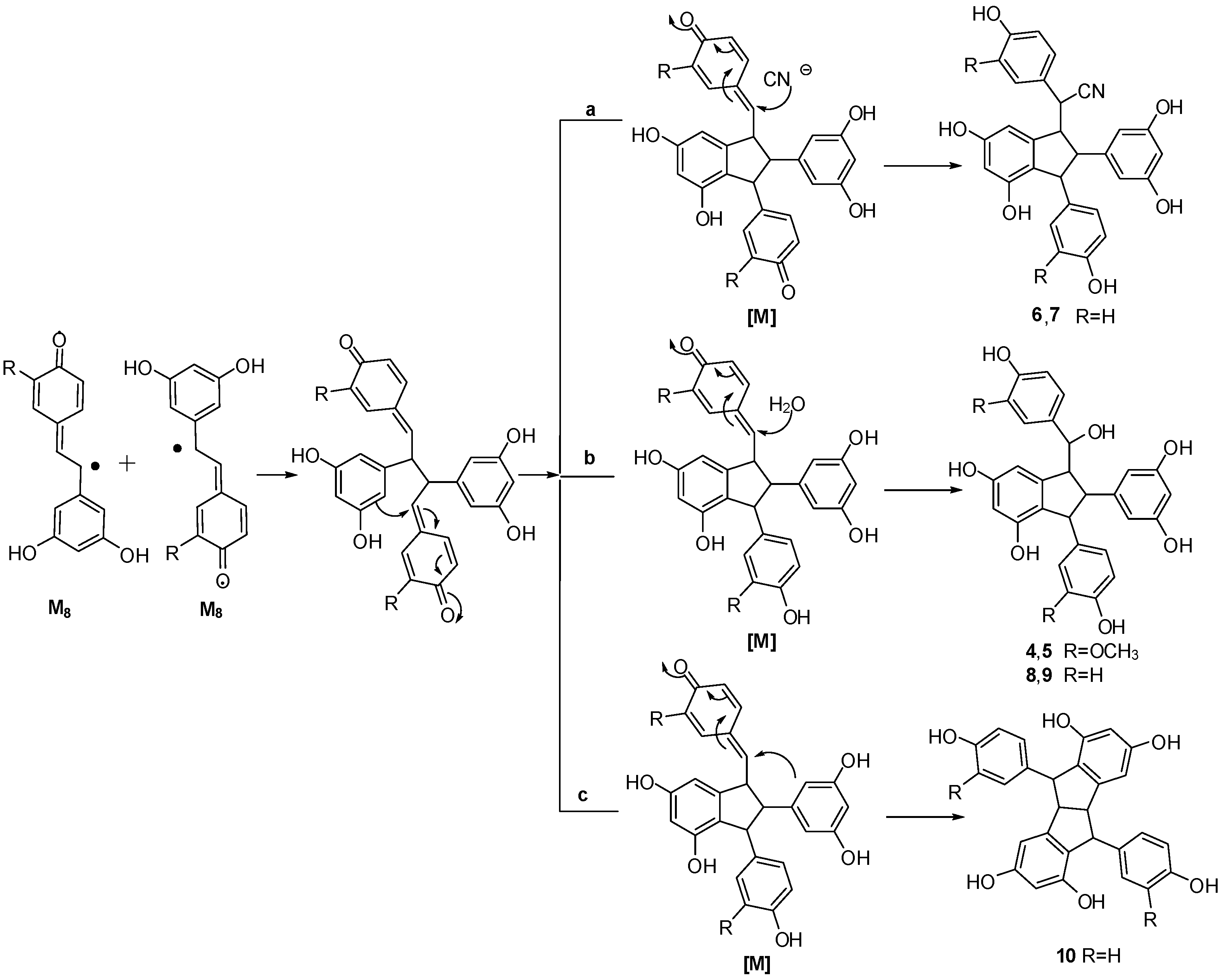
3. Experimental Section
3.1. Materials and Instrumentation
3.2. Treatment of Isorhapontigenin with Potassium Hexacyanoferrate (III)/Sodium Acetate.
3.3. Treatment of Resveratrol with Potassium Hexacyanoferrate (III)/Sodium Acetate
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yao, C.S.; Wang, X.F. Naturally Active Oligostilbenes. J. Asian Nat. Prod. Res. 2016, in press. [Google Scholar]
- Lin, M.; Yao, C.S. Natural oligostilbenes. Stud. Nat. Prod. Chem. 2006, 33, 601–644. [Google Scholar]
- Li, W.L.; Li, H.F.; Luo, Y.L.; Yang, Y.H.; Wang, N. Biosynthesis of resveratrol dimers by regioselective oxidative coupling reaction. Synlett 2010, 8, 1247–1250. [Google Scholar] [CrossRef]
- Snyder, S.A.; Zografos, A.L.; Lin, Y.Q. Total synthesis of resveratrol-based natural products: A chemoselective solution. Angew. Chem. Int. Ed. 2007, 46, 8186–8191. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.A.; Breazzano, S.P.; Ross, A.G.; Lin, Y.Q.; Zografos, A.L. Total synthesis of diverse carbogenic complexity within the resveratrol class from a common building block. J. Am. Chem. Soc. 2009, 131, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhong, C.; Lu, H.F.; Li, G.Y.; Sun, X. Toward the synthesis of caraphenol C: Substituted effect on the nazarov cyclyzation of 2-arylchalcones. Synlett 2008, 3, 458–462. [Google Scholar]
- Zhong, C.; Zhu, J.; Chang, J.; Sun, X. Concise total synthesis of (±)-isopaucifloral F, (±)-qaudranglarin A and (±)-pallidol. Tetrahedron Lett. 2011, 52, 2815–2817. [Google Scholar] [CrossRef]
- Li, W.L.; Li, H.; Li, Y.; Hou, Z.J. Total synthesis of (±)-quadrangularin A. Angew. Chem. Int. Ed. 2006, 45, 7609–7611. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.A.; Gollner, A.; Chiriac, M.I. Regioselective reaction for programmable resveratrol oligomer synthesis. Nature 2011, 474, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Velu, S.S.; Buniyamin, I.; Ching, L.K.; Feroz, F.; Noorbatcha, I.; Gee, L.C.; Awang, K.; Wahab, I.A.; Weber, J.F.F. Regio- and stereoselective biomimetic synthesis of oligostilbenoid dimers from resveratrol analogues: Influence of the solvent, oxidant, and substitution. Chem. -Eur. J. 2008, 14, 11376–11384. [Google Scholar] [CrossRef] [PubMed]
- Takaya, Y.; Terashima, K.; Ito, J.; He, Y.H.; Tateoka, M.; Yamaguchi, N.; Niwa, M. Biomimic transformation of resveratrol. Tetrahedron 2005, 61, 10285–10290. [Google Scholar] [CrossRef]
- Sako, M.; Hosokawa, H.; Ito, T.; Iinuma, M. Regioselective oxidative coupling of 4-hydroxystilbenes: synthesis of resveratrol and ε-viniferin (E)-dehydrodimers. J. Org. Chem. 2004, 69, 2598–2600. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Zhang, Y.; Lin, M.B.; Hou, Q.; Yao, C.S.; Shi, J.G. Biomimetic synthesis of active isorhapontigenin dimers. J. Asian Nat. Prod. Res. 2014, 16, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.F.; Hou, Q.; Lin, M.B.; Yao, C.S.; Shi, J.G. Preparation of active resveratrol dimeric derivatives by oxidative coupling reaction using AgOAc as oxidant. Chin. J. Org. Chem. 2014, 34, 886–892. [Google Scholar] [CrossRef]
- Yao, C.S.; Lin, M.; Yang, Q.Y. Preparation of active gnetol dimers by oxidative coupling reaction and acid-catalyzed dimerization. Chin. J. Org. Chem. 2013, 33, 312–318. [Google Scholar] [CrossRef]
- Yao, C.S.; Zhou, L.X.; Lin, M. Preparation on oligostilbenes of isorhapontigenin by oxidative coupling reaction. Chem. Pharm. Bull. 2004, 52, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.S.; Wang, Y.H.; Li, R.L.; Lin, M. Stilbene dimers from the lianas of Gnetum hainanense. Phytochemistry 2000, 54, 875–881. [Google Scholar] [CrossRef]
- Kim, H.J.; Saleem, M.; Seo, S.H.; Jin, C.; Lee, Y.S. Two new antioxidant stilbene dimers, parthenostilbenins A and B from Parthenocissus tricuspidata. Planta Med. 2005, 71, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, K.; Shimogai, N.; Takaishi, Y.; Rao, S.; Imakura, Y. Four stilbenes from Salacia lehmbachii. Phytochemistry 1997, 44, 1569–1573. [Google Scholar] [CrossRef]
- Zhou, L.X.; Lin, M. A new stilbene dimer-shegansu B from Belamcanda chinensis. J. Asian Nat. Prod. Res. 2000, 2, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, M.; Tanaka, T.; Iinuma, M. Five resveratrol oligomers from roots of Sophora Leachiana. Phytochemistry 1995, 38, 733–740. [Google Scholar] [CrossRef]
- Khan, M.; Nabi, S.G.; Prakashi, S.; Zaman, A. Pallidol, a resveratrol dimer from Cissus pallida. Phytochemistry 1986, 25, 1945–1948. [Google Scholar] [CrossRef]
- Roger, P.; Camille, P.; Junlien, B.J.; Raffaele, T.; Kartia, G.; Olivier, V. δ-viniferin, a resveratrol dehydrodimer: One of the major stilbenes synthesized by stressed grapevine leaves. J. Agric. Food Chem. 2003, 51, 5488–5492. [Google Scholar]
- Pilar, R.B.; Lorena, M.C.; Jose, M.L.; Francisco, G.C. Kinetic mechanism and product characterization of the enzymatic peroxidation of pterostilbene as model of the detoxification process of stilbene-type phytoalexins. Phytochemistry 2011, 72, 100–108. [Google Scholar]
- Ponzoni, C.; Beneventi, E.; Cramarossa, M.R.; Raimondi, S.; Trevisi, G.; Pagnoni, U.M.; Riva, S.; Forti, L. Laccase-catalyzed dimerization of hydroxystilbenes. Adv. Synth. Catal. 2007, 349, 1497–1506. [Google Scholar] [CrossRef]
- Wan, X.; Wang, X.B.; Yang, M.H.; Wang, J.S.; Kong, L.Y. Dimerization of piceatannol by momordica charantia peroxidase and α-glucosidase inhibitory activity of the biotransformation products. Bioorg. Med. Chem. 2011, 19, 5085–5092. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, J.-S.; Wen, J.; Wang, X.-F.; Zhang, J.-Q.; Zhang, J.-F.; Kang, Y.-L.; Hui, Y.-W.; Zheng, W.-S.; Yao, C.-S. Potassium Hexacyanoferrate (III)-Catalyzed Dimerization of Hydroxystilbene: Biomimetic Synthesis of Indane Stilbene Dimers. Molecules 2015, 20, 22662-22673. https://doi.org/10.3390/molecules201219872
Xie J-S, Wen J, Wang X-F, Zhang J-Q, Zhang J-F, Kang Y-L, Hui Y-W, Zheng W-S, Yao C-S. Potassium Hexacyanoferrate (III)-Catalyzed Dimerization of Hydroxystilbene: Biomimetic Synthesis of Indane Stilbene Dimers. Molecules. 2015; 20(12):22662-22673. https://doi.org/10.3390/molecules201219872
Chicago/Turabian StyleXie, Jing-Shan, Jin Wen, Xian-Fen Wang, Jian-Qiao Zhang, Ji-Fa Zhang, Yu-Long Kang, You-Wei Hui, Wen-Sheng Zheng, and Chun-Suo Yao. 2015. "Potassium Hexacyanoferrate (III)-Catalyzed Dimerization of Hydroxystilbene: Biomimetic Synthesis of Indane Stilbene Dimers" Molecules 20, no. 12: 22662-22673. https://doi.org/10.3390/molecules201219872