
The Antileishmanial Potential of C-3 Functionalized Isobenzofuranones against Leishmania (Leishmania) Infantum Chagasi

 , , and
, , and
Abstract
:
1. Introduction
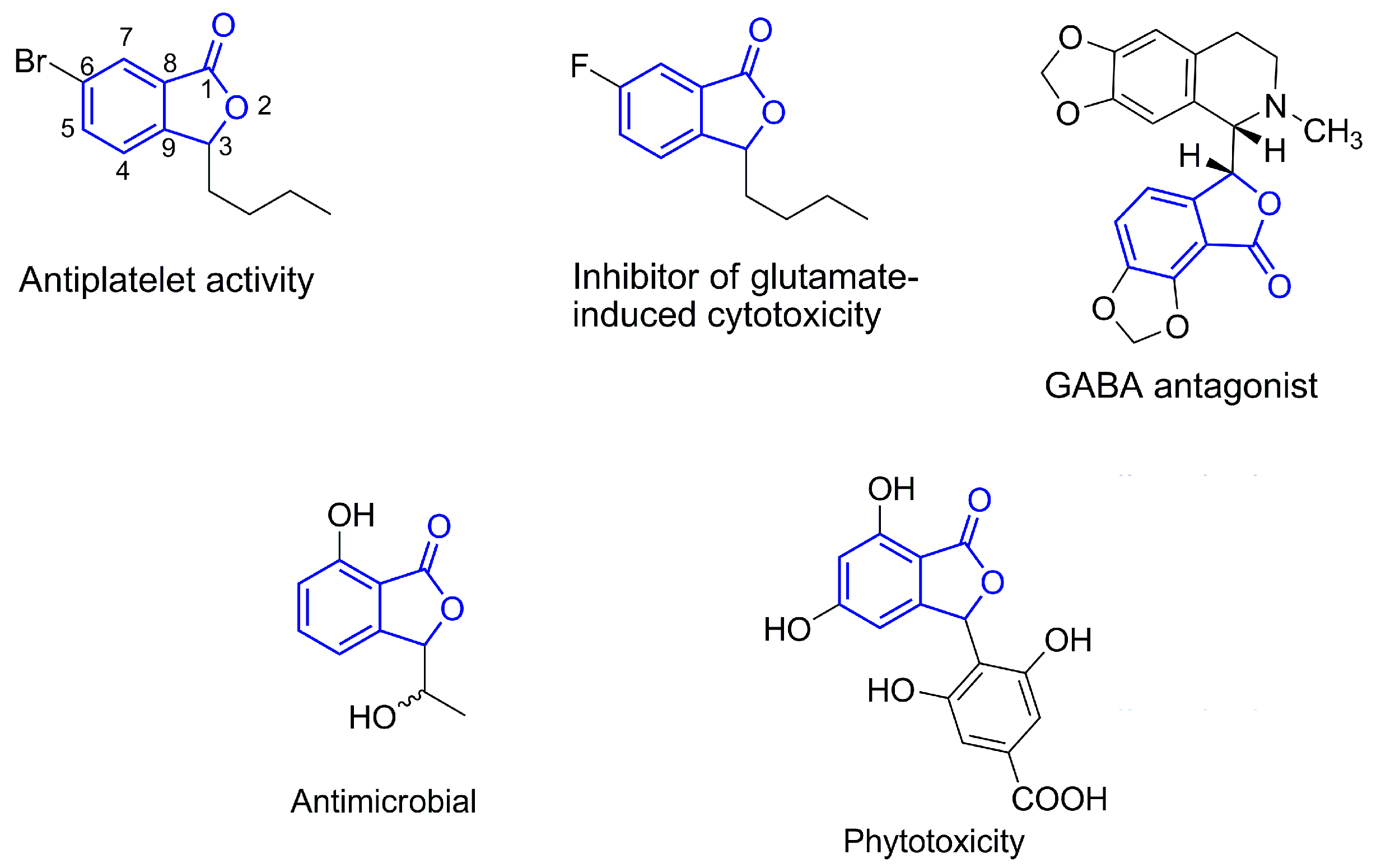
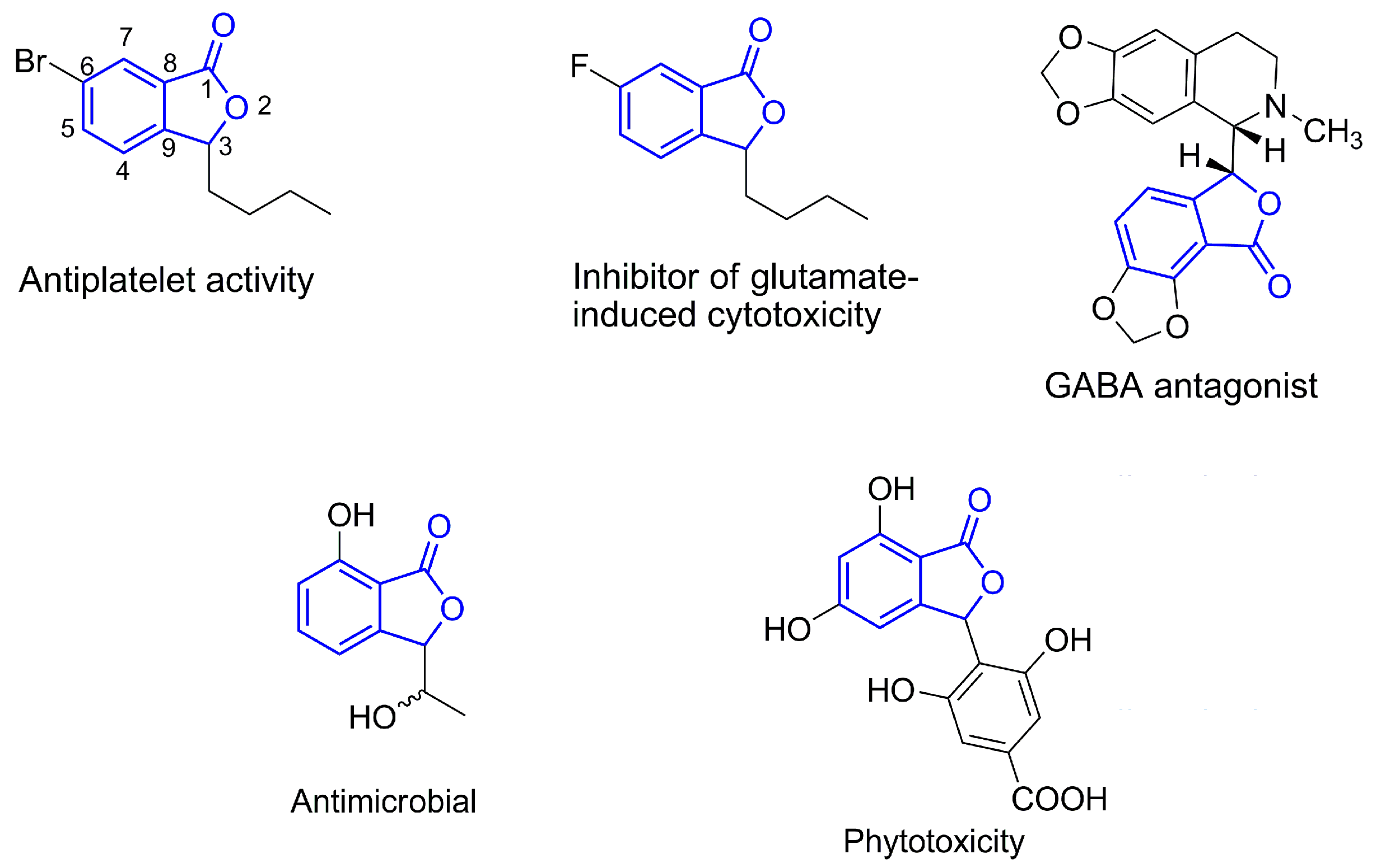
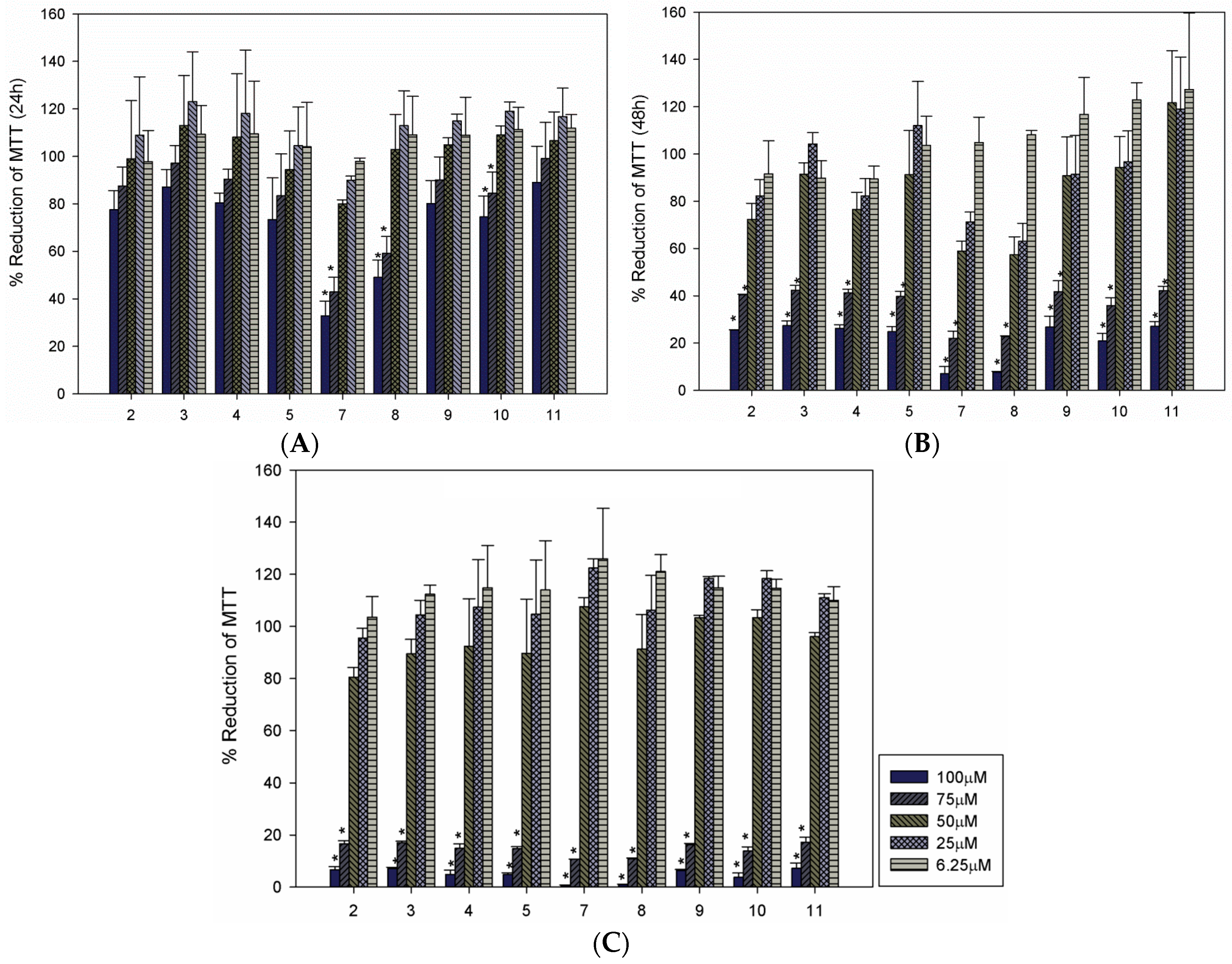
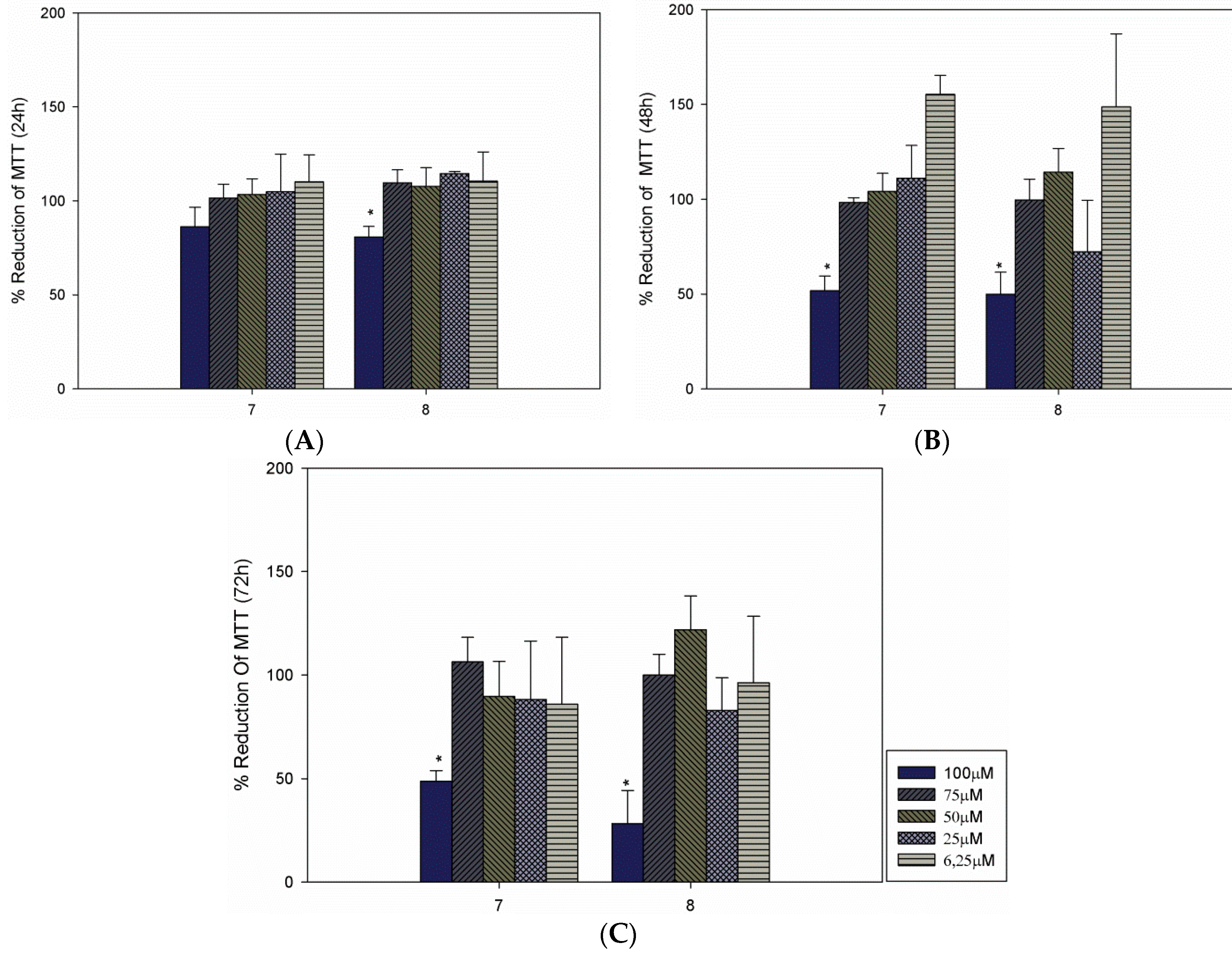
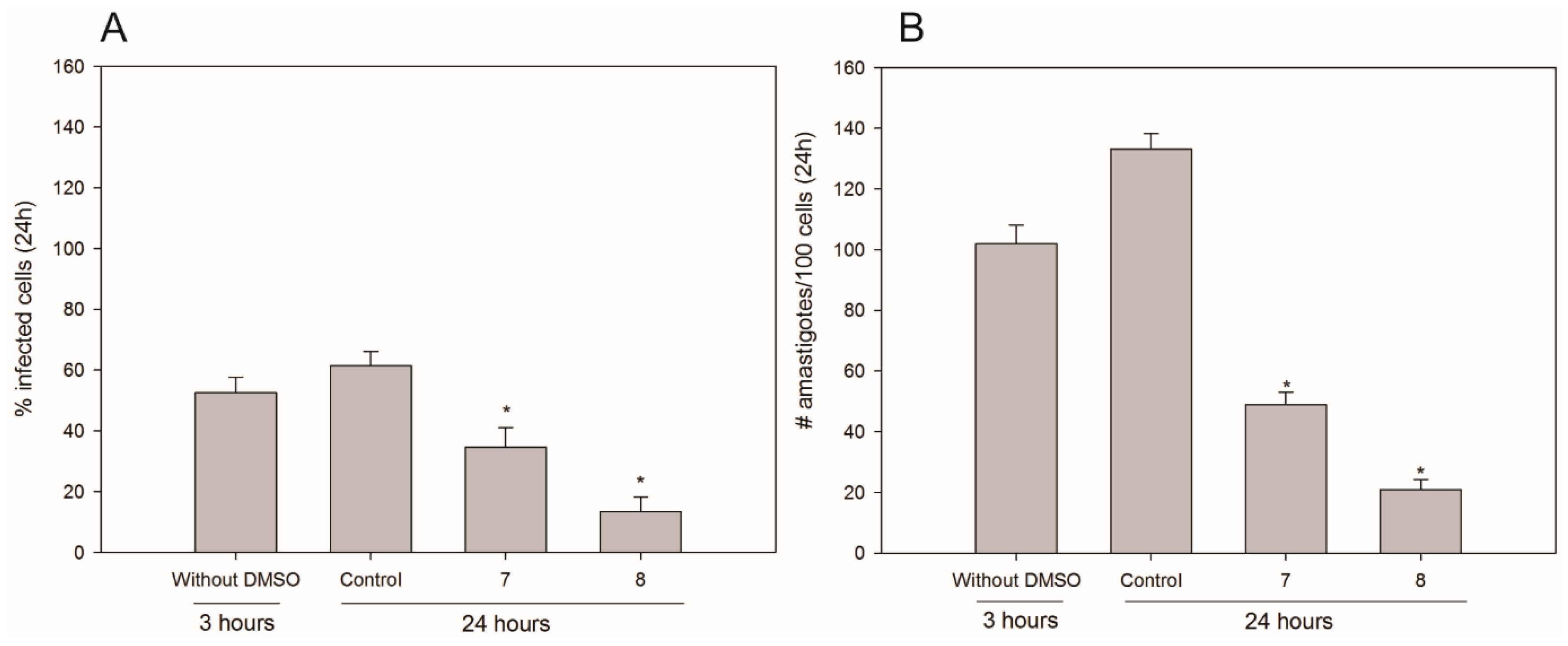
2. Results and Discussion
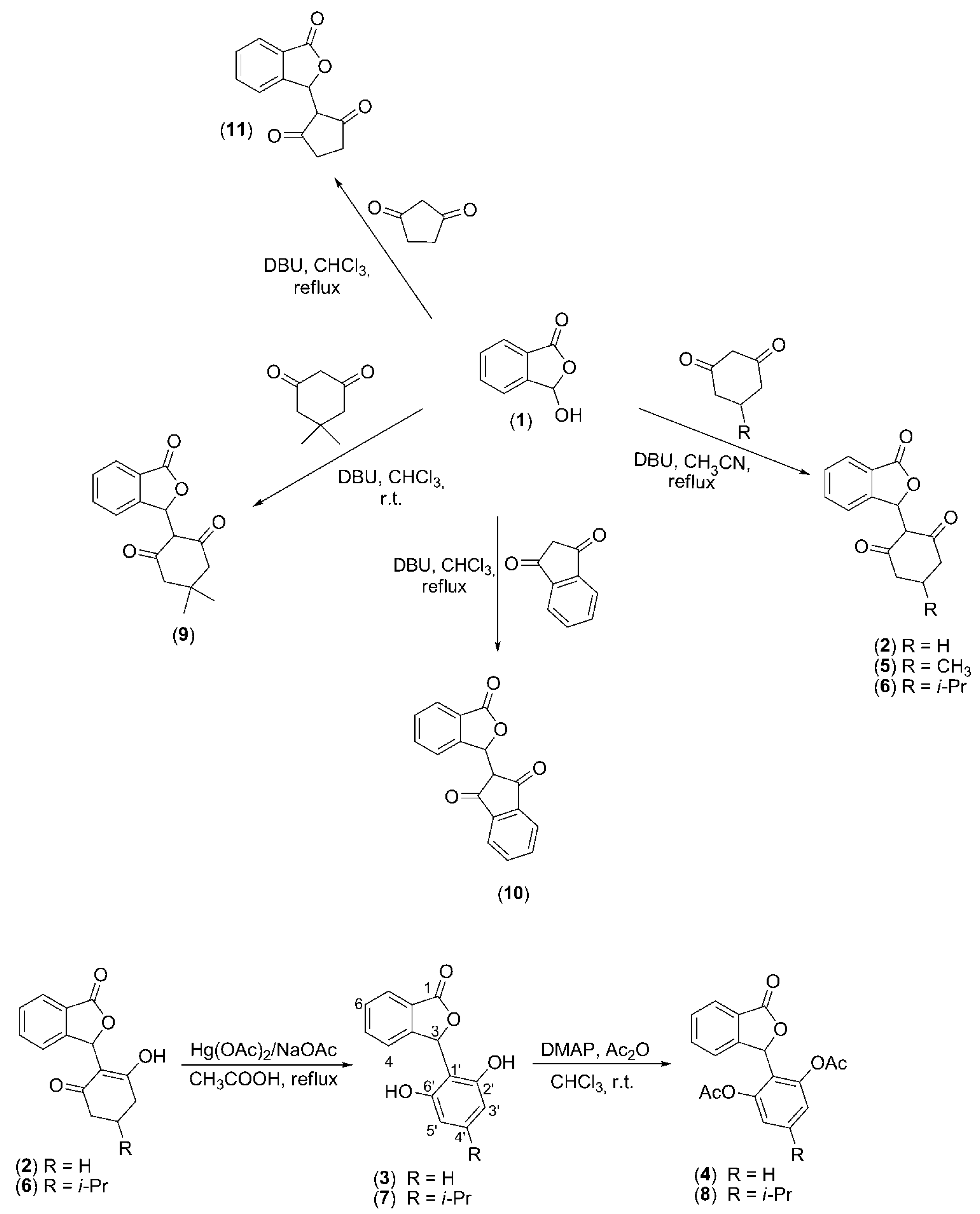
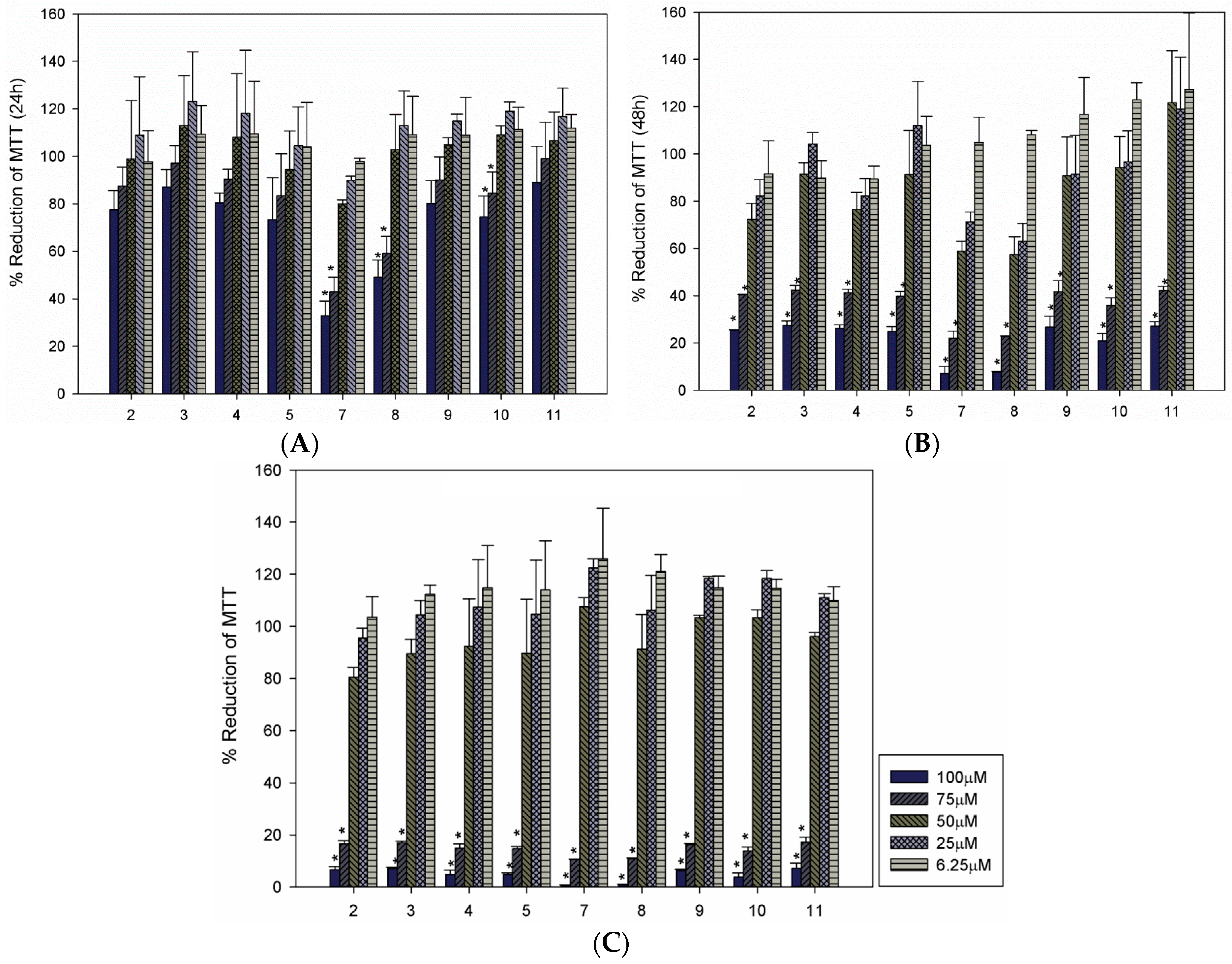

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | LC50 (µM) a | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| 2 | 74.54 | 65.55 | 58.96 |
| 3 | >100 | 65.73 | 59.08 |
| 4 | >100 | 65.69 | 59.20 |
| 5 | >100 | 61.69 | 58.96 |
| 7 | 60.48 | 62.72 | 59.75 |
| 8 | 65.93 | 63.90 | 58.46 |
| 9 | 75.01 | 54.78 | 60.51 |
| 10 | 74.27 | 61.84 | 60.53 |
| 11 | >100 | 30.79 | 60.17 |
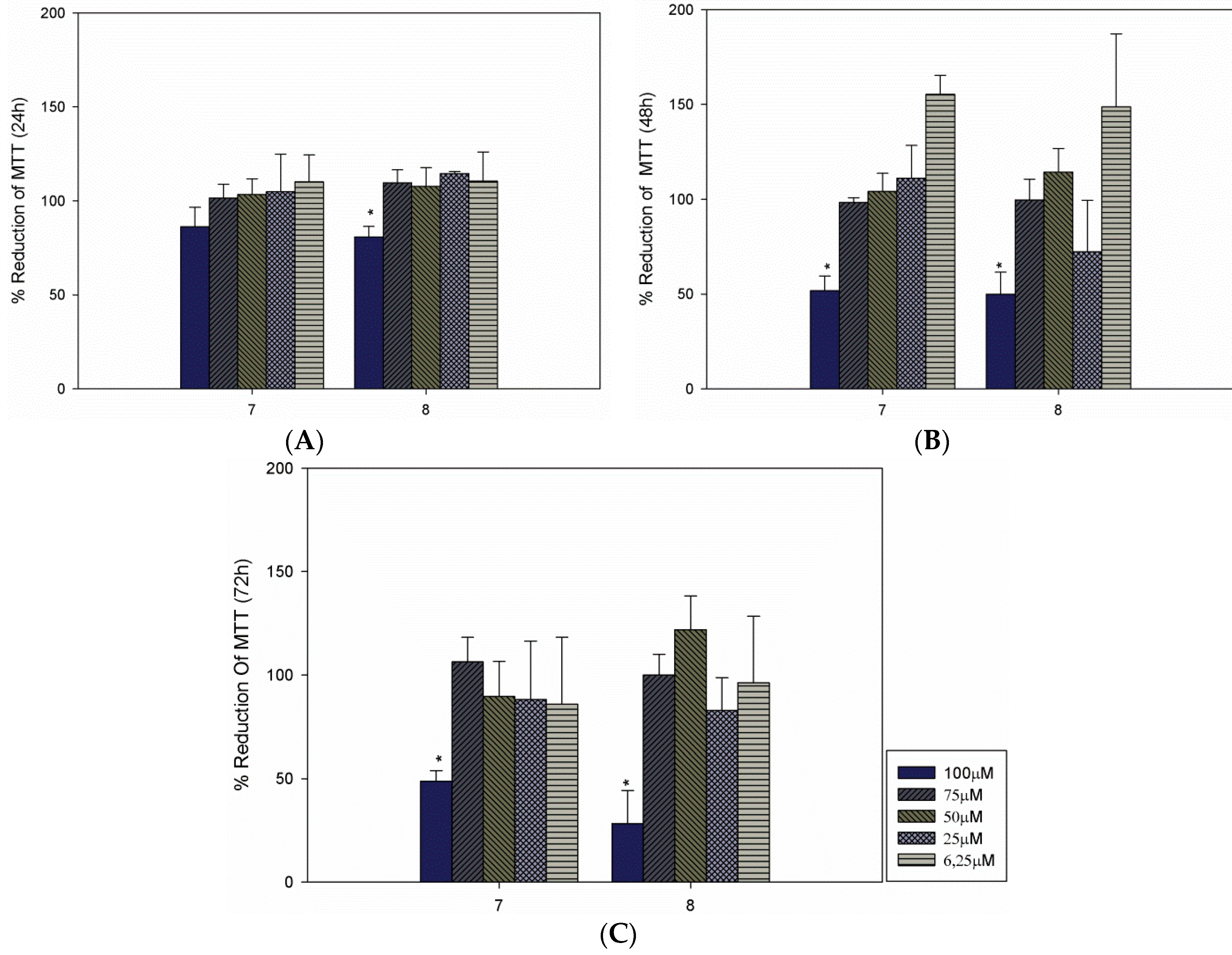
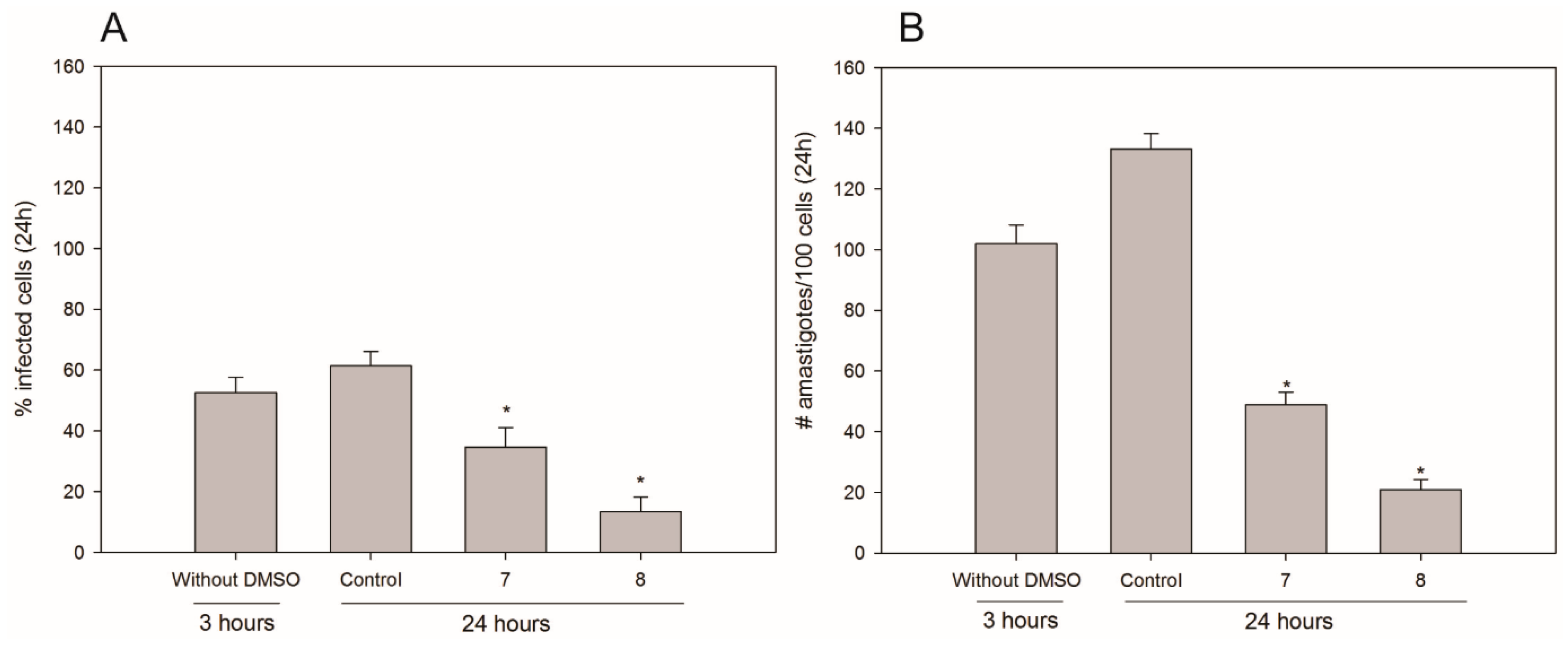
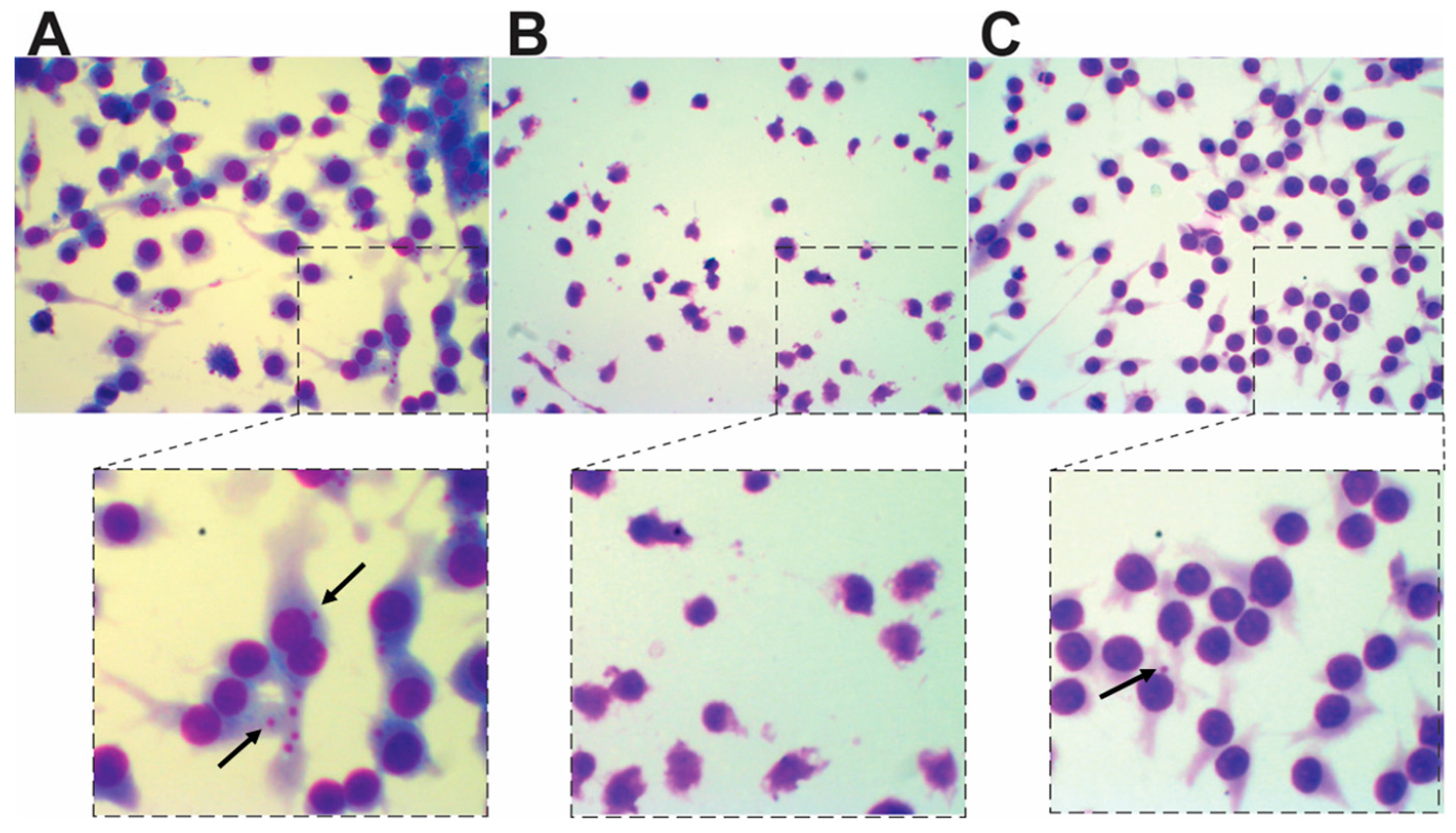
3. Experimental Section
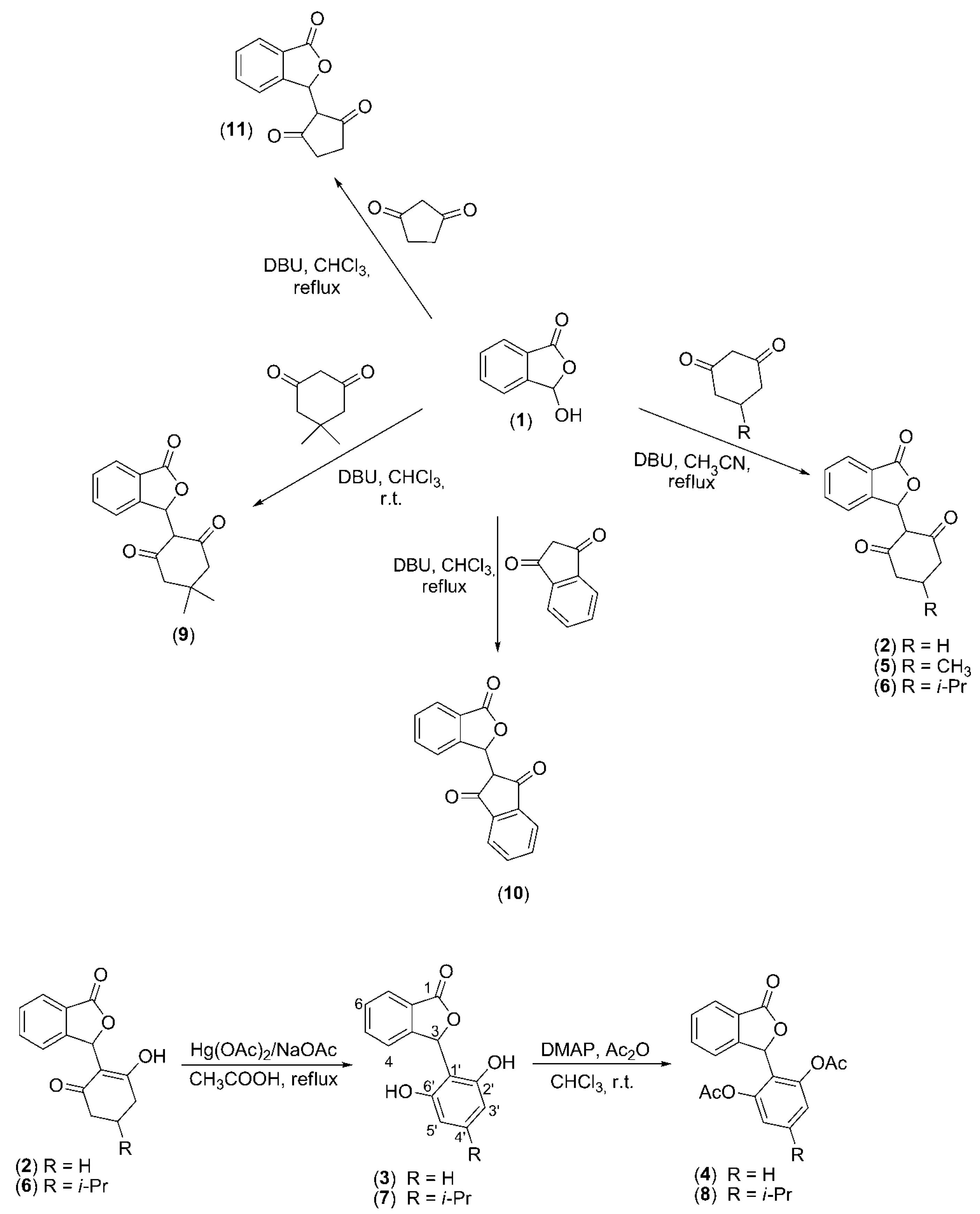
3.1. Syntheses
3.2. Obtaining of Promastigotes Parasites and Leishmanicidal Activity Evaluation
3.3. Cultivation of Macrophages and Cytotoxicity Evaluation
3.4. Infection Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Leishmaniasis; Fact Sheet No. 375; WHO: Geneva, Switzerland, 2015; Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 30 October 2015).
- WHO. Control of leishmaniases. Technical Report Series, No. 949; WHO: Geneva, Switzerland, 2010. [Google Scholar]
- Shawn, J. Further thoughts on the use of the name Leishmania (Leishmania) infantum chagasi for the aetiological agent of American visceral leishmaniasis. Mem. Inst. Oswaldo Cruz 2006, 101, 577–579. [Google Scholar] [CrossRef]
- Alvonar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J. Investigational drugs for visceral leishmaniasis. Expert Opin. Investig. Drugs 2015, 24, 43–59. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.F.; dos Santos, Y.L.; de Souza Vasconcellos, R.; Borges-Pereira, L.; Caldas, I.S.; de Almeida, M.R.; Bahia, M.T.; Fietto, J.L. Recombinant Leishmania (Leishmania) infantum Ecto-Nucleoside Triphosphate Diphosphohydrolase NTPDase-2 as a new antigen in canine visceral leishmaniasis diagnosis. Acta Trop. 2013, 125, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Gramiccia, M.; Gradoni, L. The current status of zoonotic leishmaniases and approaches to disease control. Int. J. Parasitol. 2005, 35, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Frezard, F.; Demicheli, C.; Ribeiro, R.R. Pentavalent antimonials: New perspectives for old drugs. Molecules 2009, 14, 2317–2336. [Google Scholar] [CrossRef] [PubMed]
- Garcia Bustos, M.F.; Barrio, A.; Gonzalez Prieto, G.; de Molina Raspi, E.; Cimino, R.; Cardozo, R.M.; Parada, L.A.; Yeo, M.; Soto, J.; Uncos, D.A.; et al. In Vivo Antileishmanial Efficacy of Miltefosine against Leishmania (Leishmania) Amazonensis. J. Parasitol. 2014, 100, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Coombs, G.H. Leishmaniasis—Current chemoteraphy and recent advances in the search for novel drugs. Trends Parasitol. 2003, 19, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.; Drummelsmith, J.; Papadopoulou, B. Leishmaniasis: Drugs in the clinic, resistance and new developments. Drug Resist. Update 2004, 7, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Monzote, L. Current treatment of leishmaniasis: A review. Open Antimicrob. Agents J. 2009, 1, 9–19. [Google Scholar]
- Monge-Maillo, B.; Lopez-Velez, R. Therapeutic options for visceral leishmaniasis. Drugs 2013, 73, 1863–1888. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Chan, S.-K.; Chung, H.-S.; Li, S.-L. Chemistry and biological activities of naturally occurring phthalides. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Nederland, 2005; pp. 611–669. [Google Scholar]
- Zhang, W.; Xu, L.; Yang, L.; Huang, Y.; Li, S.; Shen, Y. Phomopsidone A, a novel depsidone metabolite from the mangrove endophytic fungus Phomopsis sp. A123. Fitoterapia 2014, 96, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Gao, Y.; Qiao, H.; Hu, X.; Chang, J. Antiplatelet activity of 3-butyl-6-bromo-1(3H)-isobenzofuranone on rat platelet aggregation. J. Thromb. Thrombolysis 2012, 33, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A. Advantages of an antagonist: Bicuculline and other GABA antagonists. Br. J. Pharmacol. 2013, 169, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Ji, M.-X.; Xu, L.; Ji, B.-S. 3-butyl-6-fluoro-1(3H)-isobenzofuranone (6-F-NBP), a derivative of dl-n-butylphthalide, inhibits glutamate-induced cytotoxicity in PC12 cells. Drug Dev. Res. 2012, 73, 11–17. [Google Scholar] [CrossRef]
- Arnone, A.; Assante, G.; Nasini, G.; Strada, S.; Vercesi, A. Cryphonectric acid and other minor metabolites from a hypovirulent strain of Cryphonectria parasitica. J. Nat. Prod. 2002, 65, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Logrado, L.P.; Santos, C.O.; Romeiro, L.A.; Costa, A.M.; Ferreira, J.R.; Cavalcanti, B.C.; de Manoel Moraes, O.; Costa-Lotufo, L.V.; Pessoa, C.; dos Santos, M.L. Synthesis and cytotoxicity screening of substituted isobenzofuranones designed from anacardic acids. Eur. J. Med. Chem. 2010, 45, 3480–3489. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.R.; Bressan, G.C.; Pereira, W.L.; Ferreira, J.G.; de Oliveira, F.M.; Thomaz, D.C. Synthesis and antiproliferative activity of C-3 functionalized isobenzofuran-1(3H)-ones. Molecules 2013, 18, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Gray, A.I. A benzoisofuranone derivative and carbazole alkaloids from Murraya koenigii and their antimicrobial activity. Phytochemistry 2005, 66, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.R.; Pereira, J.L.; da Silva, S.F.; Guilardi, S.; Paixão, D.A.; Anconi, C.P.A.; de Almeida, W.B.; Ellena, J.; Forlani, G. Synthesis, characterization and phytotoxic activity of hydroxylated isobenzofuran-1(3H)-ones. J. Mol. Struct. 2014, 1061, 61–68. [Google Scholar] [CrossRef]
- Kwan, C.N.; Medoff, G.; Kobayashi, G.S.; Schlessinger, D.; Raskas, H.J. Potentiation of the antifungal effects of antibiotics by amphotericin B. Antimicrob. Agents Chemother. 1972, 2, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Strobel, G.; Ford, E.; Worapong, J.; Harper, J.K.; Arif, A.M.; Grant, D.M.; Fung, P.C.W.; Chaud, R.M.W. Isopestacin, an isobenzofuranone from Pestalotiopsis microspora, possessing antifungal and antioxidant activities. Phytochemistry 2002, 60, 179–183. [Google Scholar] [CrossRef]
- Dorosti, Z.; Yousefi, M.; Sharafi, S.M.; Darani, H.Y. Mutual action of anticancer and antiparasitic drugs: Are there any shared targets? Future Oncol. 2014, 10, 2529–2539. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.; Critselis, E.; Voutsas, I.F.; Perez, S.A.; Moschovi, M.; Baxevanis, C.N.; Chrousos, G.P. New use for old drugs? Prospective targets of chloroquines in cancer therapy. Curr. Drug Targets 2014, 15, 843–885. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, W.; Guo, J.; Jiang, X.; Lu, W.; Liu, M.; Pang, X. The antiparasitic drug, potassium antimony tartrate, inhibits tumor angiogenesis and tumor growth in nonsmall-cell lung cancer. J. Pharmacol. Exp. Ther. 2015, 352, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Vinayagam, J.; Saha, S.; Chowdhury, S.; Roychowdhury, S.; Jaisankar, P.; Majumder, H.K. Isobenzofuranone derivatives exhibit antileishmanial effect by inhibiting type II DNA topoisomerase and inducing host response. Pharmacol. Res. Perspect. 2014, 2, e00070. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 2–11 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, W.L.; De Souza Vasconcellos, R.; Mariotini-Moura, C.; Saar Gomes, R.; Firmino, R.D.C.; Da Silva, A.M.; Silva Júnior, A.; Bressan, G.C.; Almeida, M.R.; Crocco Afonso, L.C.; et al. The Antileishmanial Potential of C-3 Functionalized Isobenzofuranones against Leishmania (Leishmania) Infantum Chagasi. Molecules 2015, 20, 22435-22444. https://doi.org/10.3390/molecules201219857
Pereira WL, De Souza Vasconcellos R, Mariotini-Moura C, Saar Gomes R, Firmino RDC, Da Silva AM, Silva Júnior A, Bressan GC, Almeida MR, Crocco Afonso LC, et al. The Antileishmanial Potential of C-3 Functionalized Isobenzofuranones against Leishmania (Leishmania) Infantum Chagasi. Molecules. 2015; 20(12):22435-22444. https://doi.org/10.3390/molecules201219857
Chicago/Turabian StylePereira, Wagner Luiz, Raphael De Souza Vasconcellos, Christiane Mariotini-Moura, Rodrigo Saar Gomes, Rafaela De Cássia Firmino, Adalberto Manoel Da Silva, Abelardo Silva Júnior, Gustavo Costa Bressan, Márcia Rogéria Almeida, Luís Carlos Crocco Afonso, and et al. 2015. "The Antileishmanial Potential of C-3 Functionalized Isobenzofuranones against Leishmania (Leishmania) Infantum Chagasi" Molecules 20, no. 12: 22435-22444. https://doi.org/10.3390/molecules201219857