
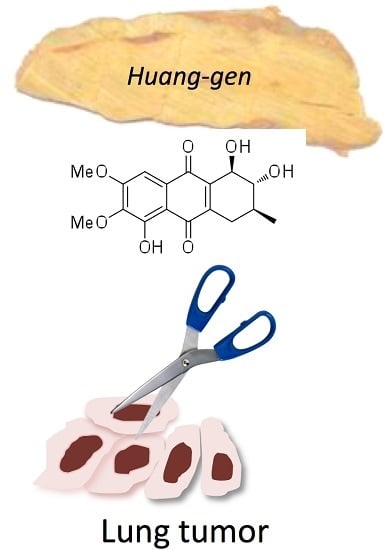
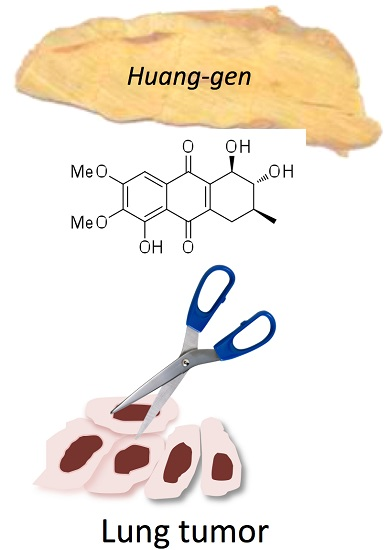
Seven New Tetrahydroanthraquinones from the Root of Prismatomeris connata and Their Cytotoxicity against Lung Tumor Cell Growth

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
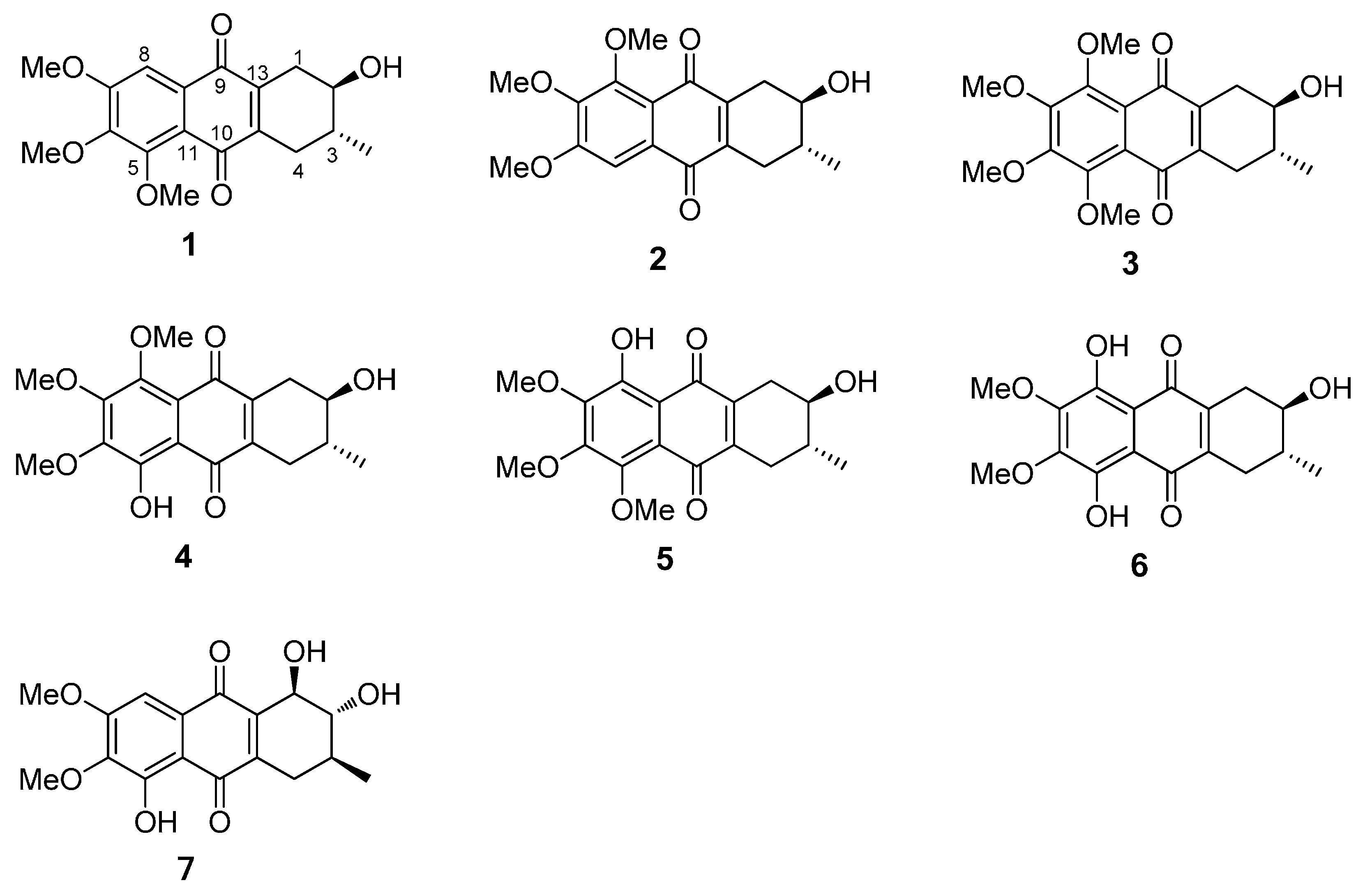
| Position | 1 b | 2 b | 3 b | 4 b | 5 b | 6 b | 7 b |
|---|---|---|---|---|---|---|---|
| δC, mult. | δC, mult. | δC, mult. | δC, mult. | δC, mult. | δC, mult. | δC, mult. | |
| 1 | 30.9, CH2 | 31.3, CH2 | 30.9, CH2 | 31.3, CH2 | 30.3, CH2 | 29.8, CH2 | 73.0, CH |
| 2 | 71.0, CH | 71.1, CH | 71.0, CH | 70.9, CH | 70.8, CH | 34.1, CH2 | 76.7, CH |
| 3 | 34.2, CH | 34.0, CH | 34.0, CH | 33.8, CH | 34.0, CH | 71.0, CH2 | 31.9, CH |
| 4 | 29.9, CH2 | 29.4, CH2 | 29.4, CH2 | 28.8, CH2 | 29.9, CH2 | 31.4, CH2 | 31.1, CH2 |
| 5 | 154.7, C | 106.1, C | 152.7, C | 154.7, C | 149.2, C | 159.5, C | 155.9, C |
| 6 | 157.3, C | 147.9, C | 151.2, C c | 146.7, C | 153.7, C | 147.9, C | 141.9, C |
| 7 | 147.9, C | 157.3, C | 151.2, C c | 153.7, C | 146.7, C | 148.0, C | 158.1, C |
| 8 | 106.1, C | 154.3, C | 152.7, C | 149.1, C | 154.6, C | 159.6, C | 104.3, CH |
| 9 | 184.2, C | 183.2, C | 183.7, C | 182.5, C | 189.3, C | 181.8, C | 185.5, C |
| 10 | 183.3, C | 184.2, C | 183.7, C | 189.4, C | 182.6, C | 181.9, C | 188.7, C |
| 11 | 129.7, C | 119.8, C | 121.8, C | 110.6, C | 118.9, C | 107.7, C | 111.0, C |
| 12 | 119.7, C | 129.6, C | 121.8, C | 119.0, C | 110.7, C | 107.8, C | 127.7, C |
| 13 | 139.8, C | 143.6, C | 141.8, C | 144.7, C | 140.3, C | 138.3, C | 145.1, C |
| 14 | 145.0, C | 141.2, C | 143.2, C | 141.8, C | 146.2, C | 137.1, C | 142.1, C |
| CH3 (3) | 17.5 | 17.4 | 17.4 | 17.4 | 17.4 | 17.6 | 17.4 |
| OCH3 (5) | 61.8 | 61.8 c | 61.6 c | ||||
| OCH3 (6) | 56.6 | 61.6 | 61.9 c | 61.9 | 62.0 | 61.3 | |
| OCH3 (7) | 61.6 | 56.6 | 61.9 c | 61.2 | 61.6 c | 61.9 c | 56.7 |
| OCH3 (8) | 61.8 | 61.8 c | 61.6 | 61.9 c |
| Position | 1 b | 2 b | 3 b | 4 b | 5 b | 6 b | 7 b |
|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | |
| 1α | 2.97ddt (19.2, 5.0, 1.8) | 2.96 br dd (19.2, 4.8) | 2.91 br dd (19.2, 4.8) | 3.04 ddt (19.2, 4.8, 1.8) | 2.96 ddt (19.2, 4.8, 2.1) | 3.01 br dd (19.0, 5.4) | 4.69 ddd (7.8, 2.6, 1.1) |
| 1β | 2.44 ddt (19.2, 7.8, 2.0) | 2.42 br dd (19.2, 7.2) | 2.39 ddt (19.2, 7.2, 3.0) | 2.52 ddt (19.2, 7.2, 2.4) | 2.45 ddt (19.2, 7.2, 1.8) | 2.32 br dd (19.0, 8.4) | |
| 2 | 3.69 dt (5.0, 7.8) | 3.66 dt (4.8, 7.2) | 3.64 dt (4.8, 7.2) | 3.76 dt (4.8, 7.2) | 3.71 dt (5.4, 7.2) | 3.5 dt (5.4, 8.4) | 3.5 dd (11.4, 7.8) |
| 3 | 1.85 m | 1.81 m | 1.80 m | 1.92 m | 1.86 m | 1.85 m | 1.85 m |
| 4α | 2.89 ddt (19.5, 6.6, 1.8) | 2.82 dd (19.8, 6.0) | 2.81 dd (19.8, 6.0) | 2.92 ddt (19.8, 6.0, 1.8) | 2.89 ddt (19.8, 5.4, 1.8) | 3.11 dd (18.6, 5.4) | 2.94 ddd (19.8, 4.8, 0.6) |
| 4β | 2.24 ddq (19.5, 7.8, 1.8) | 2.18 br dd (19.8, 8.4) | 2.18 ddq (19.8, 7.8, 1.8) | 2.28 ddt (19.8, 7.8, 2.4) | 2.26 ddt (19.8, 8.4, 1.8) | 2.55 dd (18.6, 7.8) | 2.16 ddd (19.8, 11.4, 3.6) |
| 5 | 7.39 s | ||||||
| 8 | 7.42 s | 12.9 s | 7.24 s | ||||
| CH3 (3) | 1.07 br d (7.8) | 1.03 d (6.6) | 1.01 d (6.6) | 1.12 d (6.6) | 1.07 d (7.2) | 1.07 d (6.6) | 1.22 d (6.6) |
| OH (5) | 13.2 s | 12.9 s | 12.1 s | ||||
| OCH3 (5) | 3.90 s | 3.94 s c | 3.85 s | ||||
| OCH3 (6) | 3.97 s | 3.88 s | 3.84 s c | 4.07 s | 4.01 s | 4.05 s | 3.99 s |
| OCH3 (7) | 3.93 s | 3.93 s | 3.84 s c | 4.06 s | 4.02 s | 4.04 s | 4.00 s |
| OCH3 (8) | 3.86 s | 3.94 s c | 3.90 s | ||||
| OH(8) | 13.1 s | 12. 9 s |
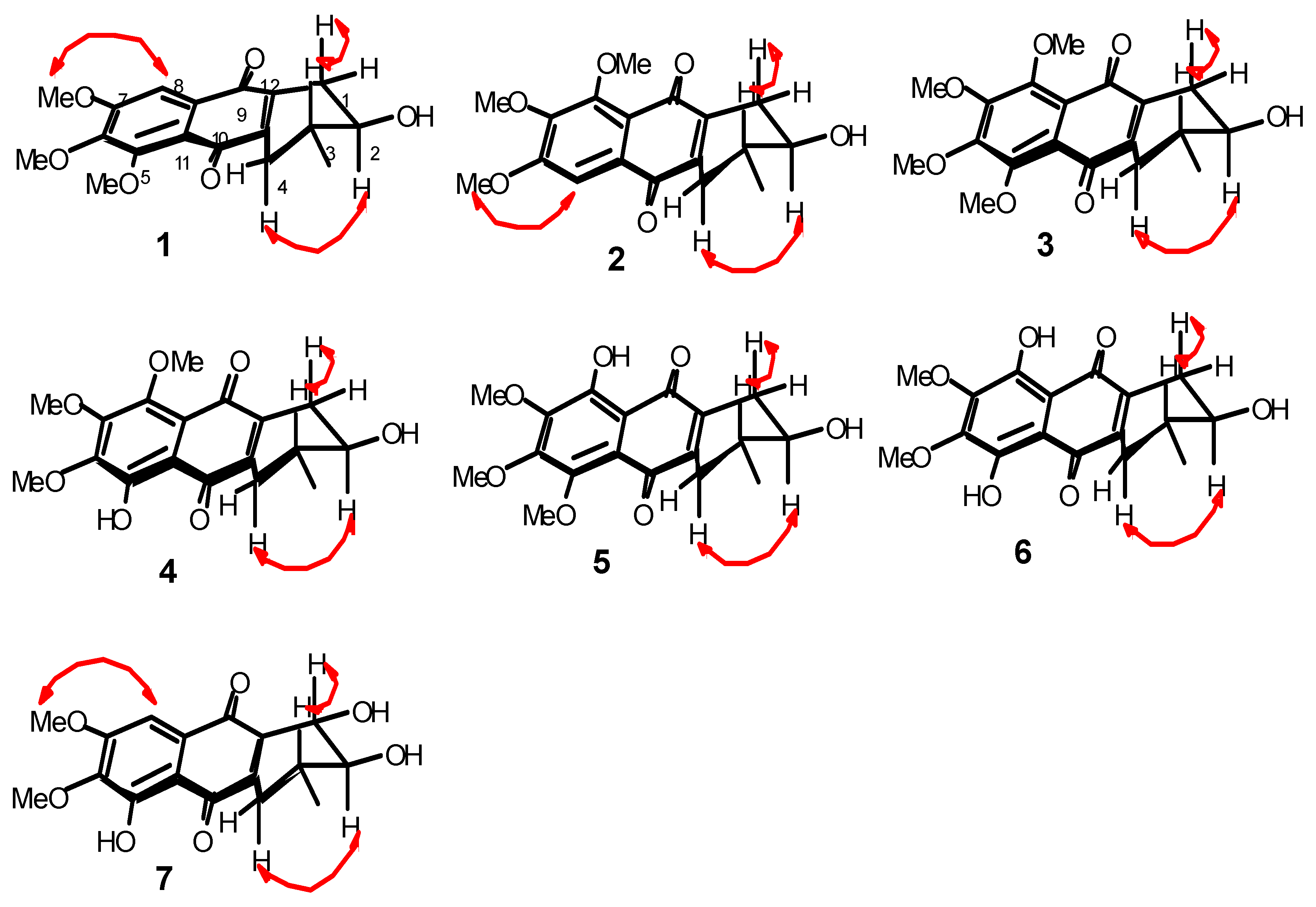
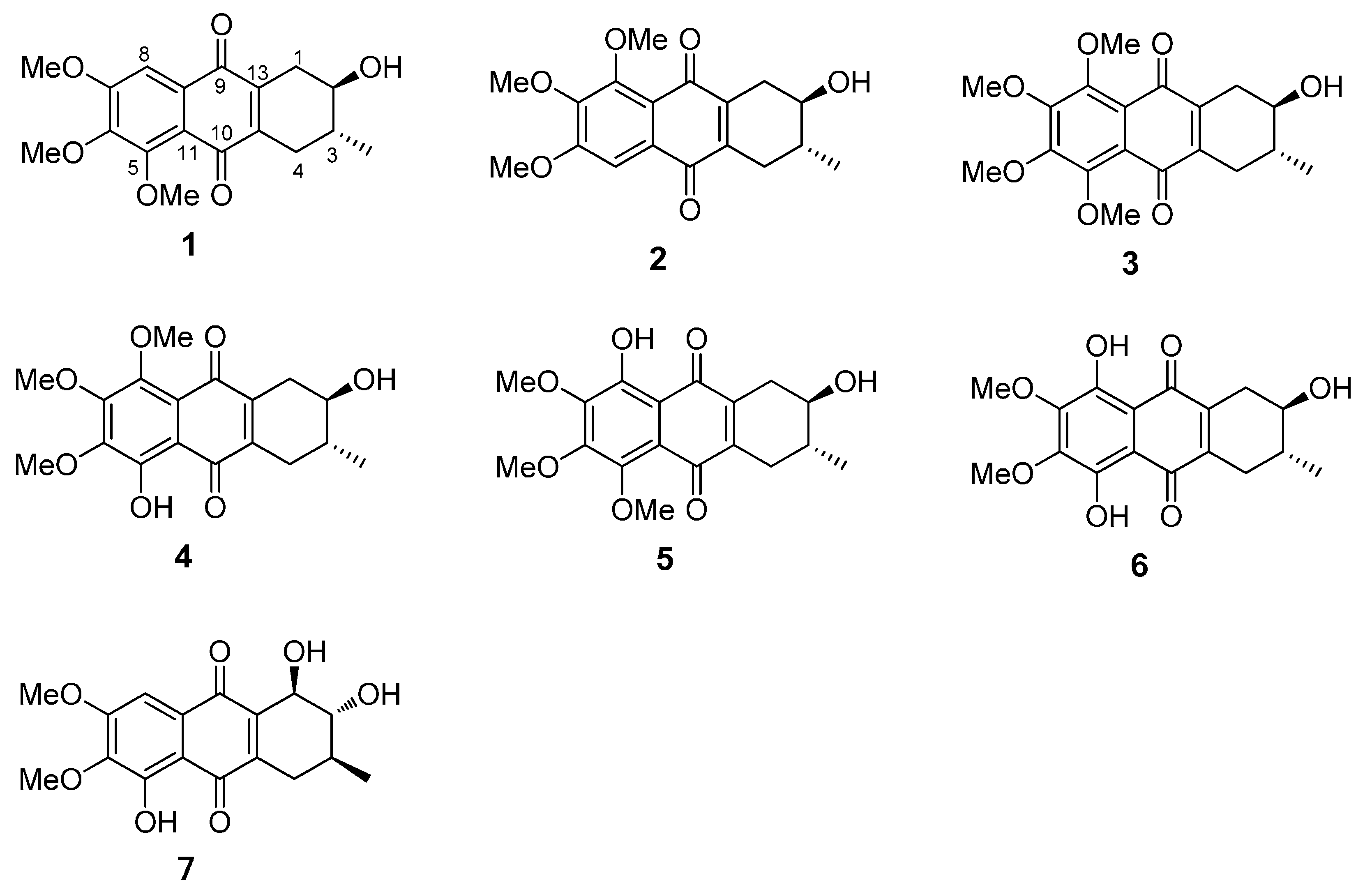
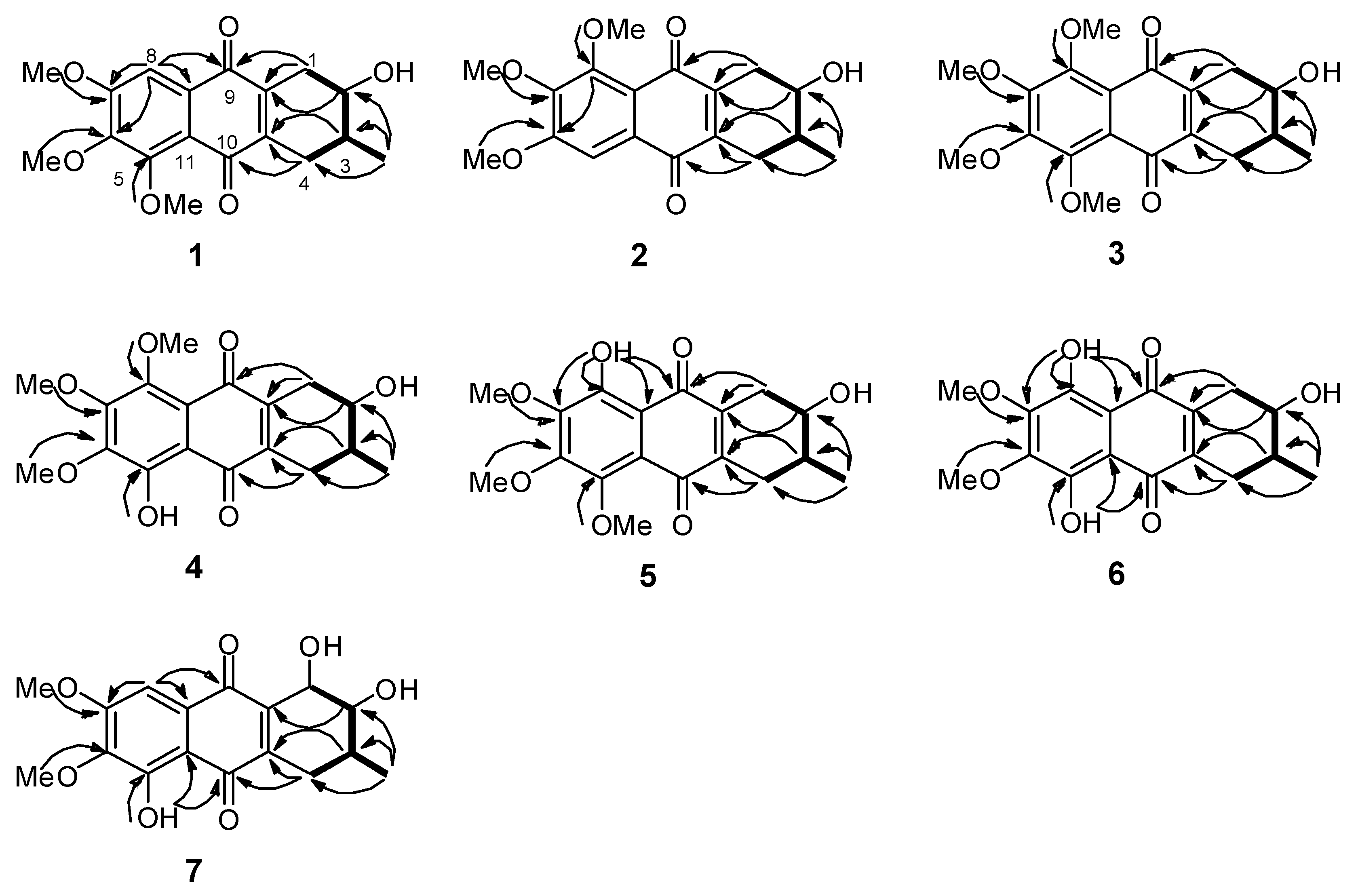
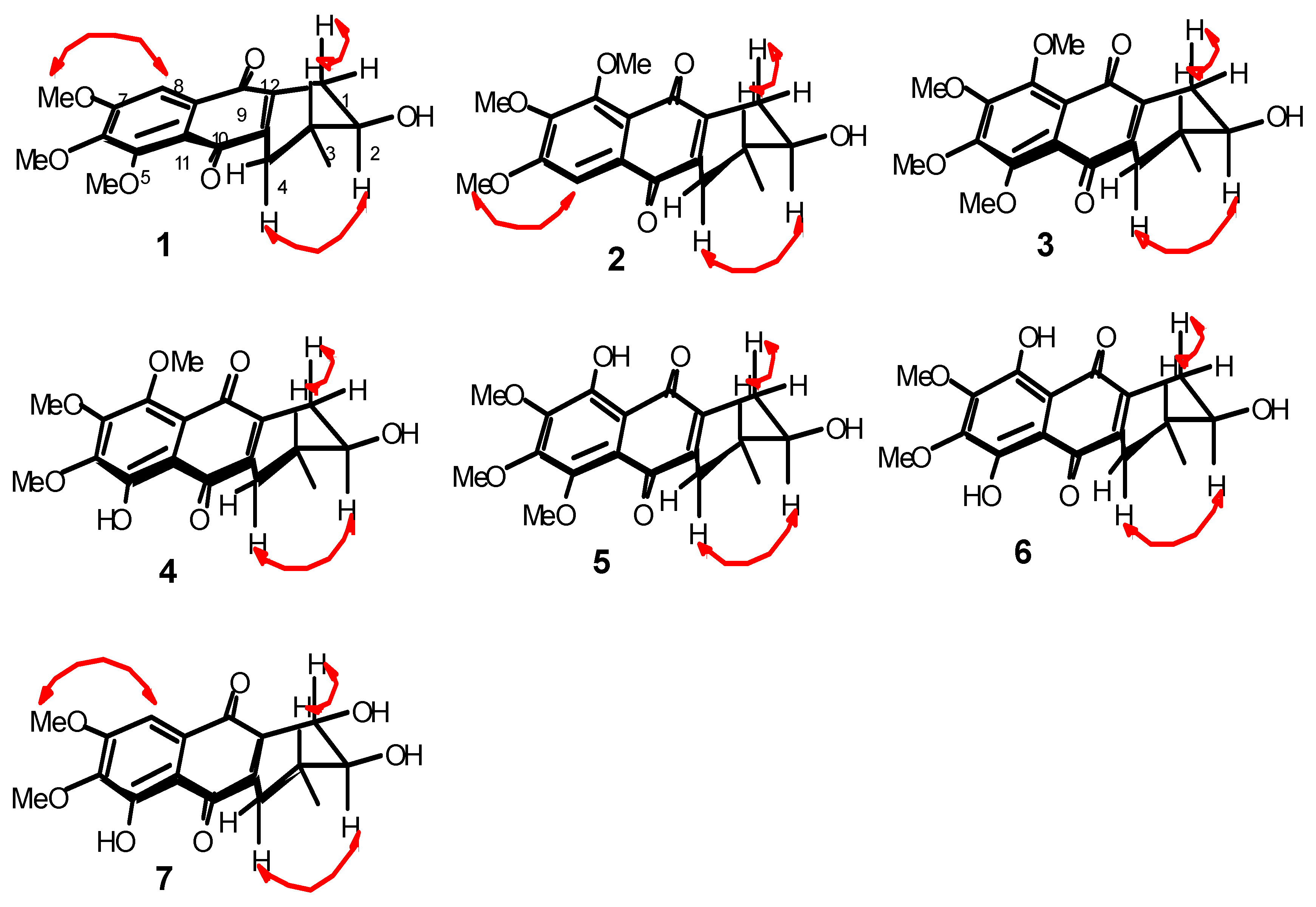
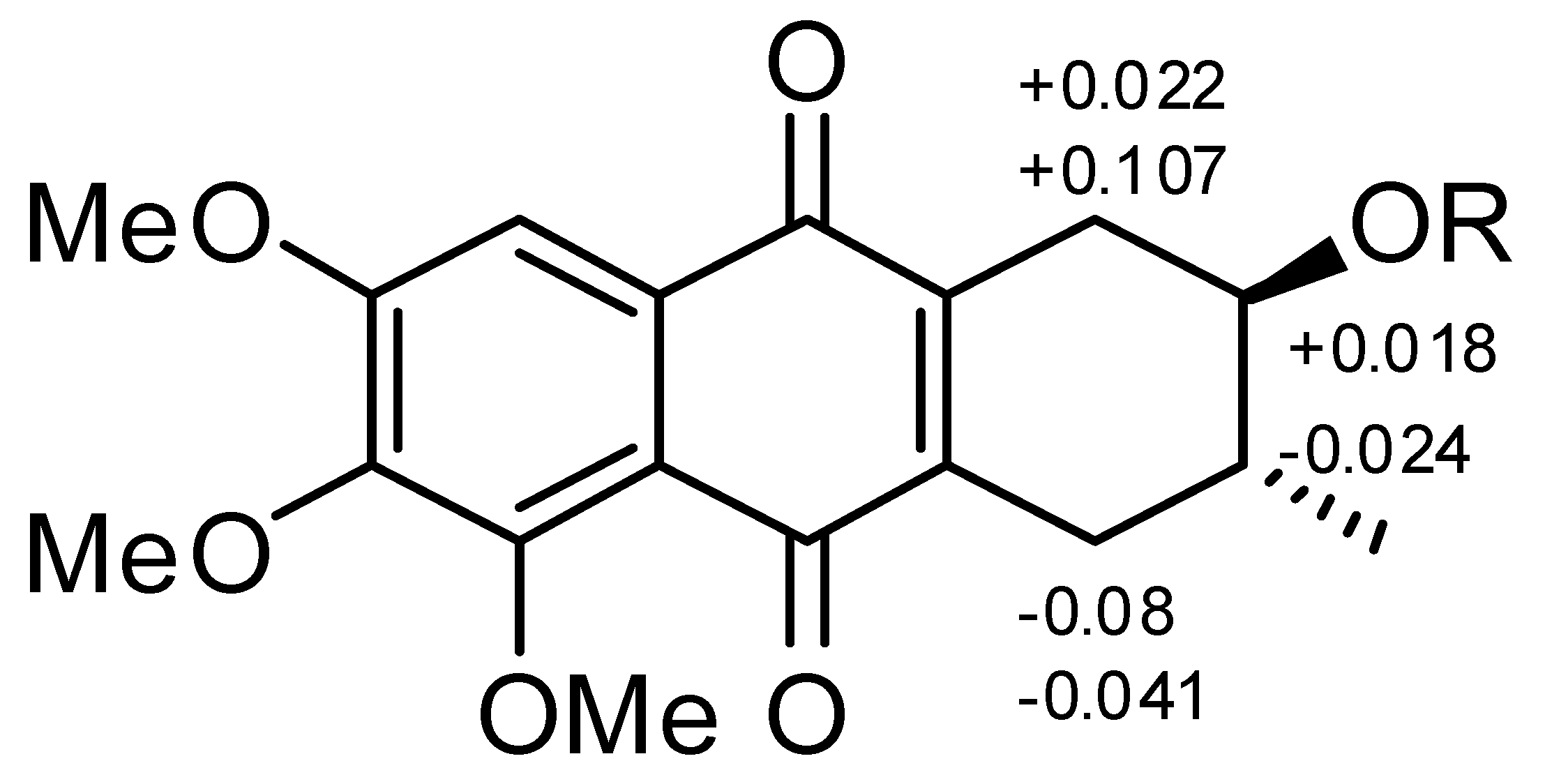
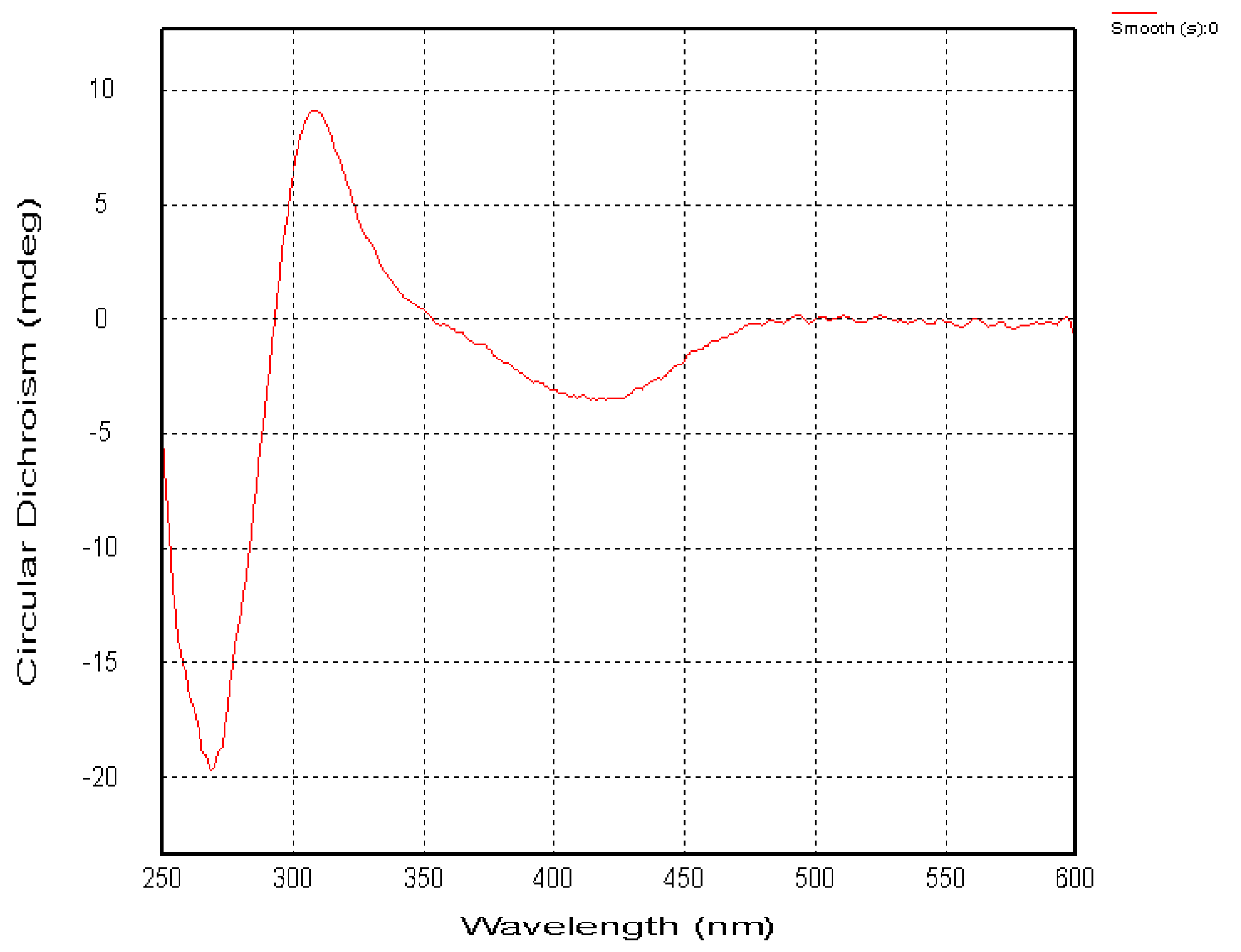
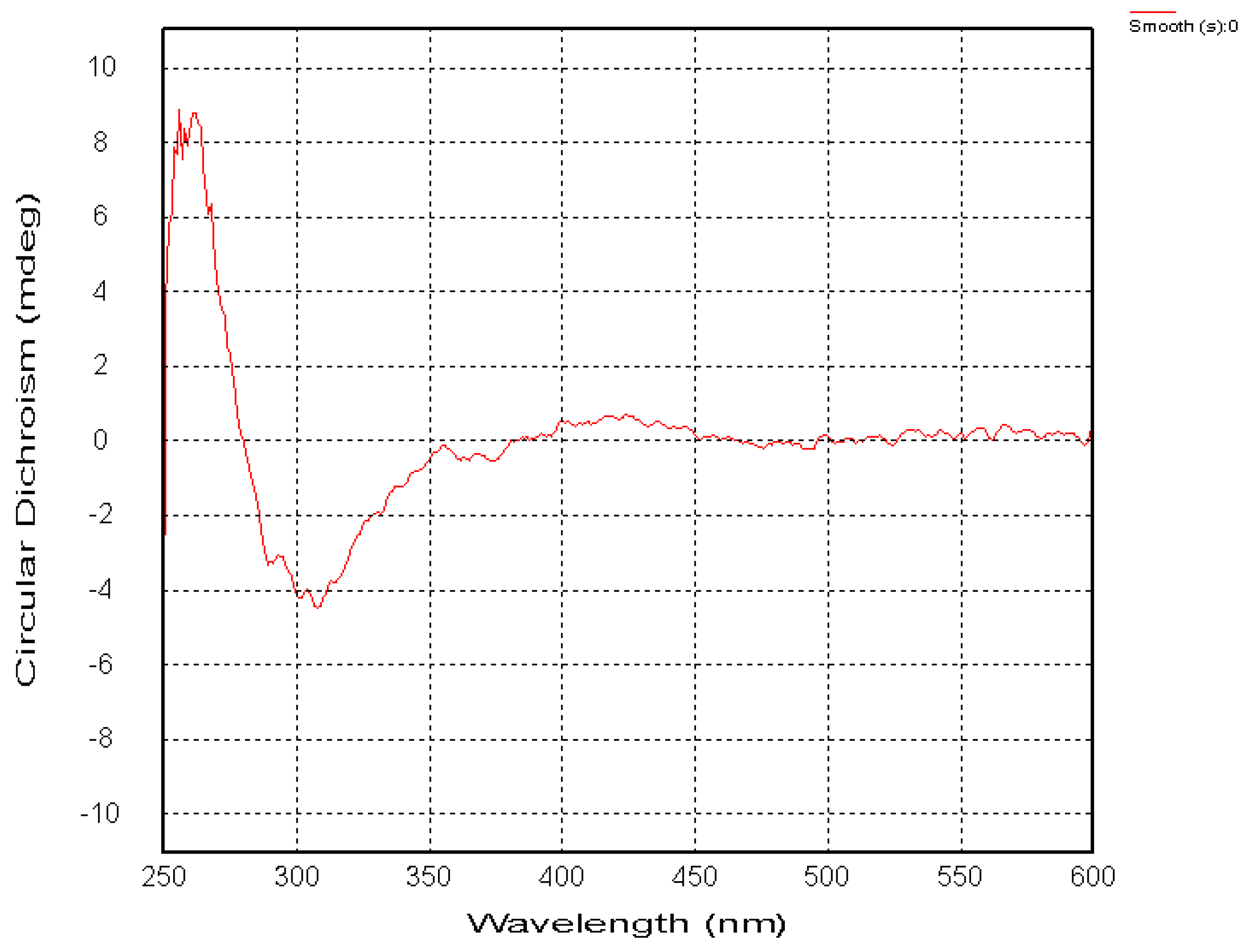
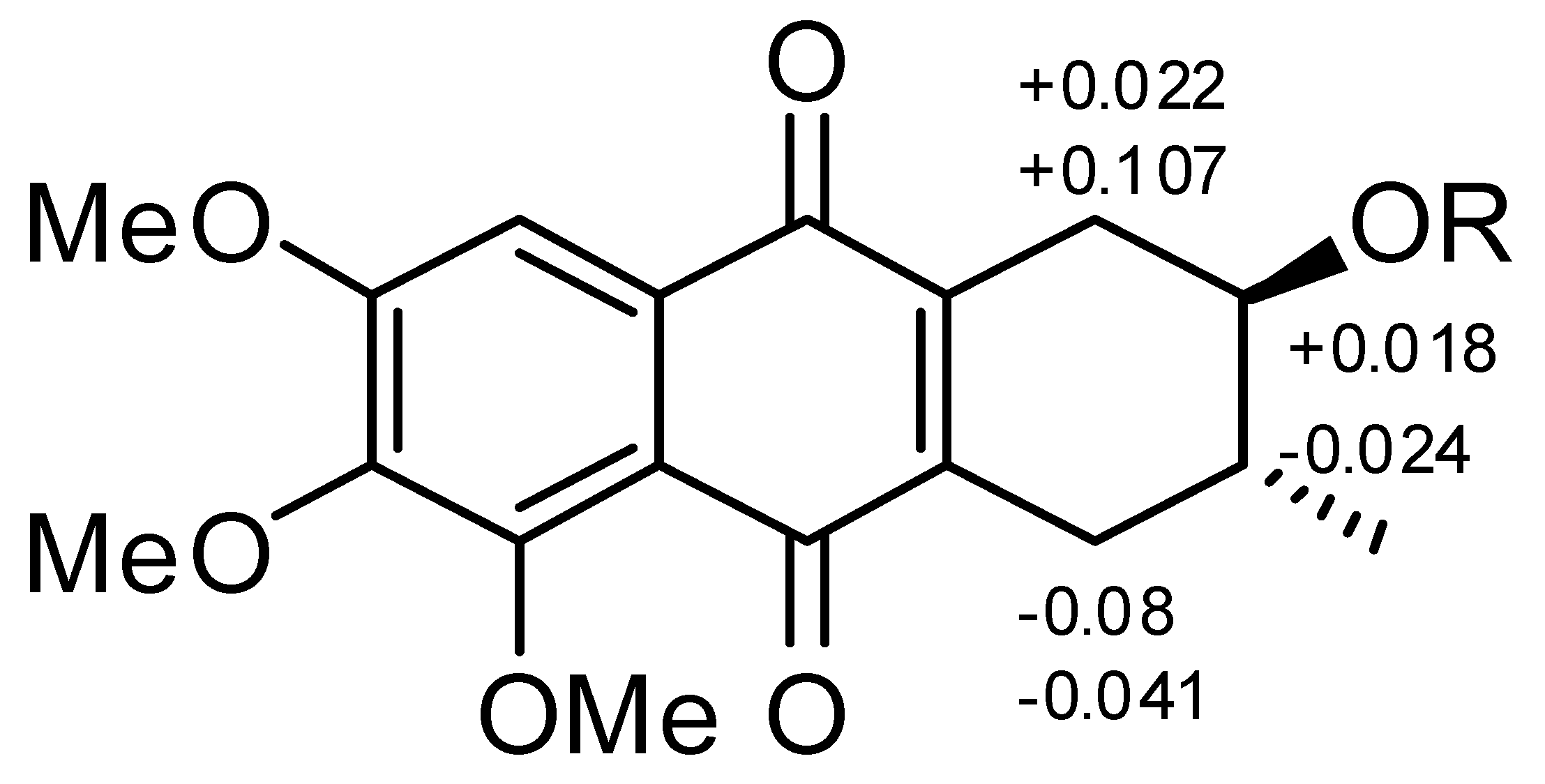
2.1. Chemical Structure of Prisconnatanones C to I (Compounds 1–7)


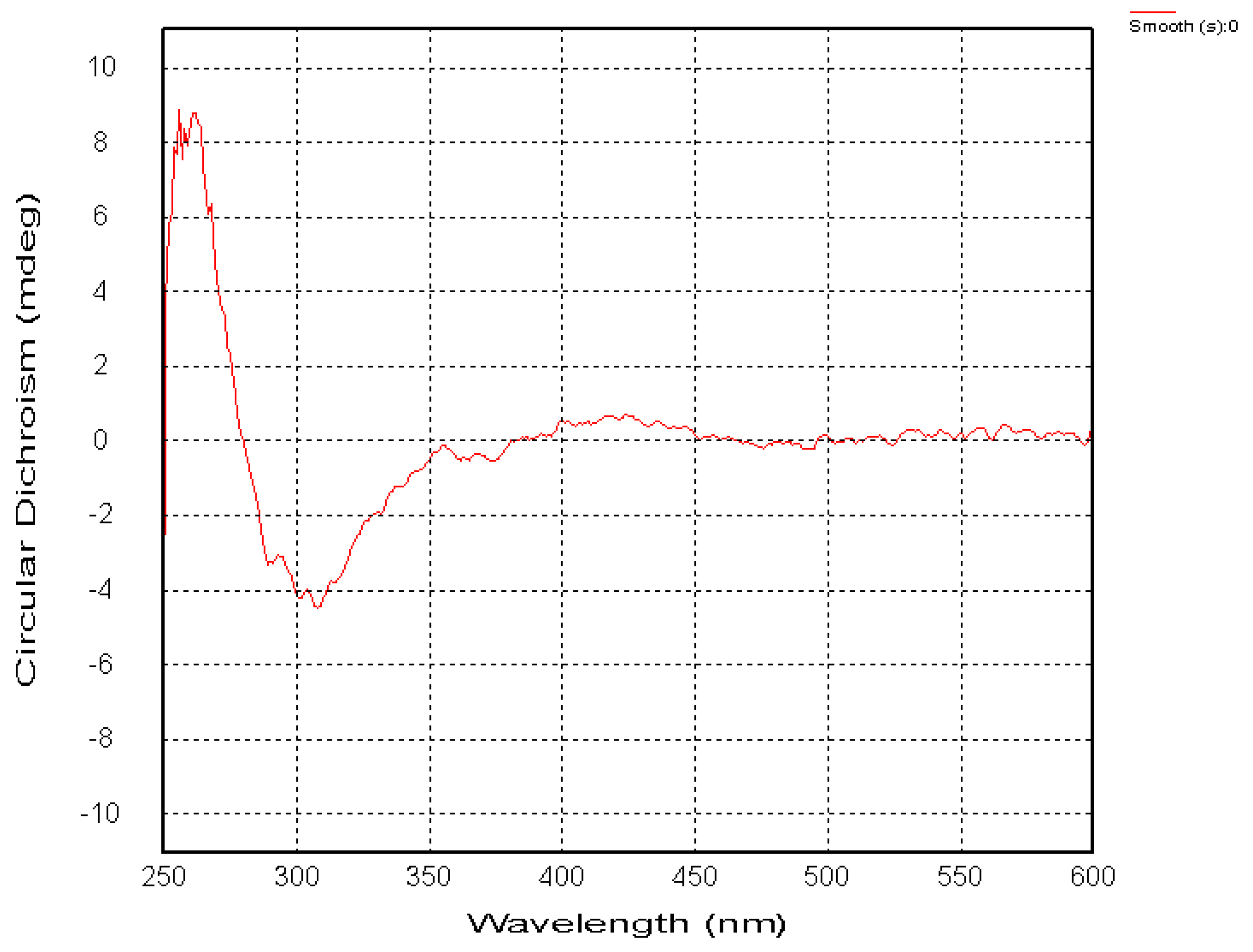
2.2. Structure-Dependent Cytotoxicity of Prisconnatanones C to I (1–7) against Tumor Cell Growth
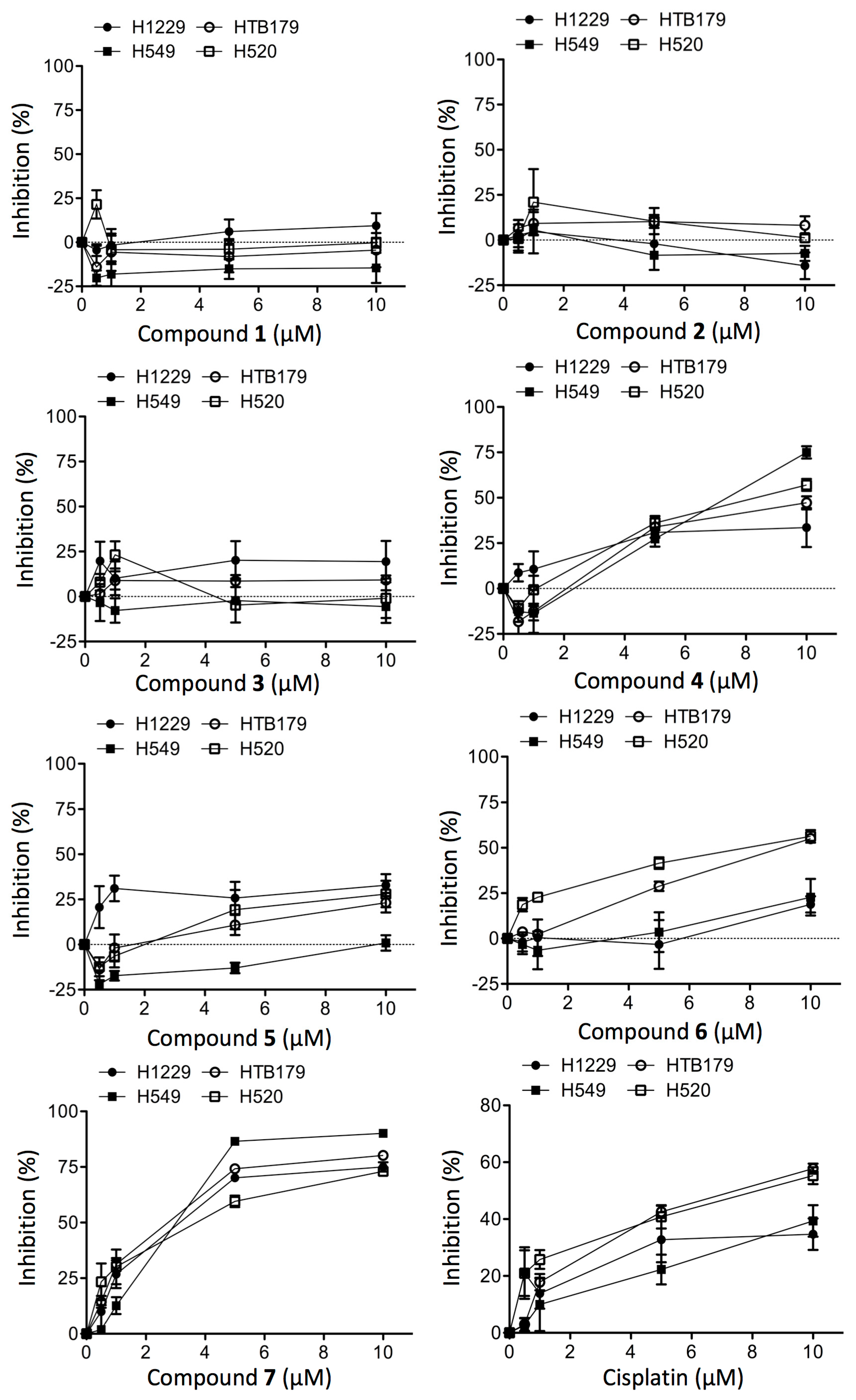
| Compound | H1229 | HTB179 | H549 | H520 |
|---|---|---|---|---|
| 1 | Nil | Nil | Nil | Nil |
| 2 | Nil | Nil | Nil | Nil |
| 3 | Nil | Nil | Nil | Nil |
| 4 | >20 | 9.8 | >20 | 9.5 |
| 5 | >20 | >20 | >20 | >20 |
| 6 | >20 | 13 | 8 | 9.5 |
| 7 | 3.0 | 2.7 | 3.0 | 3.9 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material and Drug
3.3. Extraction, Isolation and Identification
3.4. Structural Analyses
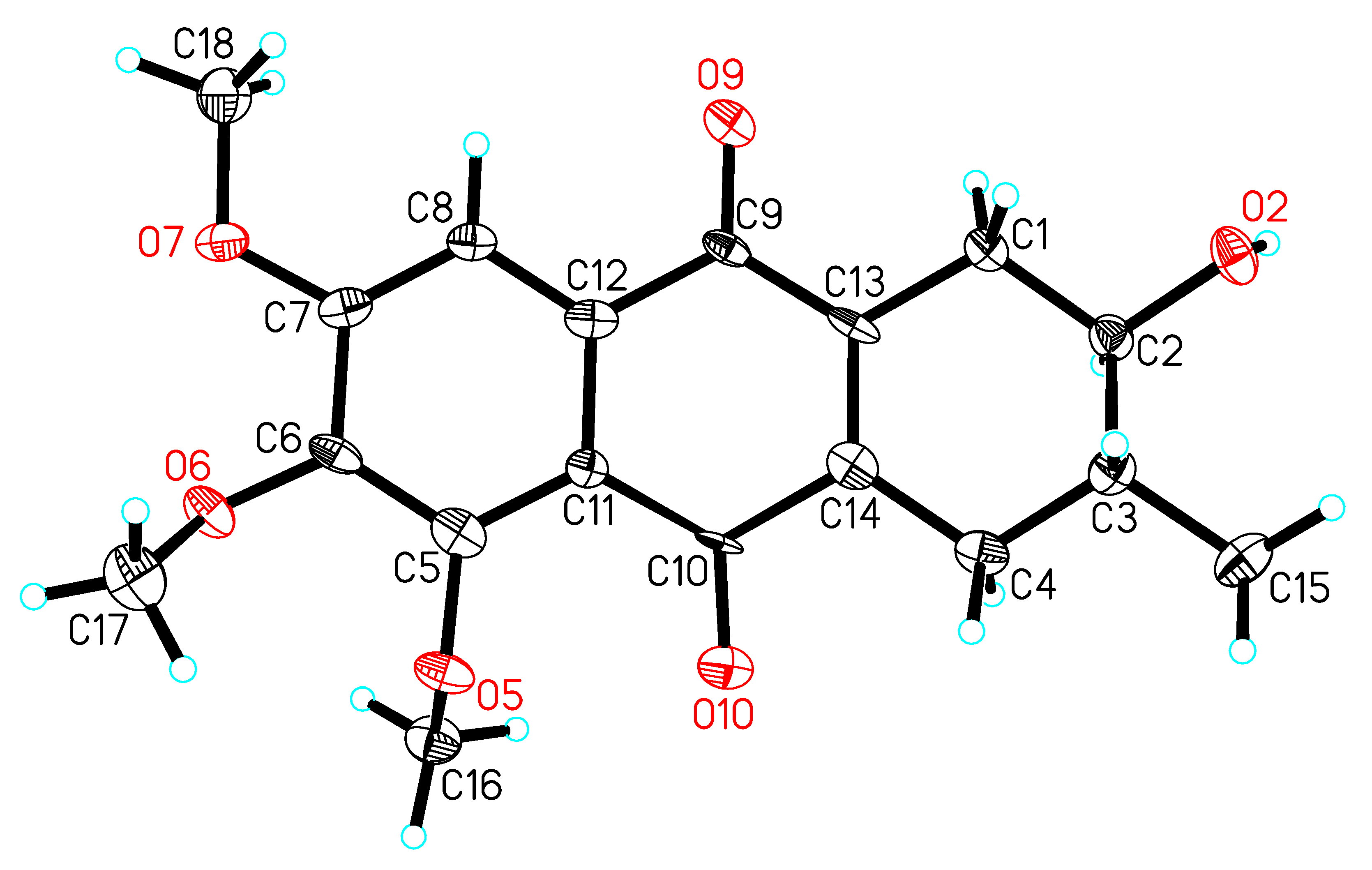
3.5. Crystal Data for 1
3.6. Esterification of Prisconnatanone C (1) with (R)- and (S)-MTPA
3.7. Tumor Cell Cultures
3.8. Cell Viability Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Compendium. Compendium of Chinese Traditional Herbal Drugs; People’s Health Press: Beijing, China, 1992; p. 772. [Google Scholar]
- Zhou, C.Y.; Deng, J.G. Progress in chemical and pharmacological studies on Prismatomeris tetrandra. J. Guangxi Tradit. Chin. Med. Univ. 2006, 9, 90–92. [Google Scholar]
- Ruan, Y.Z. Notes on the Genus Prismatomeris Thw. (Rubiaceae) of China. J. Syst. Evol. (Acta Phytotax Sin.) 1988, 26, 443–449. [Google Scholar]
- Tu, D.J.; Pang, Z.H.; Bi, N.J. Studies on constituents of Prismatomeris tetrandra (Roxb) K Schum. Yao Xue Xue Bao 1981, 16, 631–634. [Google Scholar] [PubMed]
- Jiang, J.S.; Feng, Z.M.; Zhang, P.C. Chemical constituents from root of Prismatomeris tetrandra. Zhongguo Zhong Yao Za Zhi 2005, 30, 1751–1753. [Google Scholar] [PubMed]
- Feng, Z.M.; Jiang, J.S.; Wang, Y.H.; Zhang, P.C. Anthraquinones from the roots of Prismatomeris tetrandra. Chem. Pharm. Bull. 2005, 53, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.L.; Guan, H.; Xi, P.Z.; Deng, T.; Gao, J.M. Anthraquinones from the roots of Prismatomeris tetrandra. Nat. Prod. Commun. 2010, 5, 1251–1252. [Google Scholar] [PubMed]
- Hao, J.; Feng, S.X.; Qiu, S.X.; Chen, T. Anthraquinone glycosides from the roots of Prismatomeris connata. Chin. J. Nat. Med. 2011, 8, 42–45. [Google Scholar] [CrossRef]
- Feng, S.X.; Hao, J.; Chen, T.; Qiu, S.X. A new anthraquinone and two new tetrahydroanthraquinone from the roots of Prismatomeris connata. Helv. Chim. Acta 2011, 94, 1843–1849. [Google Scholar] [CrossRef]
- Feng, S.; Bai, J.; Qiu, S.; Li, Y.; Chen, T. Iridoid and phenolic glycosides from the roots of Prismatomeris connata. Nat. Prod. Commun. 2012, 7, 561–562. [Google Scholar] [PubMed]
- Suemitsu, R.; Yamada, Y.; Sano, T.; Yamashita, K. Phytotoxic activities of altersolanol-a, Altersolanol-b and dactylariol, and activities of altersolanol-a against some microorganisms. Agric. Biol. Chem. Tokyo 1984, 48, 2383–2384. [Google Scholar] [CrossRef]
- Ardizzoni, A.; Boni, L.; Tiseo, M.; Fossella, F.V.; Schiller, J.H.; Paesmans, M.; Radosavlievic, D.; Paccaqnella, A.; Zatloukal, P.; Mazzanti, P.; et al. Cisplatin-versus carboplatin-based chemotherapy in first-line treatment of advanced non-small-cell lung cancer: An individual patient data meta-analysis. J. Natl. Cancer Inst. 2007, 99, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.X.; Guan, Q.; Chen, T.; Du, C. In vitro activities of 3-hydroxy-1,5,6-trimethoxy-2-methyl-9,10-anthraquinone against non-small cell lung carcinoma. Arch. Pharm. Res. 2012, 35, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Bliss, C. The calculation of the dose-mortality curve. Ann. Appl. Biol. 1935, 22, 134–167. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–7 are available from Dr. Tao Chen upon request and certain restrictions may apply.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Ding, X.; Feng, S.-X.; Guan, Q.; Zhang, X.-P.; Du, C.; Di, Y.-T.; Chen, T. Seven New Tetrahydroanthraquinones from the Root of Prismatomeris connata and Their Cytotoxicity against Lung Tumor Cell Growth. Molecules 2015, 20, 22565-22577. https://doi.org/10.3390/molecules201219856
Wang C, Ding X, Feng S-X, Guan Q, Zhang X-P, Du C, Di Y-T, Chen T. Seven New Tetrahydroanthraquinones from the Root of Prismatomeris connata and Their Cytotoxicity against Lung Tumor Cell Growth. Molecules. 2015; 20(12):22565-22577. https://doi.org/10.3390/molecules201219856
Chicago/Turabian StyleWang, Chunxiang, Xiao Ding, Shi-Xiu Feng, Qiunong Guan, Xiao-Ping Zhang, Caigan Du, Ying-Tong Di, and Tao Chen. 2015. "Seven New Tetrahydroanthraquinones from the Root of Prismatomeris connata and Their Cytotoxicity against Lung Tumor Cell Growth" Molecules 20, no. 12: 22565-22577. https://doi.org/10.3390/molecules201219856