
Synthesis of New 2-Halo-2-(1H-tetrazol-5-yl)-2H-azirines via a Non-Classical Wittig Reaction

 ,
, 
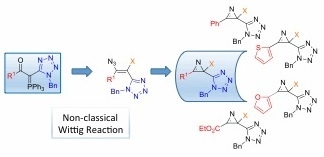
Abstract
:
1. Introduction
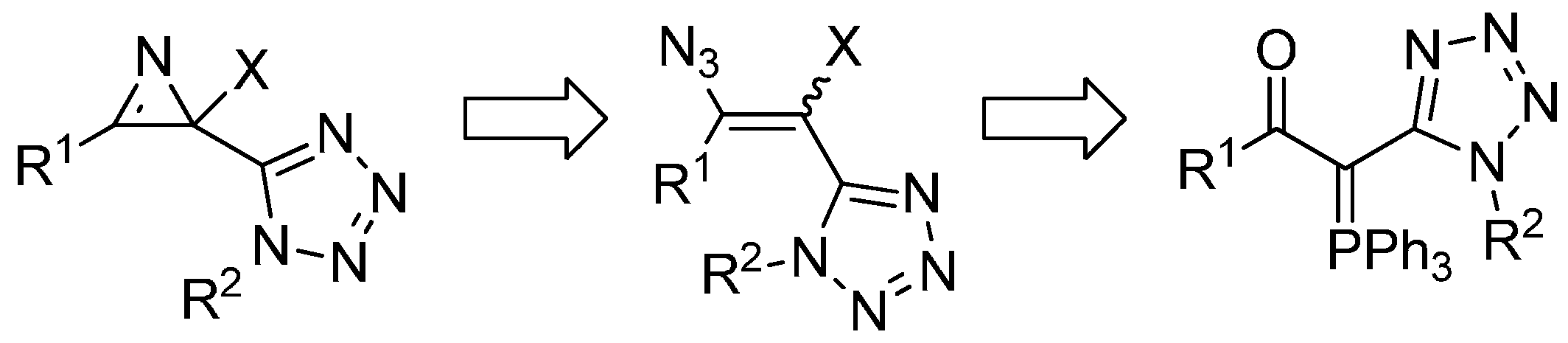
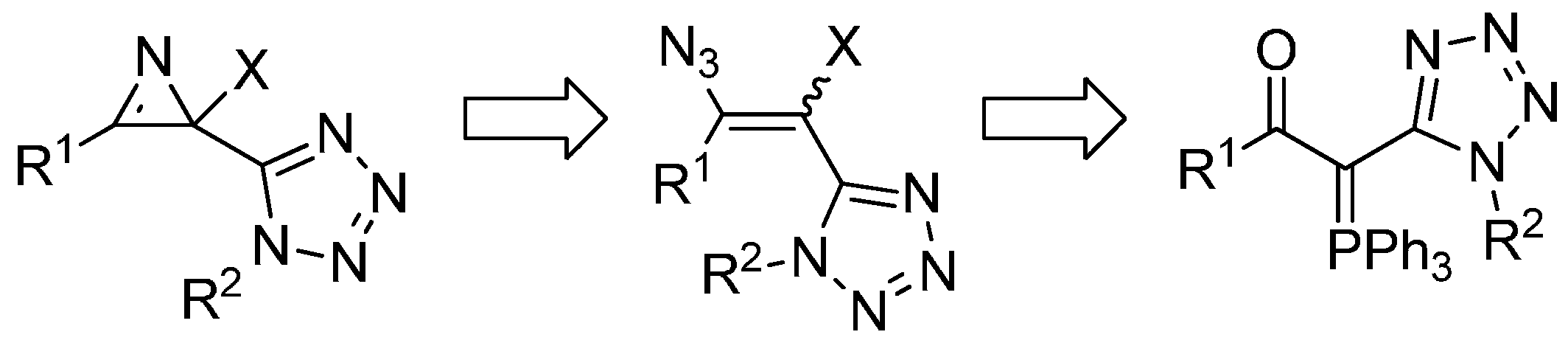
2. Results and Discussion

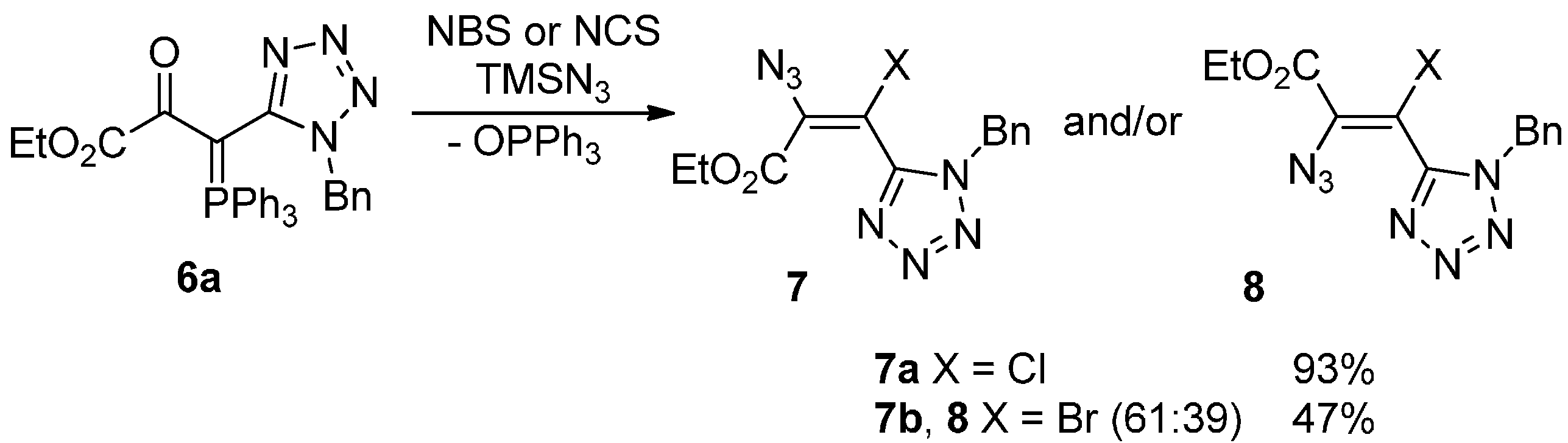
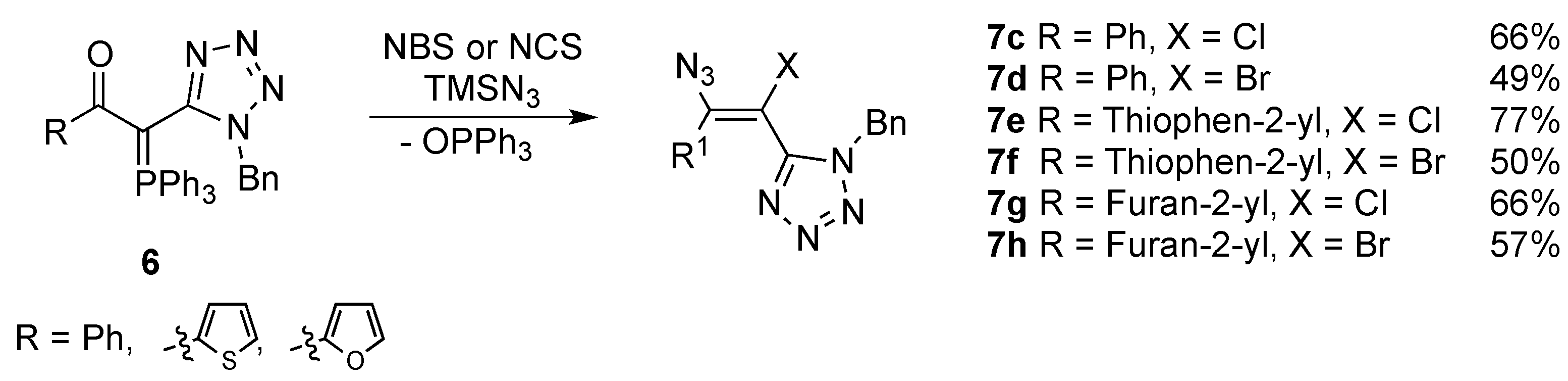
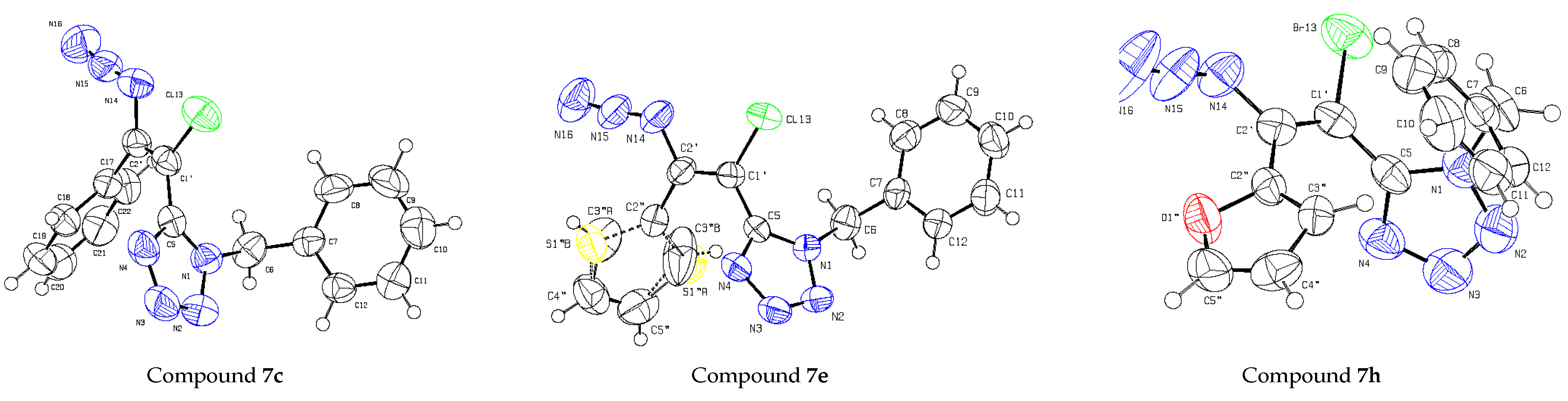
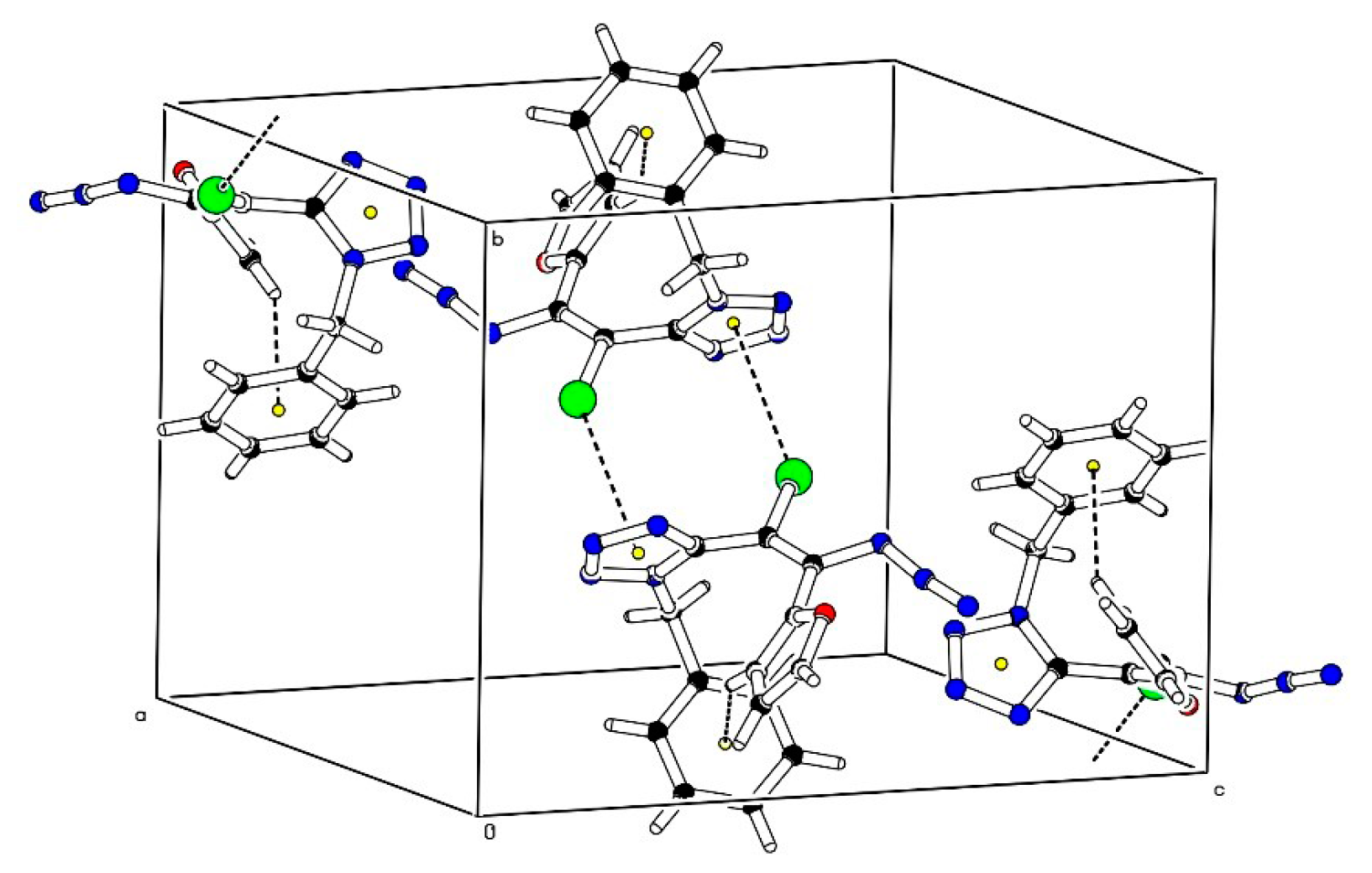

{kind=link}
{kind=link}
{kind=link}
{kind=link}
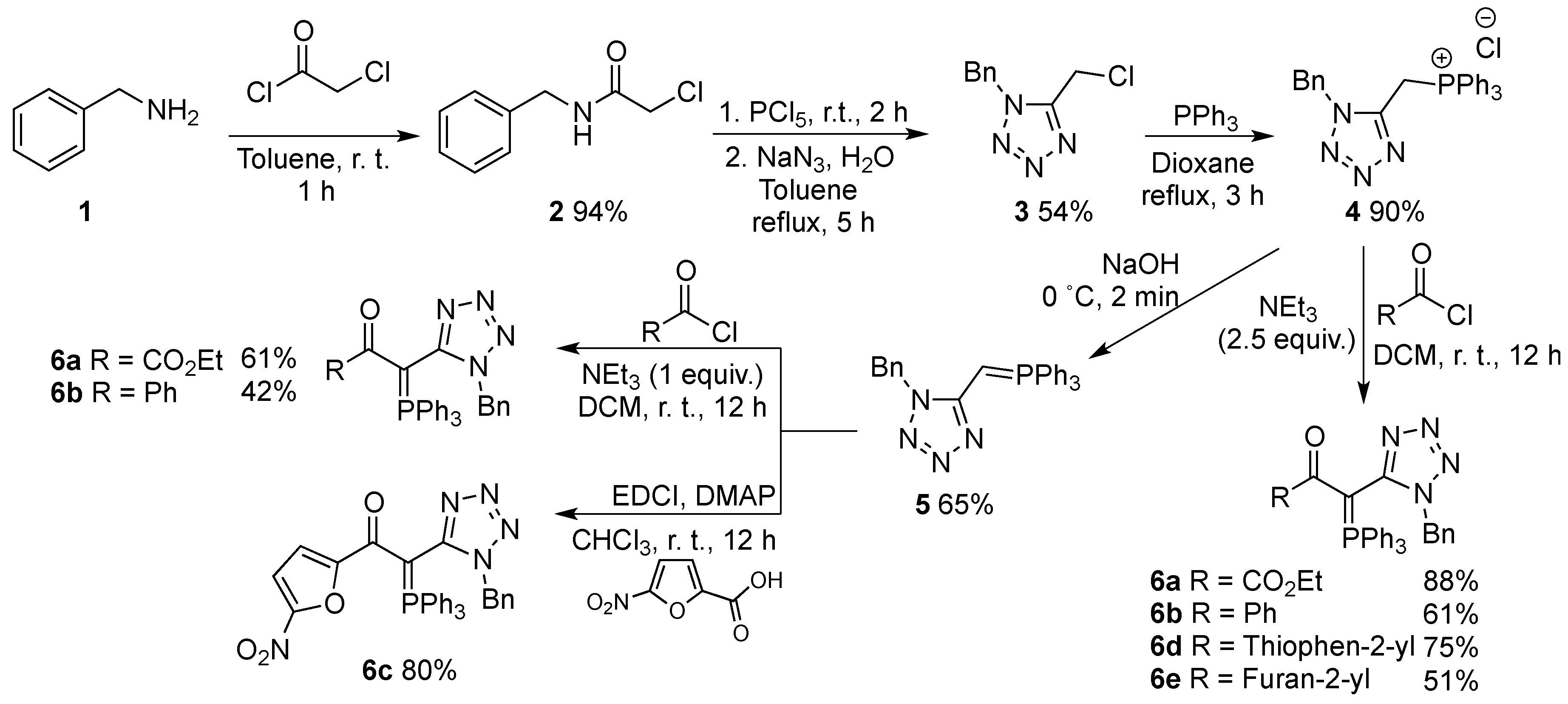{kind=link}
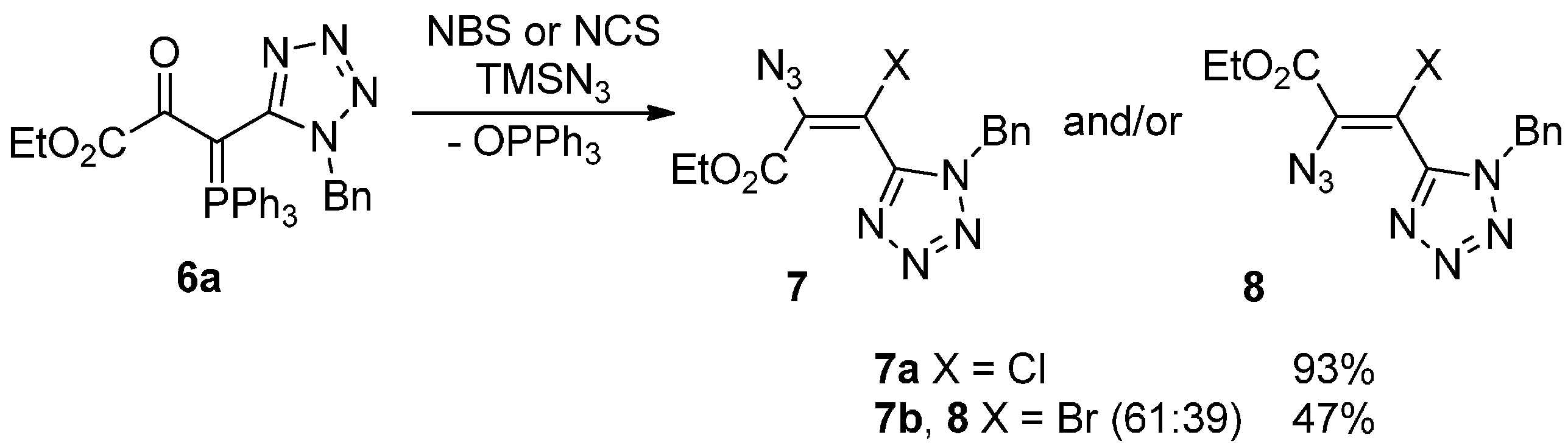{kind=link}
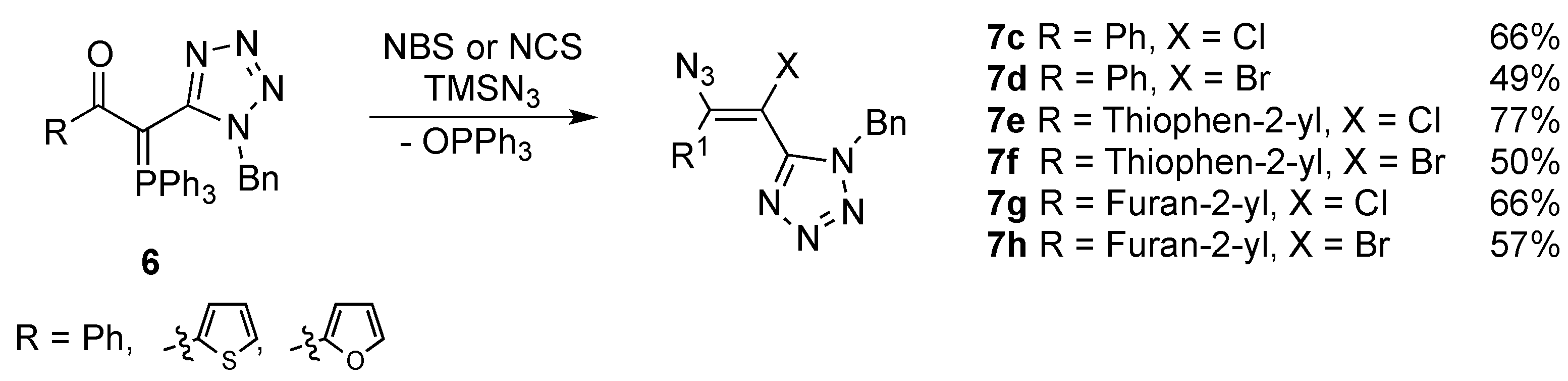{kind=link}
{kind=link}
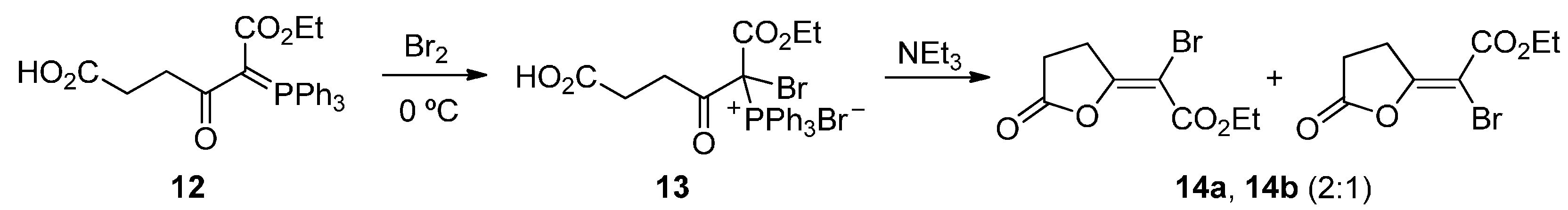{kind=link}
{kind=link}
{kind=link}
| Bond Distances or Angles | 7c | 7h | 7e |
|---|---|---|---|
| C1′–C2′ | 1.331(3) | 1.333(5) | 1.334(4) |
| C2′–N14 | 1.410(3) | 1.409(4) | 1.407(4) |
| C1′–X13 | 1.727(2) | 1.889(3) | 1.718(3) |
| C1′–C5 | 1.460(4) | 1.448(5) | 1.458(2) |
| N14–C2′–C1′ | 116.9(2) | 118.4(3) | 116.9(3) |
| C2′–C1′–X | 122.5(2) | 120.6(3) | 122.0(2) |
| C2′–C1′–C5 | 122.7(2) | 124.8(3) | 122.9(2) |
| C5–C1′–X | 114.9(2) | 114.5(2) | 114.9(2) |
| N14–C2′–C1′–C5 | −179.0(2) | −170.0(3) | 171.1(3) |
| N14–C2′–C1′–X | 0.2(3) | 5.9(4) | −3.4(4) |
| C5–N1–C6–C7 | 94.2(3) | 74.4(5) | 100.7(3) |
| N1–C6–C7–C8 | −105.3(3) | −104.6(4) | −117.7(3) |
| X–C1′–C5–N1 | 77.5(3) | 66.5(4) | −71.6(3) |
| C2′–C1′–C5–N1 | 76.1(3) | −117.4(4) | 113.4(3) |

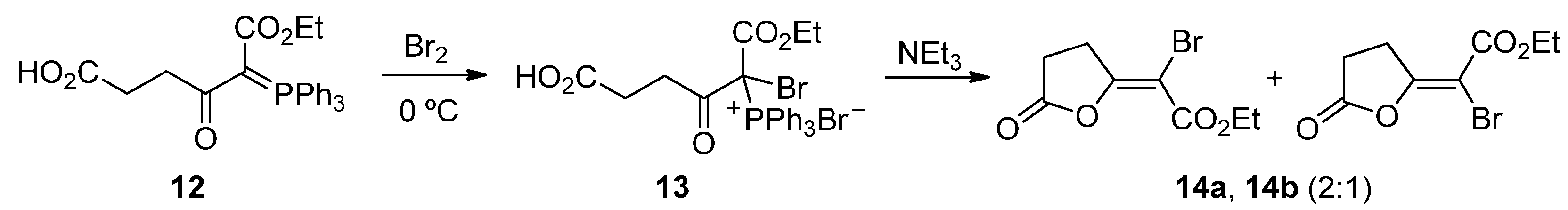
| Alkene | C–X | C–N3 | 2H-Azirine | C-2 | C-3 |
|---|---|---|---|---|---|
| 7a | 109.3 | 135.3 | 15a | 51.9 | 156.8 |
| 7b | 97.8 | 137.3 | 15b | 40.7 | 157.4 |
| 7c | 98.5 | 145.4 | 15c | 46.4 | 168.6 |
| 7d | 85.8 | 147.6 | 15d | 33.8 | 169.6 |
| 7e | 100.4 | 138.6 | 15e | 47.0 | 161.7 |
| 7f | 87.9 | 140.7 | 15f | 34.5 | 162.7 |
| 7g | 99.8 | 134.8 | 15g | 46.1 | 157.7 |
| 7h | 87.2 | 136.8 | 15h | 33.3 | 158.5 |
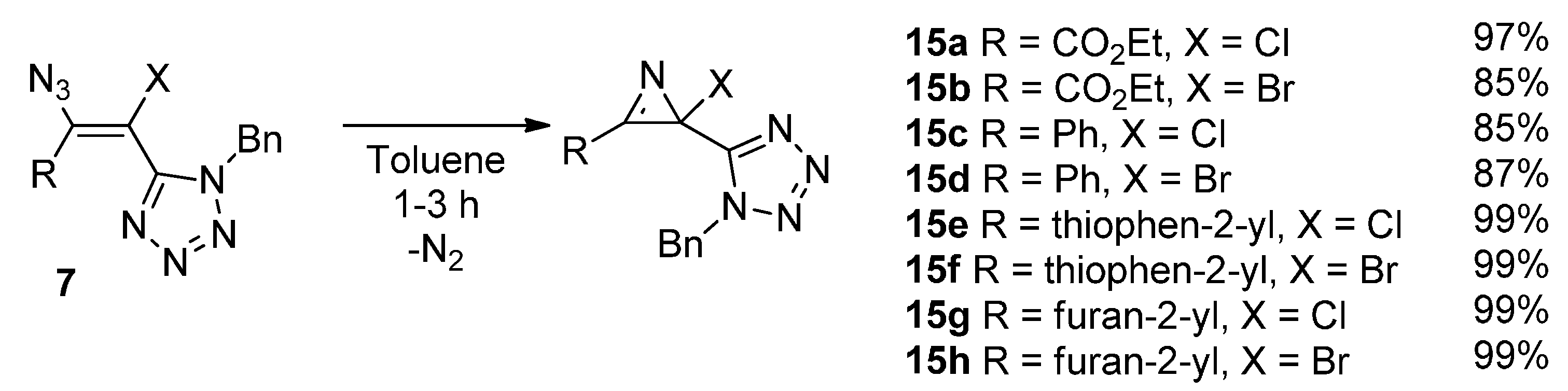
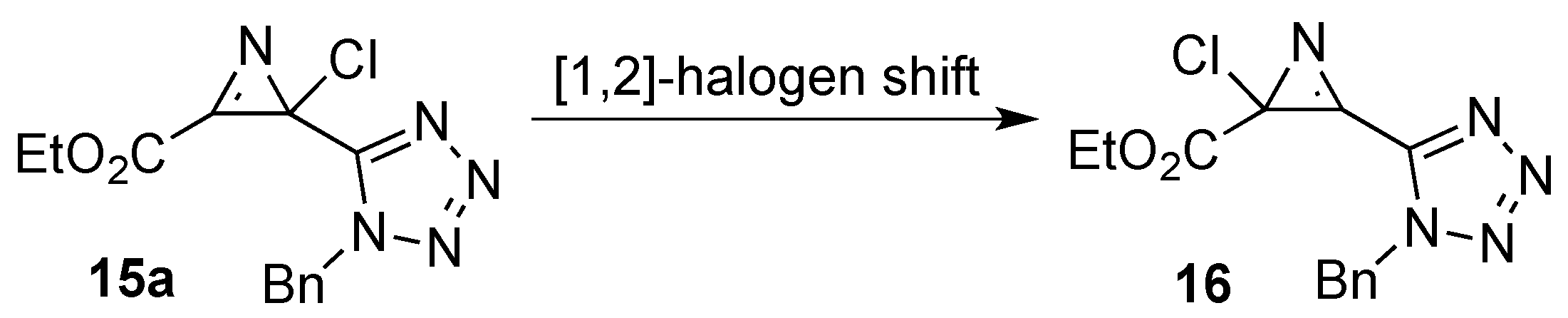
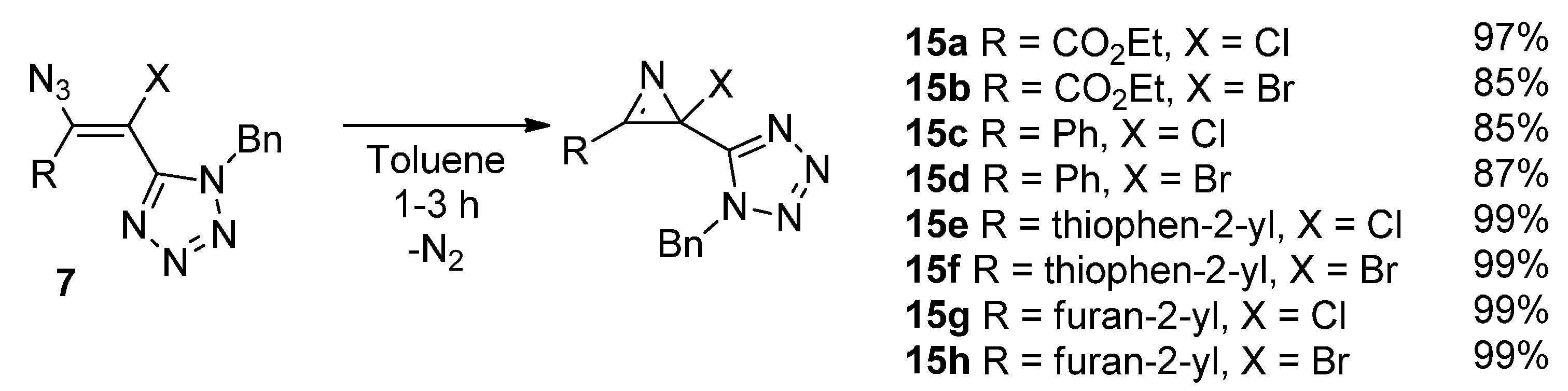
3. Experimental Section
3.1. General Information
3.2. Experimental Details
3.2.1. Synthesis of Phosphorus Ylide 5
3.2.2. General Procedure for the Synthesis of Ylides 6
3.2.3. General Procedure for the Synthesis of Haloazidoalkenes 7 and 8
3.2.4. General Procedure for the Synthesis of 2-Chloro- and 2-Bromo-2H-azirines 15
3.2.5. X-ray Crystallography Structure Determination
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khlebnikov, A.F.; Novikov, M.S. Recent advances in 2H-azirine chemistry. Tetrahedron 2013, 69, 3363–3401. [Google Scholar] [CrossRef]
- Khlebnikov, A.F.; Novikov, M.S. Ring expansions of azirines and azetines. In Topics in Heterocylicic Chemistry; Maes, B.U.W., Cossy, J., Slovenko, P., Eds.; Springer Berlin Heidelberg: Berlin, Germany, 2015; pp. 1–90. [Google Scholar]
- Pinho e Melo, T.M.V.D.; Gonsalves, A.M.R. Exploiting 2-halo-2H-azirine chemistry. Curr. Org. Synth. 2004, 1, 275–292. [Google Scholar] [CrossRef]
- Palacios, F.; de Retana, A.M.O.; de Marigorta, E.M.; de los Santos, J.M. 2H-Azirines as synthetic tools in organic chemistry. Eur. J. Org. Chem. 2001, 13, 2401–2414. [Google Scholar] [CrossRef]
- Padwa, A. Cycloaddition and cyclization chemistry of 2H-azirines. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2010; Volume 99, pp. 1–31. [Google Scholar]
- Padwa, A. Aziridines and azirines: Monocyclic. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2008; Volume 1, pp. 1–104. [Google Scholar]
- Gonsalves, A.M.d′A.R.; Cabral, A.M.T.D.P.V.; Pinho e Melo, T.M.V.D.; Gilchrist, T.L. The reaction of an α-oxophosphonium ylide with halogens: 2,3-Disubstituted diethyl butenedioates from diethyl 2-oxo-3-triphenylphosphoranylidenebutanedioate. Synthesis 1997, 673–676. [Google Scholar] [CrossRef]
- Pinho e Melo, T.M.V.D.; Gonsalves, A.M.R.; Lopes, C.S.J.; Gilchrist, T.L. Synthesis of 2-halo-2H-azirines from phosphorus ylides. Tetrahedron Lett. 1999, 40, 789–792. [Google Scholar] [CrossRef]
- Pinho e Melo, T.M.V.D.; Lopes, C.S.J.; Gonsalves, A.M.R. Synthesis and reactivity of 2-halo-2H-azirines towards nucleophiles. Tetrahedron Lett. 2000, 41, 7217–7220. [Google Scholar] [CrossRef]
- Pinho e Melo, T.M.V.D.; Lopes, C.S.J.; Cardoso, A.L.; Gonsalves, A.M.R. Synthesis of 2-halo-2H-azirines. Tetrahedron 2001, 57, 6203–6208. [Google Scholar] [CrossRef]
- Fotsing, J.R.; Banert, K. Reactions of unsaturated azides, part 17: An efficient strategy for the synthesis of small–ring heterocycles via isomerization of 2-halo-2H-azirines. Synthesis 2006, 261–272. [Google Scholar] [CrossRef]
- Alonso-Cruz, C.R.; Kennedy, A.R.; Rodríguez, M.S.; Suárez, E. Alkoxyl radical fragmentation of 3-azido-2,3-dideoxy-2-halo-hexopyranoses: A new entry to chiral polyhydroxylated 2-azido-1-halo-1-alkenes. Tetrahedron Lett. 2007, 48, 7207–7210. [Google Scholar] [CrossRef]
- Alonso-Cruz, C.R.; Kennedy, A.R.; Rodríguez, M.S.; Suárez, E. Synthesis of polyhydroxylated 2H-azirines and 2-halo-2H-azirines from 3-azido-2,3-dideoxyhexopyranoses by alkoxyl radical fragmentation. Eur. J. Org. Chem. 2008, 73, 4116–4122. [Google Scholar] [CrossRef] [PubMed]
- Banert, K.; Hagedorn, M.; Wutke, J.; Ecochard, P.; Schaarschmidt, D.; Lang, H. Elusive ethynyl azides: Trapping by 1,3-dipolar cycloaddition and decomposition to cyanocarbenes. Chem. Commun. 2010, 46. [Google Scholar] [CrossRef]
- Pinho e Melo, T.M.V.D.; Lopes, C.S.J.; Beja, A.M.; Paixão, J.A.; Silva, M.R.; Veiga, L.A.; Gonsalves, A.M.R. Reactivity of 2-halo-2H-azirines. 1. Reactions with nucleophiles. J. Org. Chem. 2002, 67, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Fotsing, J.R.; Banert, K. New way to methylene-2H-azirines and their use as powerful intermediates for the stereo- and regioselective synthesis of compounds with vinylamine substructure. Eur. J. Org. Chem. 2006, 3617–3625. [Google Scholar] [CrossRef]
- Lopes, S.; Nunes, C.M.; Fausto, R.; Pinho e Melo, T.M.V.D. 4-Halo-1,3-oxazoles: Unambiguous structural assignment of 2-halo-2-benzoyl-2H-azirine-3-carboxylates thermal ring expansion products. J. Mol. Struct. 2009, 919, 47–53. [Google Scholar] [CrossRef]
- Shah, S.R.; Navathe, S.S.; Dikundwar, A.G.; Row, T.N.G.; Vasella, A.T. Thermal rearrangement of azido ketones into oxazoles via azirines: One-pot, metal-free heteroannulation to functionalized 1,3-oxazoles. Eur. J. Org. Chem. 2013, 264–267. [Google Scholar] [CrossRef]
- Novikov, M.S.; Smetanin, I.A.; Khlebnikov, A.F.; Rostovskii, N.V.; Yufit, D.S. Synthesis of electron-poor 4-halo-2-azabuta-1,3-dienes by Rh(II)-catalyzed diazo ester-azirine coupling. 2-Azabuta-1,3-diene-2,3-dihydroazete valence isomerism. Tetrahedron Lett. 2012, 53, 5777–5780. [Google Scholar] [CrossRef]
- Smetanin, I.A.; Novikov, M.S.; Rostovskii, N.V.; Khlebnikov, A.F.; Starova, G.L.; Yufit, D.S. 4-Halo-2-azabuta-1,3-dienes as Intermediates in the rhodium carbenoid-initiated transformation of 2-halo-2H-azirines into 2,3-dihydroazetes and 2,5-dihydrooxazoles. Tetrahedron 2015, 71, 4616–4628. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Gimeno, L.; Lemos, A.; Palacios, F.; Pinho e Melo, T.M.V.D. The neber approach to 2-(tetrazol-5-yl)-2H-azirines. J. Org. Chem. 2013, 78, 6983–6991. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.L.; Lemos, A.; Pinho e Melo, T.M.V.D. Selective synthesis of tetrasubstituted 4-(tetrazol-5-yl)-1H-imidazoles from 2-(tetrazol-5-yl)-2H-azirines. Eur. J. Org. Chem. 2014, 24, 5159–5165. [Google Scholar] [CrossRef]
- Harvill, E.K.; Herbst, R.M.; Schreiner, E.G. Haloalkyltetrazole and aminoalkyltetrazole derivatives. J. Org. Chem. 1952, 17, 1597–1616. [Google Scholar] [CrossRef]
- Uchida, M.; Komatsu, M.; Morita, S.; Kanbe, T.; Nakagawa, K. Studies on gastric antiulcer active agents. II. Synthesis of tetrazole alkanamides and related compounds. Chem. Pharm. Bull. 1989, 37, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Kawano, Y.; Natsumé, S.; Tanaka, T.; Watanabe, T.; Nagano, M.; Sugawara, S.-I.; Miyadera, T. Synthesis of highly antibacterial 3-vinylcephalosporin derivatives. Chem. Pharm. Bull. 1978, 26, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Grodner, J.; Salaciński, T. Synthesis of new tetrazol-5-ylalkenyl derivatives of ferrocene by wittig olefination. Synthesis 2012, 44, 3071–3076. [Google Scholar] [CrossRef]
- Denney, D.B.; Ross, S.T. The preparation and reaction of some halophosphoranes. J. Org. Chem. 1962, 27, 998–1000. [Google Scholar] [CrossRef]
- Abell, A.D.; Murphy, P.J.; Hoult, D.A.; Morris, K.M.; Taylor, J.M.; Trent, J.O. Halogenation of keto acid phosphoranes: Synthesis of halo enol lactones and haloallenes. J. Org. Chem. 1993, 58, 1531–1537. [Google Scholar] [CrossRef]
- Abell, A.D.; Taylor, J.M. Synthesis of cyclic acylated enamino ester dipeptide analogues via the bromolactonization of a keto acid phosphorane. J. Org. Chem. 1993, 58, 14–15. [Google Scholar] [CrossRef]
- Kayser, M.M.; Zhu, J.; Hooper, D.L. On the synthesis and the mechanism of formation of halogenated enol lactones. Can. J. Chem. 1997, 75, 1322–1330. [Google Scholar] [CrossRef]
- Murphy, P.J.; Lee, S.E. Recent synthetic applications of the non-classical wittig reaction. J. Chem. Soc. Perkin Trans. 1 1999, 3049–3066. [Google Scholar] [CrossRef]
- Ciabattoni, J.; Cabell, M., Jr. 3-Chloro-azirines. Photochemical formation and thermal isomerization. J. Am. Chem. Soc. 1971, 93, 1482–1483. [Google Scholar] [CrossRef]
- Padwa, A.; Blacklock, T.J.; Carlsen, P.H.J.; Pulwer, M.J. Synthetic approaches toward the bi(2H-azirine) system. J. Org. Chem. 1979, 44, 3281–3287. [Google Scholar] [CrossRef]
- Wasserman, H.H.; Ennis, D.S.; Blum, C.A.; Rotello, V.M. The conversion of carboxylic acids to keto phosphorane precursors of 1,2,3-vicinal tricarbonyl compounds. Tetrahedron Lett. 1992, 33, 6003–6006. [Google Scholar] [CrossRef]
- Bruker, APEX2. In SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Spek, A. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, A.L.; Sousa, C.; Henriques, M.S.C.; Paixão, J.A.; Pinho e Melo, T.M.V.D. Synthesis of New 2-Halo-2-(1H-tetrazol-5-yl)-2H-azirines via a Non-Classical Wittig Reaction. Molecules 2015, 20, 22351-22363. https://doi.org/10.3390/molecules201219848
Cardoso AL, Sousa C, Henriques MSC, Paixão JA, Pinho e Melo TMVD. Synthesis of New 2-Halo-2-(1H-tetrazol-5-yl)-2H-azirines via a Non-Classical Wittig Reaction. Molecules. 2015; 20(12):22351-22363. https://doi.org/10.3390/molecules201219848
Chicago/Turabian StyleCardoso, Ana L., Carmo Sousa, Marta S. C. Henriques, José A. Paixão, and Teresa M. V. D. Pinho e Melo. 2015. "Synthesis of New 2-Halo-2-(1H-tetrazol-5-yl)-2H-azirines via a Non-Classical Wittig Reaction" Molecules 20, no. 12: 22351-22363. https://doi.org/10.3390/molecules201219848