
Stereo- and Regiocontrolled Syntheses of Exomethylenic Cyclohexane β-Amino Acid Derivatives



Abstract
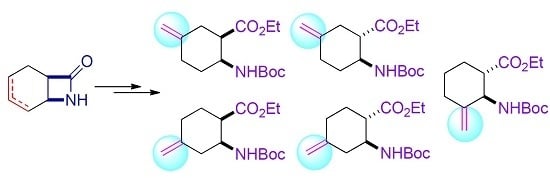
:
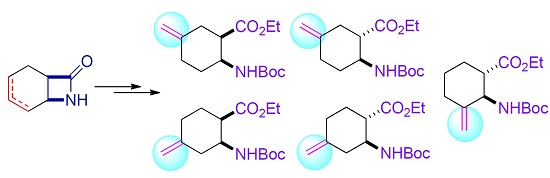{kind=link}
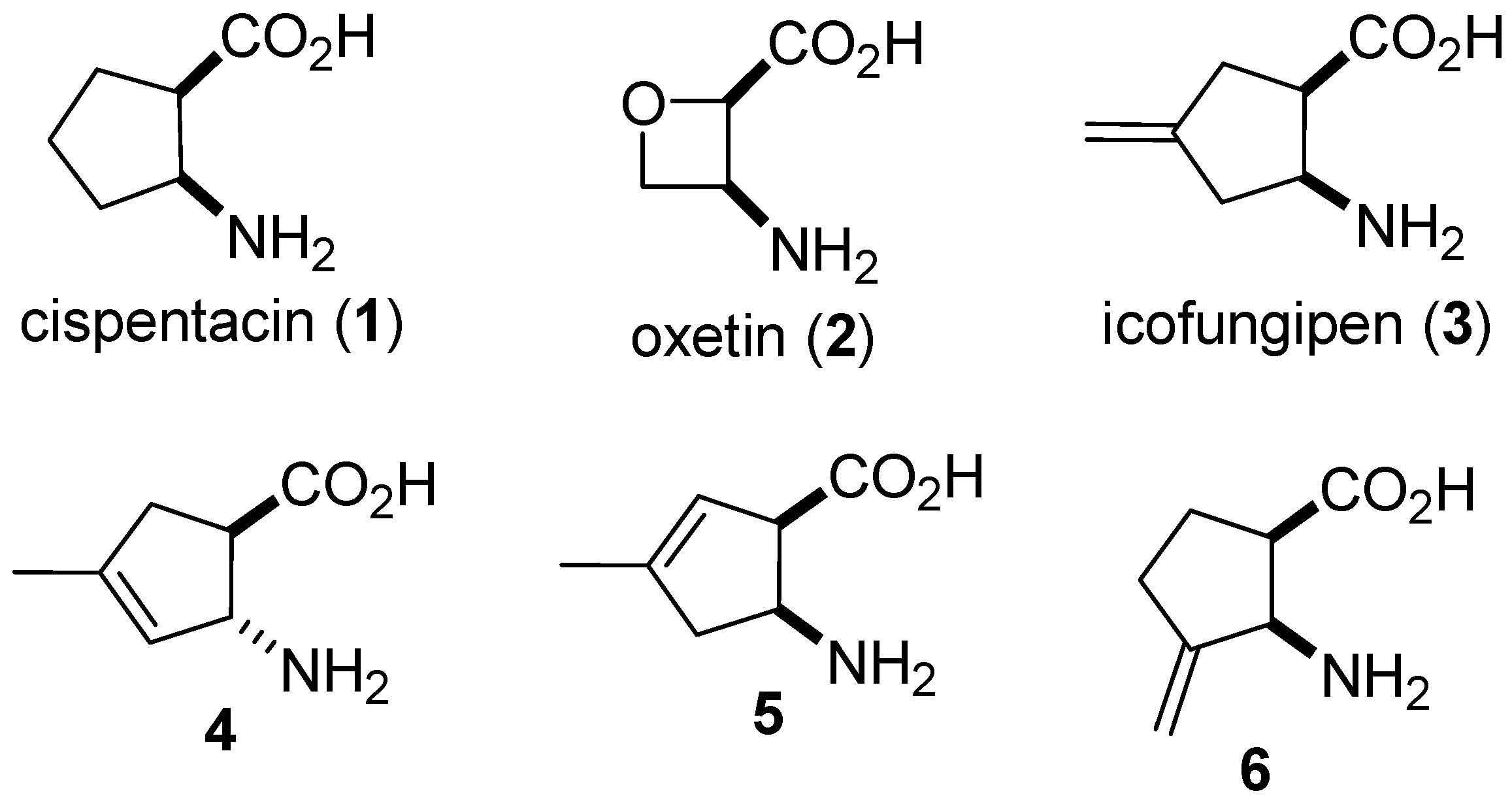{kind=link}
{kind=link}
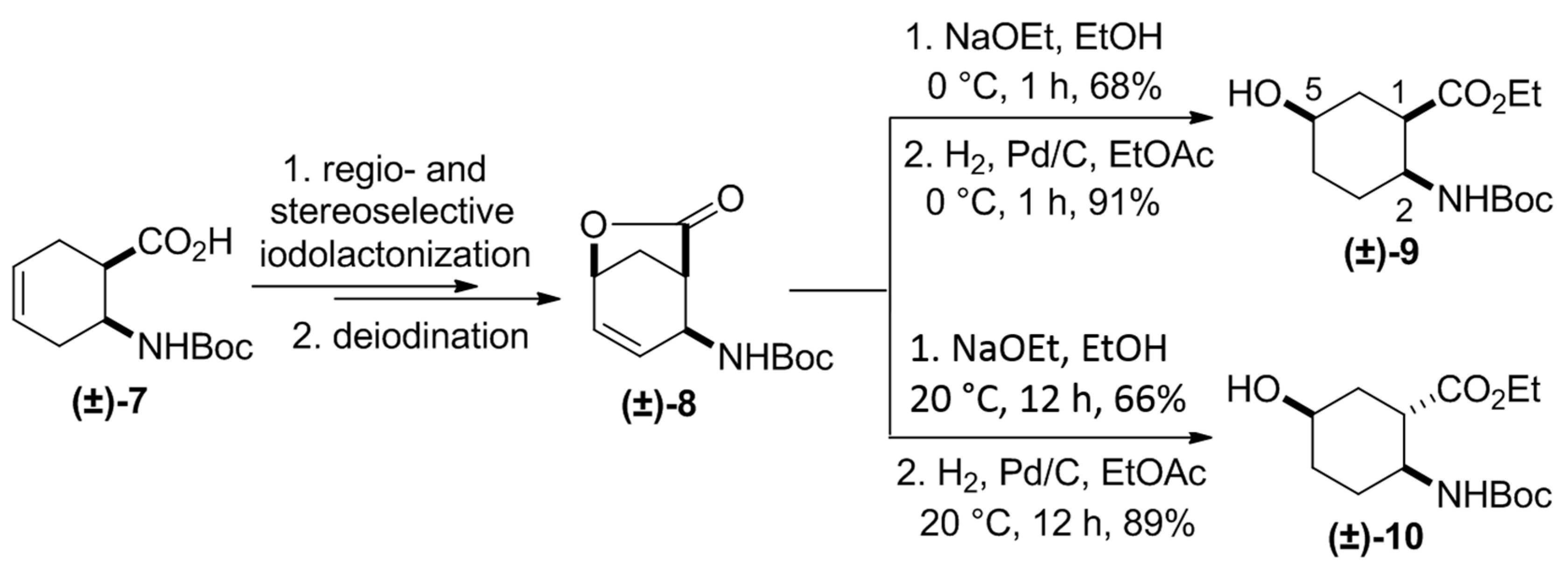{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
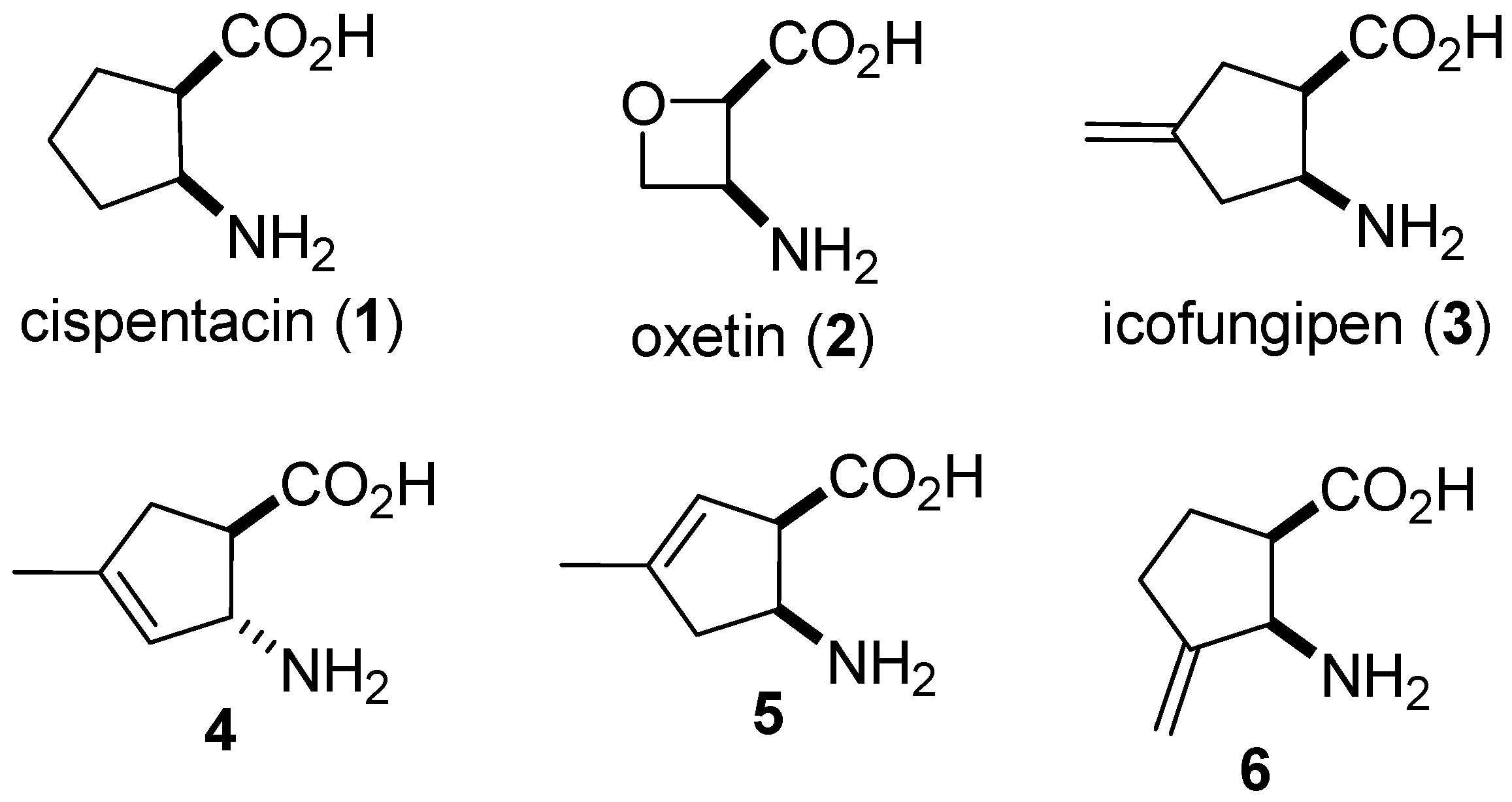
1. Introduction
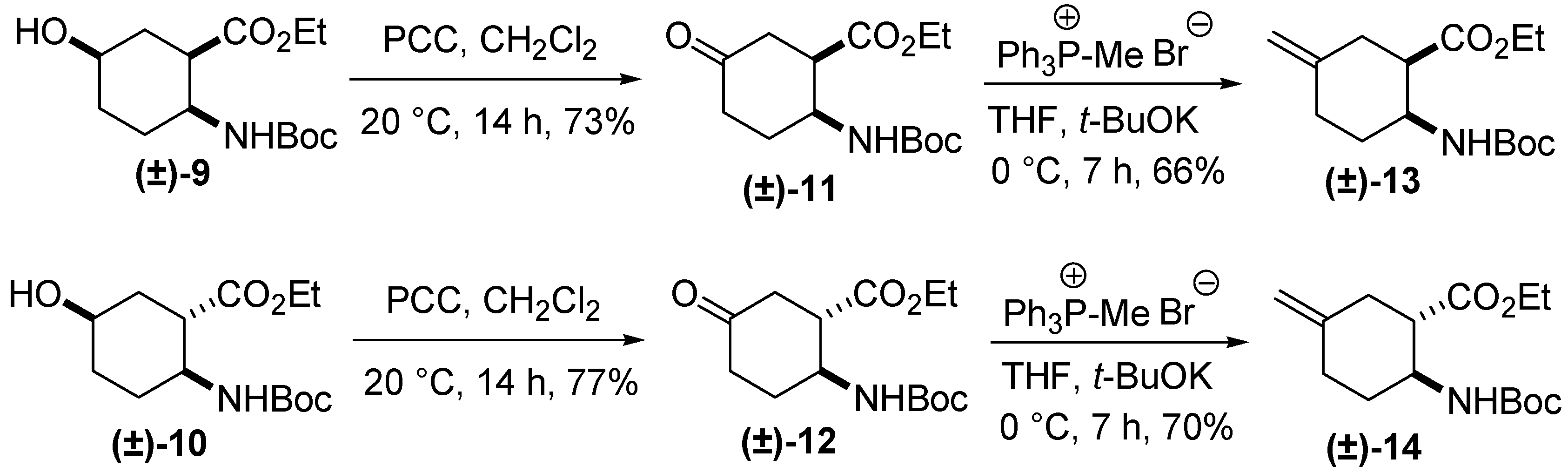
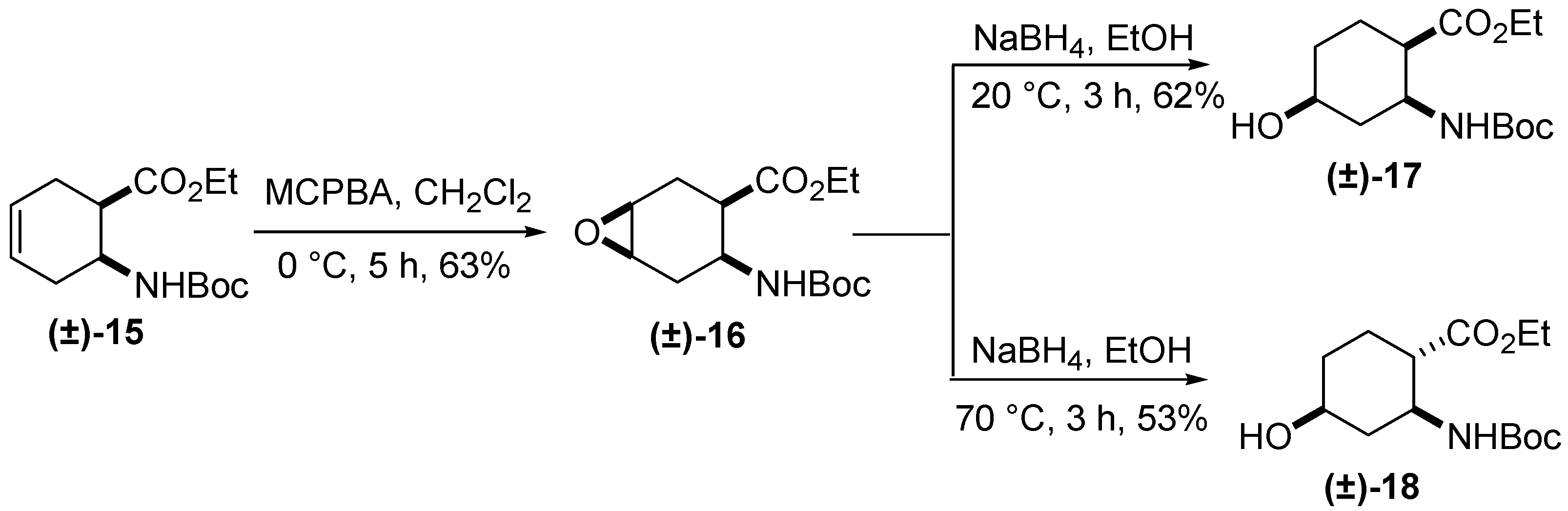
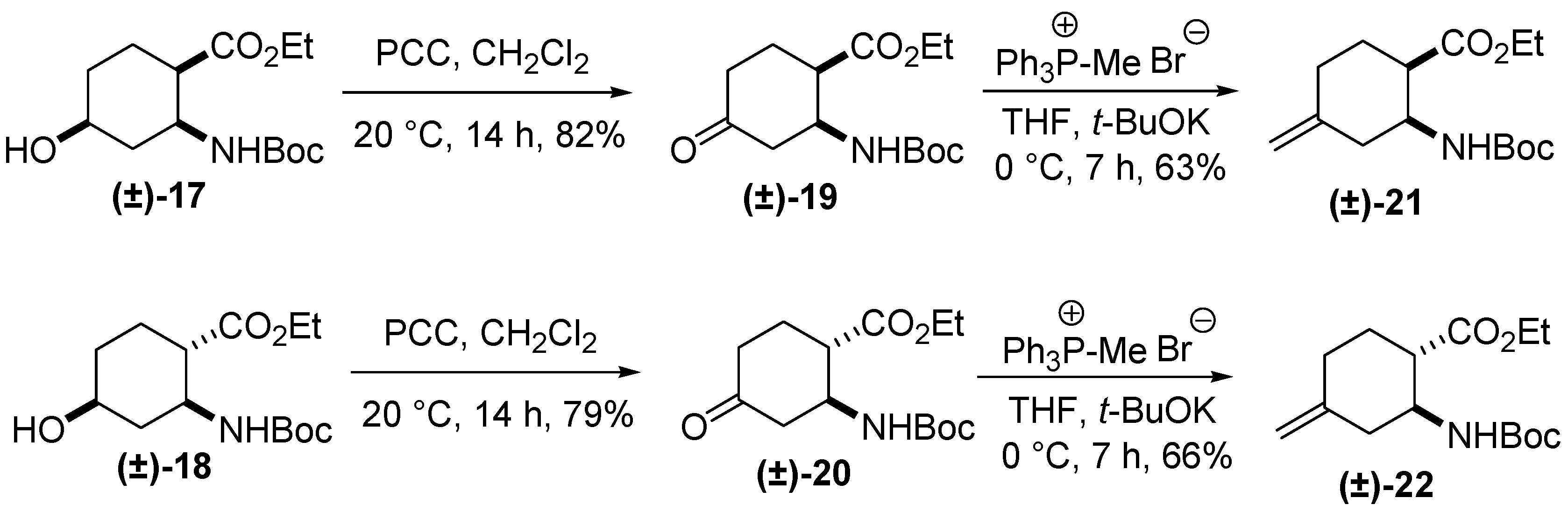
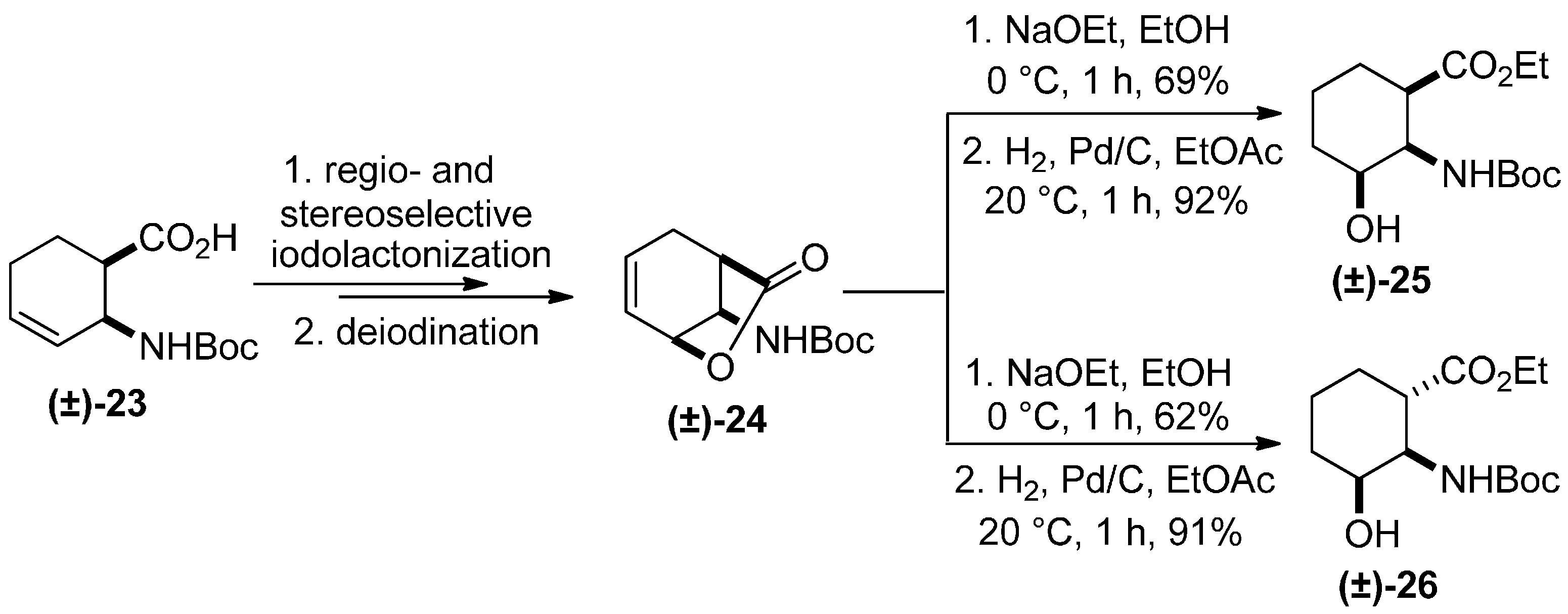
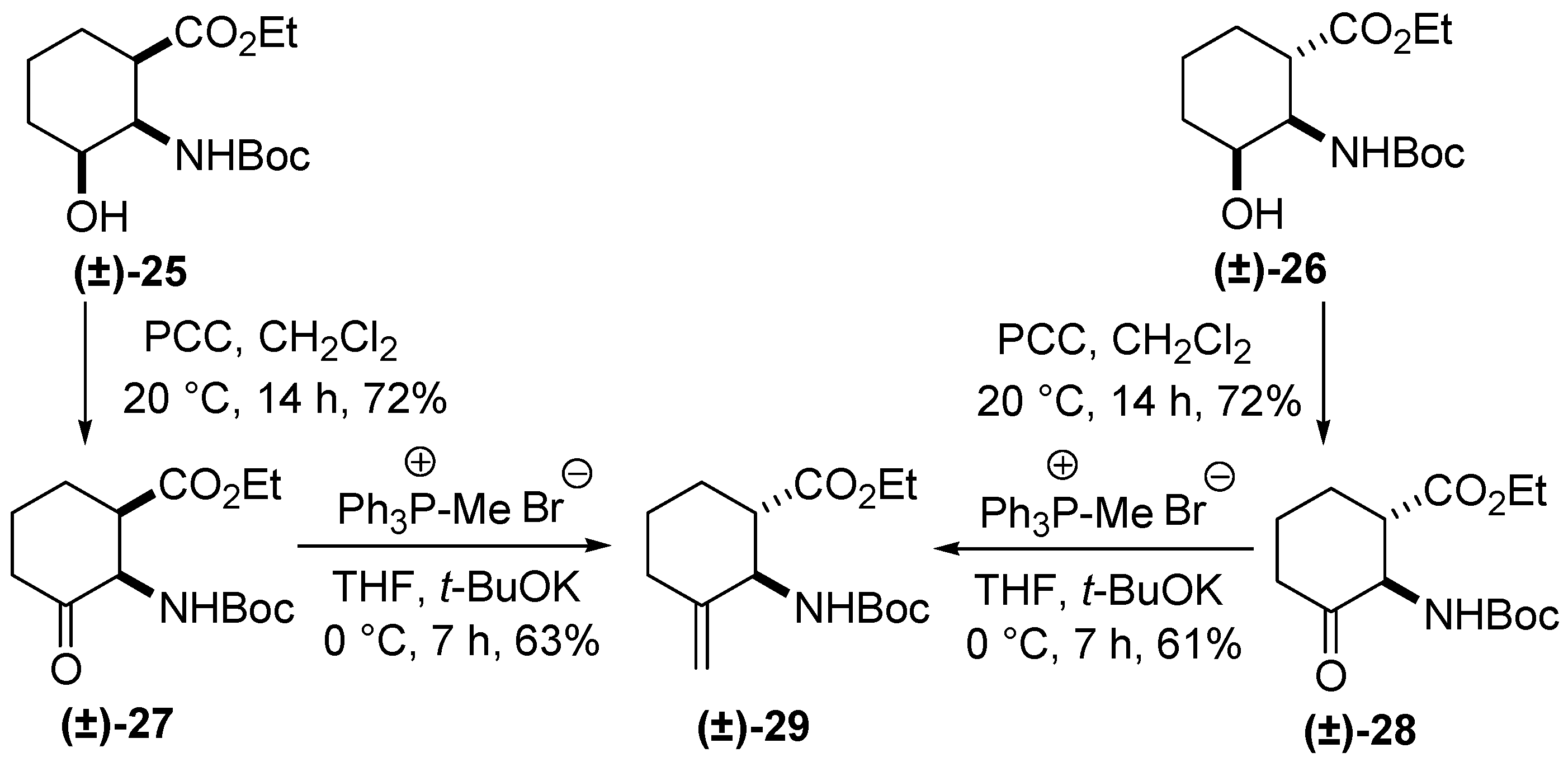
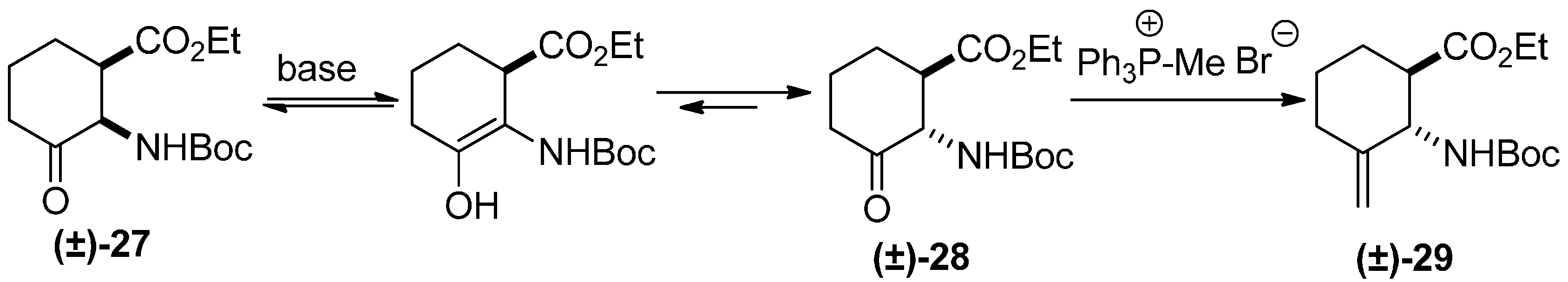
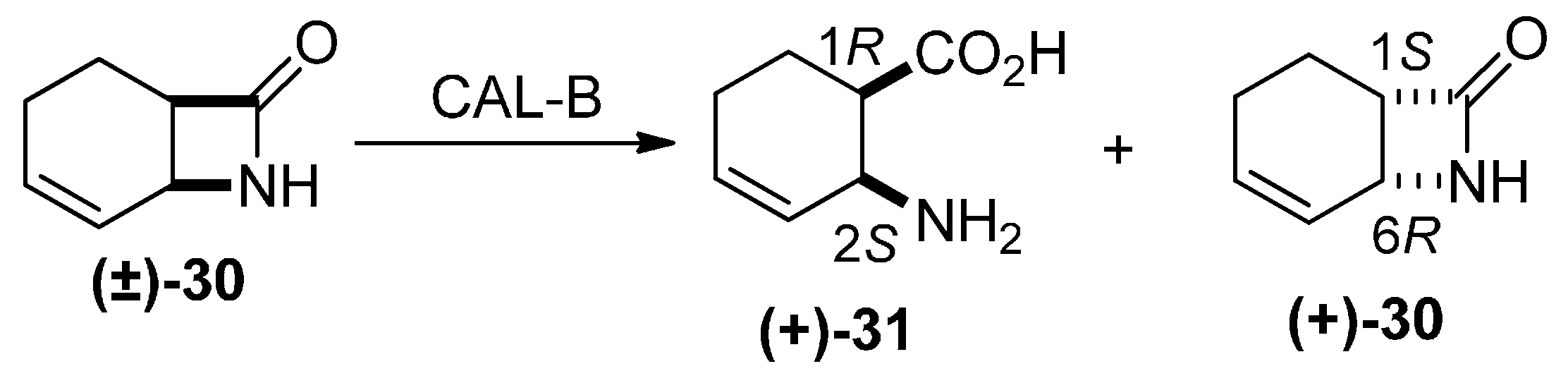
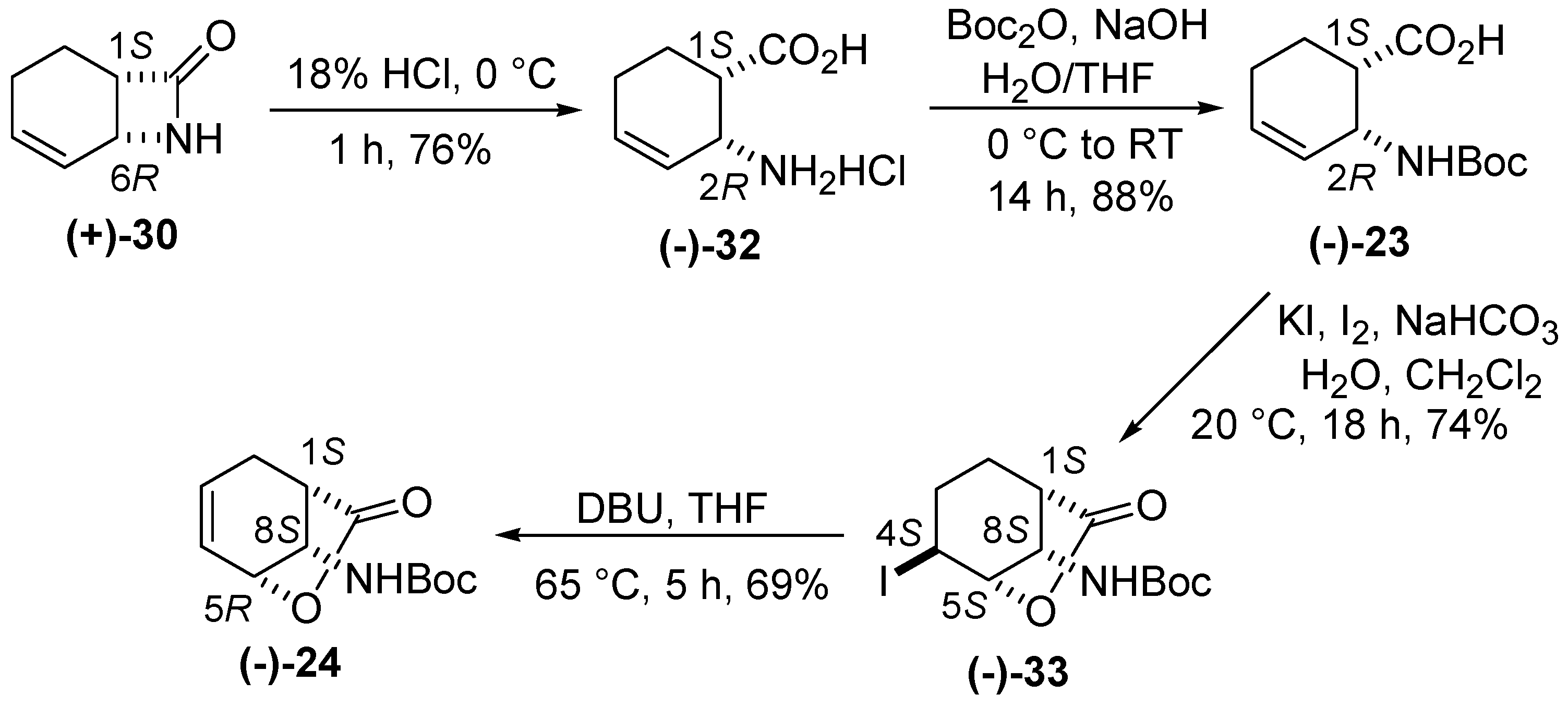
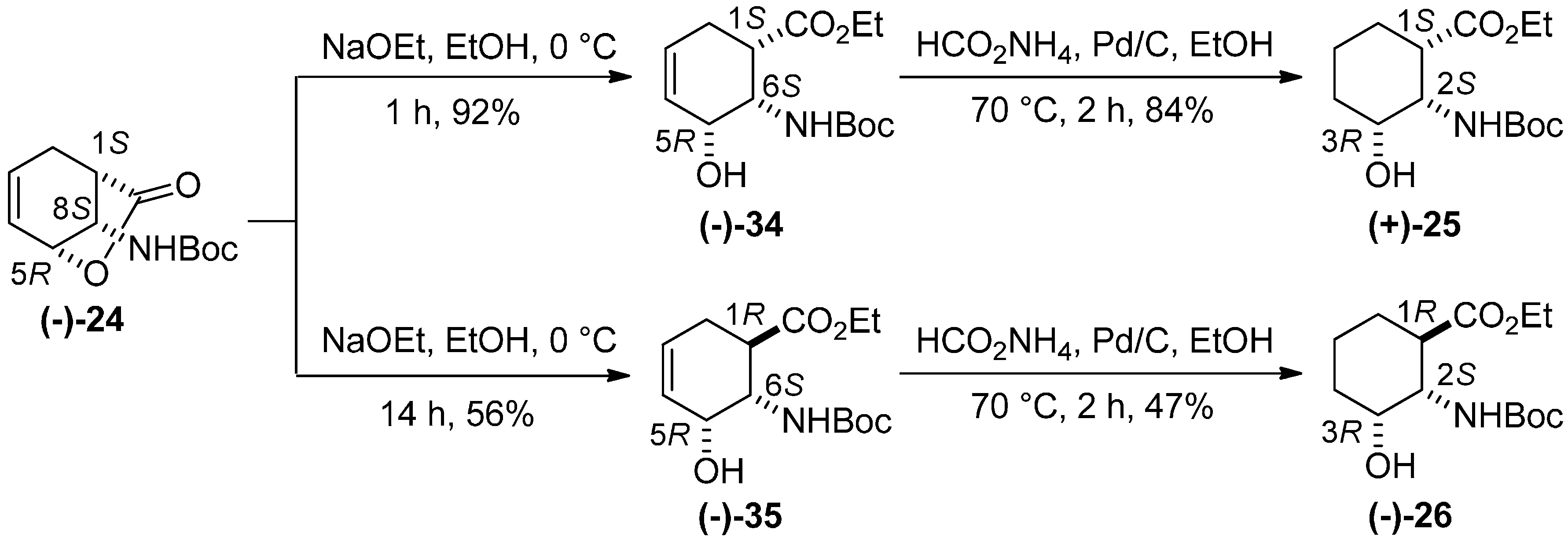
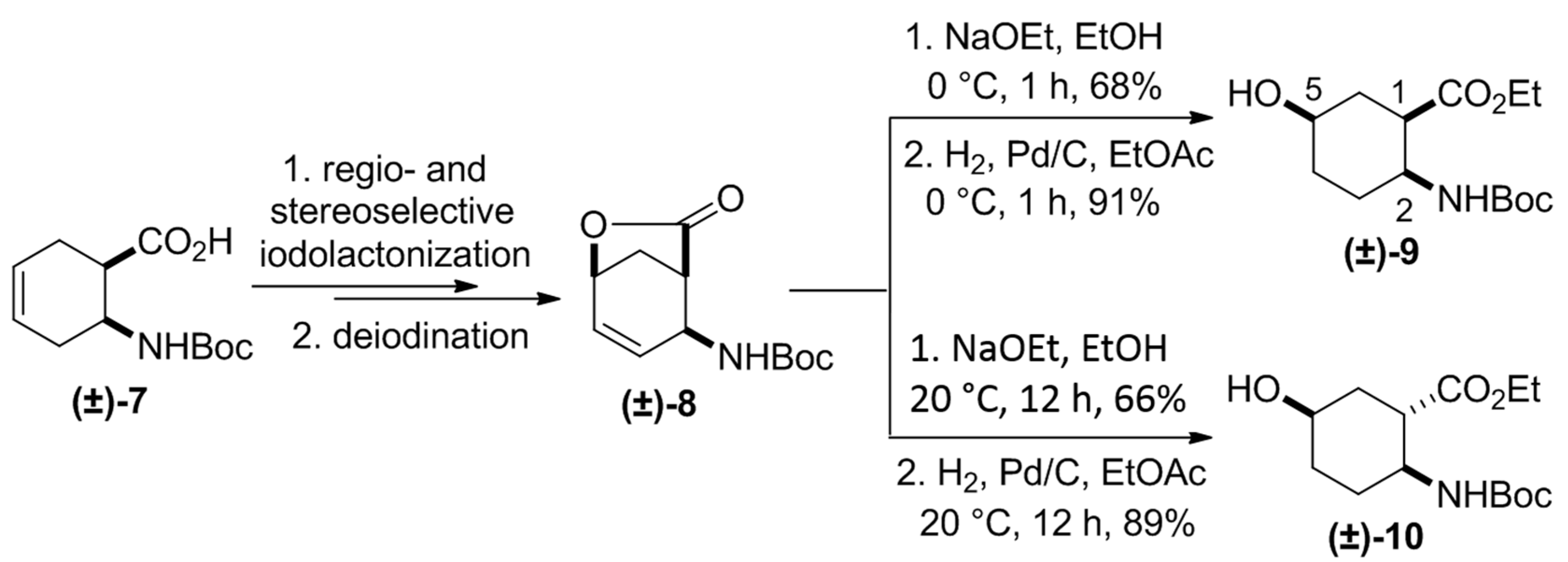
2. Results and Discussion

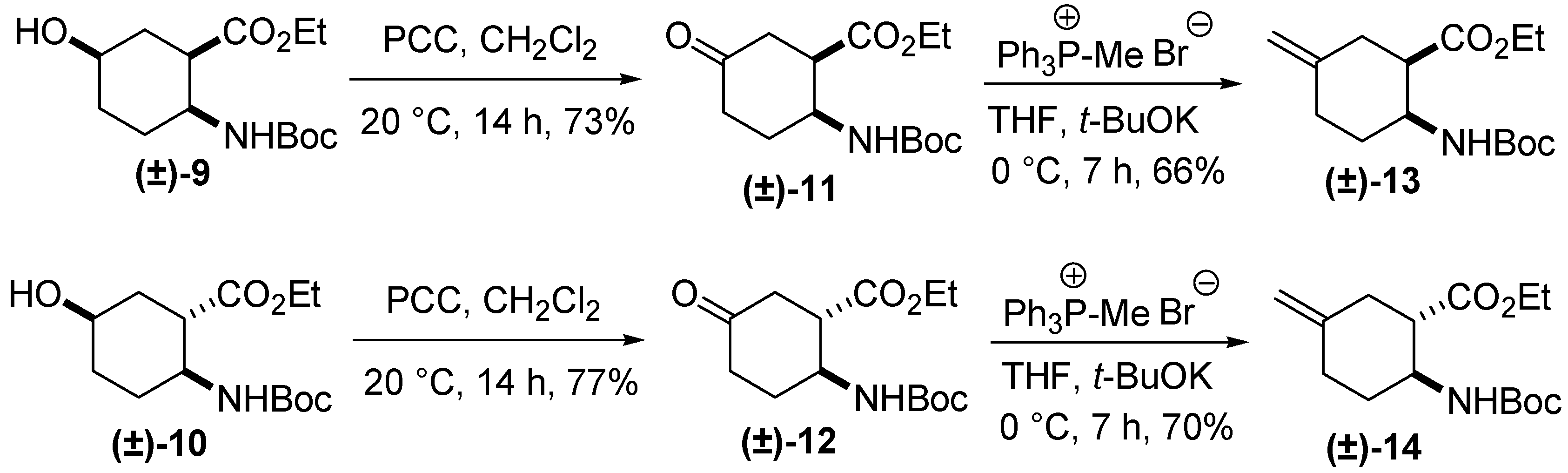
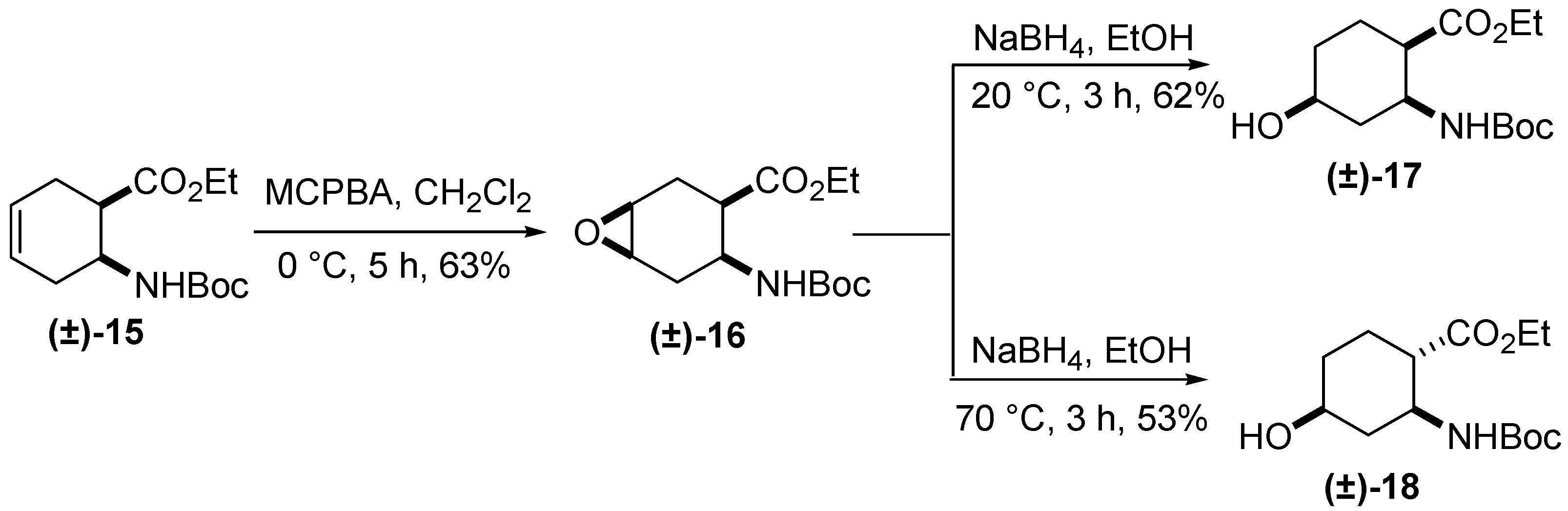

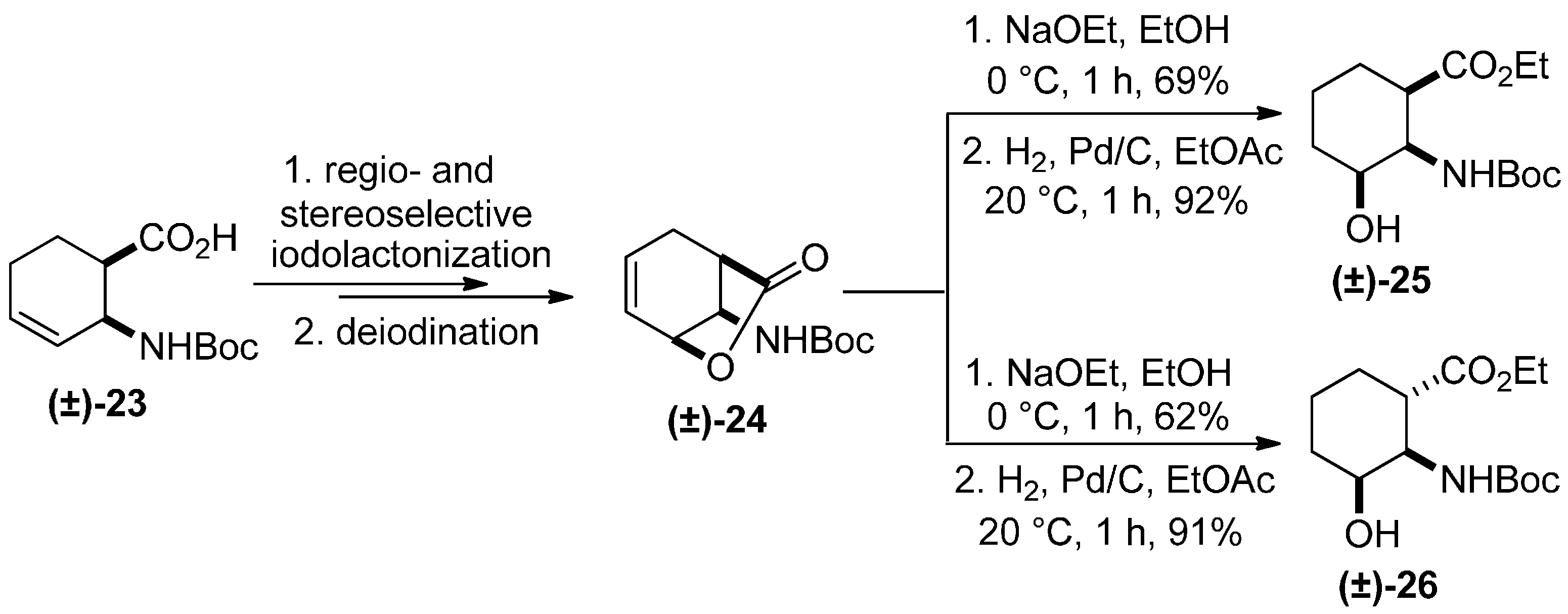
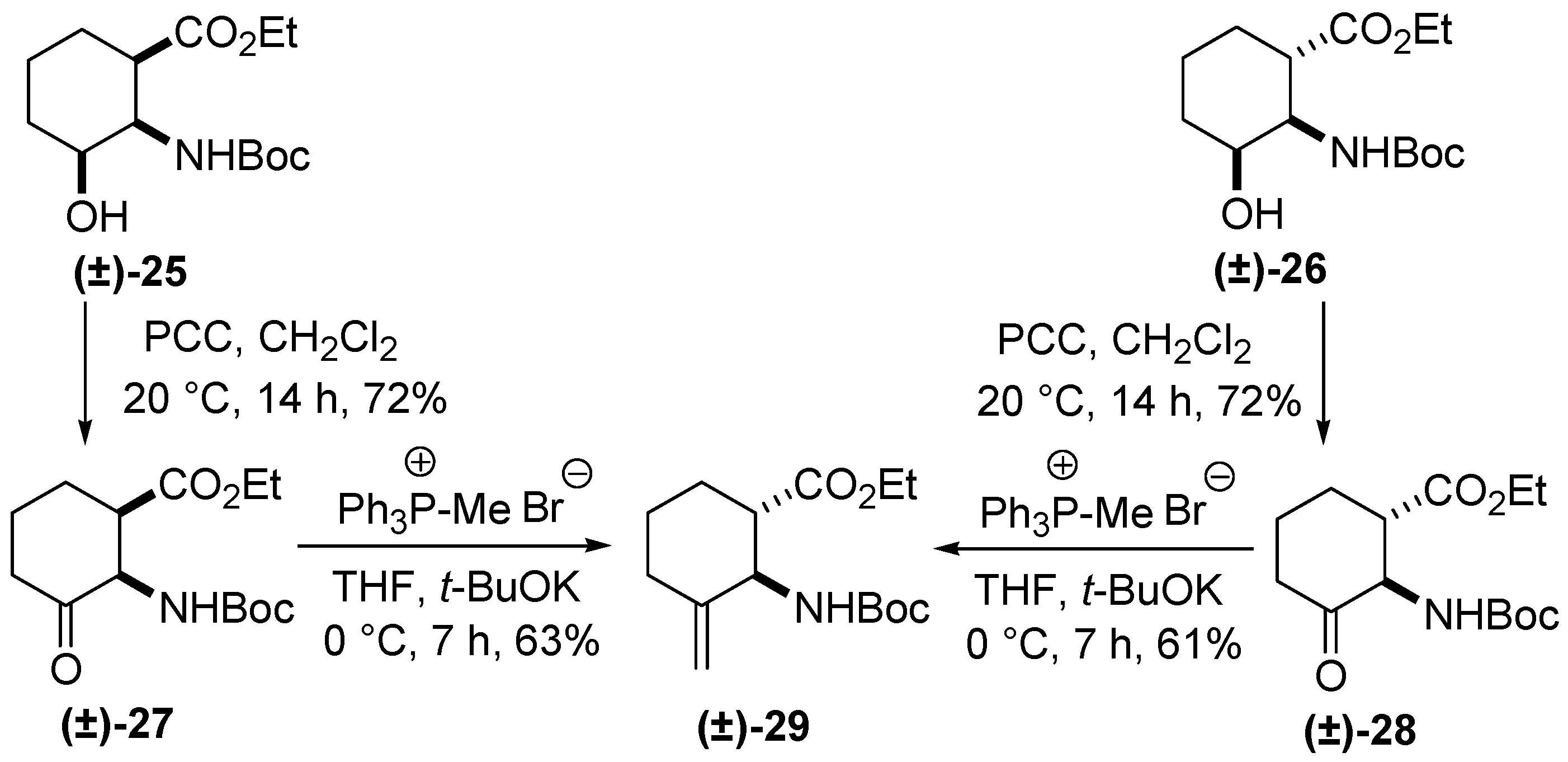




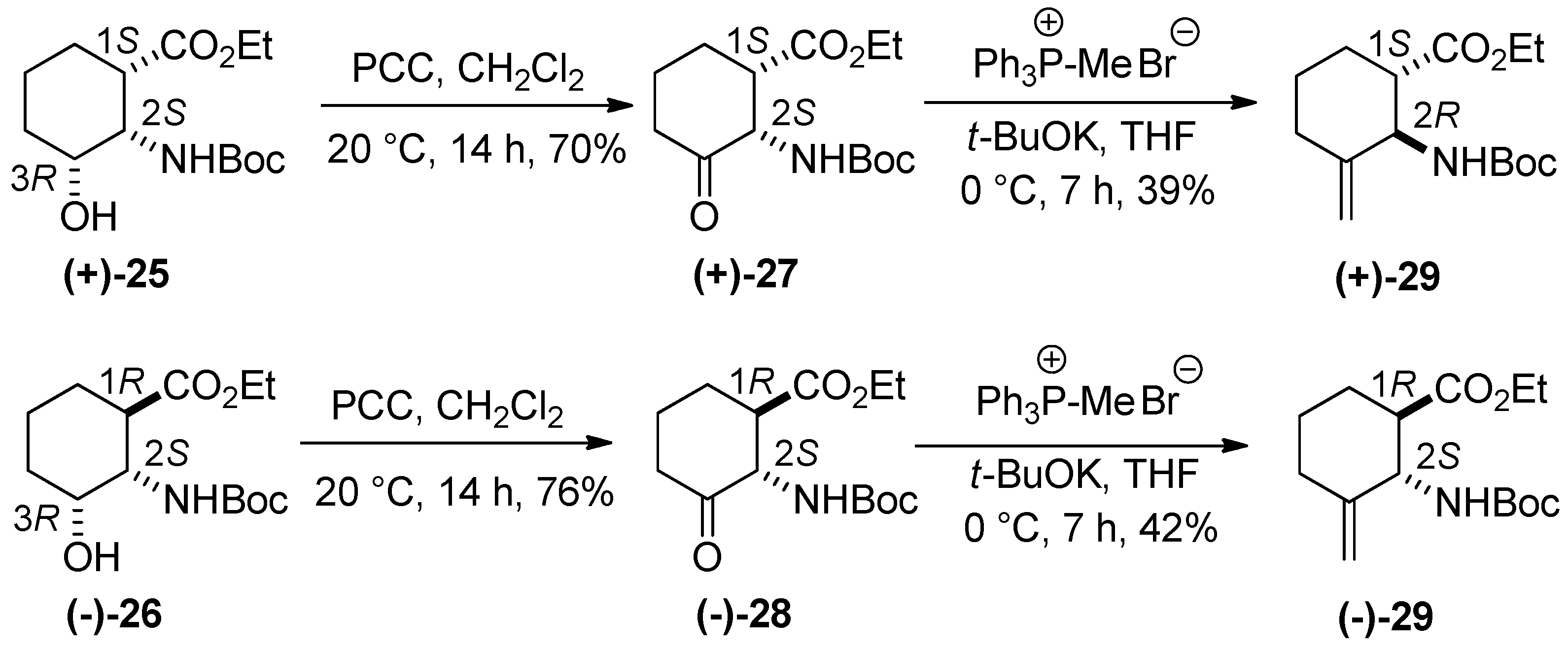
3. Experimental Section
3.1. General Procedure for the Methylenation of Oxo Esters
3.2. Characterization of the Enantiomerically Pure Substances
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kiss, L.; Fülöp, F. Synthesis of carbocyclic and heterocyclic β-aminocarboxylic acids. Chem. Rev. 2014, 114, 1116–1169. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Fülöp, F. Selective syntheses of functionalized cyclic β-amino acids via transformation of the ring C-C double bonds. Synlett 2010, 1302–1314. [Google Scholar] [CrossRef]
- Juaristi, E.; Soloshonok, V. Enantioselective Synthesis of β-Amino Acids, 2nd ed.; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Kiss, L.; Cherepanova, M.; Forró, E.; Fülöp, F. New access route to functionalized cispentacins from norbornene β-amino acids. Chem. Eur. J. 2013, 19, 2102–2107. [Google Scholar] [CrossRef] [PubMed]
- Nonn, M.; Kiss, L.; Sillanpää, R.; Fülöp, F. Selective nitrile oxide dipolar cycloaddition for the synthesis of highly functionalized β-aminocyclohexanecarboxylate stereoisomers. Tetrahedron 2012, 68, 9942–9948. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Sillanpää, R.; Fülöp, F. Diastereo- and enantioselective synthesis of orthogonally protected 2,4-diaminocyclopentanecarboxylates: A flip from α-amino- to β,γ-diaminocarboxylates. J. Org. Chem. 2007, 72, 8786–8790. [Google Scholar] [CrossRef] [PubMed]
- Cellis, S.; Gorrea, E.; Nolis, P.; Illa, O.; Ortuno, R.M. Designing hybrid foldamers: The effect on the peptide conformational bias of β- versus α- and γ-linear residues in alternation with (1R,2S)-2-aminocyclobutane-1-carboxylic acid. Org. Biomol. Chem. 2012, 10, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Mowery, B.P.; Lee, S.E.; Kissounko, D.A.; Epand, R.F.; Epand, R.M.; Weisblum, B.; Stahl, S.S.; Gellman, S.H.J. Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc. 2007, 129, 15474–14576. [Google Scholar] [CrossRef] [PubMed]
- Gorrea, E.; Nolis, P.; Torres, E.; Da Silva, E.; Amabilino, D.B.; Branchadell, V.; Ortuno, R.M. Self-assembly of chiral trans-cyclobutane-containing β-dipeptides into ordered aggregates. Chem. Eur. J. 2011, 17, 4588–4597. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.A.; Weisblum, B.; Gellman, S.H. Use of parallel synthesis to probe structure-activity relationships among 12-helical β-peptides: Evidence of a limit on antimicrobial activity. J. Am. Chem. Soc. 2005, 127, 11516–11529. [Google Scholar] [CrossRef] [PubMed]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Berlicki, L.; Pilsl, L.; Wéber, E.; Mándity, I.M.; Cabrele, C.; Martinek, T.; Fülöp, F.; Reiser, O. Unique α,β- and α,α,β,β-peptide foldamers based on cis-β-aminocyclopentanecarboxylic acid. Angew. Chem. Int. Ed. 2012, 51, 2208–2212. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Pereira, E.; Faure, S.; Aitken, D.J. Expedient preparation of all isomers of 2-aminocyclobutanecarboxylic acid in enantiomerically pure form. J. Org. Chem. 2009, 74, 3217–3220. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, J.; Kunisch, F.; Matzke, M.; Militzer, H.-C.; Schmidt, A.; Schönfeld, W. Novel antifungal β-amino acids: Synthesis and activity against Candida albicans. Bioorg. Med. Chem. Lett. 2003, 13, 433–436. [Google Scholar] [CrossRef]
- Hamersak, Z.; Roje, M.; Avdagic, A.; Sunjic, V. Quinine-mediated parallel kinetic resolution of racemic cyclic anhydride: Stereoselective synthesis, relative and absolute configuration of novel alicyclic β-amino acids. Tetrahedron Asymmetry 2007, 18, 635–644. [Google Scholar] [CrossRef]
- Petraitiene, R.; Petraitis, V.; Kelaher, A.M.; Sarafandi, A.A.; Mickiene, D.; Groll, A.H.; Sein, T.; Bacher, J.; Walsh, T.J. Efficacy, plasma pharmacokinetics, and safety of icofungipen, an inhibitor of Candida isoleucyl-tRNA synthetase, in treatment of experimental disseminated candidiasis in persistently neutropenic rabbits. Antimicrob. Agents Chemother. 2005, 49, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, A.; Hahn, M.G.; Dumic, M.; Mittendorf, J. Alicyclic β-amino acids in medicinal chemistry. Amino Acids 2005, 29, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Hasenoehrl, A.; Galic, T.; Ergovic, G.; Marsic, N.; Skerlev, M.; Mittendorf, M.; Geschke, U.; Schmidt, A.; Schoenfeld, W. In vitro activity and in vivo efficacy of antifungal agent, against the pathogenic icofungipen (PLD-118), a novel oral yeast Candida albicans. Antimicrob. Agents Chemother. 2006, 50, 3011–3018. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Forró, E.; Fustero, S.; Fülöp, F. Selective synthesis of new fluorinated alicyclic β-amino ester stereoisomers. Eur. J. Org. Chem. 2011, 2011, 4993–5001. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Fülöp, F. Selective syntheses of novel highly functionalized β-aminocyclohexanecarboxylic acids. Tetrahedron 2012, 68, 4438–4443. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Martinek, T.A.; Bernáth, G.; de Kimpe, N.; Fülöp, F. Stereoselective synthesis of hydroxylated β-aminocyclohexanecarboxylic acids. Tetrahedron 2008, 64, 5036–5043. [Google Scholar] [CrossRef]
- Kiss, L.; Nonn, M.; Sillanpaa, R.; Fustero, S.; Fülöp, F. Efficient regio- and stereoselective access to novel fluorinated β-aminocyclohexanecarboxylates. Beilstein J. Org. Chem. 2013, 9, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Forró, E.; Fustero, S.; Fülöp, F. Regio- and diastereoselective fluorination of alicyclic β-amino acids. Org. Biomol. Chem. 2011, 9, 6528–6534. [Google Scholar] [CrossRef] [PubMed]
- Forró, E.; Fülöp, F. Advanced procedure for the enzymatic ring opening of unsaturated alicyclic β-lactams. Tetrahedron Asymmetry 2004, 15, 2875–2880. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors in mg quantities.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss, L.; Forró, E.; Orsy, G.; Ábrahámi, R.; Fülöp, F. Stereo- and Regiocontrolled Syntheses of Exomethylenic Cyclohexane β-Amino Acid Derivatives. Molecules 2015, 20, 21094-21102. https://doi.org/10.3390/molecules201219749
Kiss L, Forró E, Orsy G, Ábrahámi R, Fülöp F. Stereo- and Regiocontrolled Syntheses of Exomethylenic Cyclohexane β-Amino Acid Derivatives. Molecules. 2015; 20(12):21094-21102. https://doi.org/10.3390/molecules201219749
Chicago/Turabian StyleKiss, Loránd, Enikő Forró, György Orsy, Renáta Ábrahámi, and Ferenc Fülöp. 2015. "Stereo- and Regiocontrolled Syntheses of Exomethylenic Cyclohexane β-Amino Acid Derivatives" Molecules 20, no. 12: 21094-21102. https://doi.org/10.3390/molecules201219749