
A Practical Route for the Preparation of 1,4,7-Triazacyclononanyl Diacetates with a Hydroxypyridinonate Pendant Arm

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
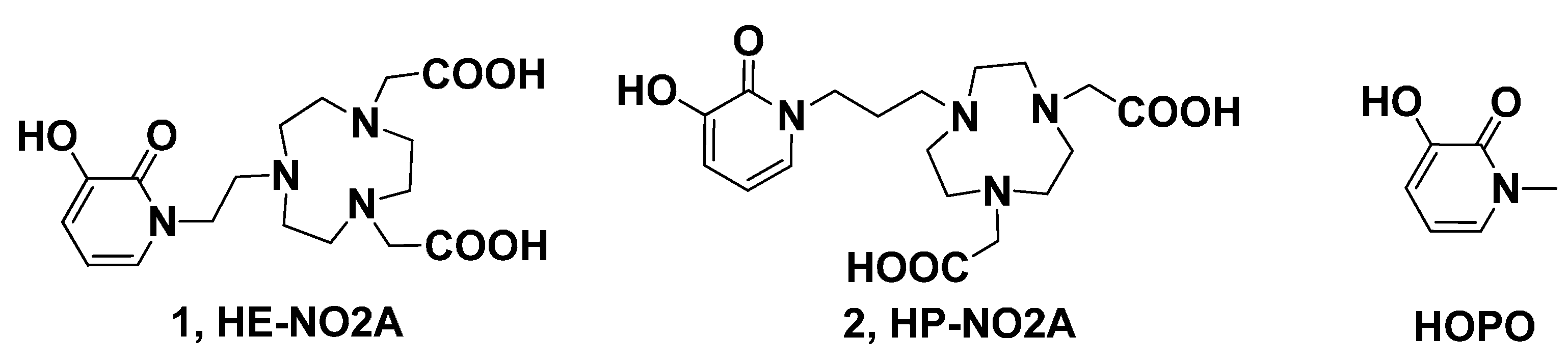
:1. Introduction
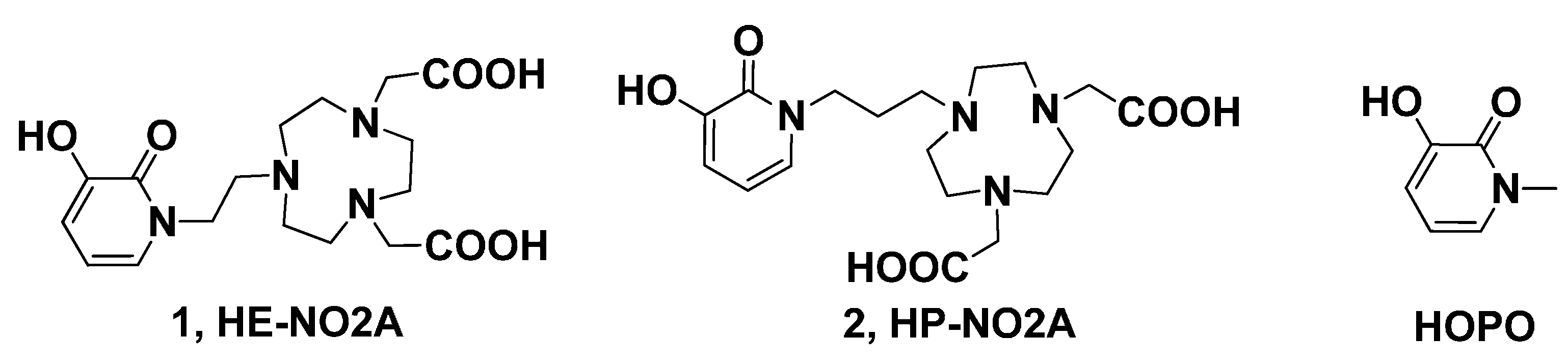
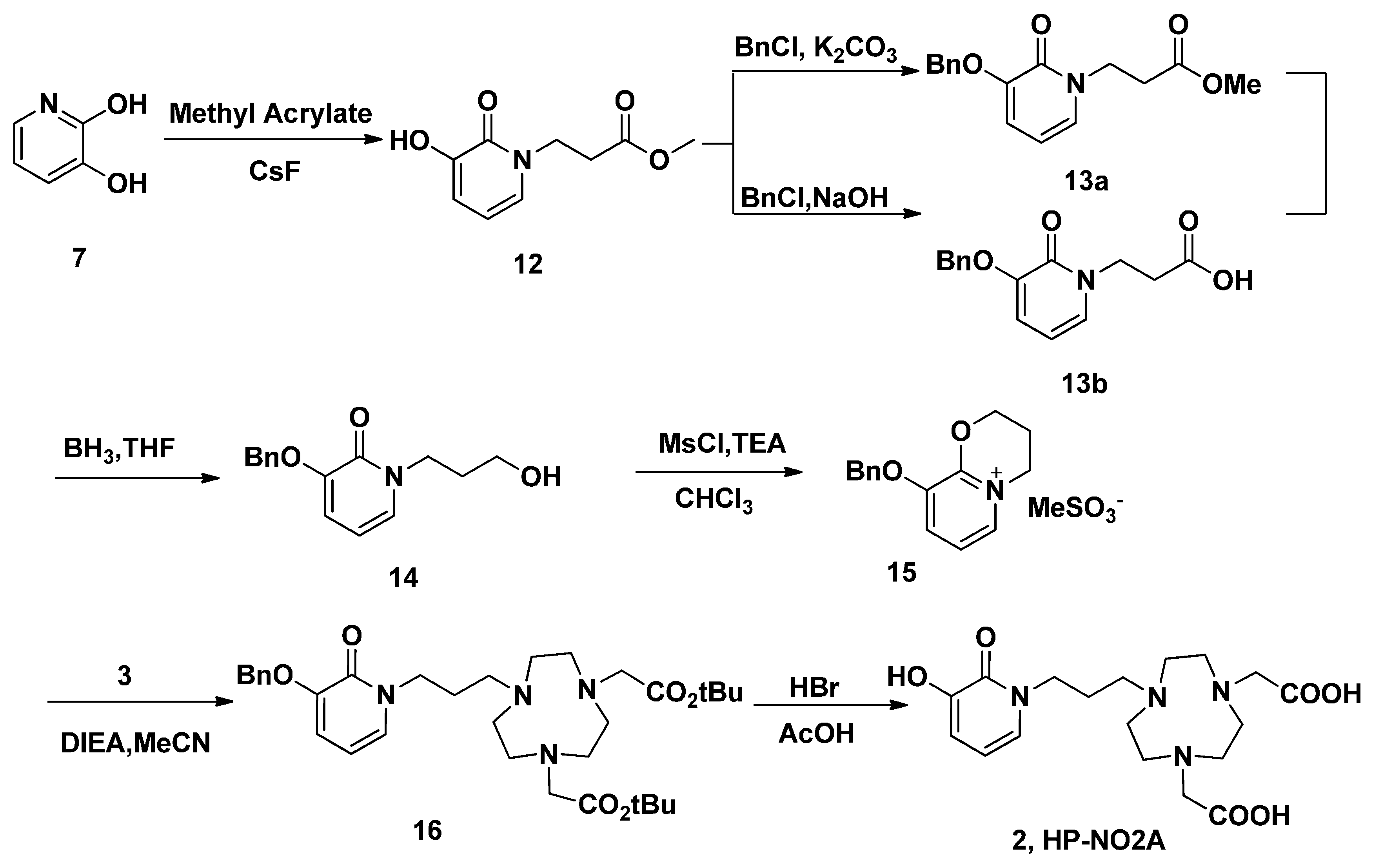
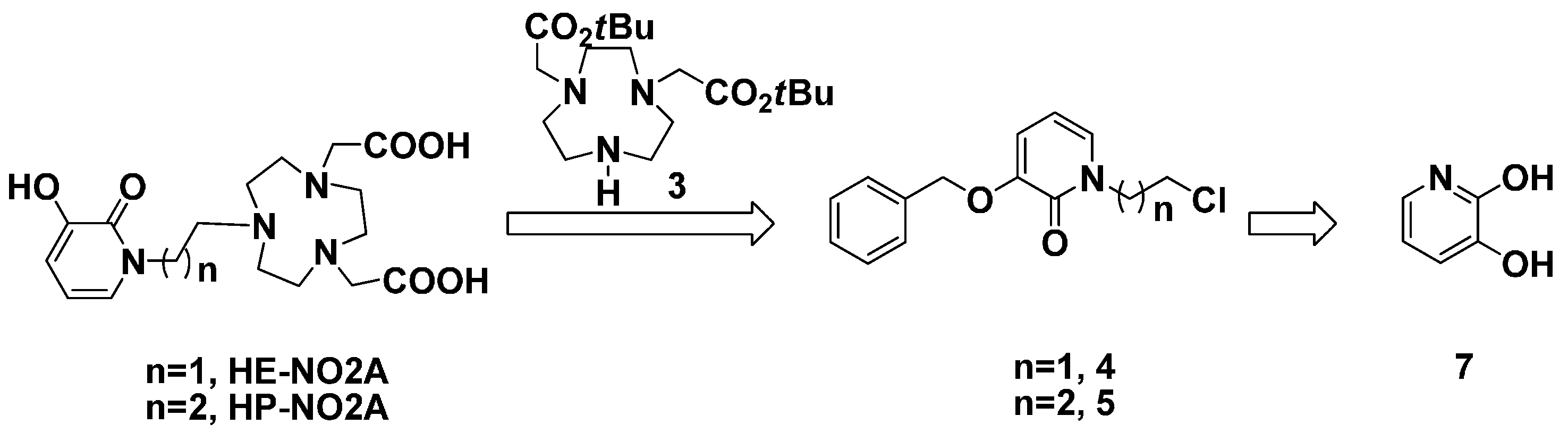
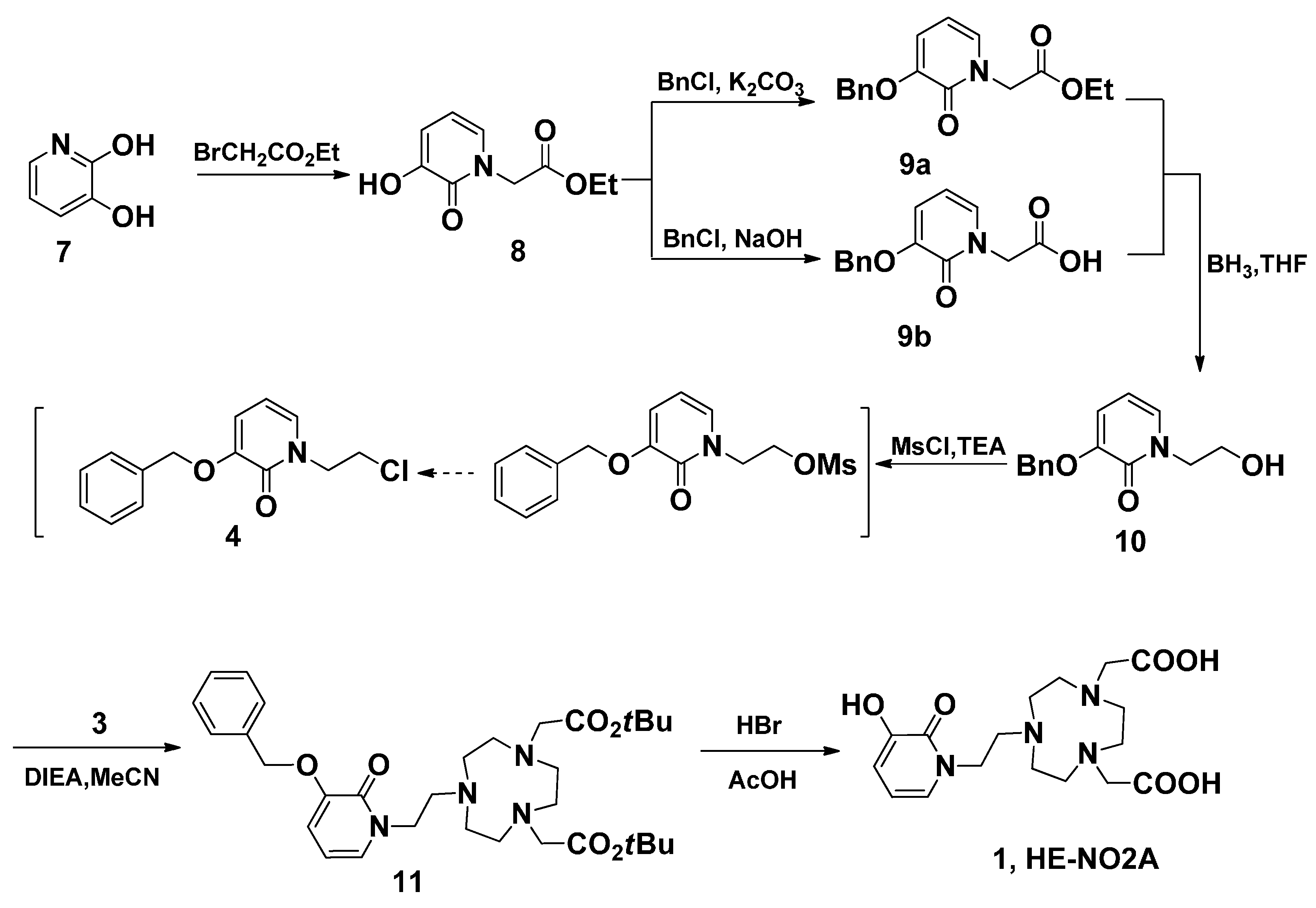
2. Results and Discussion


3. Experimental Section
3.1. General Information
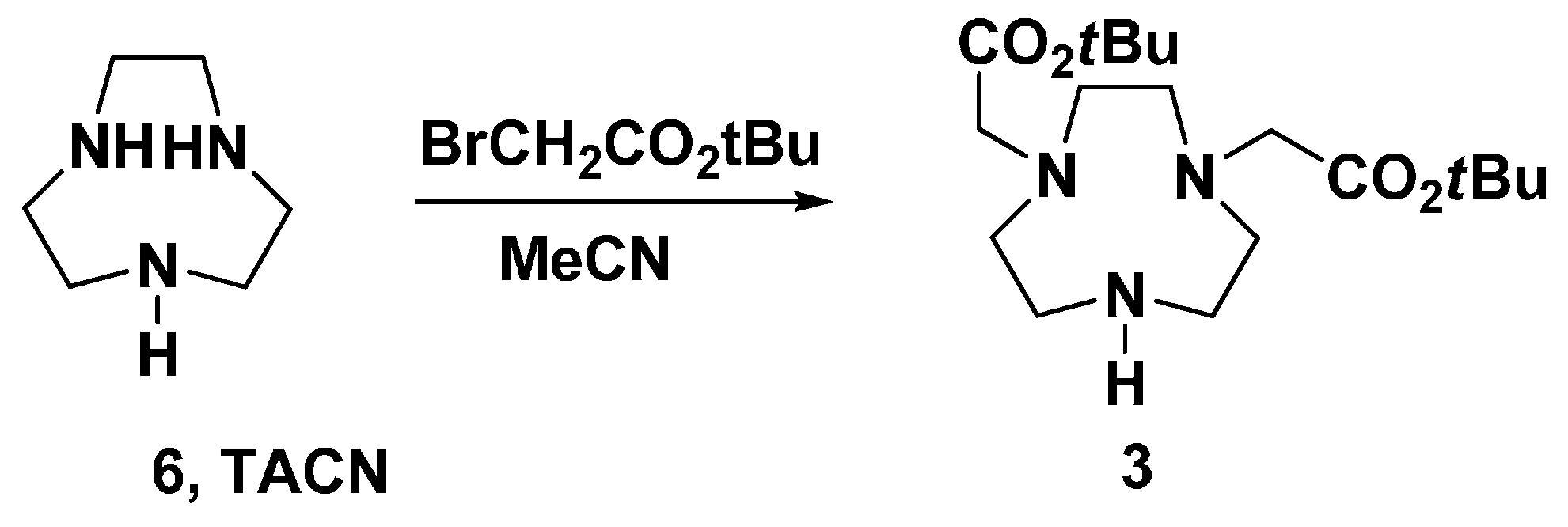
3.2. 1,4-bis(tert-Butoxycarbonylmethyl)-1,4,7-triazacyclononane (3)
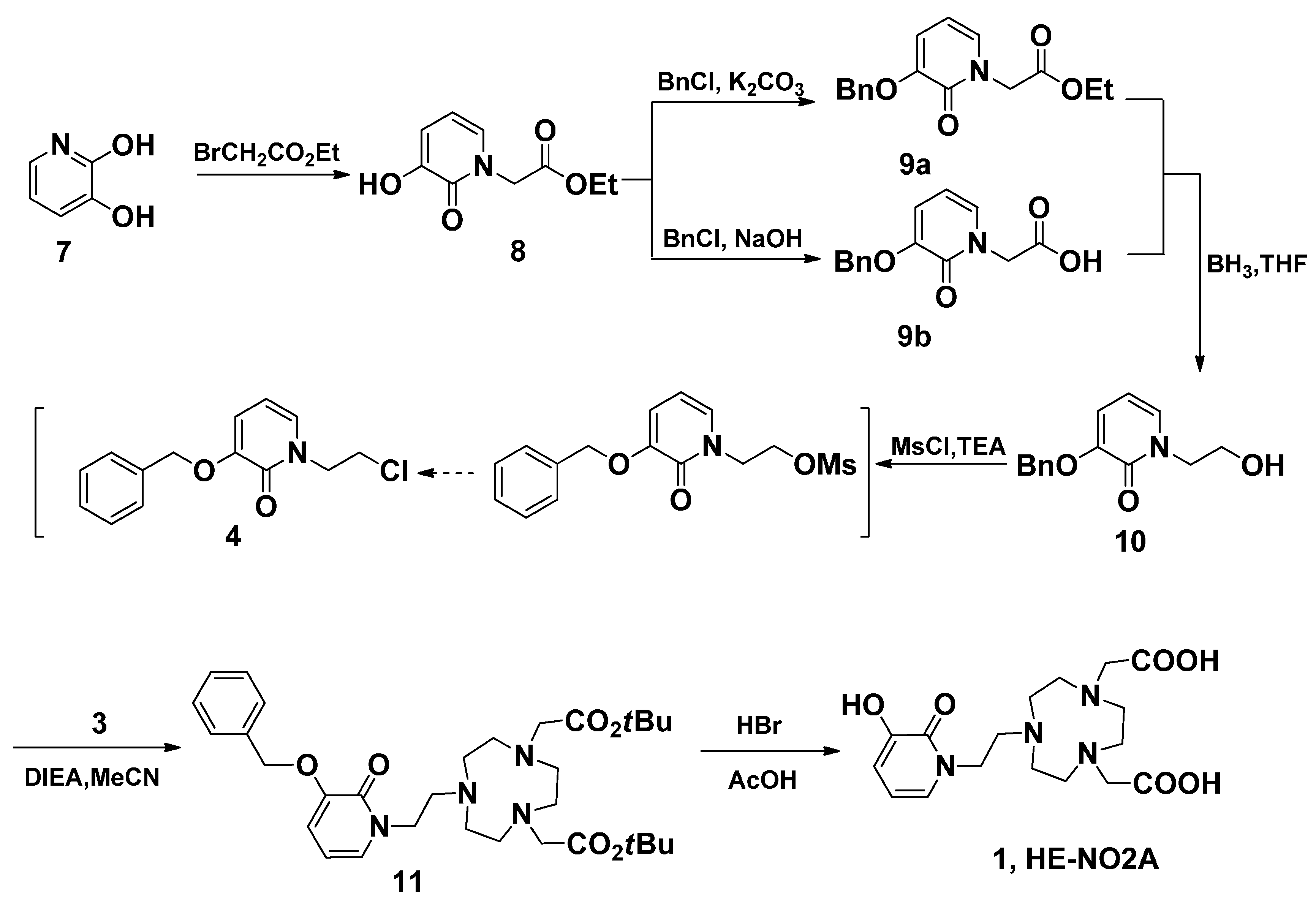
3.3. Ethyl 2-(3-Hydroxy-2-oxopyridin-1(2H)-yl) Acetate (8)
3.4. Ethyl 2-(3-(Benzyloxy)-2-oxopyridin-1(2H)-yl) Acetate (9a)
3.5. 2-(3-(Benzyloxy)-2-oxopyridin-1(2H)-yl)acetic Acid (9b)
3.6. 3-(Benzyloxy)-1-(2-hydroxyethyl)pyridin-2(1H)-one (10)
3.7. 3-(Benzyloxy)-1-(2-hydroxyethyl)pyridin-2(1H)-one (10)
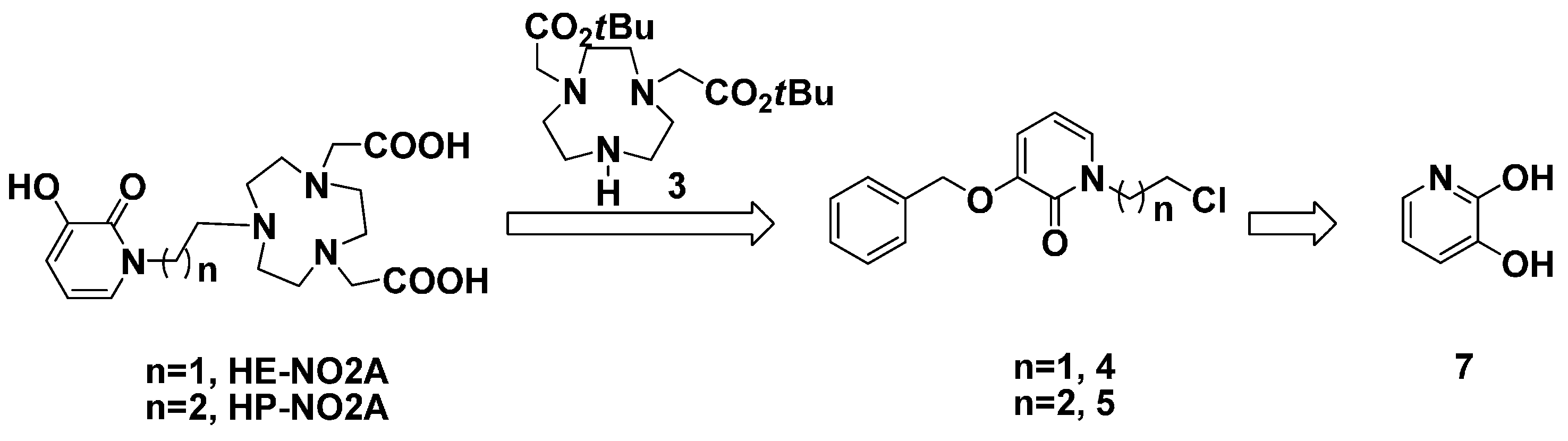
3.8. 3-(Benzyloxy)-1-(2-chloroethyl)pyridin-2(1H)-one (4)
3.9. tert-Butyl-2,2'-(7-(2-(3-(benzyloxy)-2-oxopyridin-1(2H)-yl)ethyl)-1,4,7-triazonane-1,4-diyl) Diacetate (11)
3.10. 2,2'-(7-(2-(3-Hydroxy-2-oxopyridin-1(2H)-yl)ethyl)-1,4,7-triazonane-1,4-diyl) Diacetic Acid (HE-NO2A, 1)
3.11. Methyl 3-(3-Hydroxy-2-oxopyridin-1(2H)-yl) Propanoate (12)
3.12. Methyl 3-(3-(Benzyloxy)-2-oxopyridin-1(2H)-yl) Propanoate (13a)
3.13. 3-(3-(Benzyloxy)-2-oxopyridin-1(2H)-yl) Propanoic Acid (13b)
3.14. 3-(Benzyloxy)-1-(3-hydroxypropyl) Pyridin-2(1H)-one (14)
3.15. 3-(Benzyloxy)-1-(3-hydroxypropyl) Pyridin-2(1H)-one (14)
3.16. 9-(Benzyloxy)-3,4-dihydro-2H-pyrido[2,1-b][1,3]oxazin-5-ium Methanesulfonate (15)
3.17. tert-Butyl-2,2'-(7-(3-(3-(benzyloxy)-2-oxopyridin-1(2H)-yl)propyl)-1,4,7-triazonane-1,4-diyl) Diacetate (16)
3.18. 2,2'-(7-(3-(3-Hydroxy-2-oxopyridin-1(2H)-yl)propyl)-1,4,7-triazonane-1,4-diyl) Diacetic Acid (HP-NO2A, 2)
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burke, B.P.; Archibald, S.J. Macrocyclic coordination chemistry. Annu. Rep. Prog. Chem. Sect. A: Inorg. Chem. 2013, 109, 232–253. [Google Scholar] [CrossRef]
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium(III) chelates as mri contrast agents: Structure, dynamics, and applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef] [PubMed]
- Pandya, D.N.; Bhatt, N.; An, G.I.; Ha, Y.S.; Soni, N.; Lee, H.; Lee, Y.J.; Kim, J.Y.; Lee, W.; Ahn, H.; et al. Propylene cross-bridged macrocyclic bifunctional chelator: A new design for facile bioconjugation and robust 64cu complex stability. J. Med. Chem. 2014, 57, 7234–7243. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Šimeček, J.; Wester, H.-J. Phosphinic acid functionalized polyazacycloalkane chelators for radiodiagnostics and radiotherapeutics: Unique characteristics and applications. ChemMedChem 2014, 9, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.L.; Dang, J.V.; Weitz, E.A.; Lewandowski, C.; Pierre, V.C. Effect of lanthanide complex structure on cell viability and association. Inorg. Chem. 2014, 53, 6013–6021. [Google Scholar] [CrossRef] [PubMed]
- Kharisov, B.I.; Kharissova, O.V.; Berdonosov, S.S. Radioactive nanoparticles and their main applications: Recent advances. Recent Pat. Nanotechnol. 2014, 8, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Comby, S.; Surender, E.M.; Kotova, O.; Truman, L.K.; Molloy, J.K.; Gunnlaugsson, T. Lanthanide-functionalized nanoparticles as mri and luminescent probes for sensing and/or imaging applications. Inorg. Chem. 2014, 53, 1867–1879. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, P.; Yu, B.; Chen, Y.; Wang, J.; Ji, L.; Chao, H. Targeting nucleus DNA with a cyclometalated dipyridophenazineruthenium (II) complex. J. Med. Chem. 2014, 57, 8971–8983. [Google Scholar] [CrossRef] [PubMed]
- Adams, N.; Arts, H.J.; Bolton, P.D.; Cowell, D.; Dubberley, S.R.; Friederichs, N.; Grant, C.M.; Kranenburg, M.; Sealey, A.J.; Wang, B.; et al. Imido titanium ethylene polymerization catalysts containing triazacyclic ligands. Organometallics 2006, 25, 3888–3903. [Google Scholar] [CrossRef]
- Tfouni, E.; Doro, F.G.; Figueiredo, L.E.; Pereira, J.C.M.; Metzker, G.; Franco, D.W. Tailoring no donors metallopharmaceuticals: Ruthenium nitrosyl ammines and aliphatic tetraazamacrocycles. Curr. Med. Chem. 2010, 17, 3643–3657. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, P.; Soriano, E.; Perez-Mayoral, E.; Garcia-Amo, M.; Cerdan, S. Flexible Tetraazamacrocycles as Metal Ligands. Their Relevance in Magnetic Resonance Imaging; Research Signpost: Trivandrum, India, 2005; pp. 69–86. [Google Scholar]
- Haines, R.I. Pendant-arm tri- and tetraazamacrocycles: Synthesis, structure and properties of their first-row transition metal complexes. Rev. Inorg. Chem. 2001, 21, 165–205. [Google Scholar] [CrossRef]
- Meyer, M.; Dahaoui-Gindrey, V.; Lecomte, C.; Guilard, R. Conformations and coordination schemes of carboxylate and carbamoyl derivatives of the tetraazamacrocycles cyclen and cyclam, and the relation to their protonation states. Coord. Chem. Rev. 1998, 178–180, 1313–1405. [Google Scholar] [CrossRef]
- Warden, A.; Graham, B.; Hearn, M.T.W.; Spiccia, L. Synthesis of novel derivatives of 1,4,7-triazacyclononane. Org. Lett. 2001, 3, 2855–2858. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.C.; Spiccia, L.; Hearn, M.T.W.; Boas, J.F.; Pilbrow, J.R. The synthesis, structure and properties of copper(II) complexes of asymmetrically functionalized derivatives of 1,4,7-triazacyclononane. Dalton Trans. 2005. [Google Scholar] [CrossRef] [PubMed]
- Schulz, D.; Weyhermüller, T.; Wieghardt, K.; Butzlaff, C.; Trautwein, A.X. A structural and magnetochemical study of some copper(ii) complexes containing the ligands 1,4,7-triazacyclononane-n-acetate (L1) and N-(2-hydroxybenzyl)-1,4,7-triazacyclononane (HL2). Inorg. Chim. Acta. 1996, 246, 387–394. [Google Scholar] [CrossRef]
- Malkhasian, A.Y.S.; Finch, M.E.; Nikolovski, B.; Menon, A.; Kucera, B.E.; Chavez, F.A. N,Nʹ-dimethylformamide-derived products from catalytic oxidation of 3-hydroxyflavone. Inorg. Chem. 2007, 46, 2950–2952. [Google Scholar] [CrossRef] [PubMed]
- Malkhasian, A.; Finch, M.E.; Pawlak, P.L.; Anderson, J.M.; Brennessel, W.W.; Chavez, F.A. Synthesis, structure, and characterization of dichloro-(1-benzyl-4-acetato-1,4,7-triazacyclononane) iron(III). Z. Anorg. Allg. Chem. 2008, 634, 1087–1092. [Google Scholar] [CrossRef]
- Pawlak, P.L.; Panda, M.; Li, J.; Banerjee, A.; Averill, D.J.; Nikolovski, B.; Shay, B.J.; Brennessel, W.W.; Chavez, F.A. Oxalate oxidase model studies–substrate reactivity. Eur. J. Inorg. Chem. 2015, 2015, 646–655. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Wieghardt, K. The chemistry of 1,4,7-triazacyclononane and related tridentate macrocyclic compounds. Prog. Inorg. Chem. 1987, 35, 329–436. [Google Scholar]
- Schulz, D.; Weyhermüller, T.; Wieghardt, K.; Nuber, B. The monofunctionalized 1,4,7-triazacyclononane derivatives 1,4,7-triazacyclononane-n-acetate (L1) and N-(2-hydroxybenzyl-1,4,7-triazacyclononane (HL2) and their complexes with vanadium (IV)/(V). Localized and delocalized electronic structures in compounds containing the mixed valent [OVIV-O-VVO]3+ core. Inorg. Chim. Acta 1995, 240, 217–229. [Google Scholar]
- Simecek, J.; Zemek, O.; Hermann, P.; Wester, H.J.; Notni, J. A monoreactive bifunctional triazacyclononane phosphinate chelator with high selectivity for gallium-68. ChemMedChem 2012, 7, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.-S.; Song, H.A.; Ma, X.; Milenic, D.E.; Brady, E.D.; Lim, S.; Lee, H.; Baidoo, K.; Cheng, D.; Brechbiel, M.W. Novel bimodal bifunctional ligands for radioimmunotherapy and targeted mri. Bioconjugate Chem. 2008, 19, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.-S.; Song, H.A.; Birch, N.; Le, T.; Lim, S.; Ma, X. Efficient synthesis and evaluation of bimodal ligand neta. Bioorg. Med. Chem. Lett. 2008, 18, 3436–3439. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.S.; Chen, Y.; Lee, H.; Liu, D.; Sun, X.; Kweon, J.; Lewis, M.R.; Chong, H.-S. Synthesis and evaluation of a new bifunctional NETA chelate for molecular targeted radiotherapy using 90Y or 177Lu. Nucl. Med. Biol. 2015, 42, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.S.; Song, H.A.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W.; Chong, H.-S. Preclinical evaluation of NETA-based bifunctional ligand for radioimmunotherapy applications using 212Bi and 213Bi: Radiolabeling, serum stability, and biodistribution and tumor uptake studies. Nucl. Med. Biol. 2013, 40, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.-S.; Garmestani, K.; Bryant, L.H.; Milenic, D.E.; Overstreet, T.; Birch, N.; Le, T.; Brady, E.D.; Brechbiel, M.W. Synthesis and evaluation of novel macrocyclic and acyclic ligands as contrast enhancement agents for magnetic resonance imaging. J. Med. Chem. 2006, 49, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Gorden, A.E.V.; Xu, J.; Raymond, K.N.; Durbin, P. Rational design of sequestering agents for plutonium and other actinides. Chem. Rev. 2003, 103, 4207–4282. [Google Scholar] [CrossRef] [PubMed]
- Bunin, D.I.; Chang, P.Y.; Doppalapudi, R.S.; Riccio, E.S.; An, D.; Jarvis, E.E.; Kullgren, B.; Abergel, R.J. Dose-dependent efficacy and safety toxicology of hydroxypyridinonate actinide decorporation agents in rodents: Towards a safe and effective human dosing regimen. Radiat. Res. 2013, 179, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.; Choi, S.Y.; Jeong, J.M.; Lee, J.Y.; Hoigebazar, L.; Lee, Y.-S.; Lee, D.S.; Chung, J.-K.; Lee, M.C.; Chung, Y.K. Stable aluminium fluoride chelates with triazacyclononane derivatives proved by X-ray crystallography and 18F-labeling study. Chem. Commun. 2011, 47, 9732–9734. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.S.; Sun, X.; Zhong, Y.; Bober, K.; Lewis, M.R.; Liu, D.; Ruthengael, V.C.; Sin, I.; Kang, C.S. Synthesis and evaluation of an enantiomerically enriched bifunctional chelator for 64Cu-based positron emission tomography (PET) imaging. Eur. J. Inorg. Chem. 2014, 2014, 1305–1313. [Google Scholar] [CrossRef]
- De Sá, A.; Bonnet, C.S.; Geraldes, C.F.G.C.; Tóth, É.; Ferreira, P.M.T.; André, J.P. Thermodynamic stability and relaxation studies of small, triaza-macrocyclic Mn(II) chelates. Dalton Trans. 2013, 42, 4522–4532. [Google Scholar]
- Ambrosi, G.; Dapporto, P.; Formica, M.; Fusi, V.; Giorgi, L.; Guerri, A.; Lucarini, S.; Micheloni, M.; Paoli, P.; Pontellini, R. Macrocyclic ligands bearing two 3-(hydroxy)-2-pyridinone moieties as side-arms. Conformational studies, synthesis, crystal structure, and alkali and alkaline earth complex formation. New J. Chem. 2004, 28, 1359–1367. [Google Scholar] [CrossRef]
- Lambert, T.N.; Chittamuru, S.; Jacobs, H.K.; Gopalan, A.S. New methodology for the preparation of 3-hydroxy-2-pyridinone (3,2-hopo) chelators—Reaction of amines with a novel electrophilic 3,2-hopo precursor. Tetrahedron Lett. 2002, 43, 7379–7383. [Google Scholar] [CrossRef]
- Chittamuru, S.; Lambert, T.N.; Martinez, G.; Jacobs, H.K.; Gopalan, A.S. New methodology for the preparation of 3-hydroxy-2-pyridinone (3,2-hopo) chelators and extractants. Part 2: Reactions of alcohols, phenols, and thiols with an electrophilic 3,2-hopo reagent. Tetrahedron Lett. 2007, 48, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1 and 2 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gai, Y.; Hu, Z.; Rong, Z.; Ma, X.; Xiang, G. A Practical Route for the Preparation of 1,4,7-Triazacyclononanyl Diacetates with a Hydroxypyridinonate Pendant Arm. Molecules 2015, 20, 19393-19405. https://doi.org/10.3390/molecules201019393
Gai Y, Hu Z, Rong Z, Ma X, Xiang G. A Practical Route for the Preparation of 1,4,7-Triazacyclononanyl Diacetates with a Hydroxypyridinonate Pendant Arm. Molecules. 2015; 20(10):19393-19405. https://doi.org/10.3390/molecules201019393
Chicago/Turabian StyleGai, Yongkang, Zhongping Hu, Zhao Rong, Xiang Ma, and Guangya Xiang. 2015. "A Practical Route for the Preparation of 1,4,7-Triazacyclononanyl Diacetates with a Hydroxypyridinonate Pendant Arm" Molecules 20, no. 10: 19393-19405. https://doi.org/10.3390/molecules201019393