
A Self-Adaptive Steered Molecular Dynamics Method Based on Minimization of Stretching Force Reveals the Binding Affinity of Protein–Ligand Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Models and Methods
2.1. The Self-Adaptive SMD Strategy
2.2. The Multi-Population Genetic Algorithm
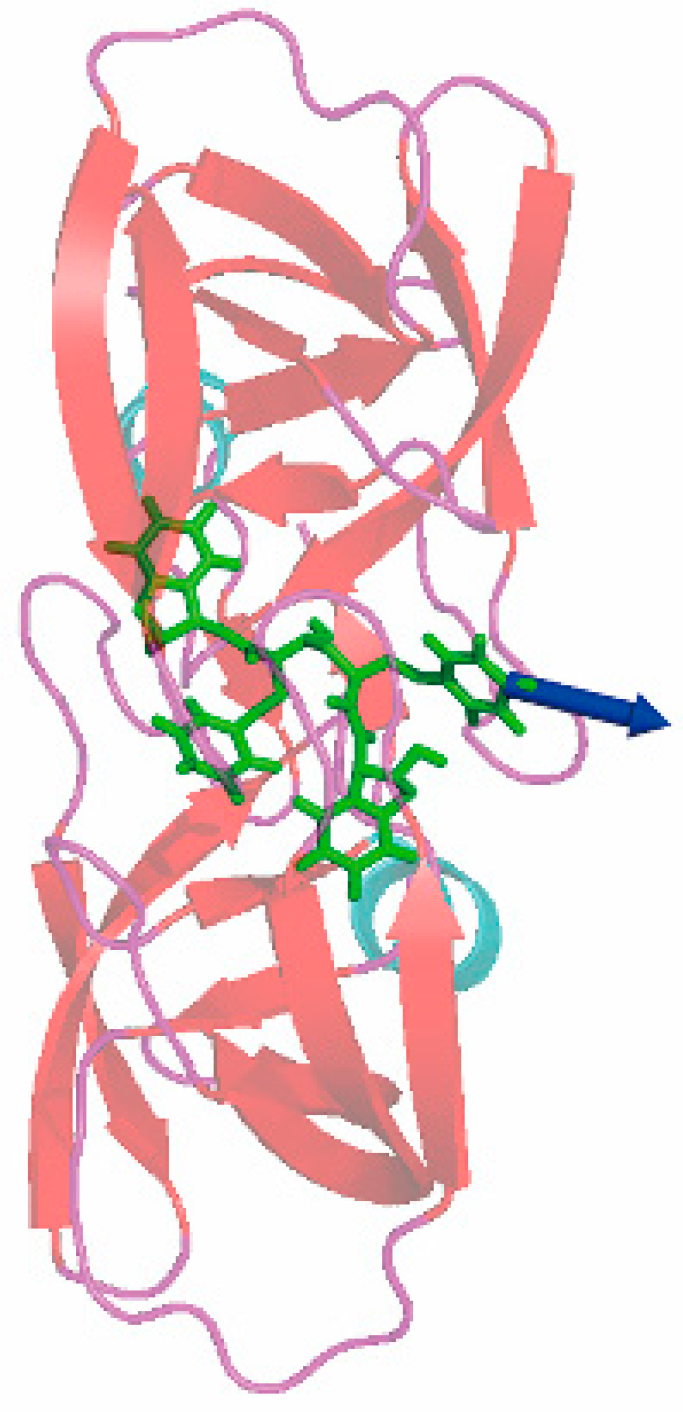
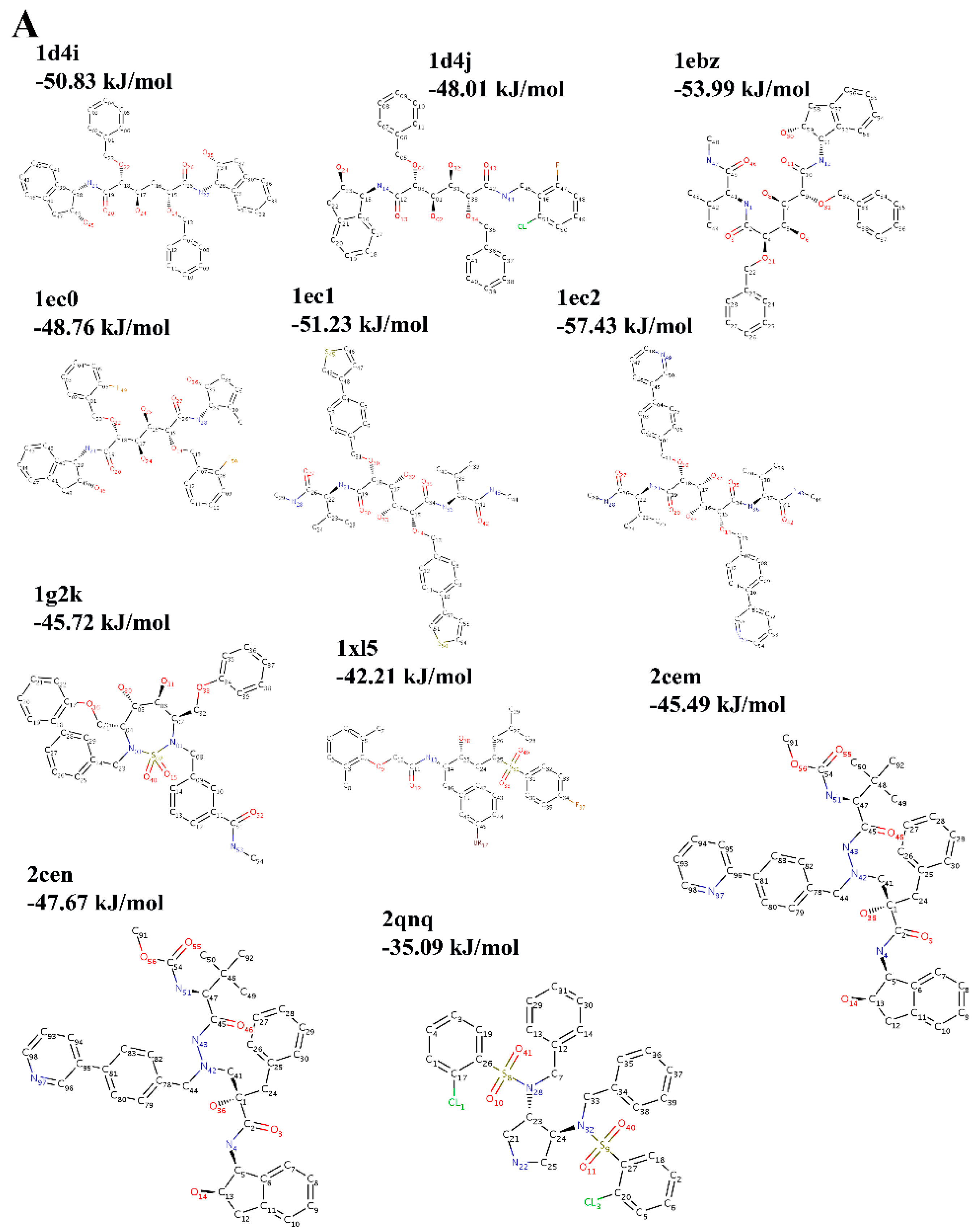
2.3. Choice of Receptor–Ligand Complexes

3. Results and Discussion
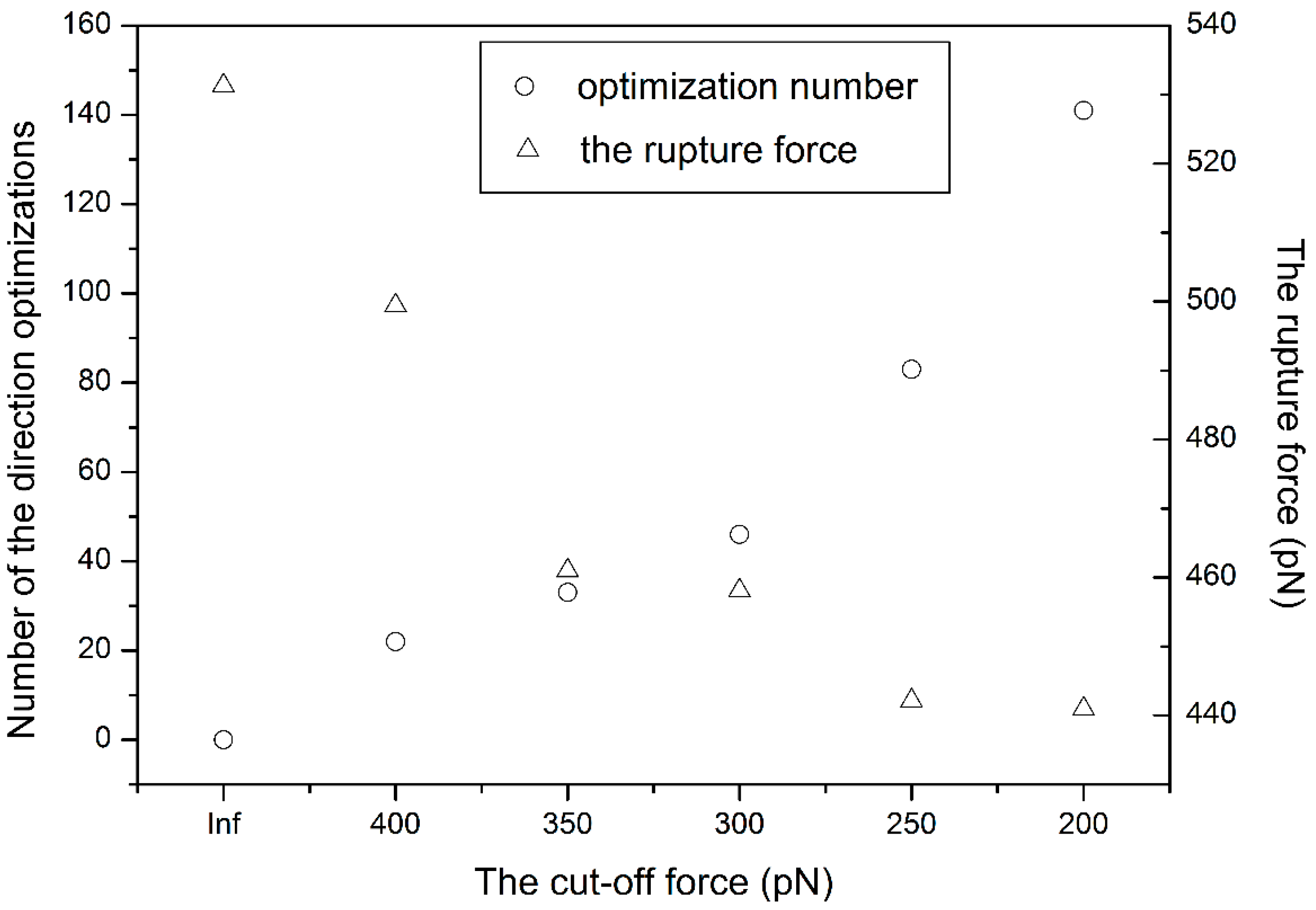
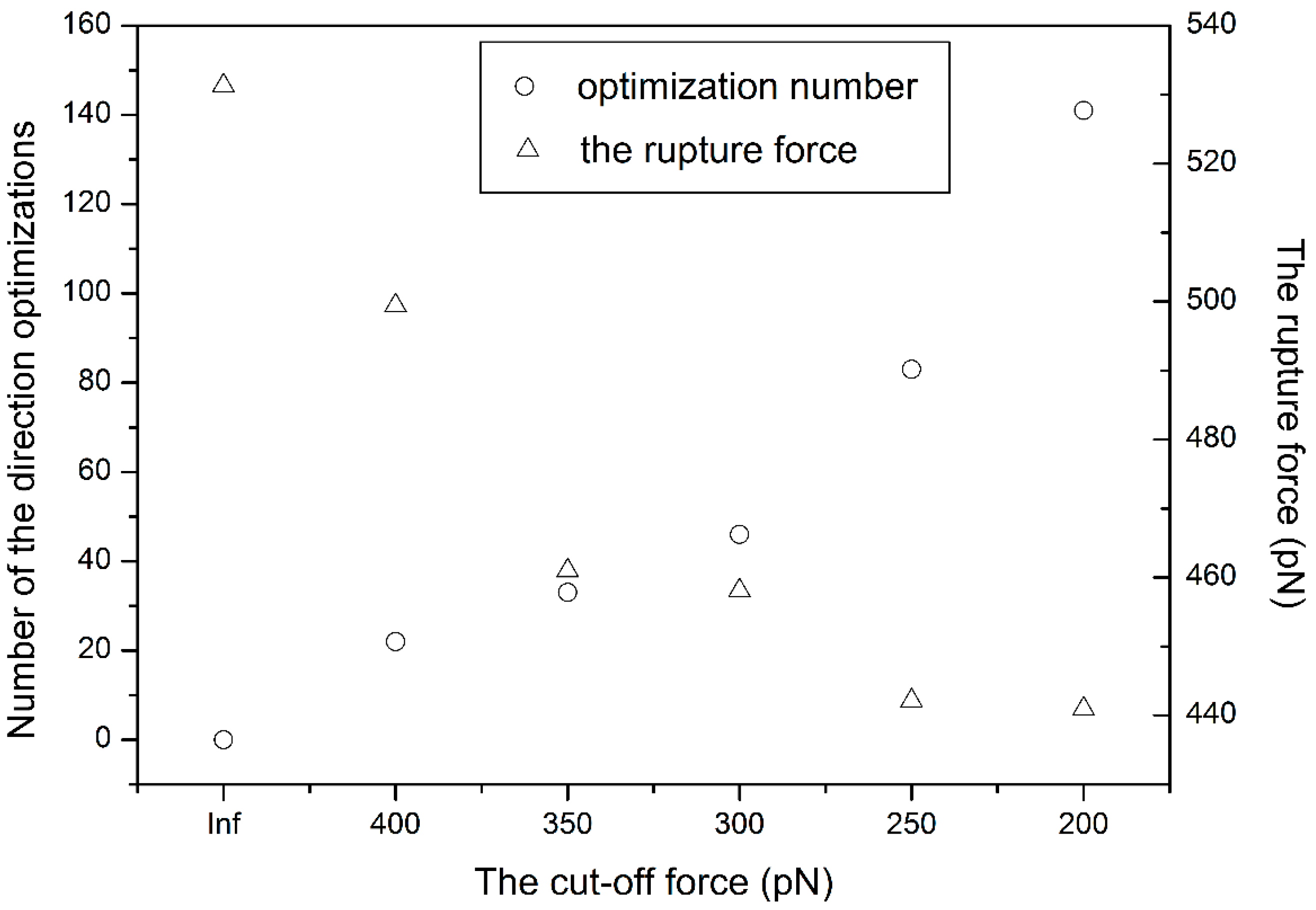
3.1. The Influence of the Cut-Off Force
3.2. The Influence of the Pulling Rate

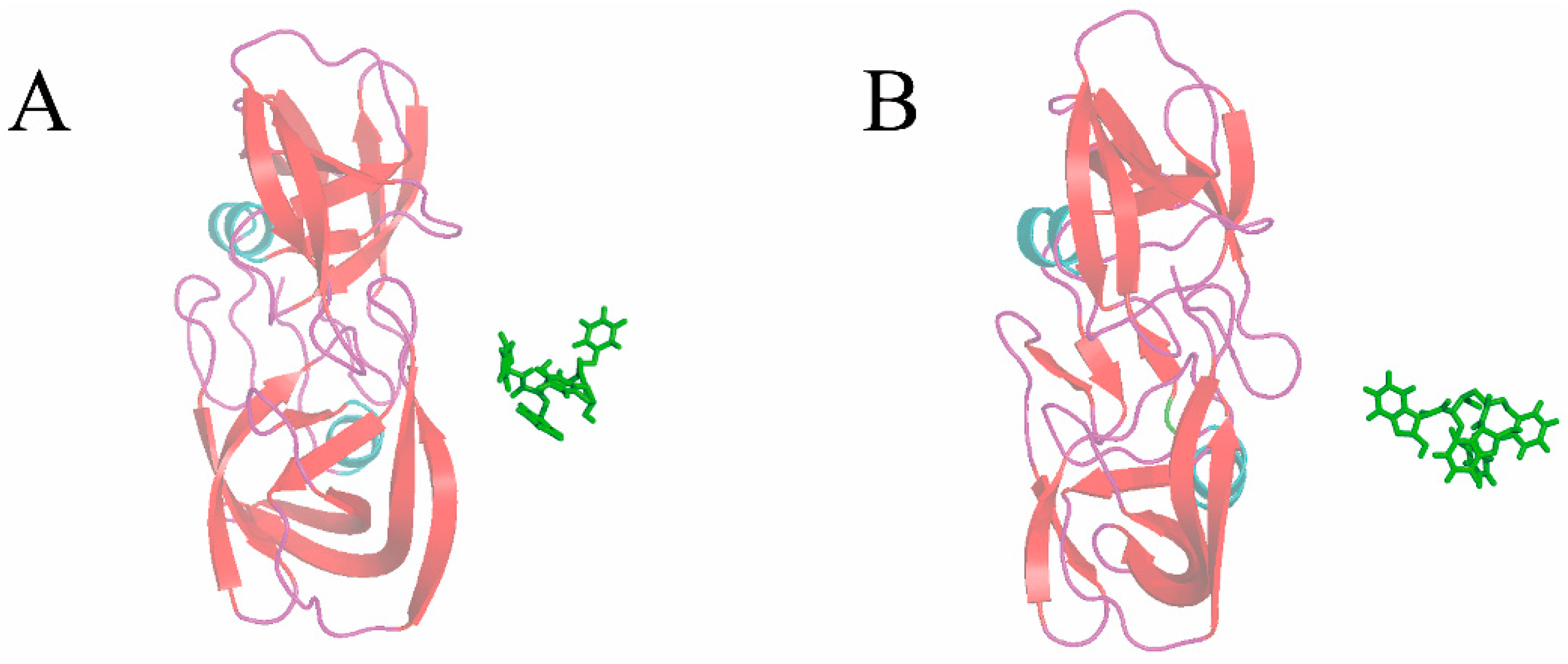
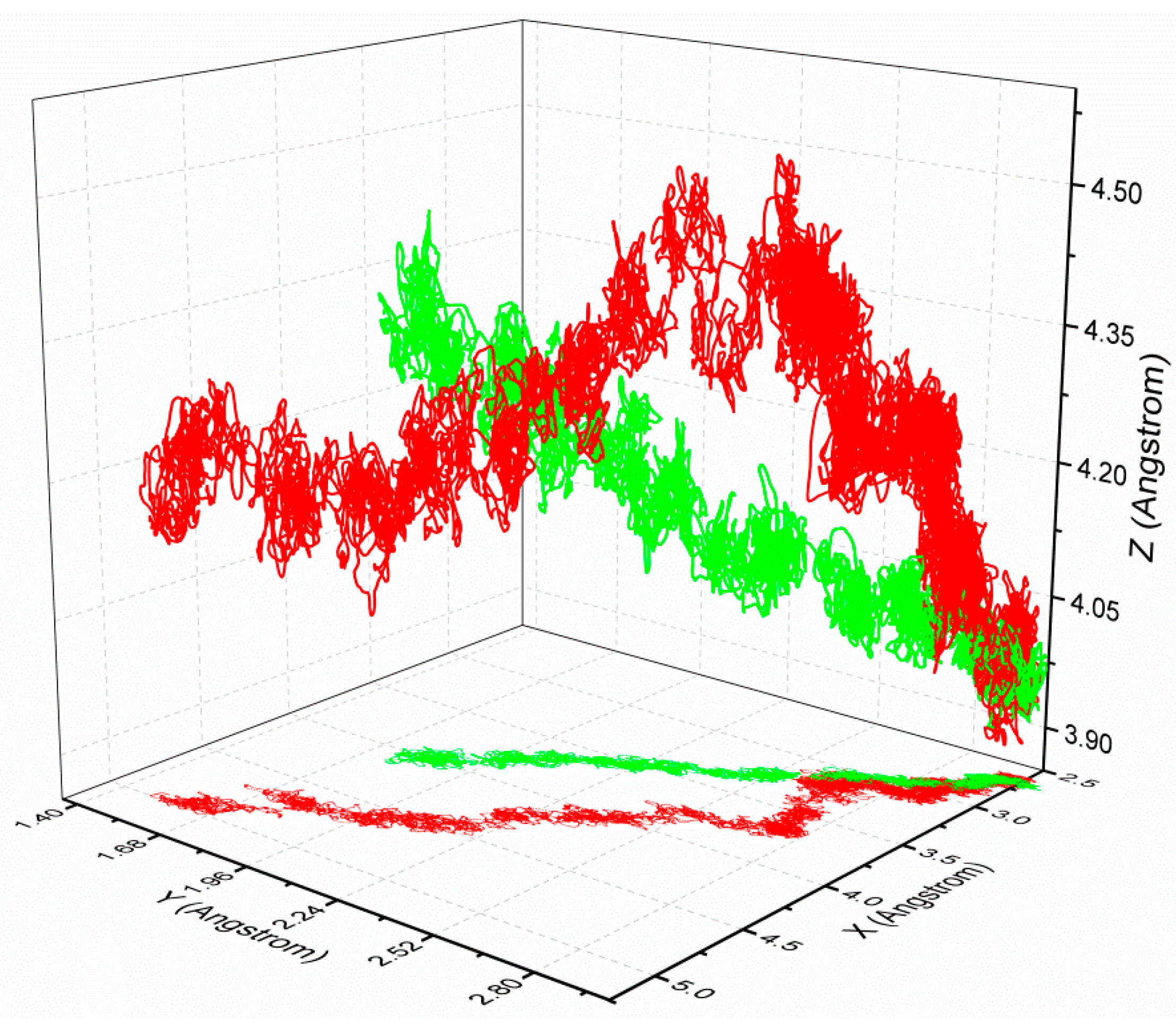
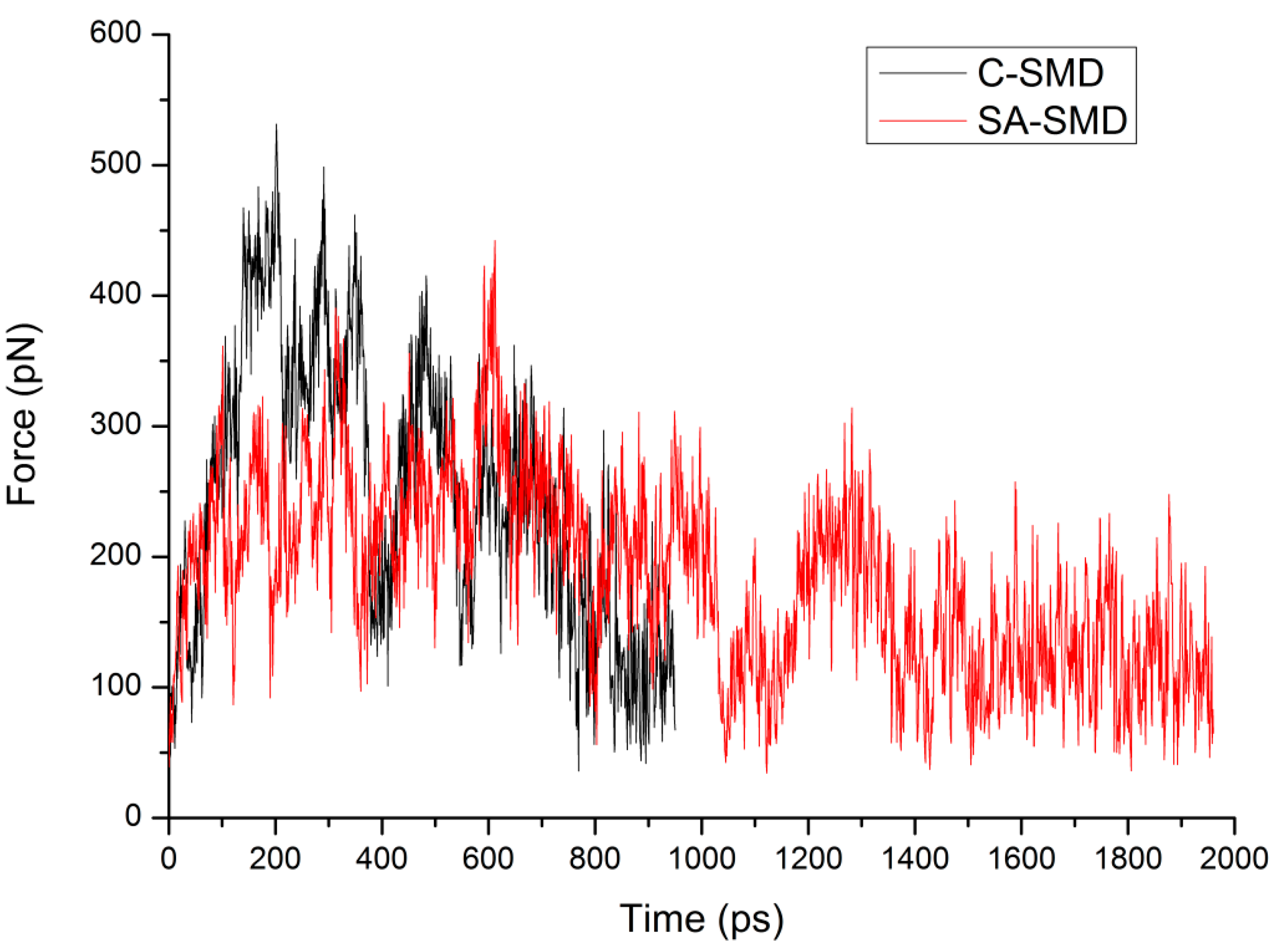
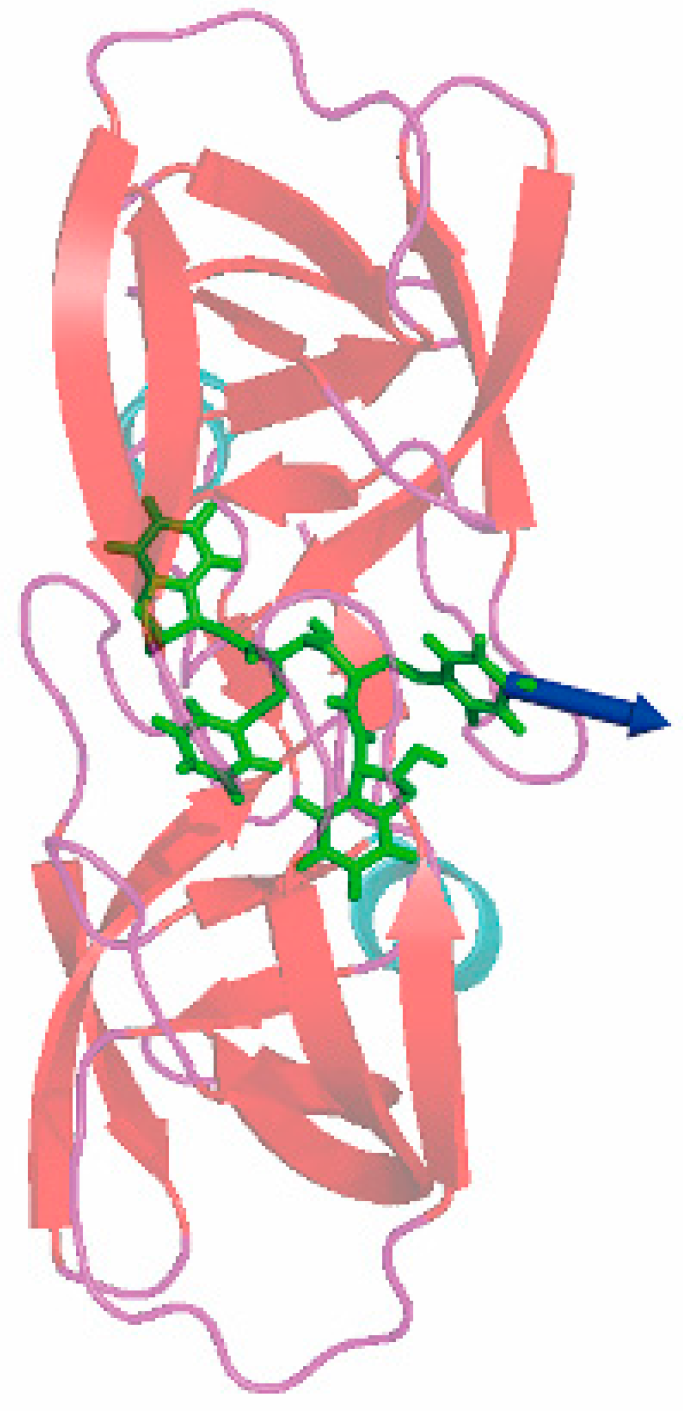
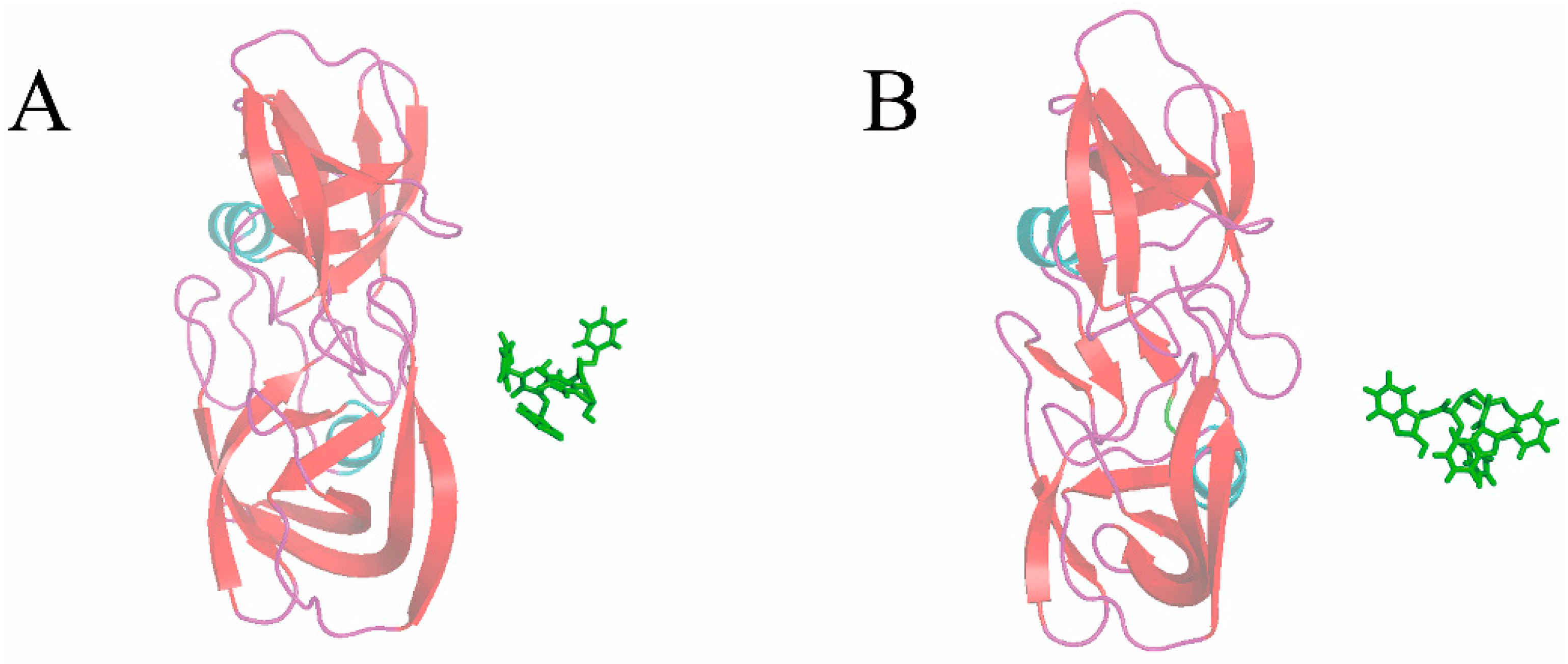
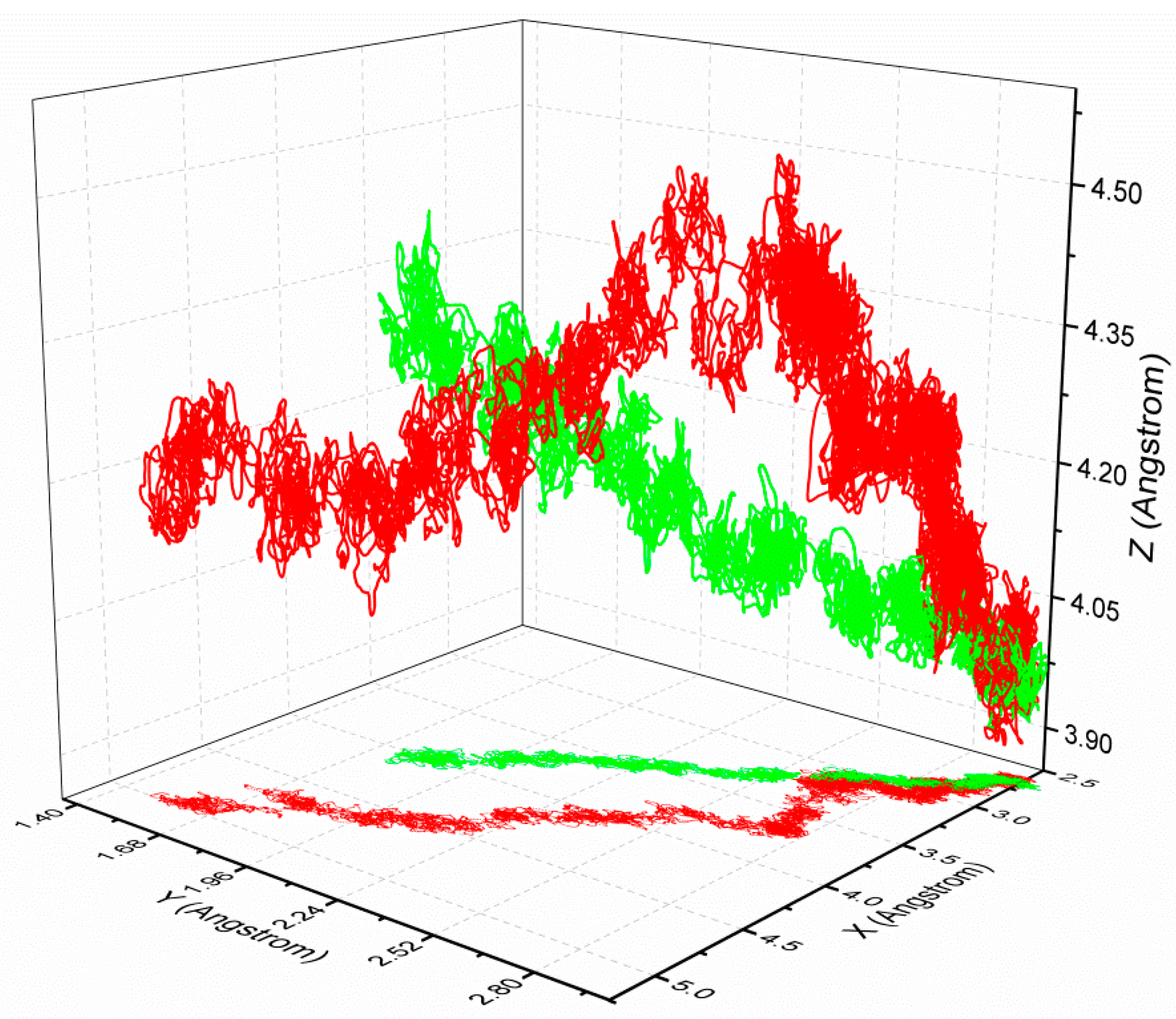
3.3. The Optimized Unbinding Process
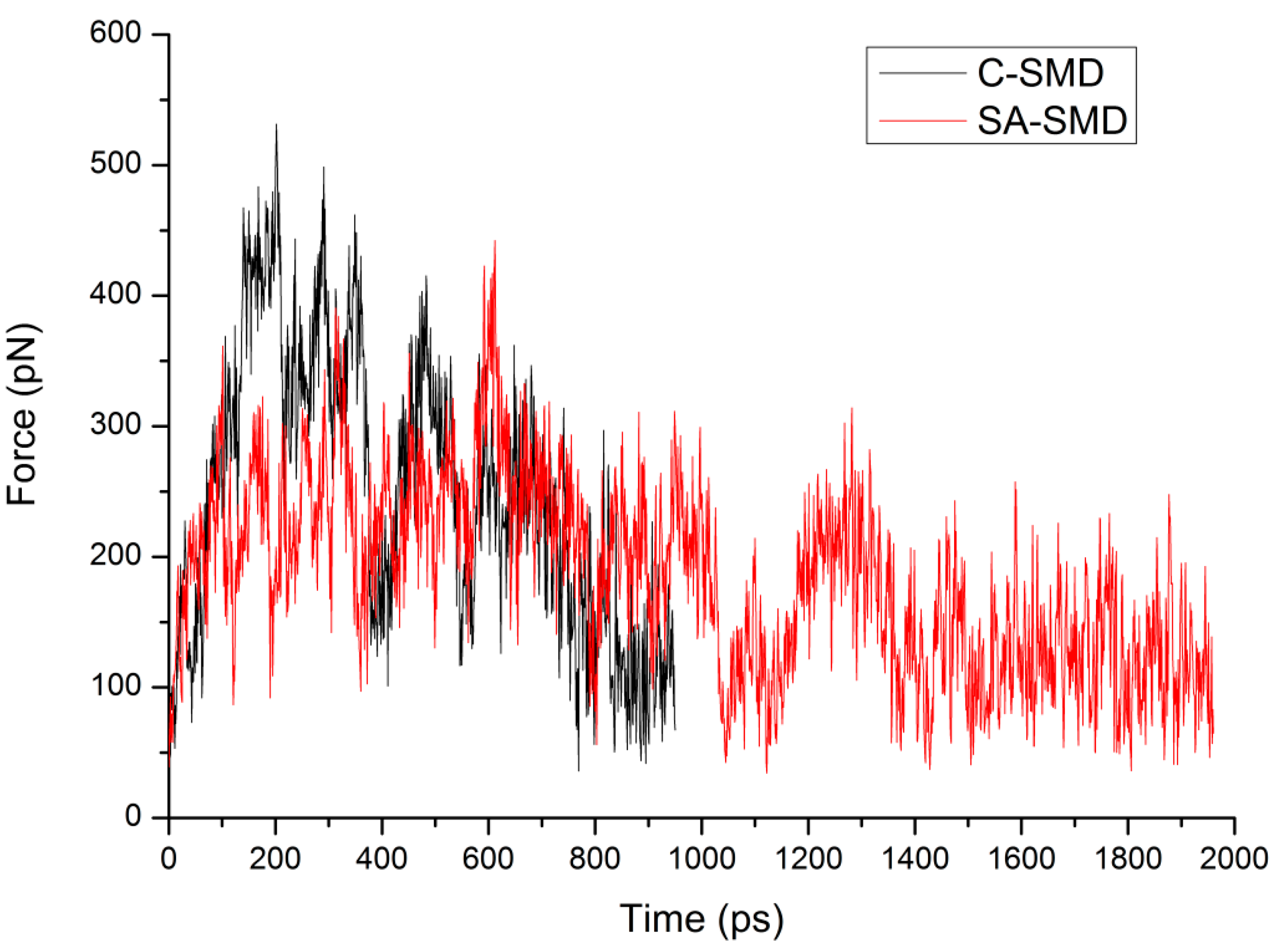
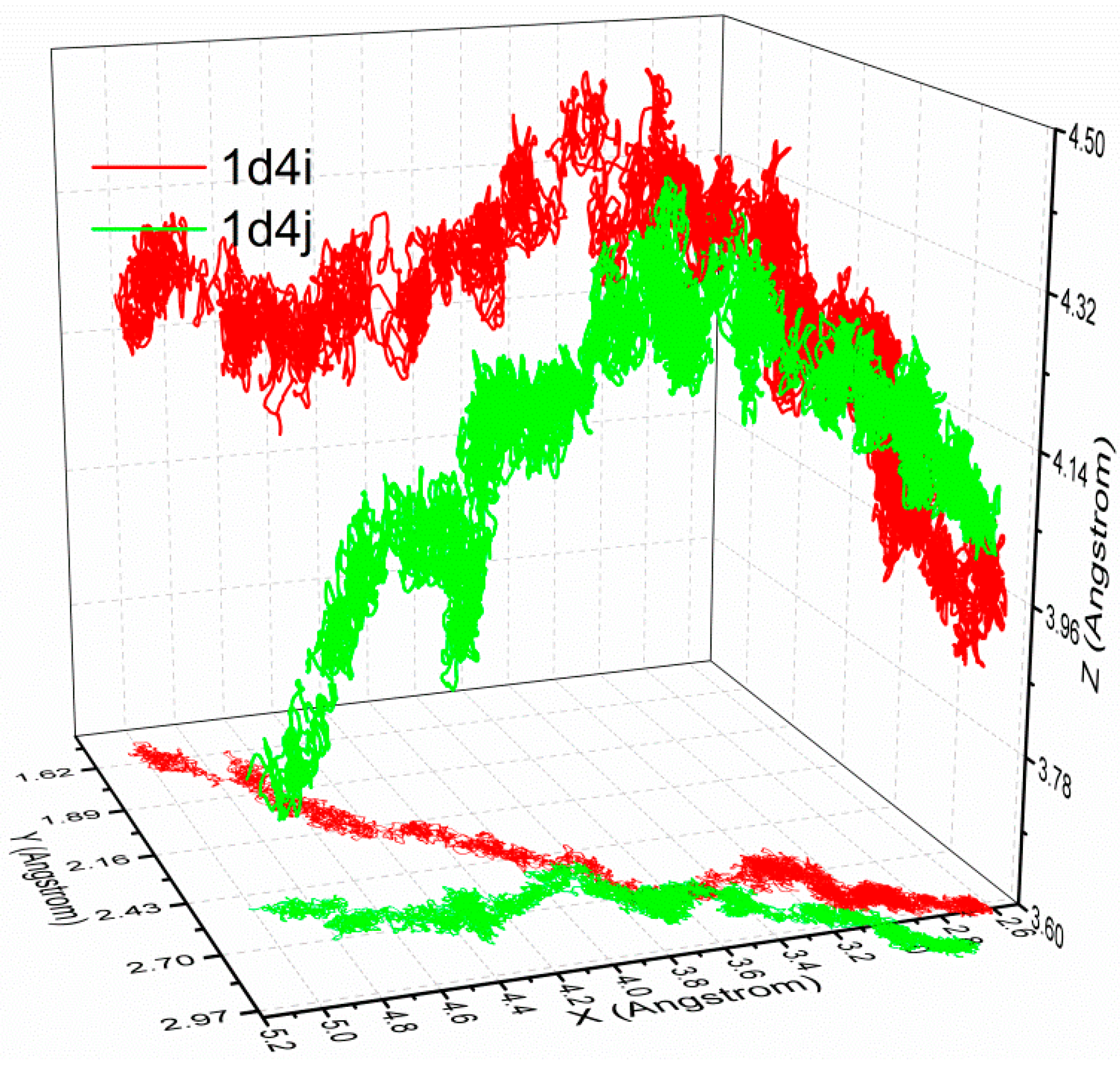
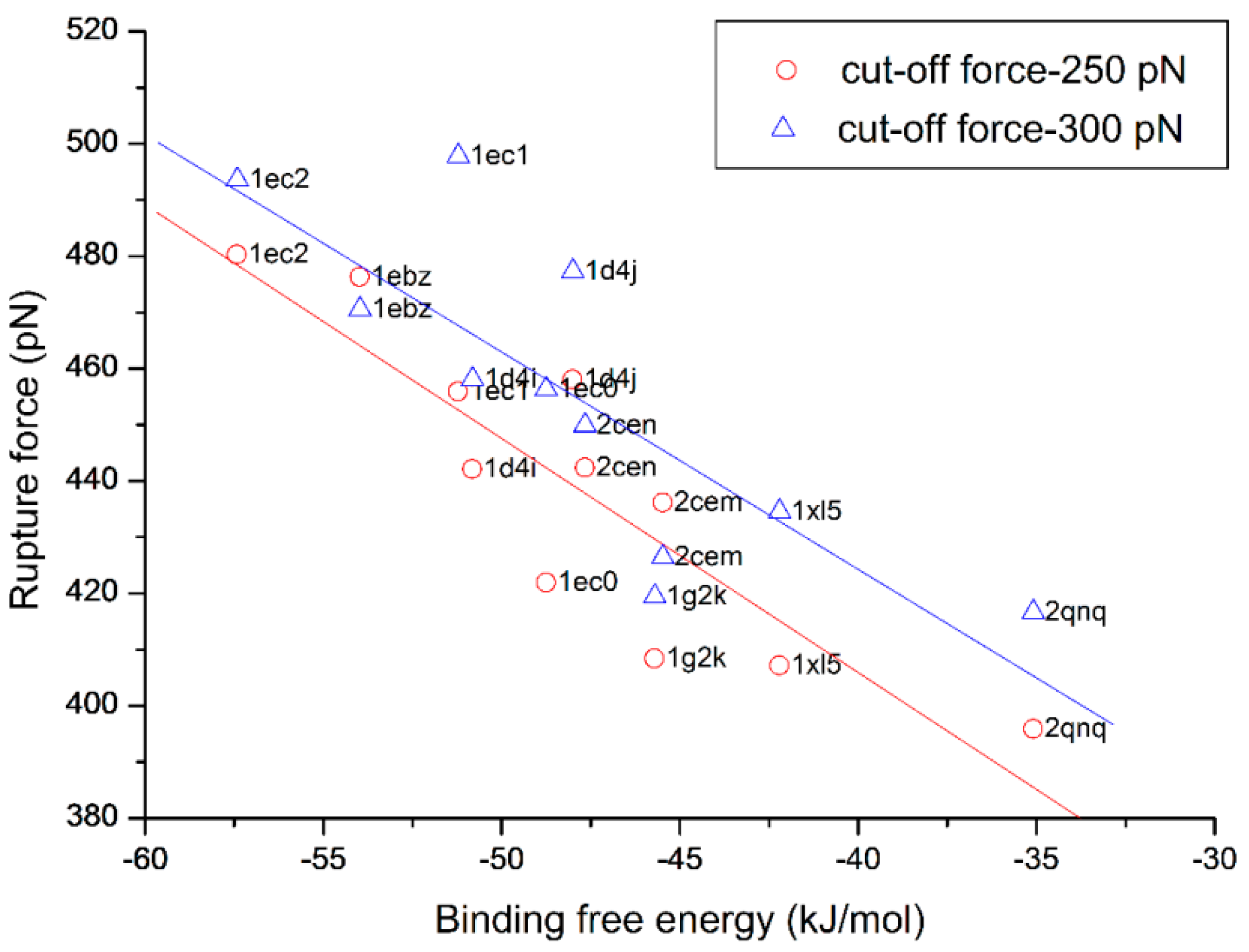
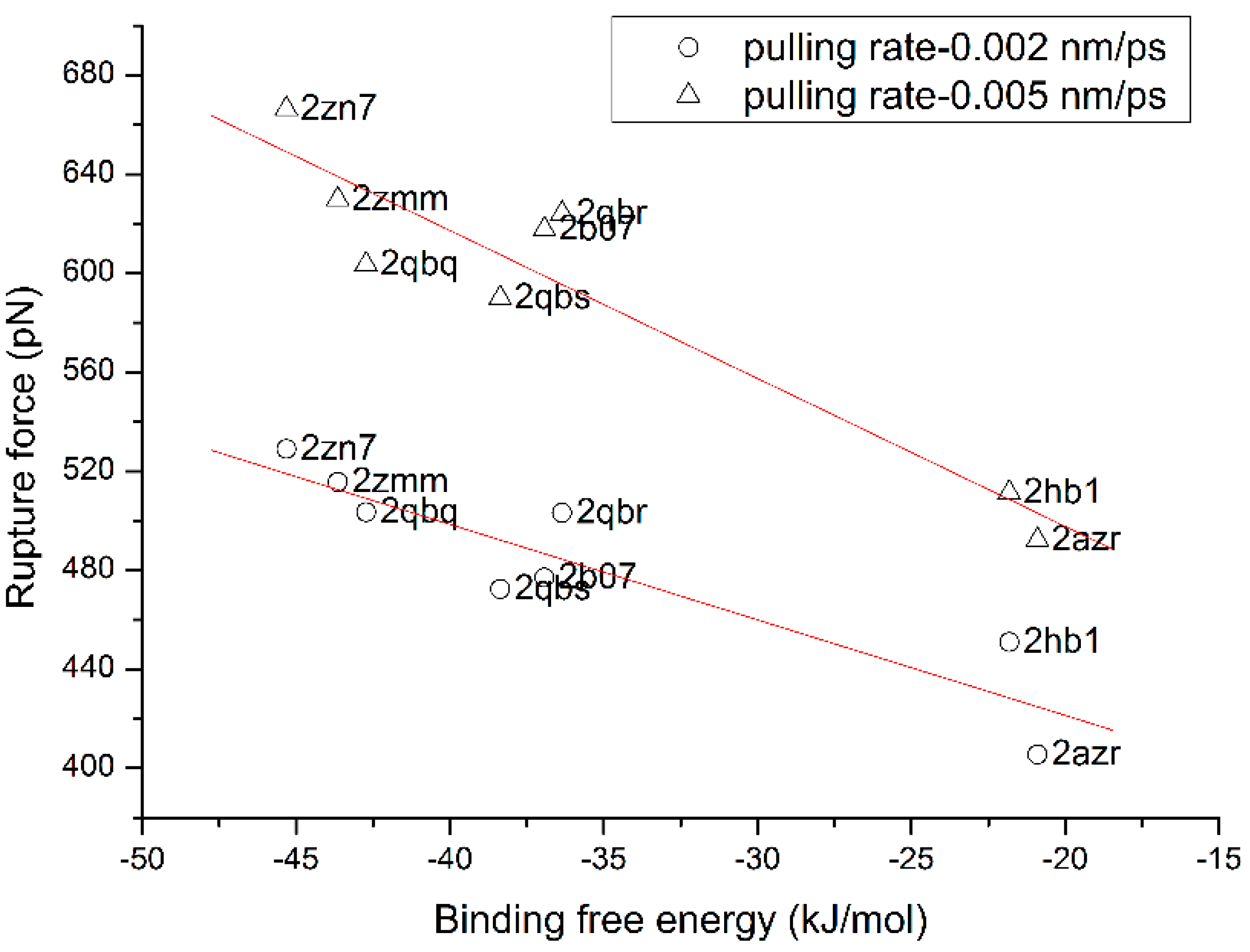
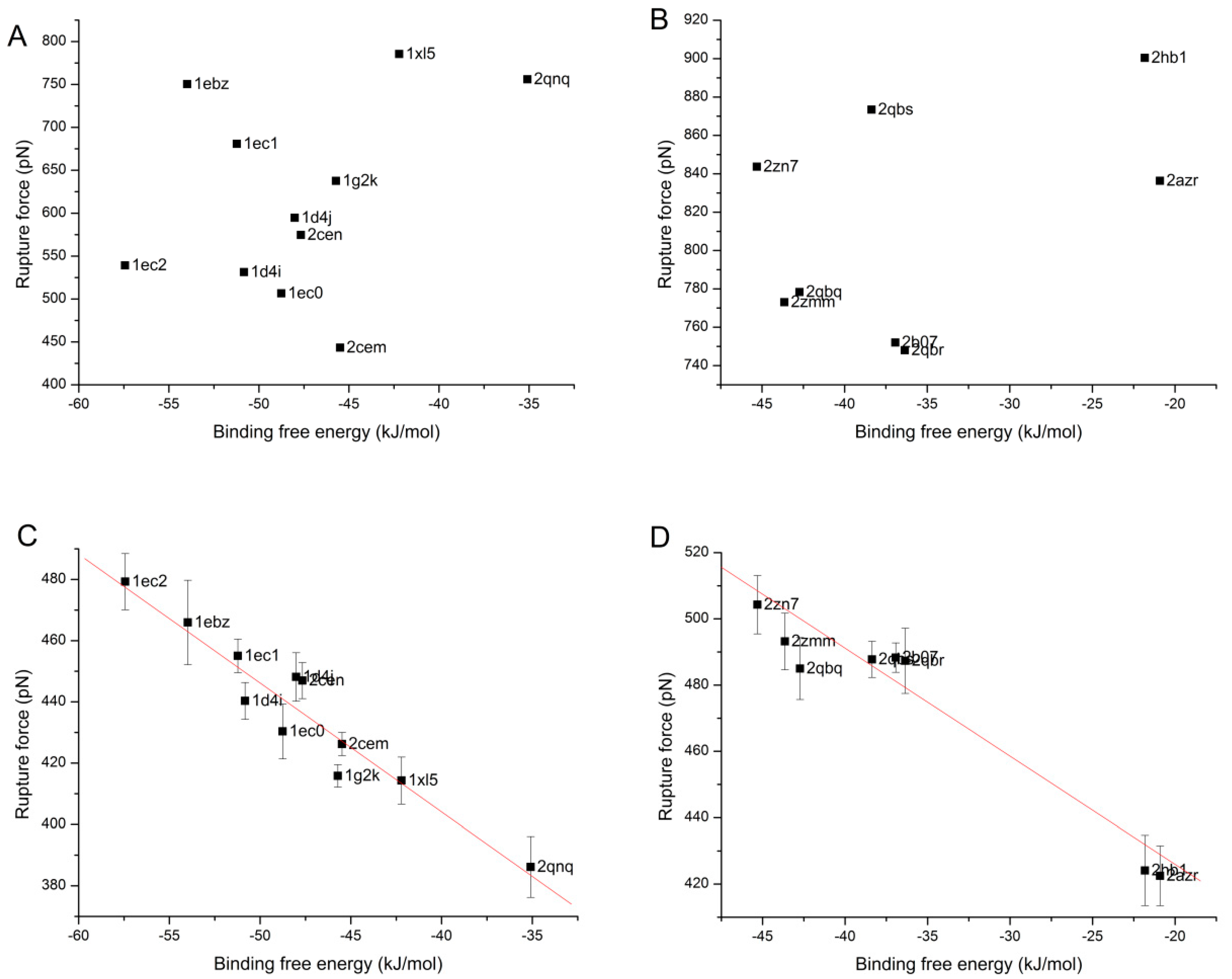
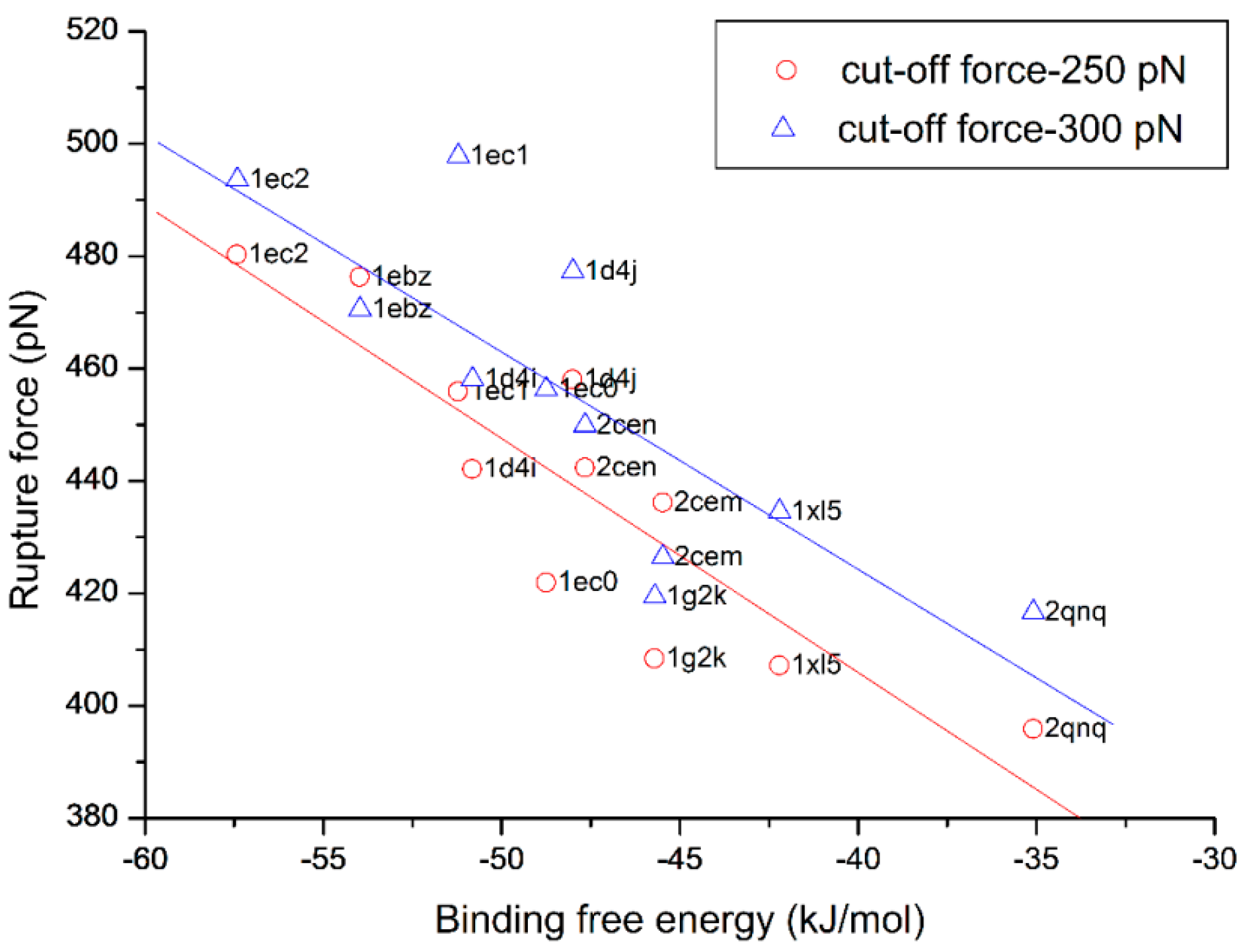
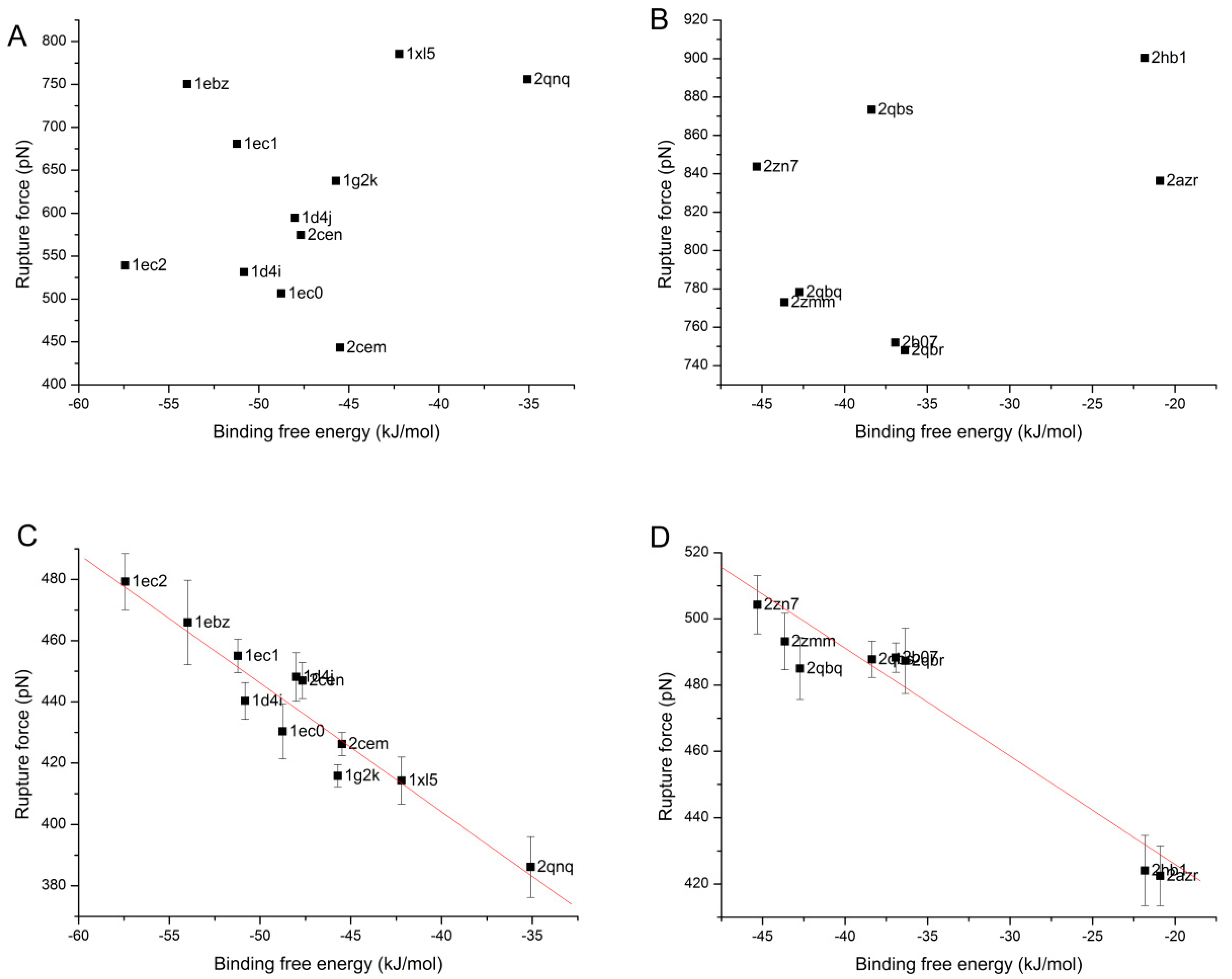
3.4. The Relationship of the Rupture Force and Binding Free Energy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Kollman, P.A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
- Schwarzl, S.M.; Tschopp, T.B.; Smith, J.C.; Fischer, S. Can the calculation of ligand binding free energies be improved with continuum solvent electrostatics and an ideal-gas entropy correction? J. Comput. Chem. 2002, 23, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics poisson-boltzmann surface area method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Wright, D.W.; Hall, B.A.; Kenway, O.A.; Jha, S.; Coveney, P.V. Computing clinically relevant binding free energies of hiv-1 protease inhibitors. J. Chem. Theory Comput. 2014, 10, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Knapp, B.; Wright, D.W.; Deane, C.M.; Coveney, P.V. Rapid, precise, and reproducible prediction of peptide-mhc binding affinities from molecular dynamics that correlate well with experiment. J. Chem. Theory Comput. 2015, 11, 3346–3356. [Google Scholar] [CrossRef]
- Straatsma, T.P.; McCammon, J.A. Multiconfiguration thermodynamic integration. J. Chem. Phys. 1991, 95, 1175–1188. [Google Scholar] [CrossRef]
- Jiang, W.; Hodoscek, M.; Roux, B. Computation of absolute hydration and binding free energy with free energy perturbation distributed replica-exchange molecular dynamics. J. Chem. Theory Comput. 2009, 5, 2583–2588. [Google Scholar] [CrossRef] [PubMed]
- Khavrutskii, I.V.; Wallqvist, A. Improved binding free energy predictions from single-reference thermodynamic integration augmented with hamiltonian replica exchange. J. Chem. Theory Comput. 2011, 7, 3001–3011. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.J.; Deng, Y.Q.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Kaus, J.W. How to deal with multiple binding poses in alchemical relative protein–ligand binding free energy calculations. J. Chem. Theory Comput. 2015, 11, 2670–2679. [Google Scholar] [CrossRef] [PubMed]
- Doudou, S.; Burton, N.A.; Henchman, R.H. Standard free energy of binding from a one-dimensional potential of mean force. J. Chem. Theory Comput. 2009, 5, 909–918. [Google Scholar] [CrossRef]
- Buch, I.; Sadiq, S.K.; Fabritiis, G.D. Optimized potential of mean force calculations for standard binding free energies. J. Chem. Theory Comput. 2011, 7, 1765–1772. [Google Scholar] [CrossRef]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Gil, V.A.; Guallar, V. Monte carlo free ligand diffusion with markov state model analysis and absolute binding free energy calculations. J. Chem. Theory Comput. 2013, 10, 282–288. [Google Scholar] [CrossRef]
- Chen, L.Y. Hybrid steered molecular dynamics approach to computing absolute binding free energy of ligand-protein complexes: A brute force approach that is fast and accurate. J. Chem. Theory Comput. 2015, 11, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Isralewitz, B.; Izrailev, S.; Schulten, K. Binding pathway of retinal to bacterio-opsin: A prediction by molecular dynamics simulations. Biophys. J. 1997, 73, 2972–2979. [Google Scholar] [CrossRef]
- Sotomayor, M.; Schulten, K. Single-molecule experiments in vitro and in silico. Science 2007, 316, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Jarzynski, C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef]
- Crooks, G.E. Nonequilibrium measurements of free energy differences for microscopically reversible markovian systems. J. Stat. Phys. 1998, 90, 1481–1487. [Google Scholar] [CrossRef]
- Vashisth, H.; Abrams, C.F. Ligand escape pathways and (un)binding free energy calculations for the hexameric insulin-phenol complex. Biophys. J. 2008, 95, 4193–4204. [Google Scholar] [CrossRef] [PubMed]
- Ytreberg, F.M. Absolute fkbp binding affinities obtained via nonequilibrium unbinding simulations. J. Chem. Phys. 2009, 130, 164906. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Li, W.; Liu, G.; Tang, Y.; Jiang, H. Computational insights into the mechanism of ligand unbinding and selectivity of estrogen receptors. J. Phys. Chem. B 2009, 113, 10436–10444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gullingsrud, J.; McCammon, J.A. Potentials of mean force for acetylcholine unbinding from the alpha7 nicotinic acetylcholine receptor ligand binding domain. J. Am. Chem. Soc. 2006, 128, 3019–3026. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, P.; Frezzato, D.; Gellini, C.; Bizzarri, M.; Chelli, R. Toward quantitative estimates of binding affinities for protein–lignad systems involving large inhibitor compounds: A steered molecular dynamics simulation route. J. Comput. Chem. 2013, 34, 1561–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedl, T.; Seifert, U. Optimal finite-time processes in stochastic thermodynamics. Phys. Rev. Lett. 2007, 98, 108301. [Google Scholar] [CrossRef] [PubMed]
- Vaikuntanathan, S.; Jarzynski, C. Escorted free energy simulations: Improving convergence by reducing dissipation. Phys. Rev. Lett. 2008, 100, 190601. [Google Scholar] [CrossRef] [PubMed]
- Ozer, G.; Keyes, T.; Quirk, S.; Hernandez, R. Multiple branched adaptive steered molecular dynamics. J. Chem. Phys. 2014, 141, 064101. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, E.; Gellini, C.; Pietraperzia, G.; Cardini, G.; Chelli, R. Combining path-breaking with bidirectional nonequilibrium simulations to improve efficiency in free energy calculations. J. Chem. Phys. 2014, 140, 064104. [Google Scholar] [CrossRef] [PubMed]
- Chelli, R.; Gellini, C.; Pietraperzia, G.; Giovannelli, E.; Cardini, G. Path-breaking schemes for nonequilibrium free energy calculations. J. Chem. Phys. 2013, 138, 214109. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Pande, V.S. Comparison of efficiency and bias of free energies computed by exponential averaging, the bennett acceptance ratio, and thermodynamic integration. J. Chem. Phys. 2005, 122, 144107. [Google Scholar] [CrossRef] [PubMed]
- Chelli, R.; Procacci, P. A potential of mean force estimator based on nonequilibrium work exponential averages. Phys. Chem. Chem. Phys. 2009, 11, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, P.; Procacci, P.; Chelli, R. Hummer and szabo-like potential of mean force estimator for bidirectional nonequilibrium pulling experiments/simulations. J. Phys. Chem. B 2010, 114, 9546–9554. [Google Scholar] [CrossRef] [PubMed]
- Chelli, R.; Marsili, S.; Procacci, P. Calculation of the potential of mean force from nonequilibrium measurements via maximum likelihood estimators. Phys. Rev. E 2008, 77, 031104. [Google Scholar] [CrossRef]
- Minh, D.D.L.; Adib, A.B. Optimized free energies from bidirectional single-molecule force spectroscopy. Phys. Rev. Lett. 2008, 100, 180602. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Bair, E.; Hooker, G.; Pande, V.S. Equilibrium free energies from nonequilibrium measurements using maximum-likelihood methods. Phys. Rev. Lett. 2003, 91, 140610. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Drug discovery: Pulled from a protein’s embrace. Nature 2010, 466, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, F.; Perozzo, R.; Scapozza, L.; Recanatini, M.; Cavalli, A. Single-molecule pulling simulations can discern active from inactive enzyme inhibitors. J. Am. Chem. Soc. 2010, 132, 7361–7371. [Google Scholar] [CrossRef] [PubMed]
- Mai, B.K.; Viet, M.H.; Li, M.S. Top leads for swine influenza a/h1n1 virus revealed by steered molecular dynamics approach. J. Chem. Inf. Model. 2010, 50, 2236–2247. [Google Scholar] [CrossRef] [PubMed]
- Mai, B.K.; Li, M.S. Neuraminidase inhibitor r-125489—A promising drug for treating influenza virus: Steered molecular dynamics approach. Biochem. Biophys. Res. Commun. 2011, 410, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Jiang, H. A steered molecular dynamics method with direction optimization and its applications on ligand molecule dissociation. J. Biochem. Biophys. Methods 2008, 70, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Liu, X.; Wang, X.; Jiang, H. A steered molecular dynamics method with adaptive direction adjustments. Biochem. Biophys. Res. Commun. 2009, 379, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wang, X.; Yang, Y. A steered molecular dynamics method for receptor-ligand unbinding based on genetic algorithm. In Proceedings of the 2013 International Conference on Information Science and Cloud Computing Companion, Guangzhou, China, 7–8 December 2013; pp. 698–703.
- Kang, L.; Li, H.; Jiang, H.; Wang, X. An improved adaptive genetic algorithm for protein–ligand docking. J. Comput. Aided Mol. Des. 2009, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Van Buuren, A.R.; Marrink, S.J.; Berendsen, H.J.C. A molecular dynamics study of the decane/water interface. J. Phys. Chem. 1993, 97, 9206–9212. [Google Scholar] [CrossRef]
- Mark, A.E.; van Helden, S.P.; Smith, P.E.; Janssen, L.H.M.; van Gunsteren, W.F. Convergence properties of free energy calculations: .Alpha.-cyclodextrin complexes as a case study. J. Am. Chem. Soc. 1994, 116, 6293–6302. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Pullman, B., Ed.; Springer Netherlands: Dordrecht, The Netherlands, 1981; Volume 14, pp. 331–342. [Google Scholar]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, J.; Li, H.; Wang, X. A Self-Adaptive Steered Molecular Dynamics Method Based on Minimization of Stretching Force Reveals the Binding Affinity of Protein–Ligand Complexes. Molecules 2015, 20, 19236-19251. https://doi.org/10.3390/molecules201019236
Gu J, Li H, Wang X. A Self-Adaptive Steered Molecular Dynamics Method Based on Minimization of Stretching Force Reveals the Binding Affinity of Protein–Ligand Complexes. Molecules. 2015; 20(10):19236-19251. https://doi.org/10.3390/molecules201019236
Chicago/Turabian StyleGu, Junfeng, Hongxia Li, and Xicheng Wang. 2015. "A Self-Adaptive Steered Molecular Dynamics Method Based on Minimization of Stretching Force Reveals the Binding Affinity of Protein–Ligand Complexes" Molecules 20, no. 10: 19236-19251. https://doi.org/10.3390/molecules201019236