
Six-Membered Aromatic Polyazides: Synthesis and Application

Institute of Problems of Chemical Physics, Russian Academy of Sciences, Chernogolovka 142432, Moscow Region, Russian
Molecules 2015, 20(10), 19142-19171; https://doi.org/10.3390/molecules201019142
Submission received: 11 September 2015
/
Revised: 30 September 2015
/
Accepted: 13 October 2015
/
Published: 21 October 2015
(This article belongs to the Special Issue Organic Azides)
Abstract
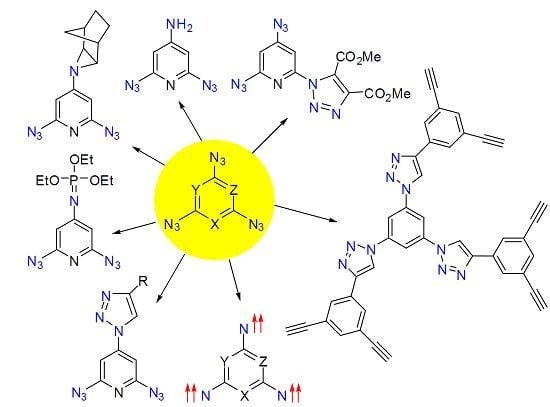
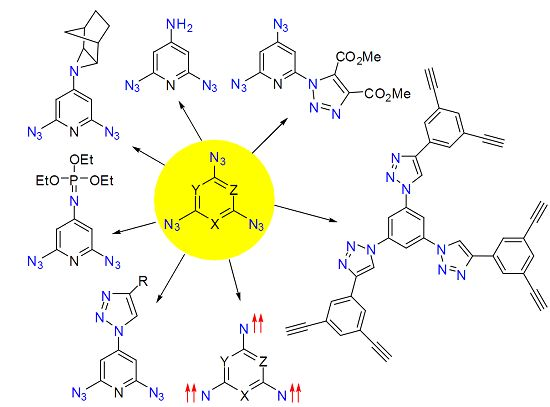
:Aromatic polyazides are widely used as starting materials in organic synthesis and photochemical studies, as well as photoresists in microelectronics and as cross-linking agents in polymer chemistry. Some aromatic polyazides possess high antitumor activity, while many others are of considerable interest as high-energy materials and precursors of high-spin nitrenes and C3N4 carbon nitride nanomaterials. The use of aromatic polyazides in click-reactions may be a new promising direction in the design of various supramolecular systems possessing interesting chemical, physical and biological properties. This review is devoted to the synthesis, properties and applications of six-membered aromatic compounds containing three and more azido groups in the ring.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
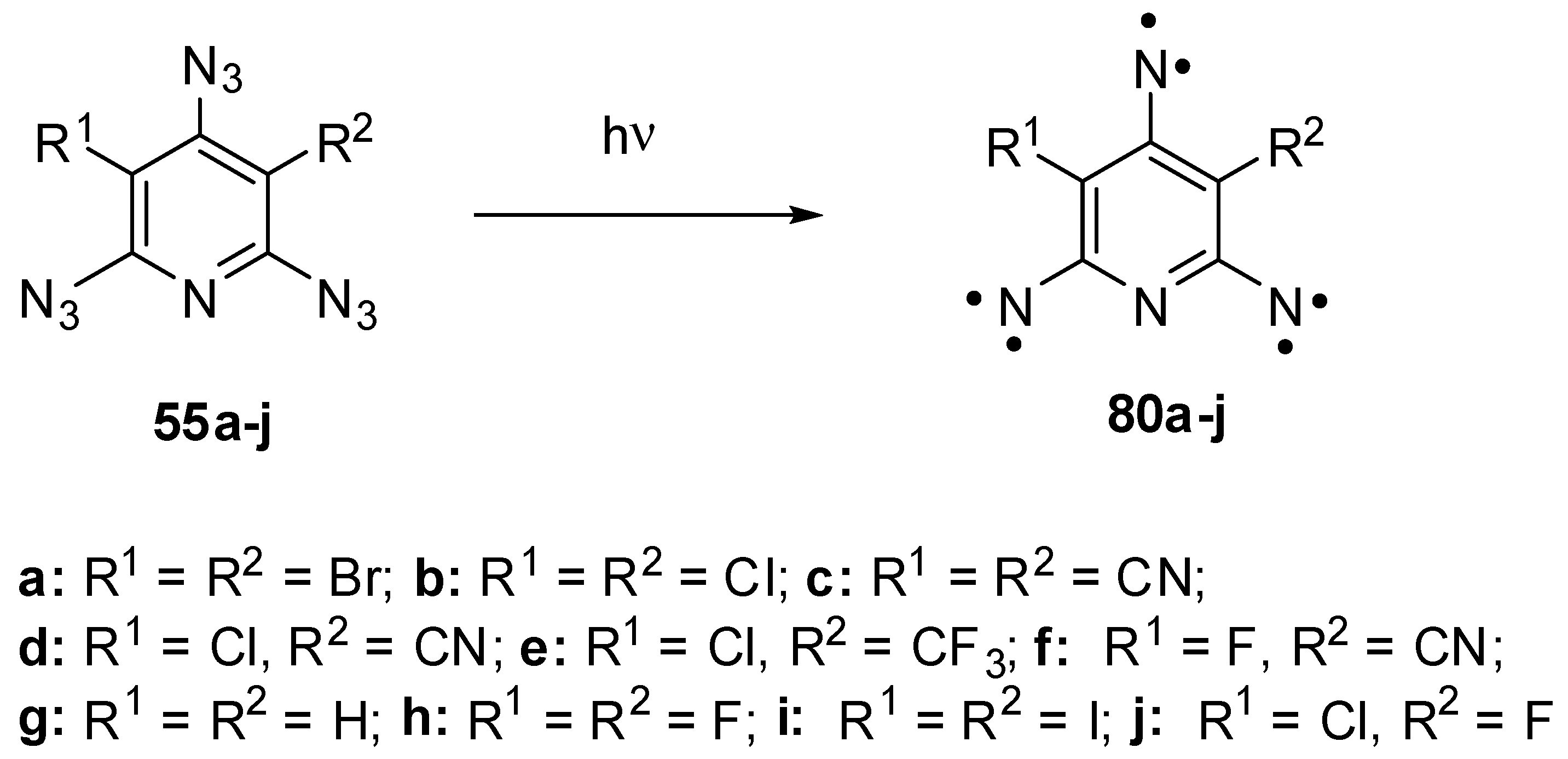{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
To date, just about two dozen six-membered aromatic compounds containing three and more azido groups in the ring are known. Nevertheless, investigations of these polyazides have played an important role in the development of chemistry. Thus, the first data on the linear structure of the azido groups were obtained due to X-ray diffraction studies of crystalline 2,4,6-triazido-1,3,5-triazine [1,2,3,4,5]. The thermal and detonative decomposition of this triazide allowed as well the first preparation of diverse C3N4 carbon nitride nanomaterials and carbon nanotubes [6,7,8,9]. The first organic septet hexaradical was prepared by the photolysis of 1,3,5-triazido-2,4,6-tricyanobenzene [10]. Later, a great variety of high-spin nitrenes possessing unusual magnetic characteristics were obtained by the photolysis of various 1,3,5-triazidobenzenes, 2,4,6-triazidopyridines, 2,4,6-triazidopyrimidines and 2,4,6-triazido-1,3,5-triazine [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]. These studies of high-spin nitrenes opened up new prospects in design of new organic magnetic materials and provided important information about UV-vis, IR and EPR spectra of organic molecules with the ground quintet and septet spin-state. In particular, it was found that the placement of heavy bromine atoms in molecules of high-spin nitrenes can strengthen several times the magnetic properties of organic polyradicals [26,33]. Six-membered aromatic triazides have also played an important role in investigations of the reactivity of the azido groups. Thus, it was found that the more electron-deficient γ-azido groups of 2,4,6-triazidopyridines selectively react with electron-rich dipolarophiles, phosphines, phosphites and reductants [34,35]. By contrast, the more electron-rich α-azido groups of the same triazides are the most reactive toward electron-poor dipolarophiles. Since 2002, organic azides are widely used as starting materials in the copper-catalyzed azide-alkyne cycloadditions or so-called click-reactions to prepare organic compounds with interesting chemical, physical and biological properties [36,37]. Recent examples on the use of aromatic diazides in such reactions demonstrate a huge synthetic potential of aromatic polyazides in design of new polyfunctional organic materials [38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60]. The present review provides concise and precise updates on the latest progress in the synthesis and investigation of six-membered aromatic polyazides made by July 2015.
2. Cyanuric Triazide
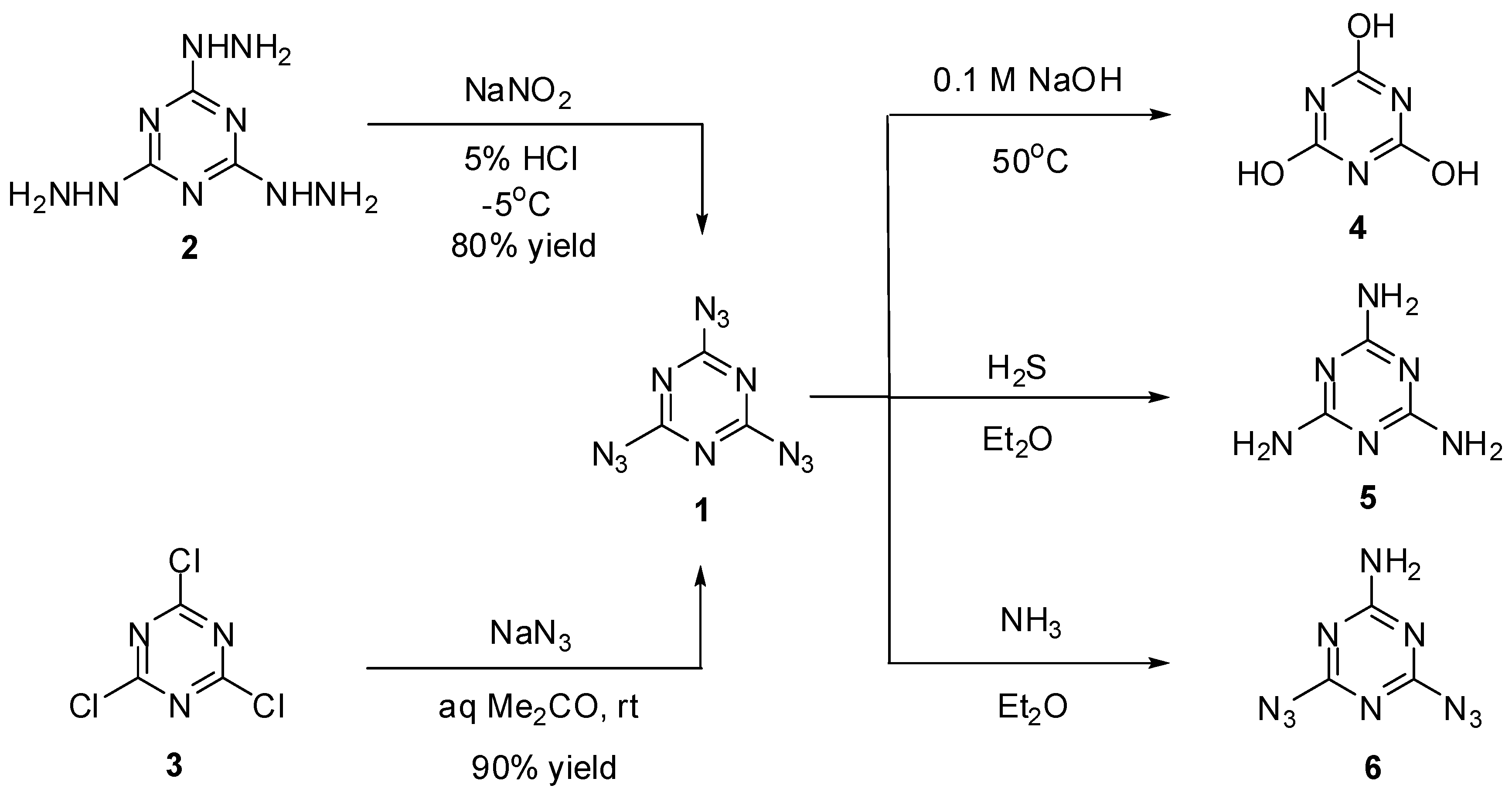
2,4,6-Triazido-1,3,5-triazine or cyanuric triazide (1) was prepared for the first time by H. Finger in 1907, using diazotization of tris-hydrazine 2 with NaNO2 in acidic media (Scheme 1) [61]. Several years later, a more convenient way to prepare 1 based on triazidation of cyanuric chloride 3 with sodium azide in aqueous acetone was developed by Ott and Ohse [62]. Currently, triazide 1 is readily prepared in almost quantitative yield by addition of small portions of solid sodium azide to solutions of chloride 3 in acetone at room temperature [63,64]. On crystallization from ethanol, triazide 1 forms dense colorless crystals with a melting point of 94 °C [61,62,63,64]. Due to its high nitrogen content (82.35%), triazide 1 is very sensitive to impact, friction and electrostatic discharge. On working with it, one should always handle 1 only with plastic spatulas and use thick gloves behind a blast shield.
Scheme 1.
Synthesis and some transformations of triazide 1.
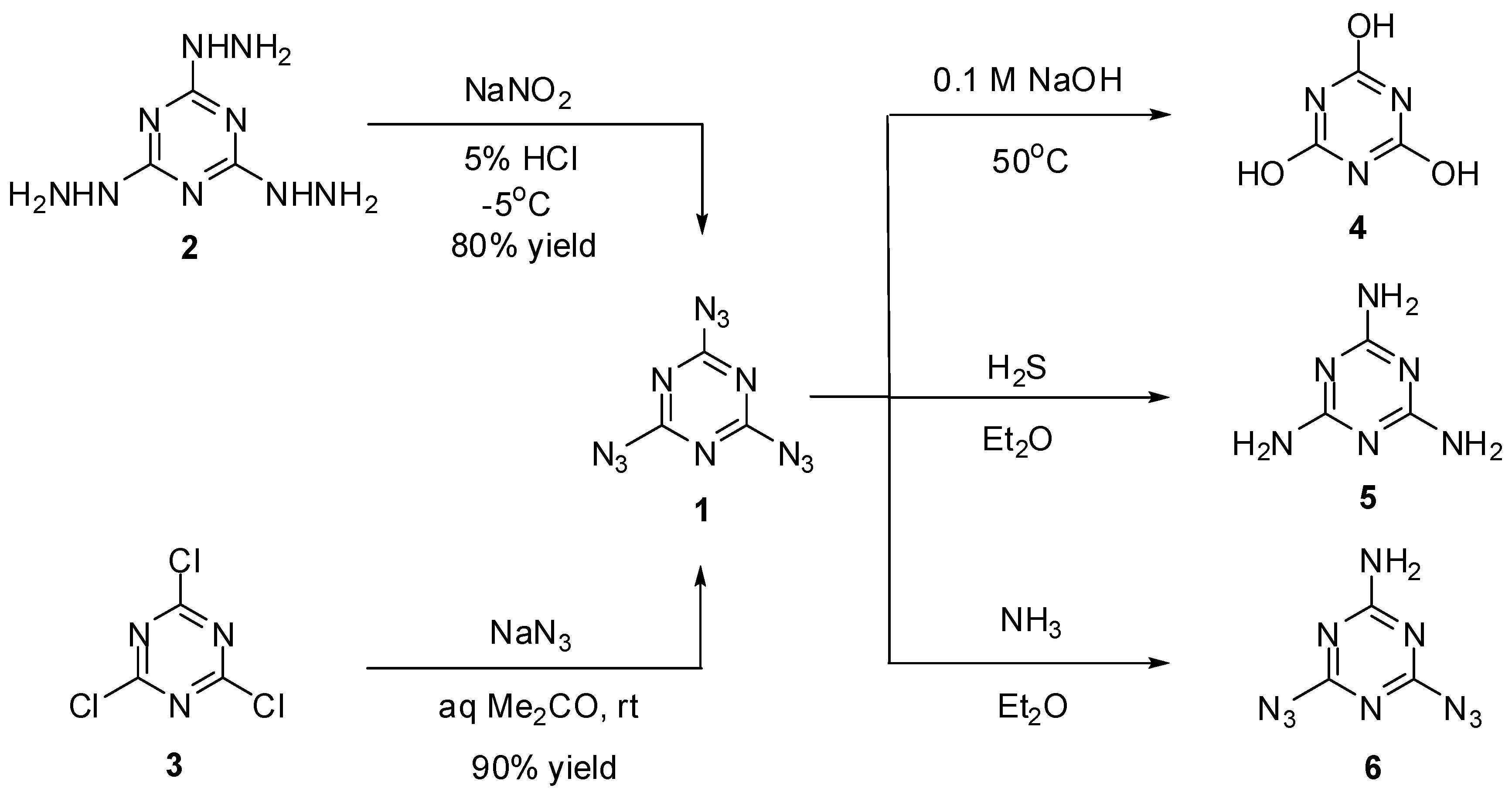
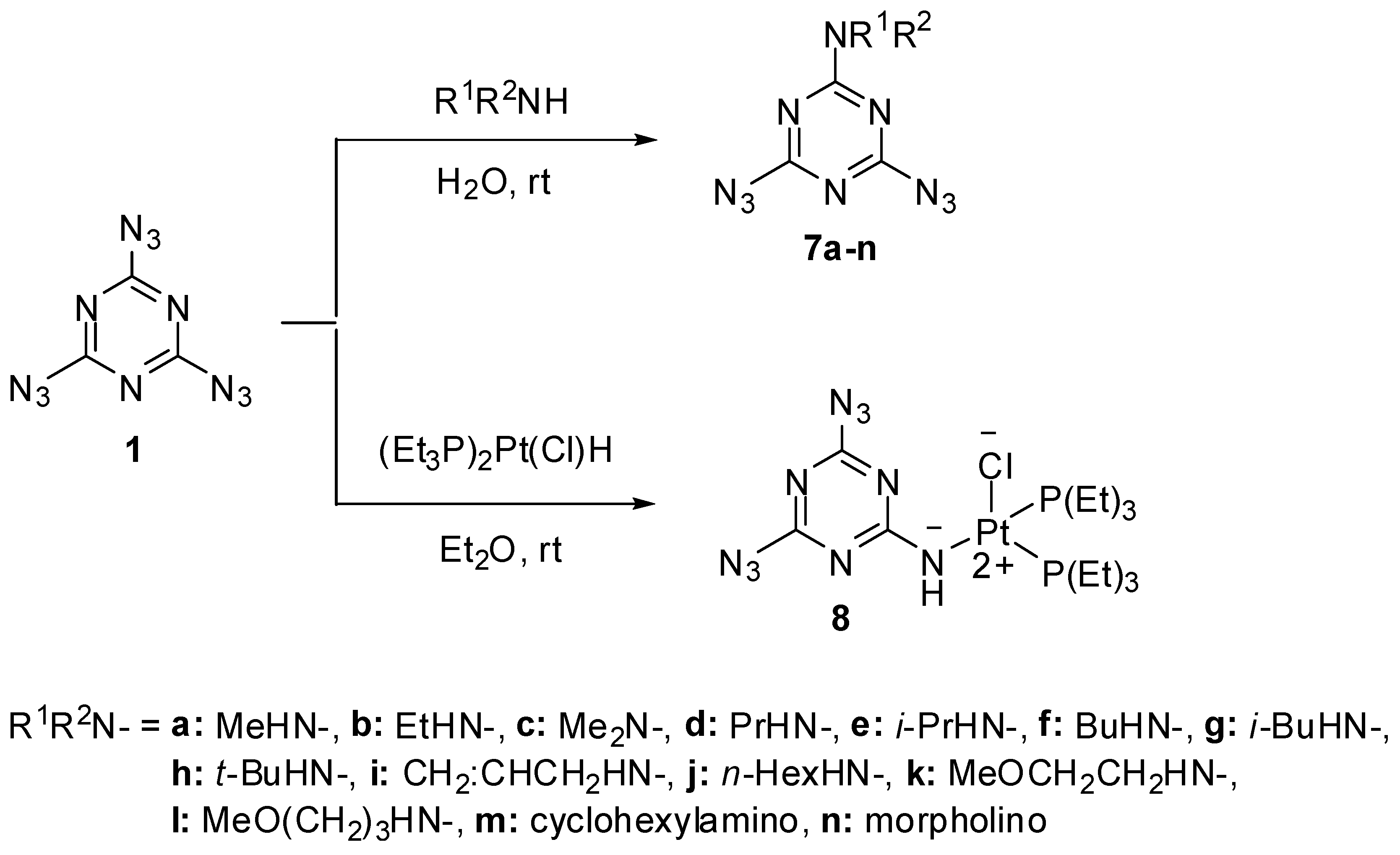
Early studies have shown that triazide 1 was quickly hydrolyzed to cyanuric acid (4) on heating in 0.1 M solutions of sodium hydroxide at 50 °C (Scheme 2) [62]. By action of gaseous hydrogen sulfide, triazide 1 was reduced to melamine (5) [65]. Similar reaction of 1 with gaseous ammonia afforded aminodiazide 6 in almost quantitative yield [65]. In the last case, the reaction can formally be considered as nucleophilic substitution reaction of the azido group in 1 for the amino group. Thus, this triazide reacts in the same manner with various alkyl amines to give aminodiazides 7a–n in high yields [66]. All diazides 5 and 7a–n present as white solids with a melting point ranging from 44 °C for 7c till 200 °C for 6 and are almost insensitive to impact, friction and electrostatic discharge. These compounds may be of practical interest as insensitive high-energy additives and reactants for click-reactions. The reduction of triazide 1 with hydridoplatinum complex (Et3P)2Pt(Cl)H gives air-stable amidoplatinum complex 8 in 100% yield (Scheme 2) [67]. The latter can be considered as an analog of aminodiazides 6 and 7a–n.
Scheme 2.
Reactions of triazide 1 with alkyl amines and (Et3P)2Pt(Cl)H.

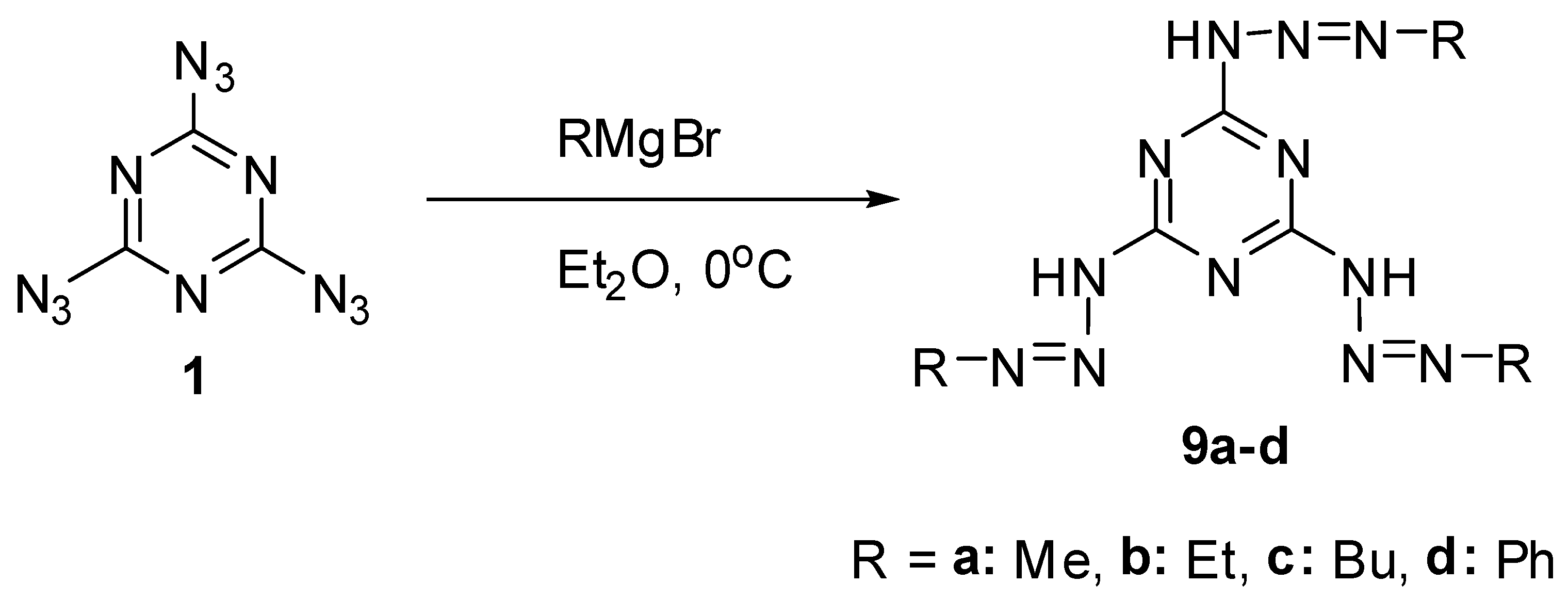
All three azido groups of 1 react with Grignard reagents to give tris-triazenes 9a–d in 47%–70% yield (Scheme 3) [68]. Triazenes are of considerable interest as anticancer drugs and powerful carcinogens [69,70,71].
Scheme 3.
Reactions of triazide 1 with Grignard reagents.

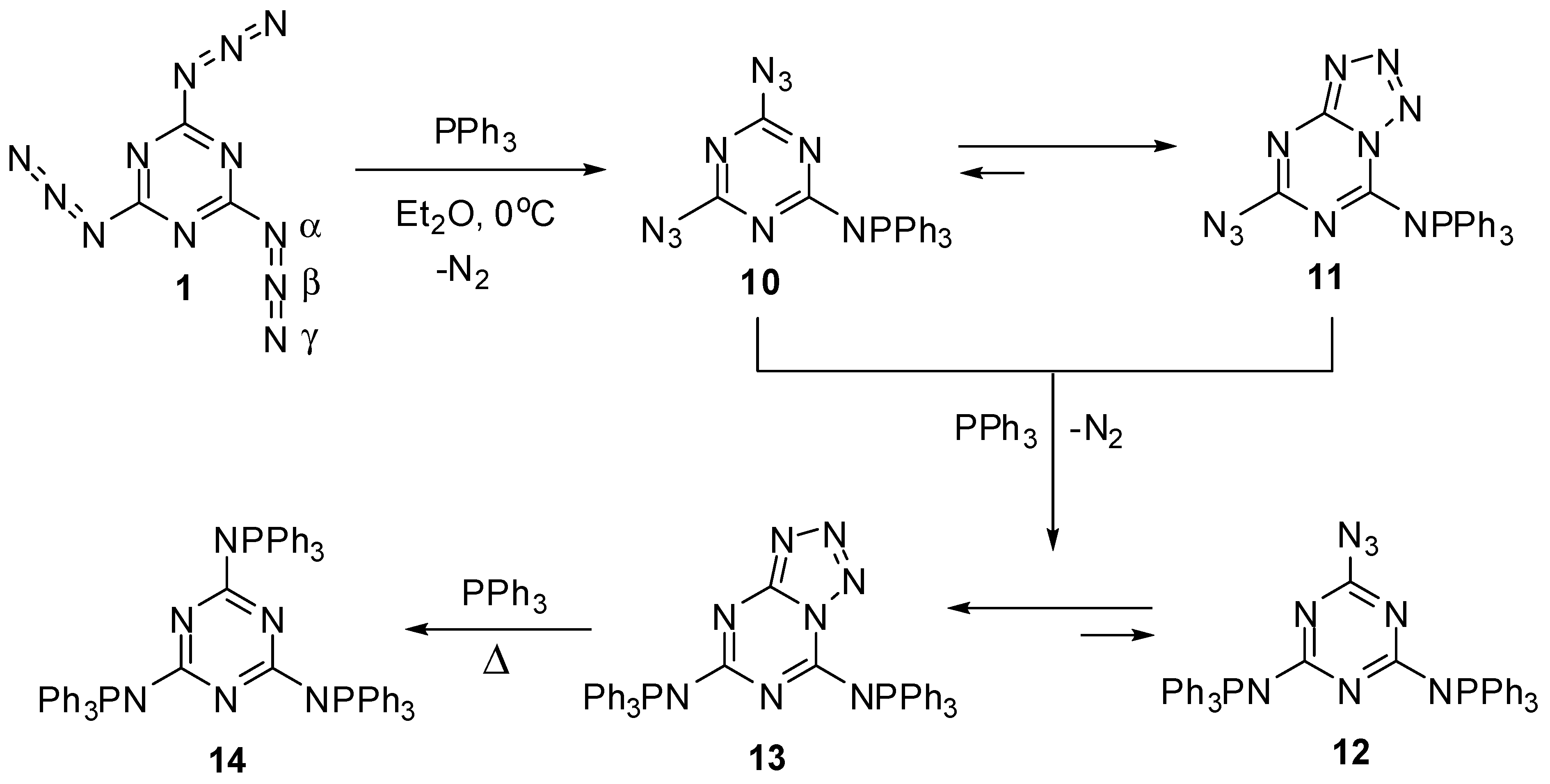
The first attempt to investigate the Staudinger-Meyer reaction of triazide 1 was undertaken by Kesting in 1923 [72]. The reaction of this triazide with an excess of triphenylphosphine in ether gave a single product that was assigned to azide 12, based on the elemental analysis data (Scheme 4). However, recent studies showed that this product really corresponds to tetrazole 13 [63,64]. The first stage of the reaction involves the formation of diazide 10 that exists in organic solutions in equilibrium with azidotetrazole 11. The ratio of 10 to 11 in chloroform and dimethylsulfoxide solutions is ~1:3 and 1:50, respectively [64]. The addition of two equivalents of triphenylphosphine to triazide 1 gives tetrazole 13 as a single product. The structures of tetrazoles 11 and 13 were unambiguously proved by using 13C-, 15N- and 31P-NMR spectroscopy and X-ray diffraction analysis [63,64]. Tetrazole 13 does not react with triphenylphosphine in solution. The tris-adduct 14 can only be obtained by melting 13 with an excess of triphenylphosphine [64].
Scheme 4.
Reactions of triazide 1 with triphenylphosphine.
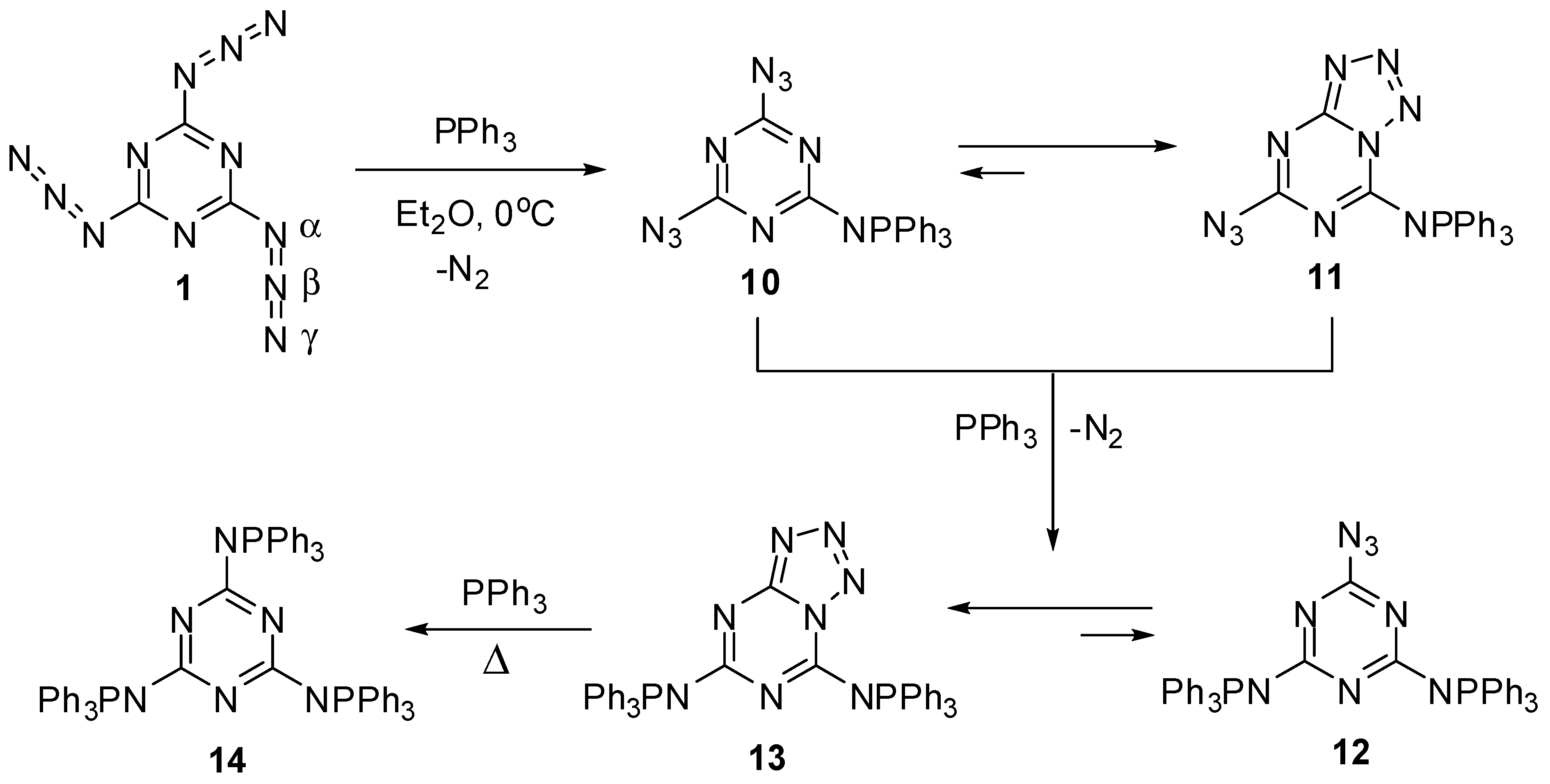
In contrast to azides 10 and 12 containing strong electron-donating aza-ylide substituents on the triazine ring, triazide 1 itself does not isomerize to tetrazolo[5,1-a]-[1,3,5]triazines in organic solutions due to insufficiently high nucleophilicity of the endo-cyclic nitrogen atoms [64]. This triazide has become the first organic azide characterized with X-ray diffraction analysis [1,2,3,4,5]. These structural studies proved that organic azides have a linear structure of the azido groups. The molecule of 1 is planar with bent azido groups giving rise to C3h symmetry (Scheme 4). The azido groups show π bond localization with a short Nβ–Nγ bond distance of 1.1156 Å (bond order ≈ 2.5) and a longer Nα–Nβ bond distance of 1.2658 Å (bond order ≈ 1.5) [63]. The C–Nα–Nβ angle is 112.5°, and the Nα–Nβ–Nγ angle is slightly bent at 172.0°. The distance between the layers is 2.947 Å, and the crystal density is 1.736 g/cm3 [63]. In IR spectra, the azido groups of 1 show four strong bands at ν 2192, 2158, 2115 and 1198 cm−1 [63]. The 13C-NMR spectrum of 1 in acetone-d6 displays a signal of the carbon atoms at δ 171.9 ppm [63]. The 15N-NMR spectrum of 1 in CDCl3 shows four signals of the nitrogen atoms at δ −131.3 (Nγ), −142.6 (Nβ), −160.2 (N-ring) and −257.3 (Nα) ppm [73].
In material chemistry, triazide 1 is often used as a reference compound in studies of new high-energy nitrogen-rich organic compounds [74,75,76,77,78,79,80]. Its sensitivity to impact, friction and spark is estimated as 6.2 cm (H50), <0.5 kg and <0.36 J, respectively [77]. This triazide has also one of the highest positive heats of formation (ΔHf = 1053 kJ/mol) among aromatic polyazides [78]. The thermal decomposition and detonation of 1 are used nowadays for preparation of C3N4 and C3N5 carbon nitrides and carbon nanotubes [6,7,8,9]. These materials possess unique mechanical, physical and chemical properties and are of considerable interest for industrial applications.
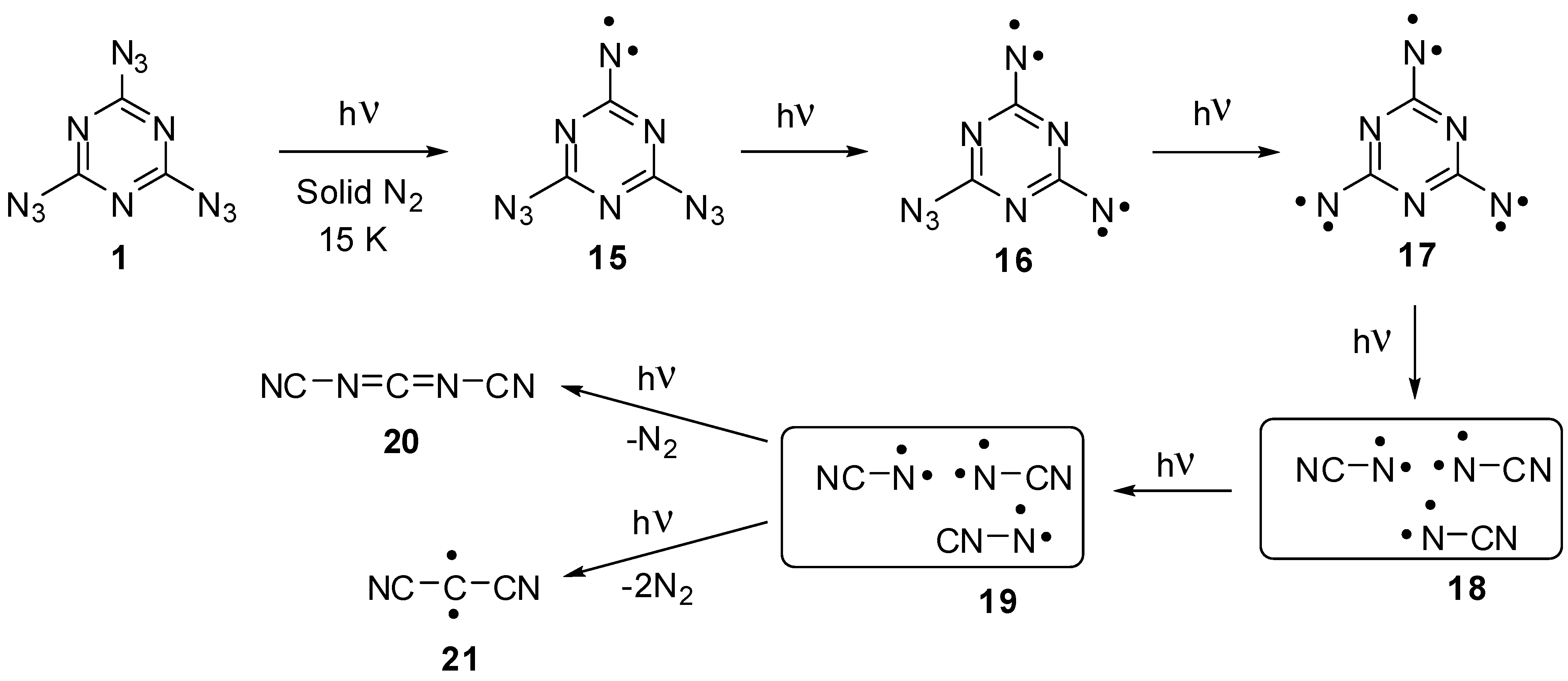
The first attempt to investigate the photochemistry of triazide 1 was undertaken by Moriarty and co-workers in 1966 [81]. It was found that low-temperature photolysis of crystalline 1 produces the stable at room temperature triplet nitrene 15 showing the largest zero-field splitting (zfs) parameter of |D| = 1.445 cm−1 among all triplet arylnitrenes (Scheme 5). More recent studies of the photolysis of crystalline 1 at 4 K allowed the EPR detection of quintet dinitrene 16 with magnetic parameters |D| = 0.28 cm−1 and |E| = 0.058 cm−1, rather unusual for quintet dinitrenes [82]. A few years later, Sato and colleagues succeeded in EPR detection of septet trinitrene 17 formed during the photolysis of triazide 1 in solid nitrogen matrices [14,15]. It was found that three endo-cyclic nitrogen atoms of the triazine ring favor the extremely high localization of spins on the nitrene units of trinitrene 17. As a result, this trinitrene showed the largest negative value of D = −0.123 cm−1 among all organic septet hexaradicals. Trinitrene 17 was photochemically unstable and decomposed to form triplet cyanonitrenes 18, that underwent further photochemical rearrangements to form triplet isocyanonitrene 19, dicyanocarbodiimide 20 and triplet dicyanocarbene 21. Overall, the photochemical studies showed that trinitrene 17 generated by the photolysis of triazide 1 possesses the record value of magnetic anisotropy among all organic hexaradicals, but is extremely reactive and photochemically unstable. Its decomposition leads to carbodiimide 20, similarly to the formation of C3N4 carbon nitrides during the thermal and detonative decomposition of triazide 1.
Scheme 5.
Photolysis of triazide 1.

3. Triazidopyrimidines
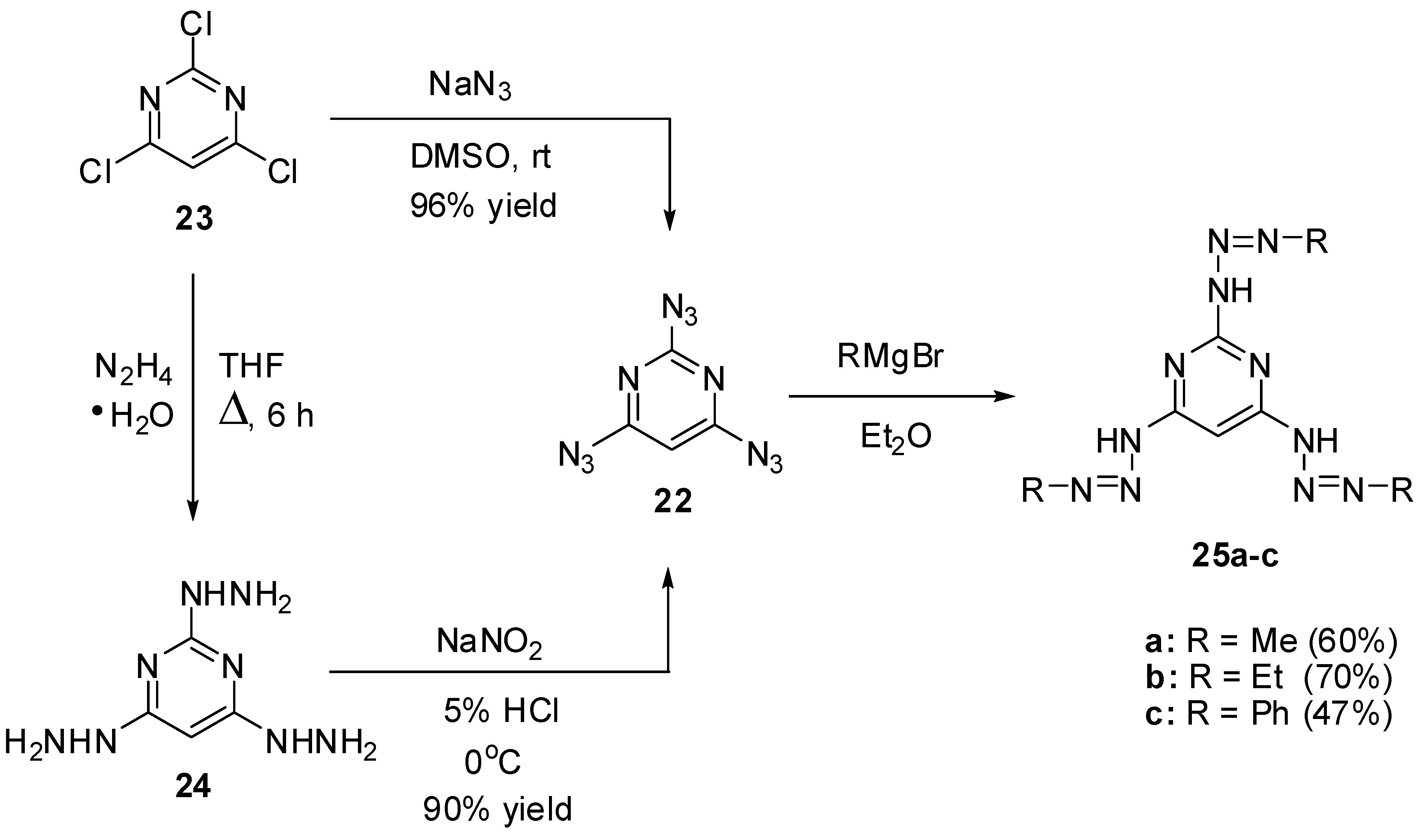
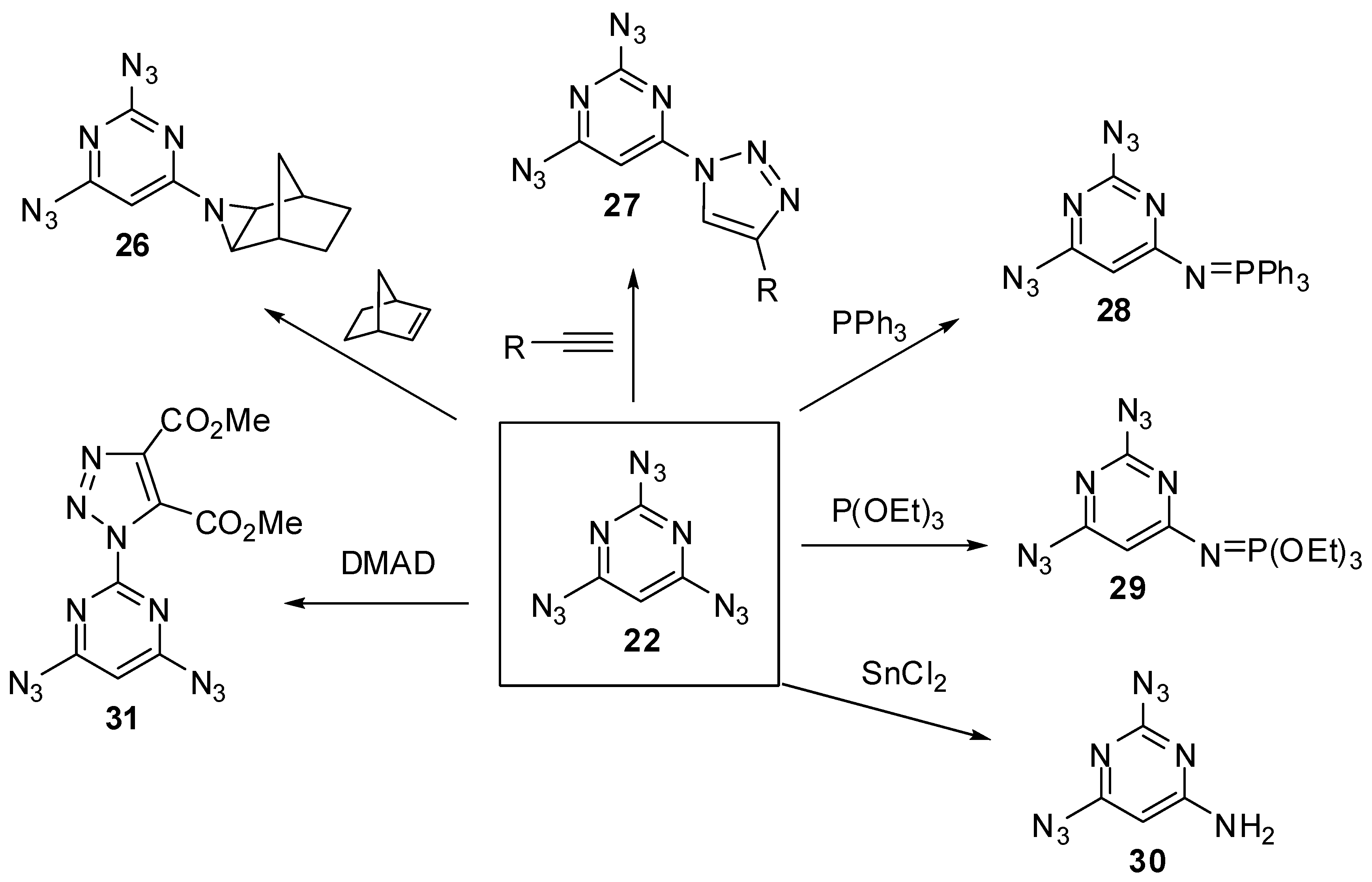
At the moment, there are known just five triazidopyrimidines. The first of them, 2,4,6-triazidopyrimidine (22), was prepared by Pochinok and co-workers in 1979 (Scheme 6) [68]. The azidation of commercially available trichloride 23 with excess sodium azide in dimethylsulfoxide at room temperature gave triazide 22 in 96% yield. On the other hand, the diazotization of tris-hydrazine 24 with NaNO2 in acidic media afforded triazide 22 in 90% yield. This triazide was used as a starting material to prepare tris-triazenes 25a–c (Scheme 6) [68]. Recently, triazide 22 was obtained by azidation of trichloride 23 with excess sodium azide in acetone at room temperature in 91% yield [78].
Scheme 6.
Synthesis and reactions of triazide 22 with Grignard reagents.

On crystallization from ethanol, triazide 22 forms dense colorless crystals with a melting point of 98 °C [68,78]. In comparison with cyanuric triazide, triazide 22 is much less sensitive to impact and friction. It may explode only under a very strong blow. Nevertheless, on working with 22, it is better to handle it only with plastic spatulas and use thick gloves behind a blast shield. In IR spectra, the azido groups of 22 show two weak bands at ν 2177 and 2100 cm−1 and two bands of medium intensity at ν 2144 and 1161 cm−1 [78]. The 1H-NMR spectrum of 22 in CDCl3 shows a signal of the proton at δ 6.88 ppm [73]. The 13C-NMR spectrum of 22 in CDCl3 displays three signals of the carbons at δ 94.3 (C-5), 161.5 (C-2) and 164.9 (C-4, C-6) ppm [73]. The 15N-NMR spectrum of 22 in CDCl3 shows seven signals of the nitrogen atoms at δ −134.5 (Nγ, 4,6-N3), −135.1 (Nγ, 2-N3), −140.0 (Nβ, 2-N3), −141.0 (Nβ, 4,6-N3), −145.6 (N-ring), −262.7 (Nα, 2-N3) and −264.8 (Nα, 4,6-N3) ppm [73]. The 13C- and 15N-NMR spectra can be very helpful in predicting the chemical reactivity of nonequivalent azido groups of 2,4,6-triazidopyrimidines. Thus, quantum-chemical studies showed that the azido groups in positions 4 and 6 of triazide 22 are the most electron-deficient [73]. In 15N-NMR spectra, these groups display the most shielded signal of the Nα atoms and should selectively react with electron-rich dipolarophiles, phosphines, phosphites and reductants to give diazides 26–30 (Scheme 7) [73]. On the other hand, the reaction of 22 with electron-deficient dimethyl acetylenedicarboxylate (DMAD) should selectively occur on the electron-rich azido group in position 2 of the pyrimidine ring to give adduct 31.
Scheme 7.
Theoretically predicted reactions for triazide 22.

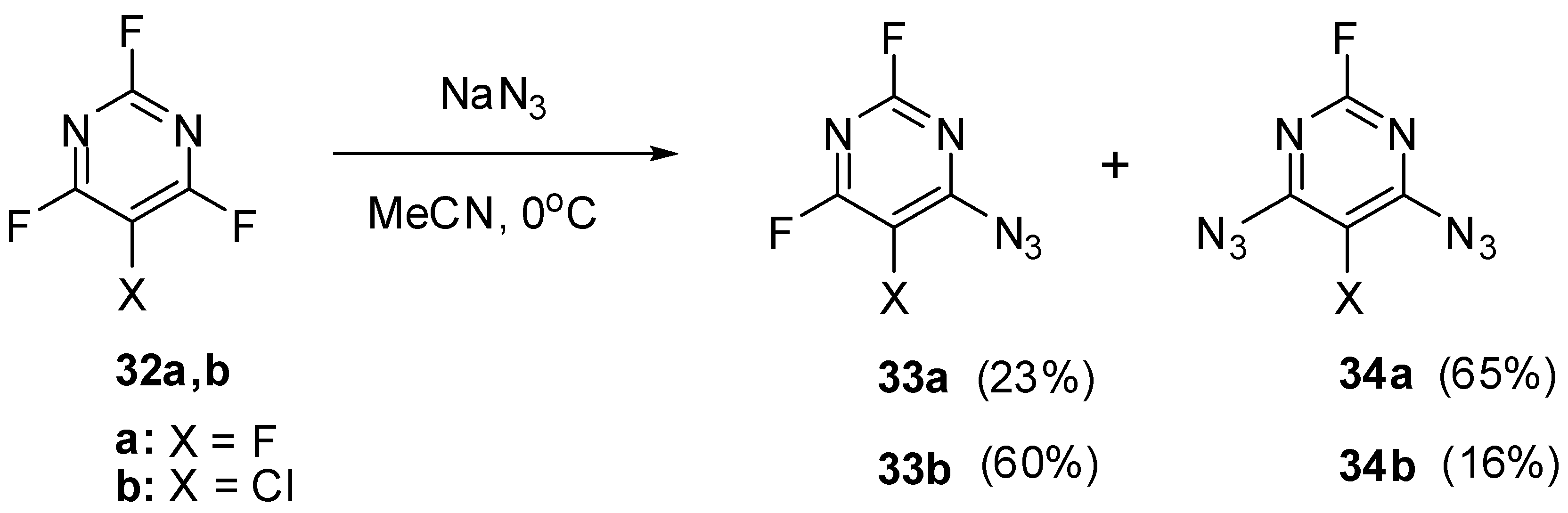
The high electron-deficiency of the azido groups in positions 4 and 6 of triazide 22 is also manifested in the most deshielded signals of the C-4 and C-6 atoms in the 13C-NMR spectrum of this triazide. As a rule, the first stage of azidation of 2,4,6-trihalopyrimidines is the replacement of the halogen atom for the azido group in position 4 of the pyrimidine ring. Thus, trihalides 32a,b reacted with sodium azide in actetonitrile at room temperature to give only monoazides 33a,b and diazides 34a,b (Scheme 8) [83].
Scheme 8.
Mono- and diazidation of fluorides 32a,b.
Similarly to triazide 22, four other 2,4,6-triazidopyrimidines were synthesized (Scheme 9) [78]. The azidation of tetrachloride 35a with sodium azide in acetone at room temperature in the presence of tert-butylammonium bromide (TBAB) gave a mixture of triazide 36 and diazide 37 in 55% and 21% yields, respectively. The reactions of trichlorides 35b,c with sodium azide in tetrahydrofuran (THF) at room temperature in the presence of TBAB gave triazide 38 and tetraazide 39 in 80% and 99% yields, respectively. Finally, the reaction of trichloride 35d with sodium azide in THF at room temperature in the presence of TBAB followed by addition of stannous chloride and trimethylsilylazide (TMSA) to intermediate products gave pentaazide 40 in 35% yield. Despite the high-nitrogen content, tetraazide 39 and pentaazide 40 are not very sensitive to impact and friction since their low melting points (22.5 and −48 °C, respectively) [78].
The structure of triazide 38 was studied by X-ray diffraction analysis [78]. It was found that the least electron-deficient azido group in position 2 of 38 has a somewhat elongated C–Nα bond distance (1.412 Å), a shortened Nα–Nβ bond distance (1.251 Å) and the least bent Nβ–Nβ–Nγ angle (172.9°). Similar parameters for the azido group in position 4 of 38 equal to 1.406 Å, 1.257 Å and 172.4°, respectively. At the same time, all three azido groups of 38 have the same Nβ–Nγ bond distances (1.119 Å) and nearly the same C–Nα–Nβ angles (~113.5°). The more bent Nα–Nβ–Nγ angles and more elongated Nα–Nβ bond distances in the azido groups in positions 4 and 6 of 38 suggest that these groups should be the least stable during thermolysis and decompose first to form triplet nitrenes and then amines.
Triazides 22, 38, 39 and 40 are typical high-energy nitrogen-rich compounds, which thermal and detonative decomposition is used for preparation of C3N4 carbon nitrides and carbon nanotubes [78]. Triazide 22 is also of interest as an antitumor agent showing high activity against Sarcoma 180, Piss lymphosarcoma and Guerin carcinoma, along with low toxicity (>1000 mg/kg) [84].
Scheme 9.
Synthesis of triazides 36, 38, 39 and 41.

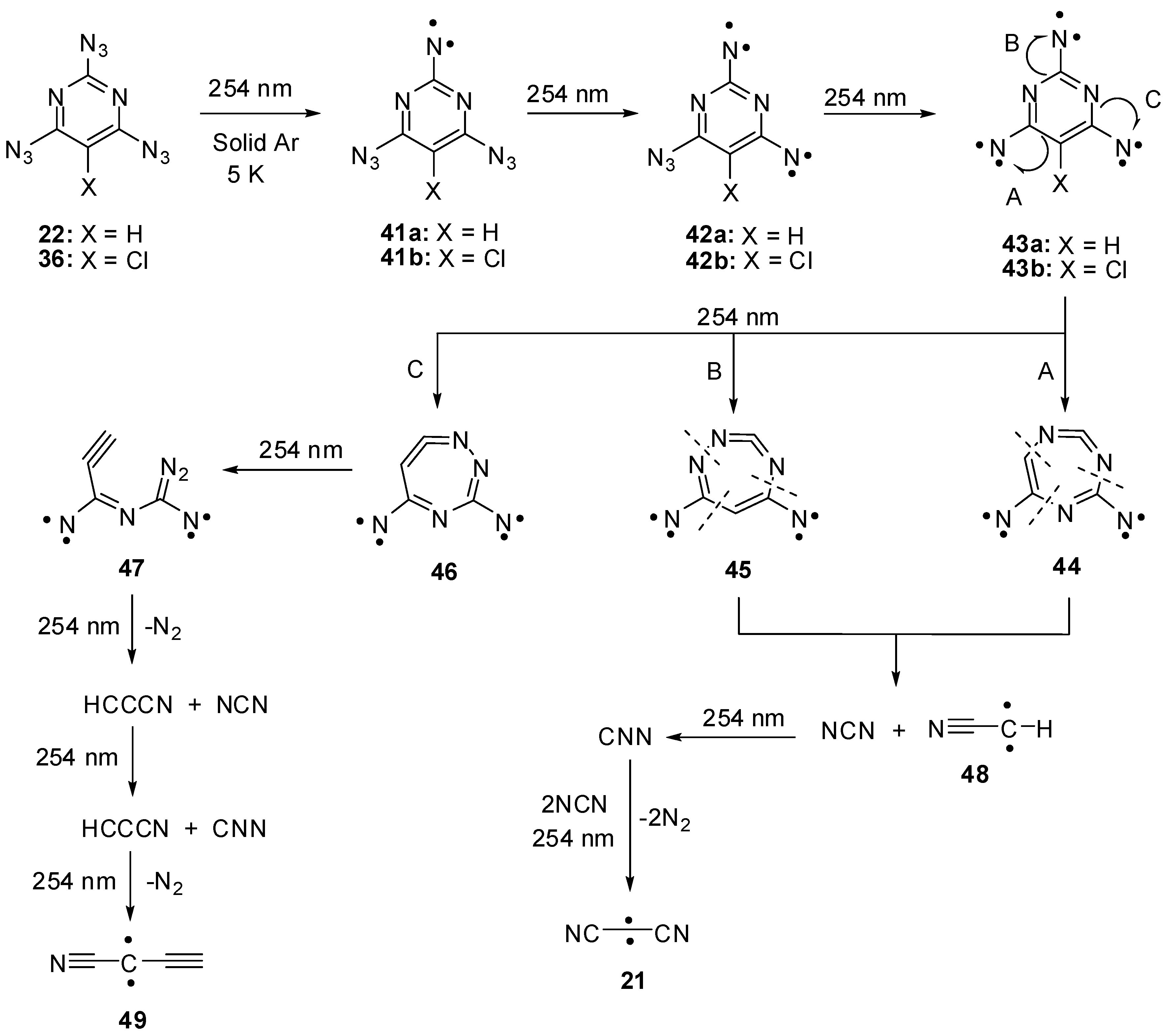
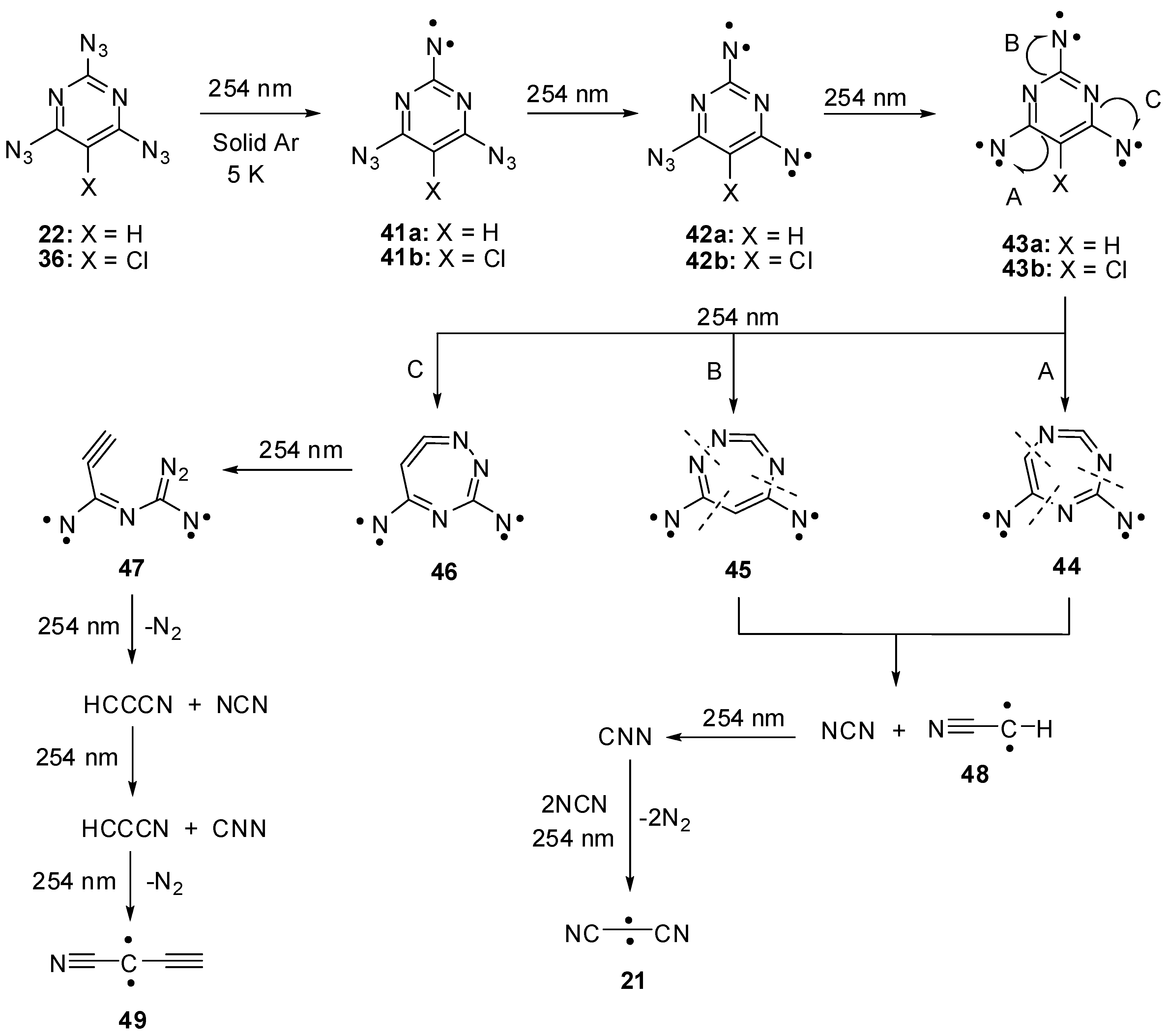
Triazides 22 and 36 were used as starting materials in several photochemical studies. First attempts to detect nitrenes during the photolysis of triazides 22 and 36 in frozen organic solutions at 77 K were unsuccessful [85]. Recently, the photolysis of 22 and 36 was studied in argon matrices at 5 K [30]. Using this technique, it was found that photochemical decompositions of 22 and 36 occur selectively to subsequently give triplet nitrenes 41a,b, quintet dinitrenes 42a,b and septet trinitrenes 43a,b (Scheme 10). Since the presence of only two endo-cyclic nitrogen atoms in the ring, trinitrene 43a showed D = −0.1122 cm−1 that was by ~10% smaller than the D-value of trinitrene 17. As in the case of trinitrene 17, trinitrenes 43a,b were photochemically unstable and decomposed to form triplet nitrenes NCN and NNC as well as triplet carbenes NCCCN, HCCN and HCCCCN. Nitrenes NCN and NNC and carbene NCCCN were previously also observed during the photolysis of cyanuric triazide [15]. The formation of the same reactive intermediates during the photolysis of triazides 17 and 22 explained well the formation of the same C3N4 carbon nitrides and carbon nanotubes during thermal and detonative decomposition of both triazides. At the same time, the photolysis of triazide 22 additionally produces carbenes HCCN and HCCCCN that are typical only for decomposition of azidopyrimidines. Of these two carbenes, the last one has never previously been generated in a laboratory, but was known as a component of interstellar matter [86].
Scheme 10.
Photolysis of triazides 22 and 36.
4. Triazidopyridazines
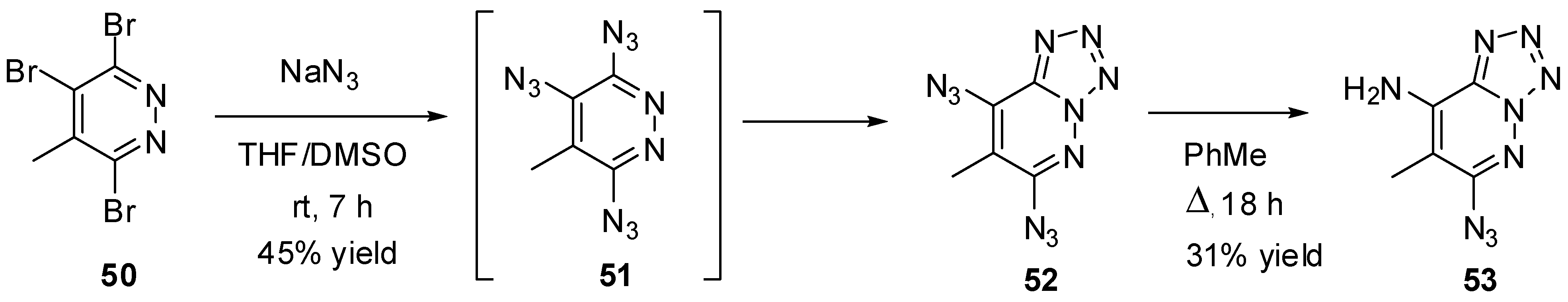
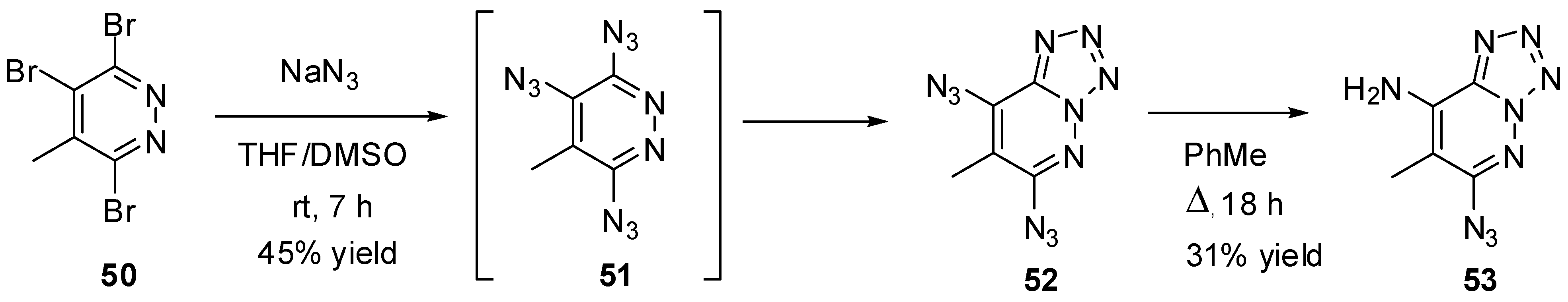
Due to azide-tetrazole isomerization, triazidopyrazines and triazidopyridazines may exist only as short-living intermediates formed during the azidation of the corresponding trihalides. Thus, recent studies have shown that azidation of tribromide 50 with sodium azide gave diazidotetrazole 52 in 45% yield (Scheme 11) [87]. It was postulated that this tetrazole is formed from intermediate triazide 51. The structure of 52 was confirmed by X-ray diffraction analysis. According to 1H-NMR studies, compound 52 exists in organic solutions exclusively as a tetrazole. On boiling in toluene, diazide 52 partially decomposed to form aminoazide 53 [87]. The latter also exists in organic solutions exclusively as a tetrazole.
Scheme 11.
Triazidation of tribromide 50.
5. Triazidopyridines
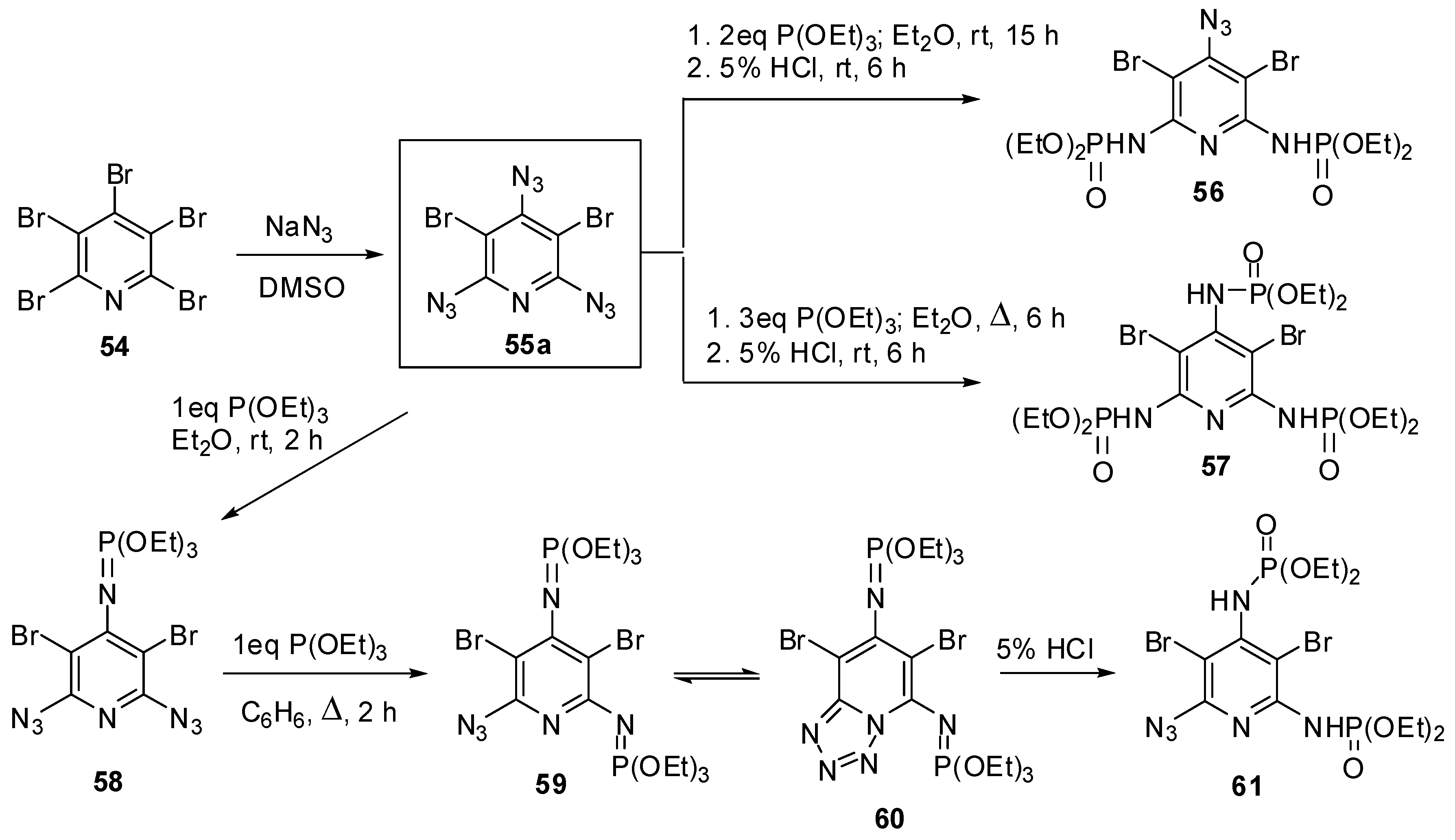
2,4,6-Triazido-3,5-dibromopyridine (55a) was the first triazidopyridine, obtained by Moshchitskii and co-workers in 1979, using the reaction of pentabromopyridine (54a) with sodium azide (Scheme 12) [88]. The reaction was carried out in DMSO at room temperature for 15 h and gave triazide 55a (a brown solid with a melting point 85–86 °C) in 97% yield. The reactions of 55a with two and three molar equivalences of triethyl phosphite followed by acidic hydrolysis of the products with 5% HCl gave bis- and tris-adducts that were assigned to compounds 56 and 57, respectively [88].
Scheme 12.
Synthesis and Staudinger-phosphite reactions of triazide 55a.
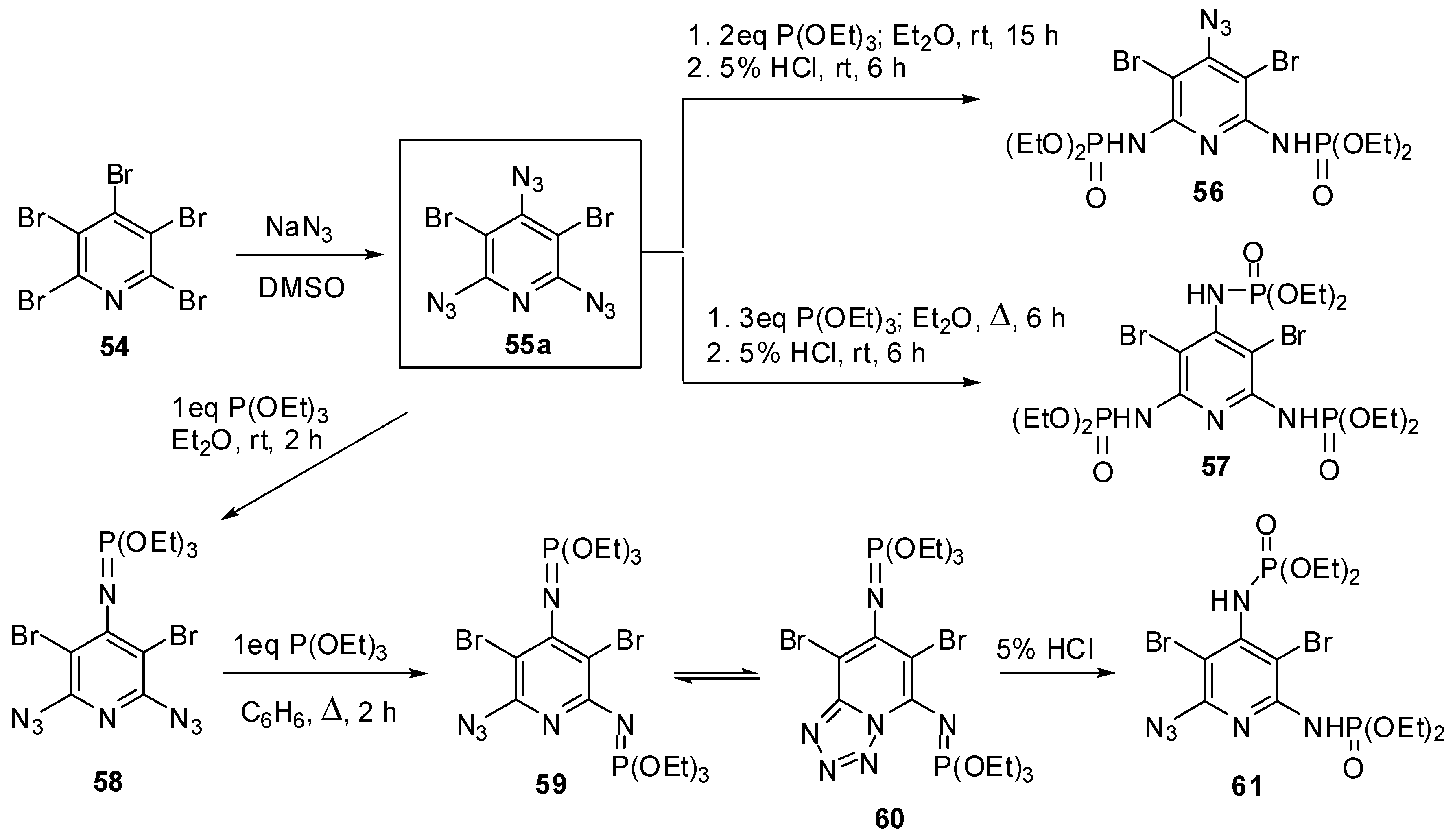
Recently, triazide 55a has been prepared by the reaction of 54a with sodium azide in DMSO at 80 °C just for 30 min in 97% yield [89]. The addition of an equimolar amount of triethyl phosphite to 55a resulted in the formation of diazide 58 as a single product (Scheme 12) [89]. The latter reacted with an equimolar amount of triethyl phosphite on boiling in benzene to form a mixture of compounds 59 and 60 in a nearly equal ratio. The hydrolysis of the mixture of 59 and 60 with 5% HCl afforded azide 61 as a single product [89]. The structures of compounds 58–60 were unambiguously confirmed by the data of 1H-, 13C- and 31P-NMR spectroscopic studies. According to these studies, the γ-azido group of triazide 55a is the most electron-deficient and, as the result, the most reactive in the Staudinger reactions. After triazide 55a, nine 2,4,6-triazidopyridines 55b–j have been synthesized (Scheme 13) [90,91,92,93,94,95,96,97,98,99,100].
Scheme 13.
Synthesis of triazides 55b–j.
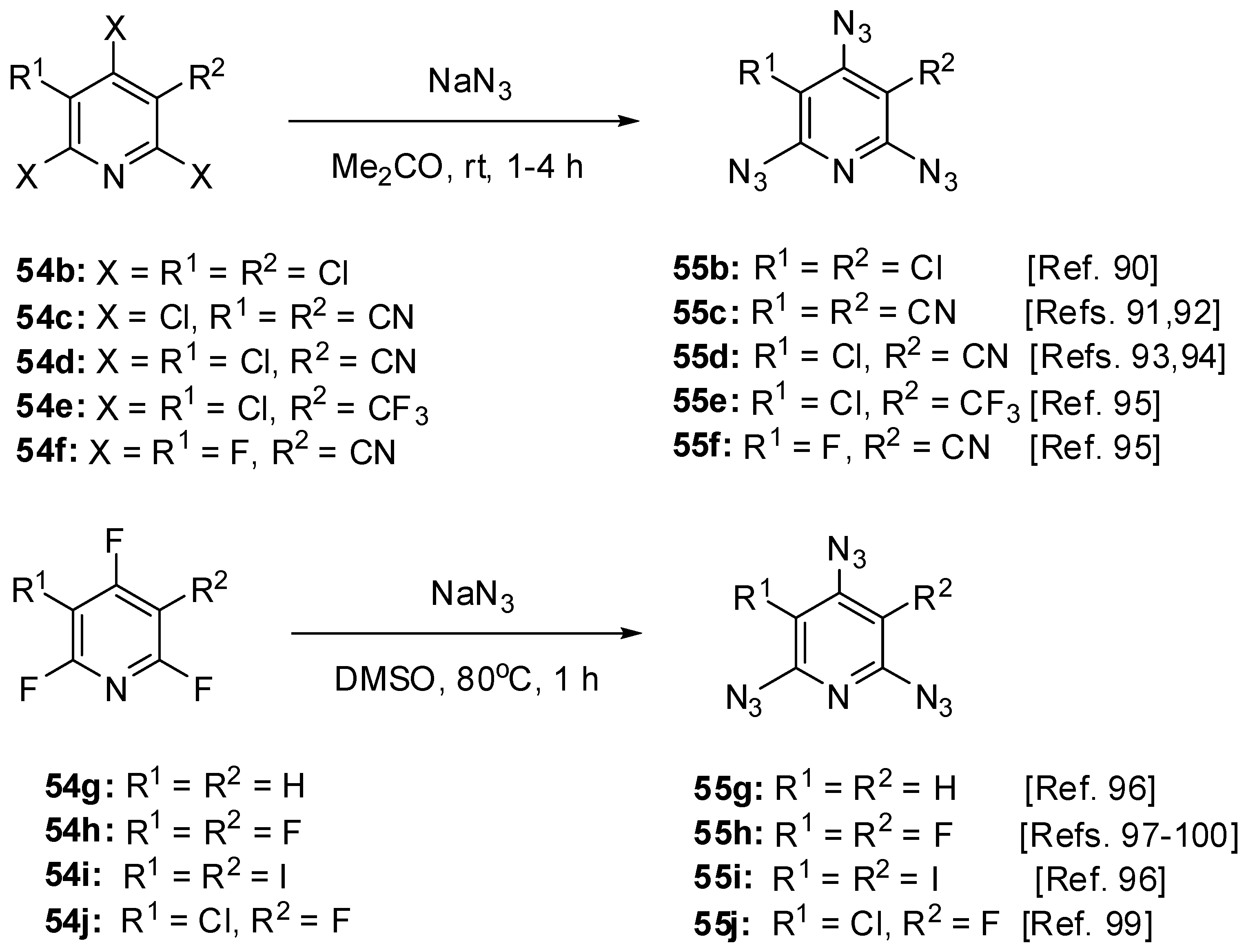
Owing to the presence of strong electron-withdrawing cyano- and trifluoromethyl-substituents on the pyridine ring, halides 54c–f readily react with sodium azide in aqueous acetone at room temperature to form triazides 55c–f in 86%–98% yields [91,92,93,94,95]. The least activated halide 54b formed triazide 55b (98%) on interaction with sodium azide in aqueous acetone only at temperature above 30 °C [90]. Similar reactions of trifluoropyridines 54g–j with sodium azide in boiling aqueous acetone stopped, as a rule, on the stage of formation of the corresponding 2,4-diazidopyridines [97,98]. At the same time, fluorides 54g–j readily underwent triazidation in hot DMSO to form triazides 55g–j in 86%–96% yield [96,99]. In all cases, the first stage of the reaction was the replacement of the halogen atom in position 4 of the pyridine ring.
Triazides 55a–j are solid materials (mp 63–135 °C) that are almost insensitive to impact, friction, and electrostatic discharge. Only triazides 55c and 55g may explode under a very strong blow [92,101]. In the IR spectra, the azido groups of 55a–j show two strong bands in the ν 2180–2120 cm−1 region and a weak band about ν 2100 cm−1 [90,91,92,93,94,95,96,97,98,99,100]. In 15N-NMR spectra, the most electron-deficient γ-azido groups of 55a–j display the most shielded Nα signals [73]. Thus, the 15N-NMR spectrum of triazide 55a shows seven signals of the nitrogen atoms at δ −129.0 (N-ring), −134.8 (Nγ, α-N3), −139.8 (Nβ, α-N3), −140.8 (Nγ, γ-N3), −144.8 (Nβ, γ-N3), −265.8 (Nα, α-N3) and −276.2 ppm (Nα, γ-N3) [89]. The high electron-deficiency of the γ-azido groups of triazides 55a–j is also manifested in the more shielded signals of the C-4 atoms in the 13C-NMR spectrum of these triazides. Thus, the 13C-NMR spectrum of triazide 55a shows three signals of the carbon atoms at δ 98.2 (Cβ), 147.4 (Cγ) and 151.0 ppm (Cα) [89]. In crystals, the α-azido groups of triazides 55b [102], 55c [103], 55d [95], 55e [104], 55f [105] and 55h [100] show typical for aryl azides values of the C–Nα (~1.40 Å), Nα–Nβ (~1.26 Å) and Nβ–Nγ (~1.12 Å) bond distances and the C–Nα–Nβ (~113°) and Nα–Nβ–Nγ (~172°) angles. Almost the same values of the C–Nα (~1.39 Å), Nα–Nβ (~1.25 Å) and Nβ–Nγ (~1.12 Å) bond distances are observed as well in the γ-azido groups of triazides 55b–f and 55h. However, these groups have essentially larger the C–Nα–Nβ (~120°) angles and smaller the Nα–Nβ–Nγ (~169°) angles. In addition, the C–C–Nα–Nβ torsion angles of these groups vary from 2 (55h) till 40° (55e), depending on the bulkiness of the substituents in positions 3 and 5 of the pyridine ring. According to theory, the smaller the Nα–Nβ–Nγ angle and the larger the C–Nα–Nβ angle in azides, the higher the reactivity of azides in reactions with dipolarophiles, phosphines, phospites and reductans [105,106,107].
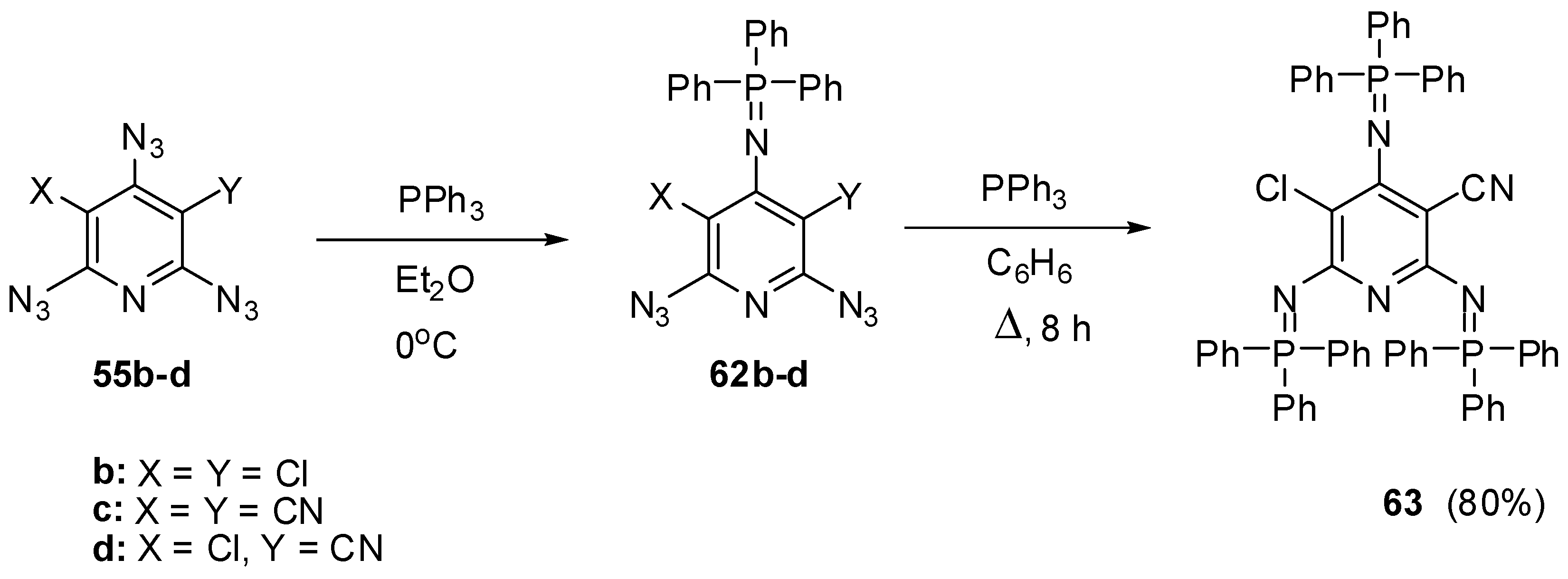
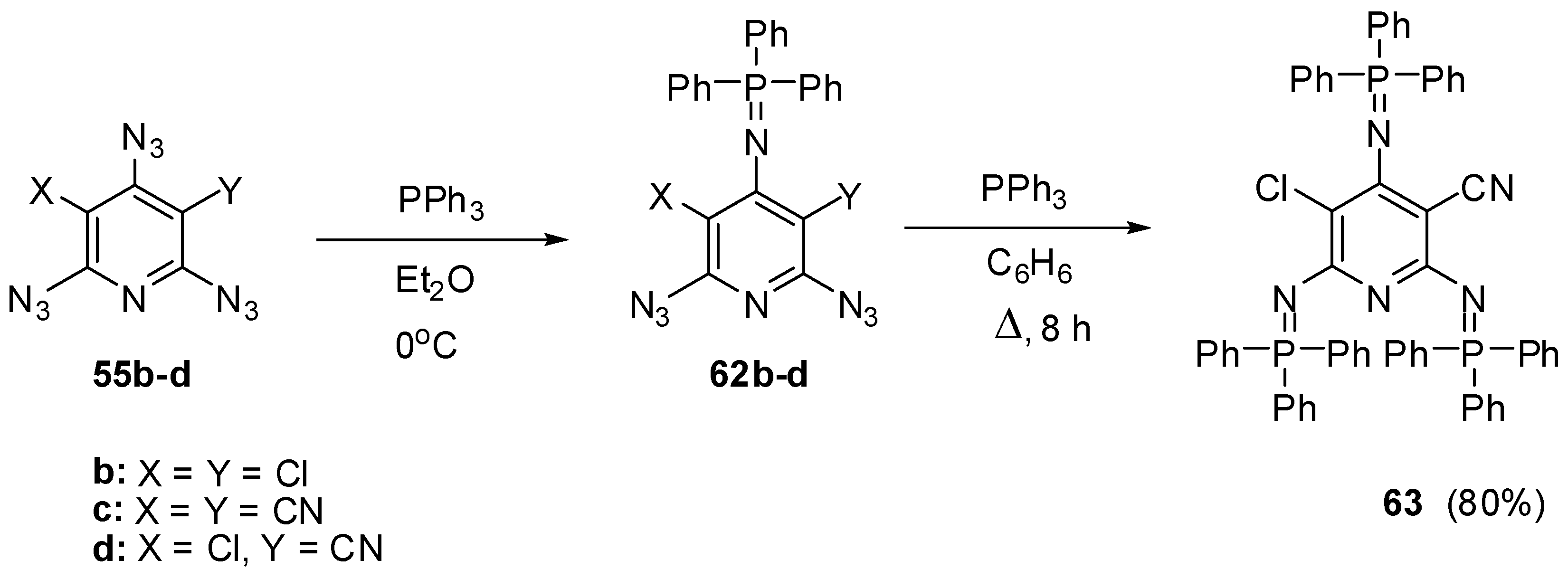
Owing to the presence of nonequivalent azido groups on the pyridine ring, triazides 55a–d were used as model compounds to investigate selective reactions on the azido groups of aromatic polyazides [34,35]. It was found that the Staudinger phosphorylation of triazides 55b–d in ether at 0 °C occurs selectively on the most electron-deficient γ-azido groups to give iminophosphoranes 62b–d in 93%–97% yields (Scheme 14) [93,108]. Similarly to preparation of tris-adduct 57 from triazide 55a (Scheme 12), diazide 62c reacted with an excess of triphenylphosphine on boiling in benzene to give tris-adduct 63 in 80% yield [93].
Scheme 14.
Selective Staudinger reactions of triazides 55b–d.
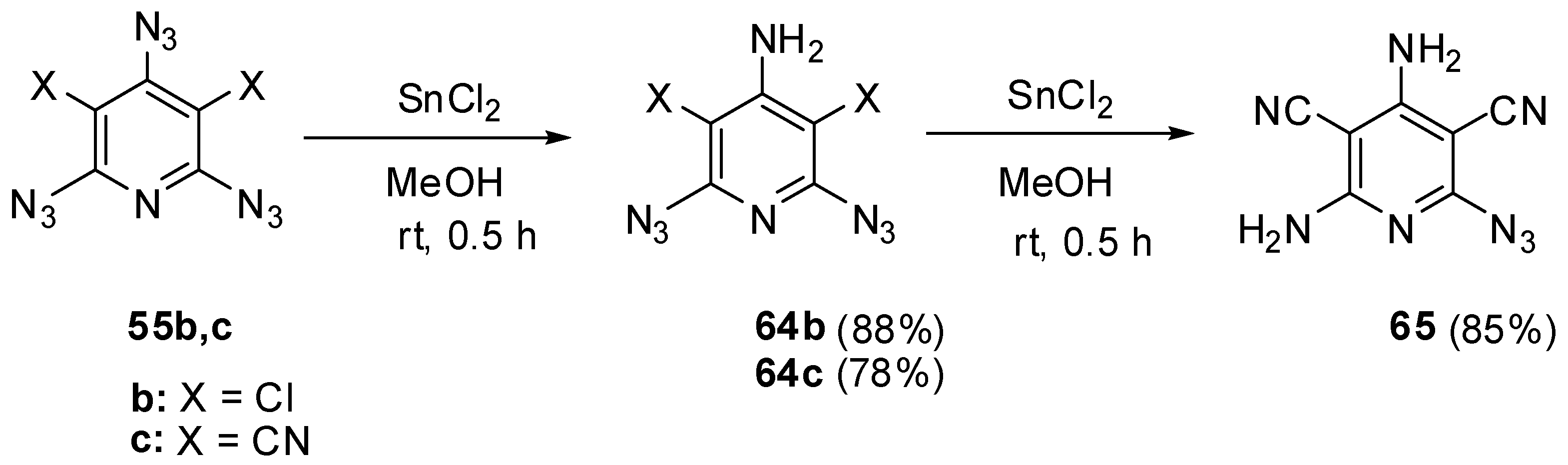
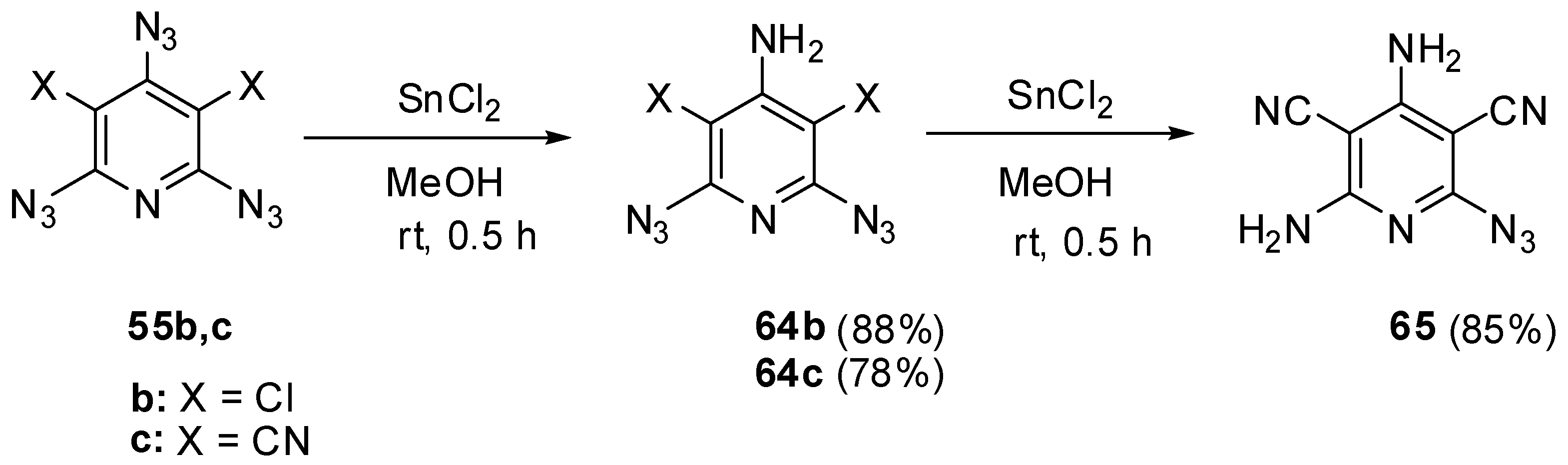
Beside selective phosphorylation, the γ-azido groups of triazides 55b–d are selectively reduced to amines. Thus, the reactions of triazides 55b,c with stannous chloride in methanol at room temperature afforded aminodiazides 64b,c (Scheme 15) [109]. The more electron-deficient diazide 64c was further reduced with stannous chloride in methanol at room temperature till diamine 65.
Scheme 15.
Selective reduction of triazides 55b,c.
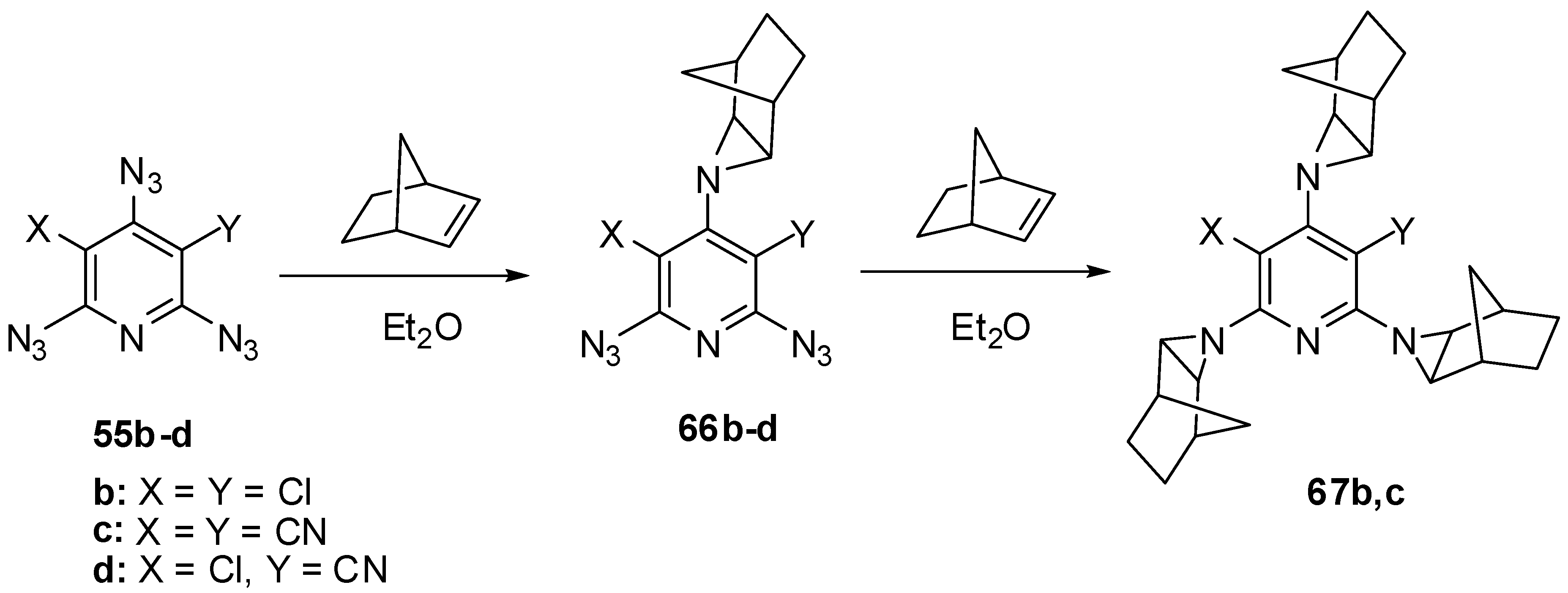
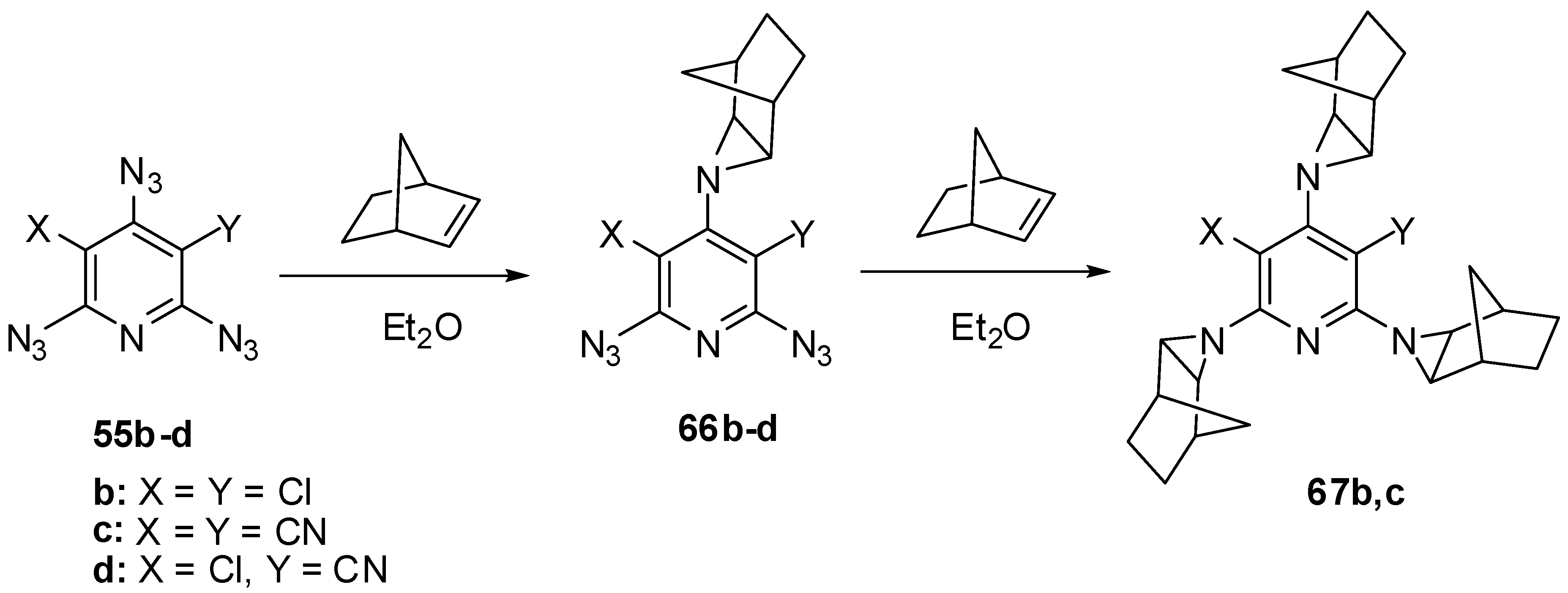
The electron-deficient γ-azido groups of triazides 55b–d are also the most reactive toward electron-rich dipolarophiles [90,110]. Thus, the reaction of triazide 55c with norbornene in ether at 0 °C occurred regio- and stereoselectively to give exo-adduct 66c in high yield (Scheme 16) [110]. At room temperature, triazide 66c readily reacted with an excess of norbornene to form tris-adduct 67c in almost quantitative yield. Similar reactions of less electron-deficient triazides 55b,d stopped at the stage of formation of mono-adducts 66b,d [90,110]. The boiling of mono-adduct 66b with norbornene in benzene gave tris-adduct 67b in 56% yield [110]. On the other hand, the reactions of mono-adduct 66b with norbornene in ether at room temperature in the presence of Rh2(OAc)4 as a catalyst gave tris-adduct 67b in 90% yield [110].
Scheme 16.
Selective addition of norbornene to triazides 55b–d.
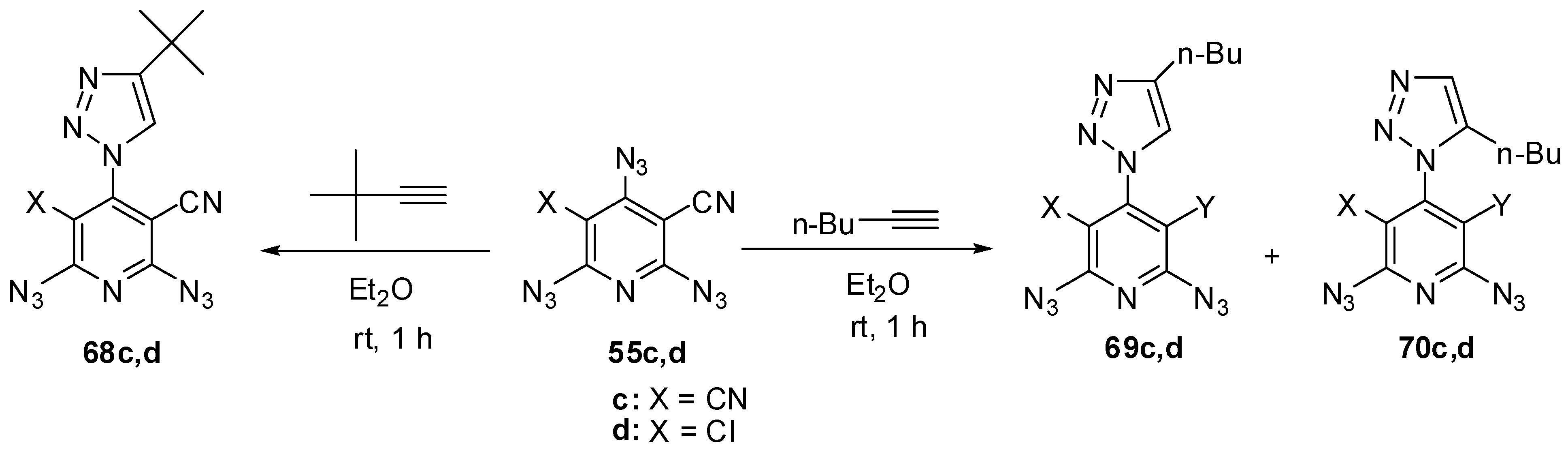
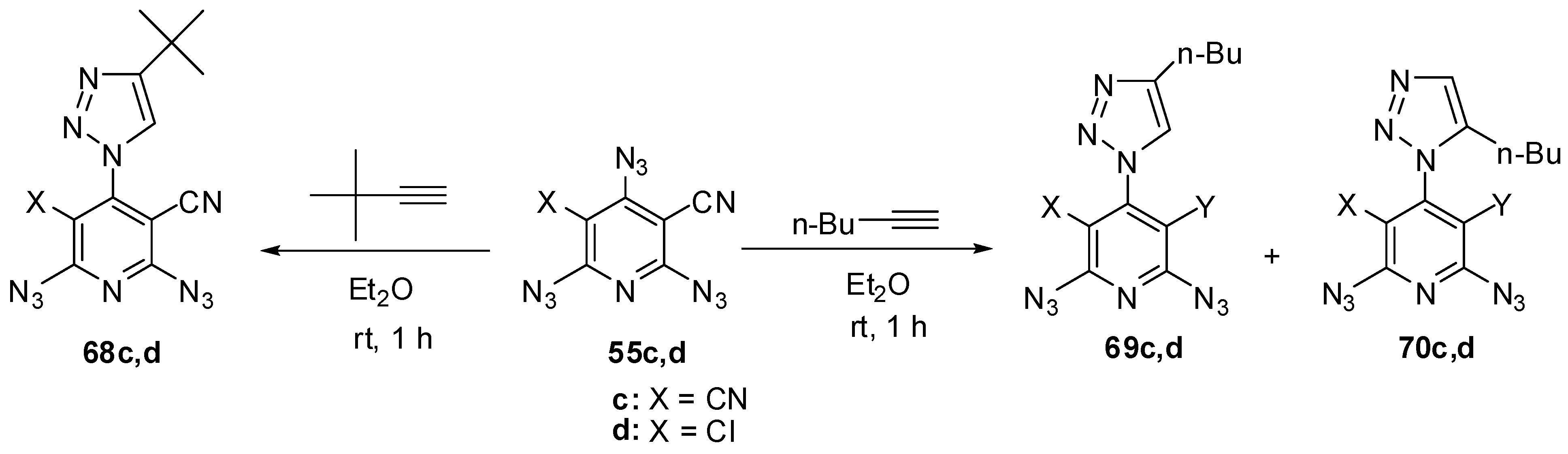
The reactions of triazides 55c,d with electron-rich tert-butylacetylene in ether at room temperature also occurred regioselectively on the γ-azido groups to form mono-adducts 68c,d in ~85% yield (Scheme 17) [111]. Similar reactions of triazides 55c,d with less sterically hindered n-butylacetylene gave mixtures of diazides 69c,d (~82%) and 70c,d (~8% yield) [111].
Scheme 17.
Selective addition of butylacetylenes to triazides 55c,d.
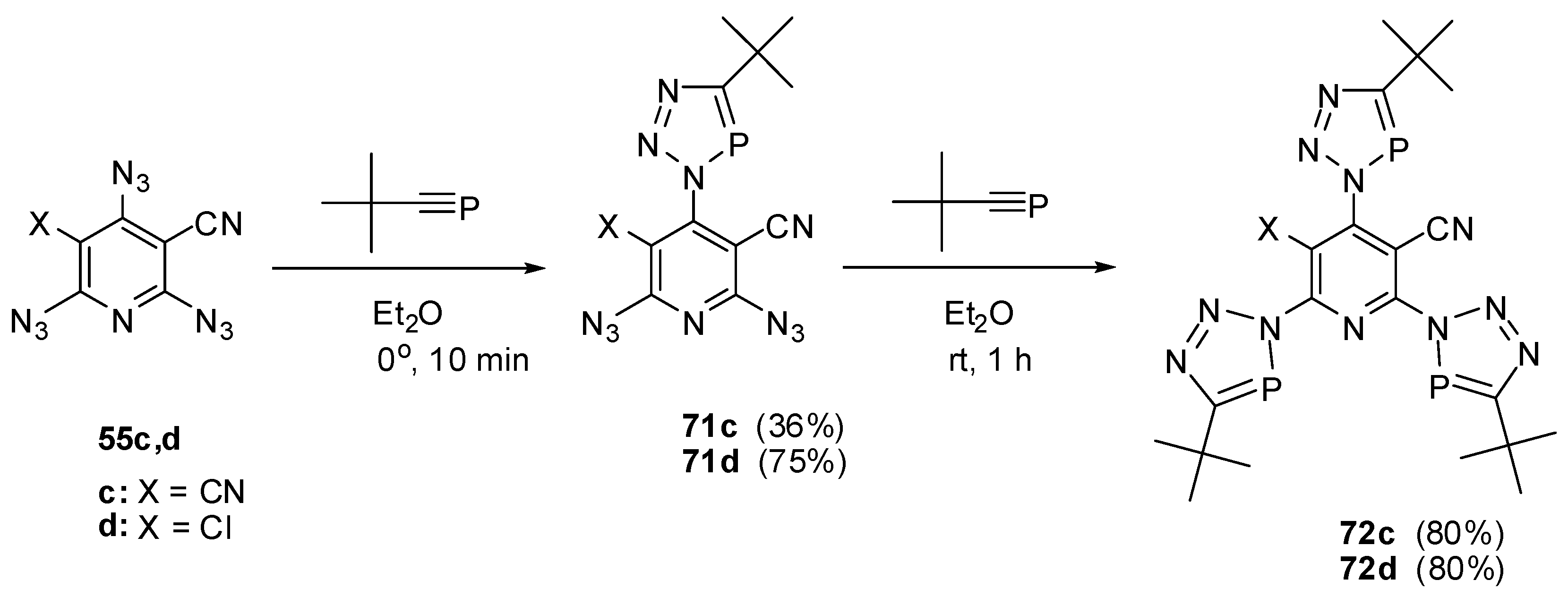
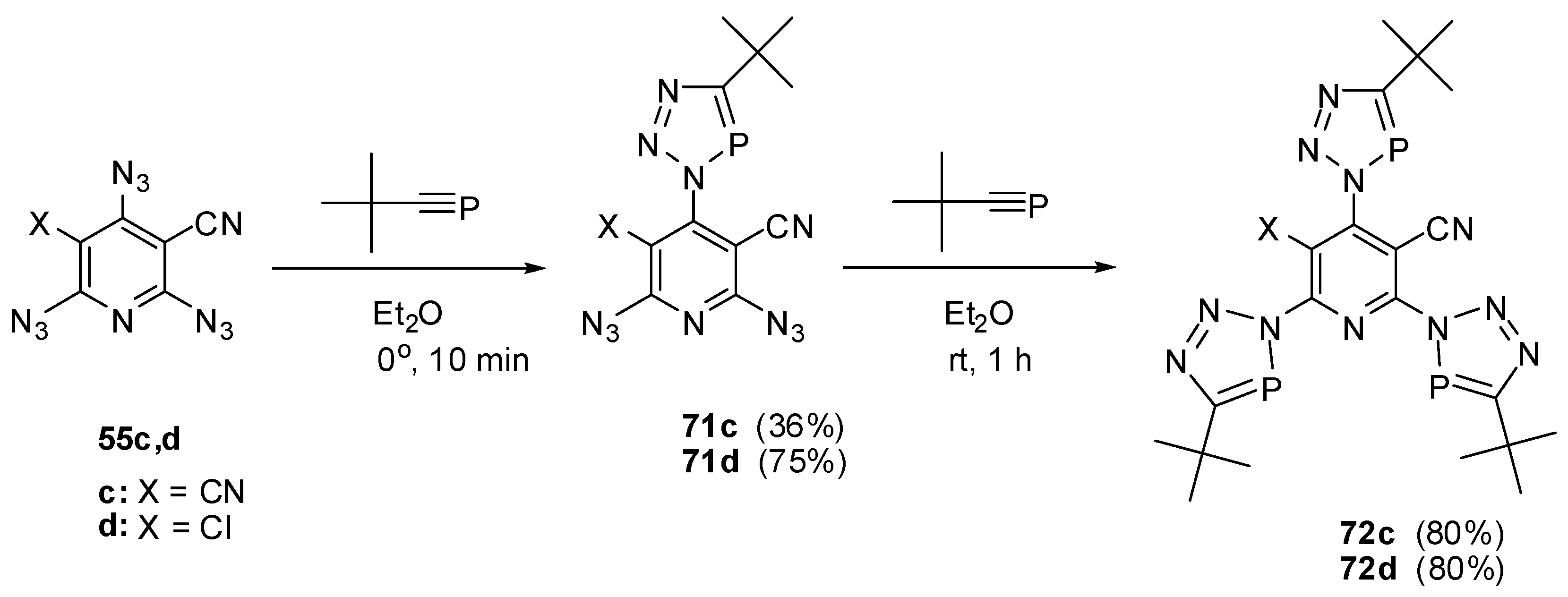
In ether at 0 °C, the extremely reactive tert-butylphosphaacetylene reacted with triazides 55c,d to form mono-adducts 71c,d (Scheme 18) [91,112,113]. The latter reacted with an excess tert-butylphosphaacetylene in ether at room temperature to give tris-adducts 72c,d [91,112,113].
Scheme 18.
Selective addition of tert-butylphosphaacetylene to triazides 55c,d.
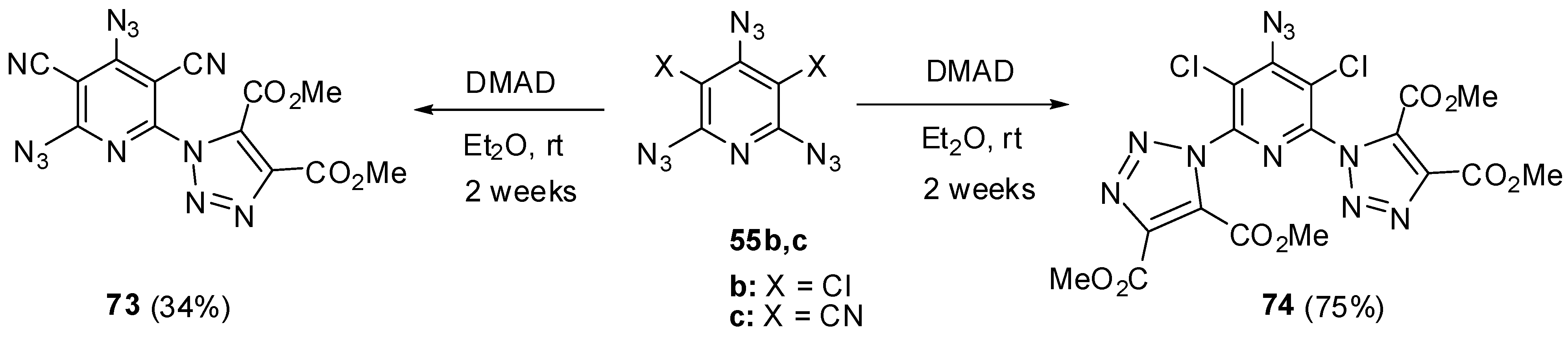
Since high electron-deficiency, triazides 55a–j are not much reactive toward electron-deficient dipolarophiles. The reaction of triazide 55c with DMAD in ether at room temperature for 2 weeks occurred selectively on the least electron-deficient α-azido group to give triazole 73 in 34% yield (Scheme 19) [114]. Similar reaction of less electron-deficient triazide 55b with DMAD led to the formation of bis-adduct 74 in 75% yield [114].
Scheme 19.
Reactions of triazides 55b,c with dimethyl acetylenedicarboxylate.
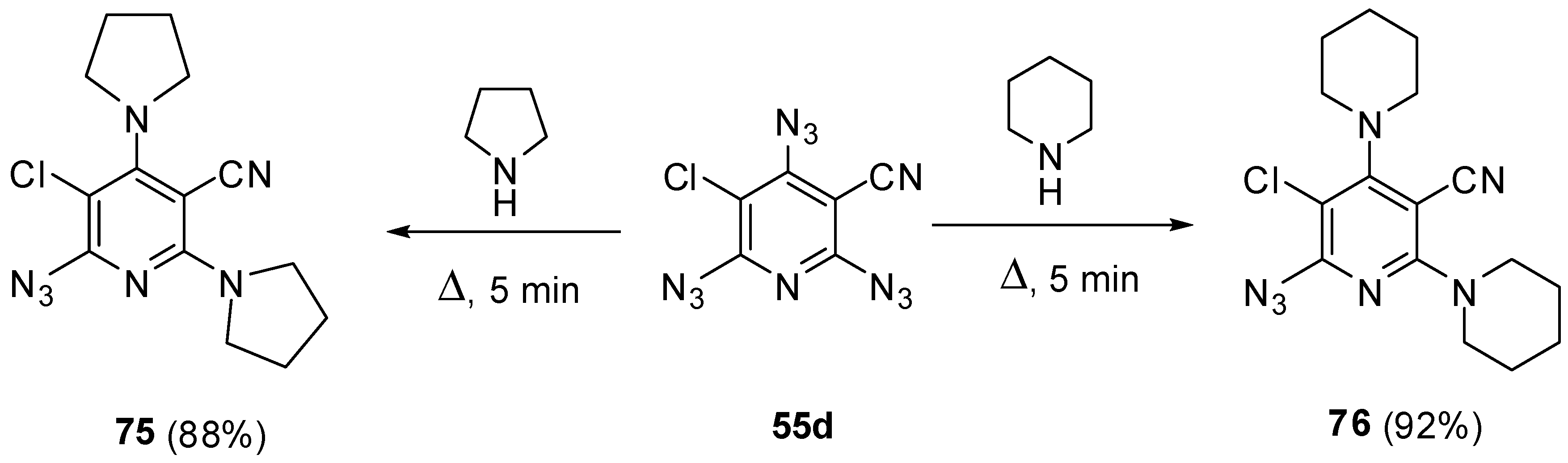
Owing to high electron-deficiency, triazides 55a–j readily react with aliphatic amines. Thus, a brief (5 min) boiling of triazide 55d in pyrrolidine or piperidine gave diamines 75 and 76 in high yields (Scheme 20) [115]. Most probably, these reactions occur by a radical mechanism involving electron transfer from amines to triazides followed by the collapse of diradical amine-azide pairs into aminopyridine and gaseous HN3. The reactions stop at the stage of bis-amination since low electron-deficiency of azides 75 and 76 [115]. Formally, these reactions are identical to amination of cyanuric triazide with aliphatic amines (see Scheme 2).
Scheme 20.
Reactions of triazide 55d with aliphatic amines.

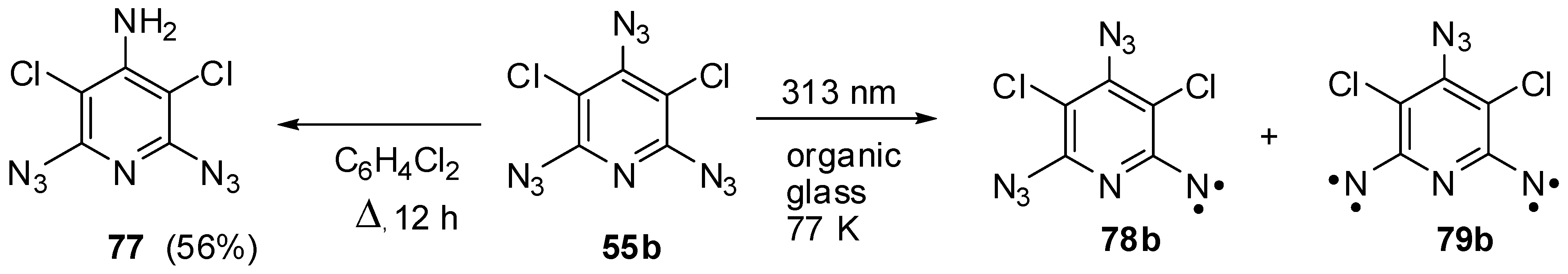
On thermolysis, triazides 55a–j undergo selective cleavage of the least durable N–N2 bonds of the γ-azido groups. Thus, the reflux of triazide 55b in p-dichlorobenzene at 160 °C for 12 h gave aminodiazide 77 in 56% yield (Scheme 21) [116]. On the other hand, the photolysis of triazide 55b in frozen at 77 K organic solutions occurred selectively on the α-azido groups to give triplet nitrene 78b and quintet dinitrene 79b as the major paramagnetic products (Scheme 21) [12,117]. This selectivity was explained by extremely low stability of the excited states of 55b arising after local excitation of the α-azido groups [117].
Scheme 21.
Selective thermolysis and photolysis of triazide 55b.

Triazides 55a–j have played an important role in investigations of high-spin organic compounds. Before that, nothing was known about the UV-vis and IR spectra of organic hexaradicals with the ground septet spin-state. Even EPR spectra of septet trinitrenes were never reported. It was surmised that such trinitrenes should show the magnetic anisotropy of D ≈ −0.055 cm−1 [10,118]. The photolysis of triazides 55b and 55h in cryogenic matrices allowed the first registration of IR and UV-vis spectra of septet trinitrenes 80b and 80d (Scheme 22) [11,13]. It was found that these trinitrenes are photochemically very stable and do not rearrange into low-molecular-weight products. Moreover, high-spin nitrenes generated by the photolysis or γ-radiolysis of triazide 55b were stable in crystals of the host triazide even at 230 K [102,103]. EPR studies of the photolysis of triazides 55a–j showed that containing light atoms trinitrenes 55b–h have nearly the same magnetic parameters of D ≈ −0.102 cm−1 and E ≈ 0.003 cm−1, independent on substituents in positions 3 and 5 of the pyridine ring [20,21,22,23,119]. Only trinitrene 80a was an exception. Due to the presence of two heavy bromine atoms on the pyridine ring, this trinitrene showed the record value of |D| = 0.297 cm−1 among all organic septet hexaradicals [26]. Recent high-level ab initio calculations have proved that trinitrene 80a has the negative sign of D and can be assigned to strong organic magnetic molecules [120]. The finding that heavy atoms drastically strengthen the magnetic properties of organic polyradicals opened up principally new prospects in design of organic magnetic materials.
Scheme 22.
Photochemical generation of septet trinitrenes from triazides 55a–j.
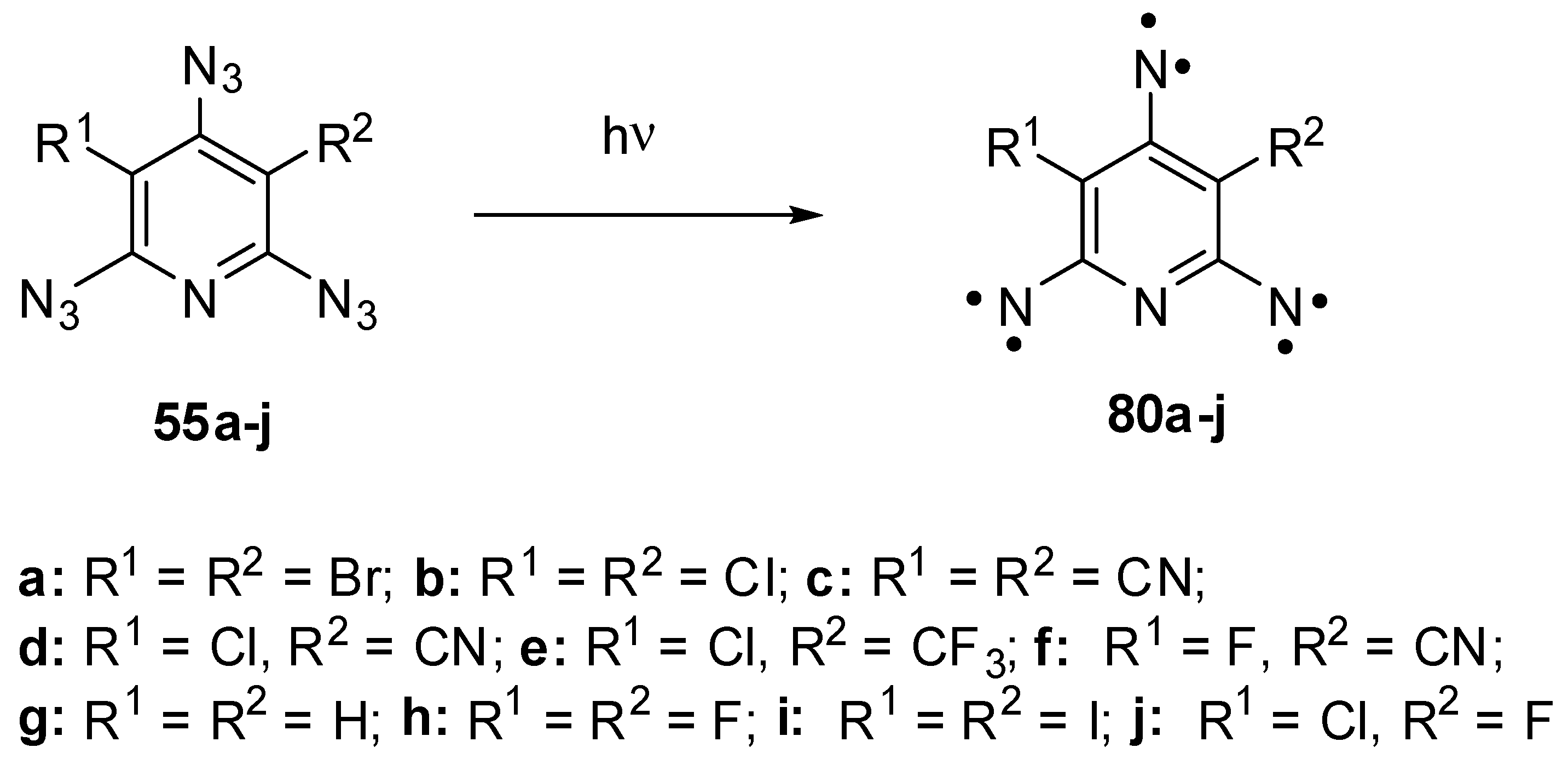
6. Triazidobenzenes
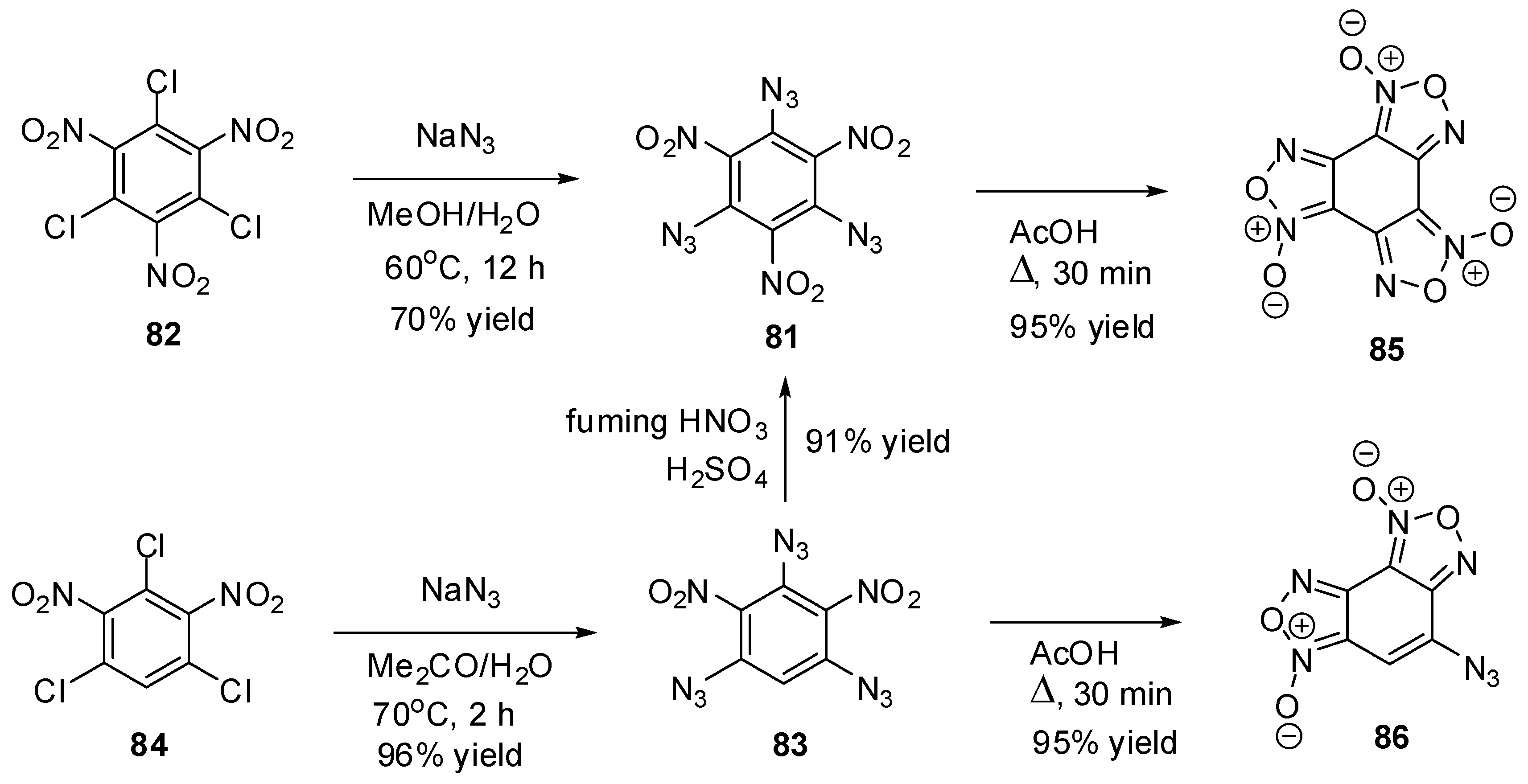
To the moment, there are known just nine triazidobenzenes. The first of them, 1,3,5-triazido-2,4,6-trinitrobenzene (81), was prepared by Turek in 1931, using the azidation of trichloride 82 with sodium azide in hot methanol (Scheme 23) [121]. Later, a more efficient way to preparation of 81, based on nitration of triazide 83, was developed [122,123,124]. The intermediate triazide 83 is readily obtained by refluxing trichloride 84 with sodium azide in the mixture of acetone and methanol [122]. Triazides 81 and 83 can also be obtained in 65%–90% yields by azidation of the corresponding chlorides 82 and 84 with sodium azide in aqueous acetone, methanol or dimethylsulfoxide solutions [125,126]. On boiling in acetic acid, both triazides 81 and 83 form benzofuroxans 85 and 86 in almost quantitative yields (Scheme 23) [122,126].
Scheme 23.
Synthesis of triazides 81 and 83.
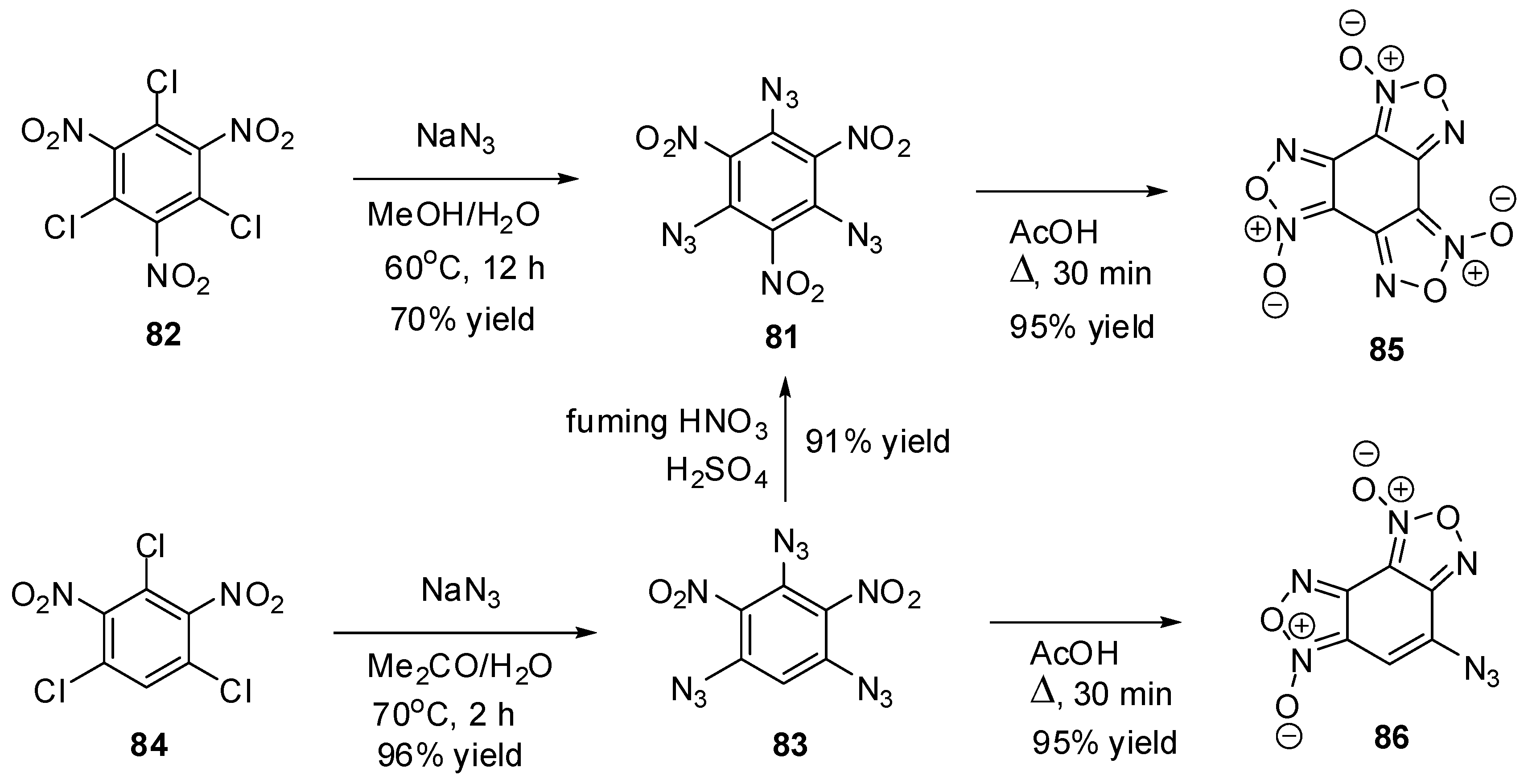
Triazides 81 and 83 are bright yellow solids with melting points of 107–108 and 128–130 °C, respectively [121,122,123,124,125,126]. Both triazides are typical high energy density organic materials that are rather sensitive toward impact and friction and very sensitive toward electrostatic discharge [74,124].
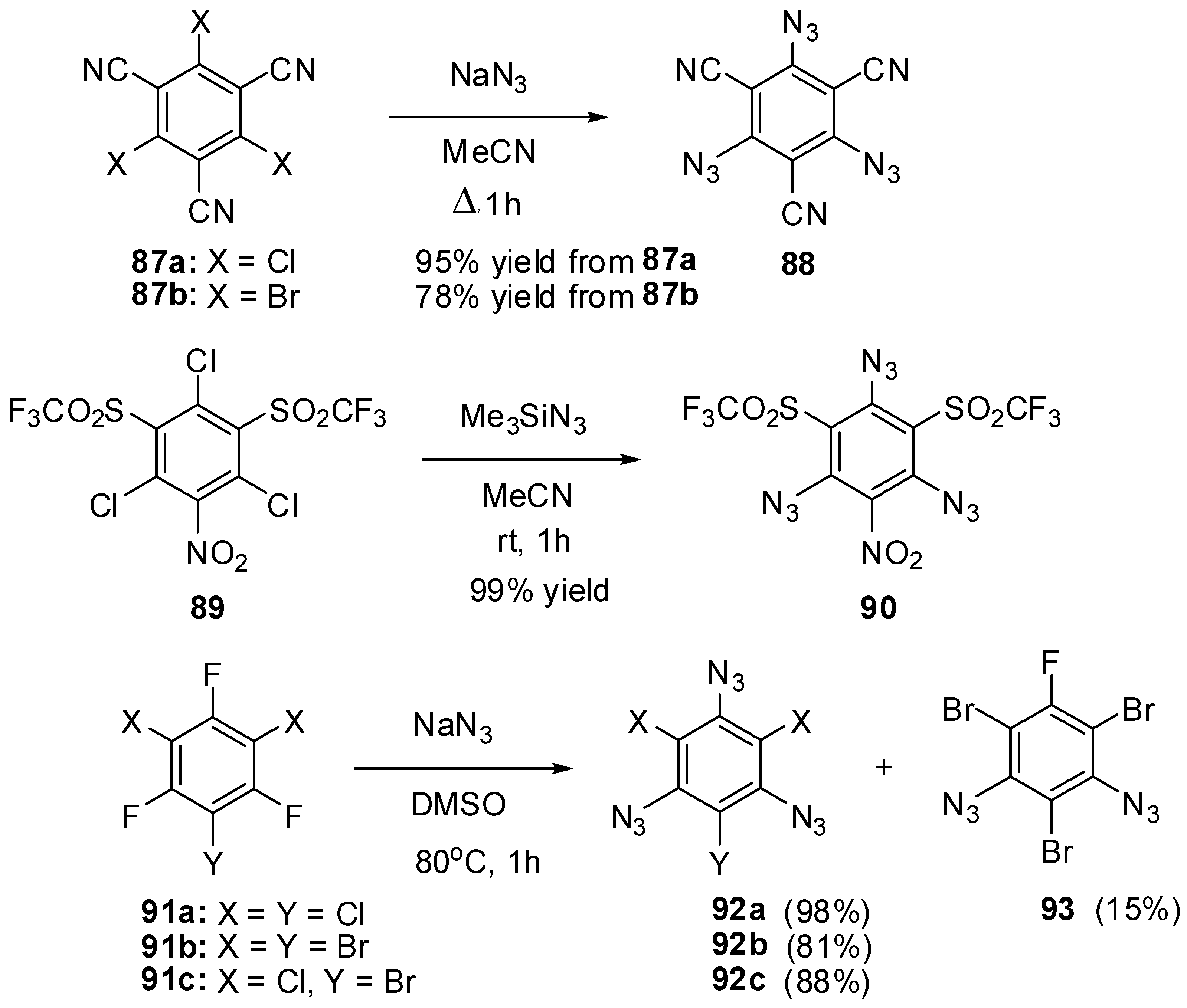
The nucleophilic substitution reactions were also used to prepare triazidobenzenes 88, 90 and 92a–c from the corresponding halides (Scheme 24) [127,128,129,130]. The azidation of halides 87a,b with sodium azide in boiling acetonitrile gave triazide 88 in high yield [127,128]. Similarly, the azidation of trichloride 89 with trimethylsilylazide in acetonitrile at room temperature afforded triazide 90 in quantitative yield [129]. Unfortunately, halides 87a,b and 89 are hardly available compounds what seriously limits the use of triazides 88 and 90 in practice. Thus, tribromide 87b is prepared from mesitylene in six steps [128]. Both triazides 88 (m.p. 161 °C) and 90 (m.p. 113–114 °C) are solids that are almost insensitive toward impact, friction and electrostatic discharge. Triazide 88 has the highest melting point among all six-membered aromatic triazides [128]. Recently, a simple and efficient method of preparation of triazidobenzenes 92a–c from commercially available trifluorides 91a–c has been developed (Scheme 24) [130]. Trifluorides 91a and 91c readily underwent chemoselective defluorination on heating in dimethylsulfoxide solutions with sodium azide to form triazides 92a and 92c in high yields. However, a similar reaction of trifluoride 91b, containing the least electron-withdrawing substituents on the benzene ring, stopped at the stage of formation of a mixture of diazide 93 and triazide 92b. The latter was obtained in 81% yield by refluxing the mixture of 93 and 92b with sodium azide in aqueous acetone, from which triazide 92b precipitated. All triazides 92a (m.p. 94 °C), 92b (m.p. 105 °C) and 92c (m.p. 83 °C) are colorless solids that are insensitive toward impact, friction and electrostatic discharge. Owing to their simple and efficient preparation from commercially available starting materials, triazides 92a–c may be of considerable interest as cross-linking agents for polymer chemistry and as starting compounds in synthetic chemistry.
Scheme 24.
Synthesis of triazides 88, 90 and 92a–c.

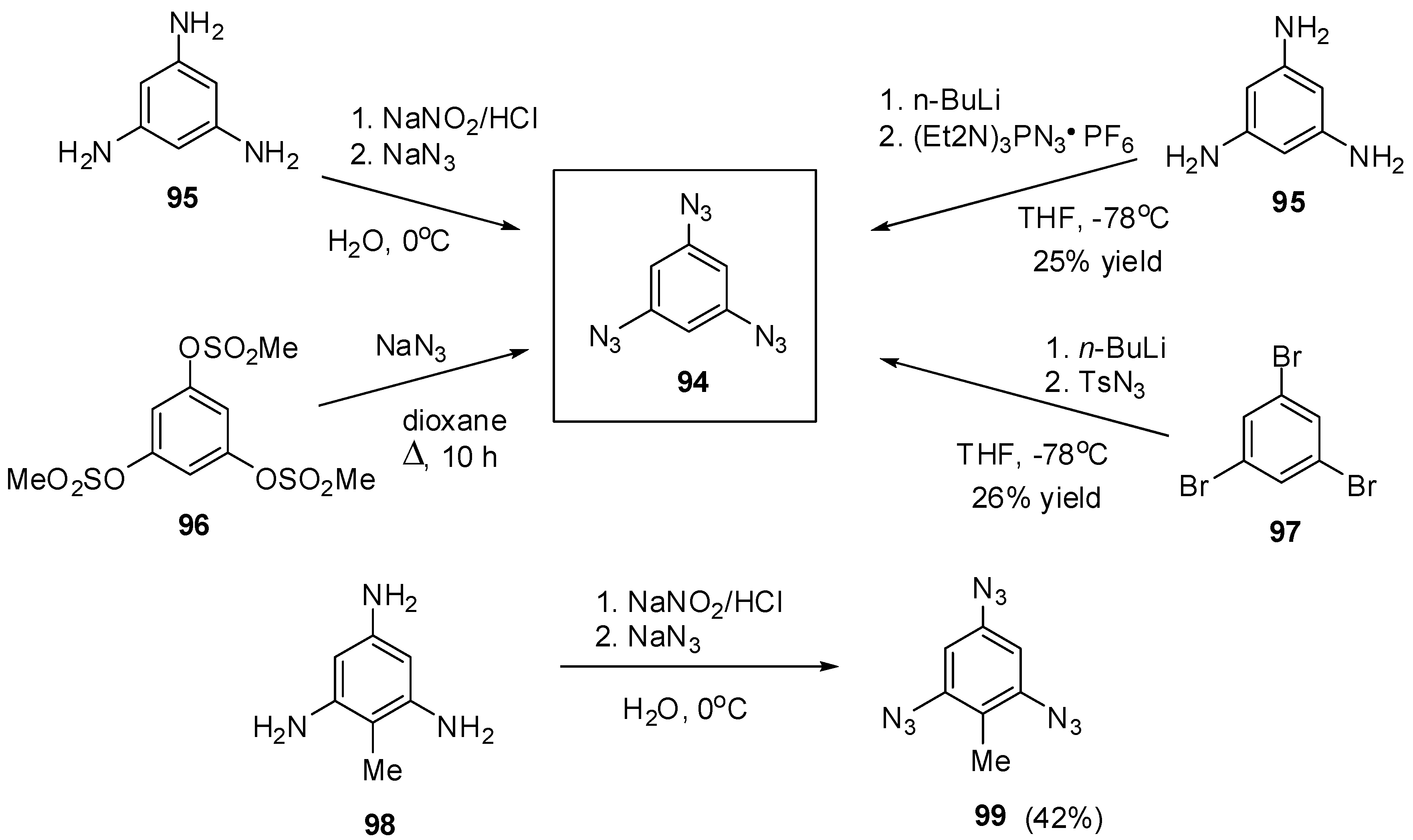
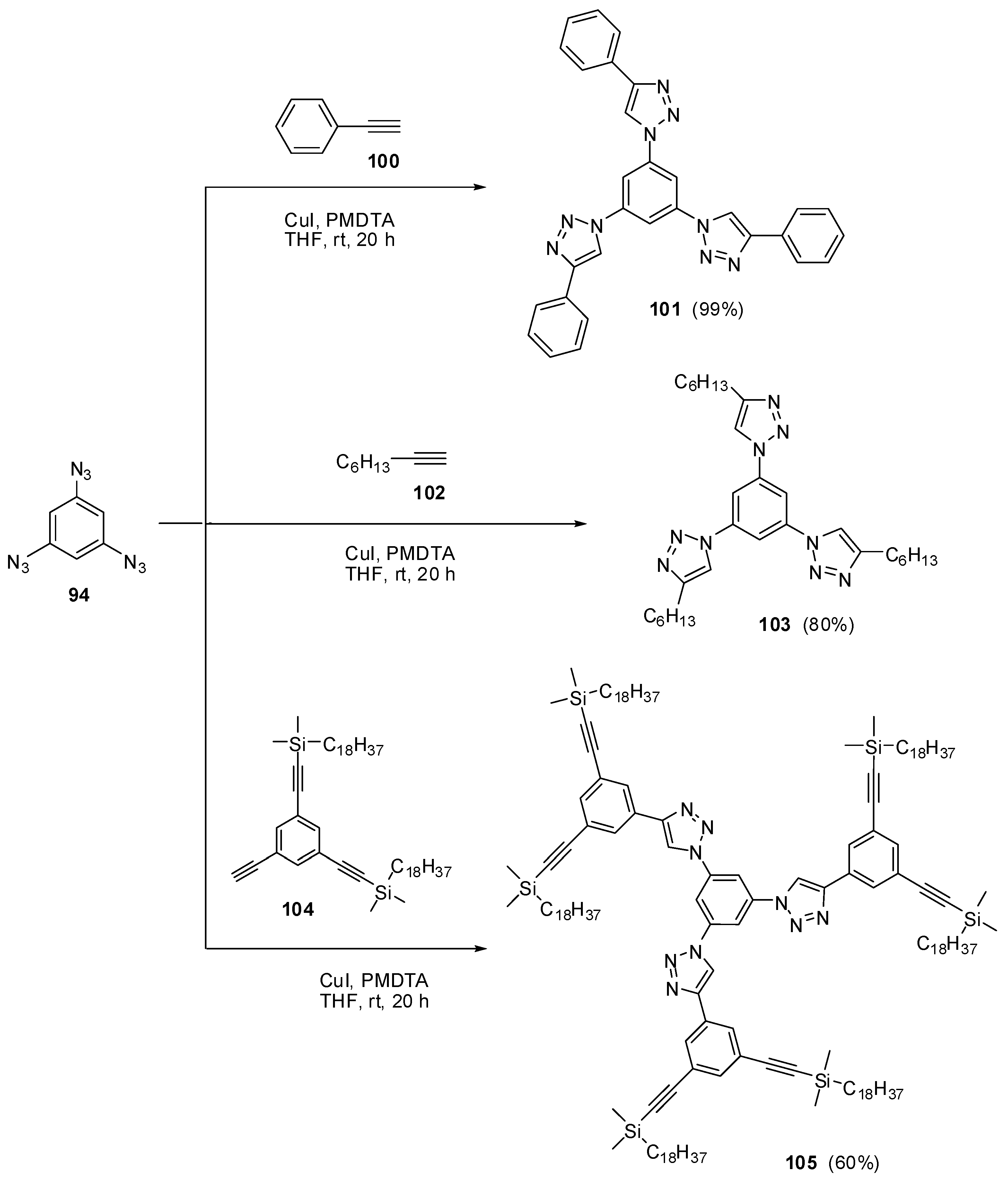
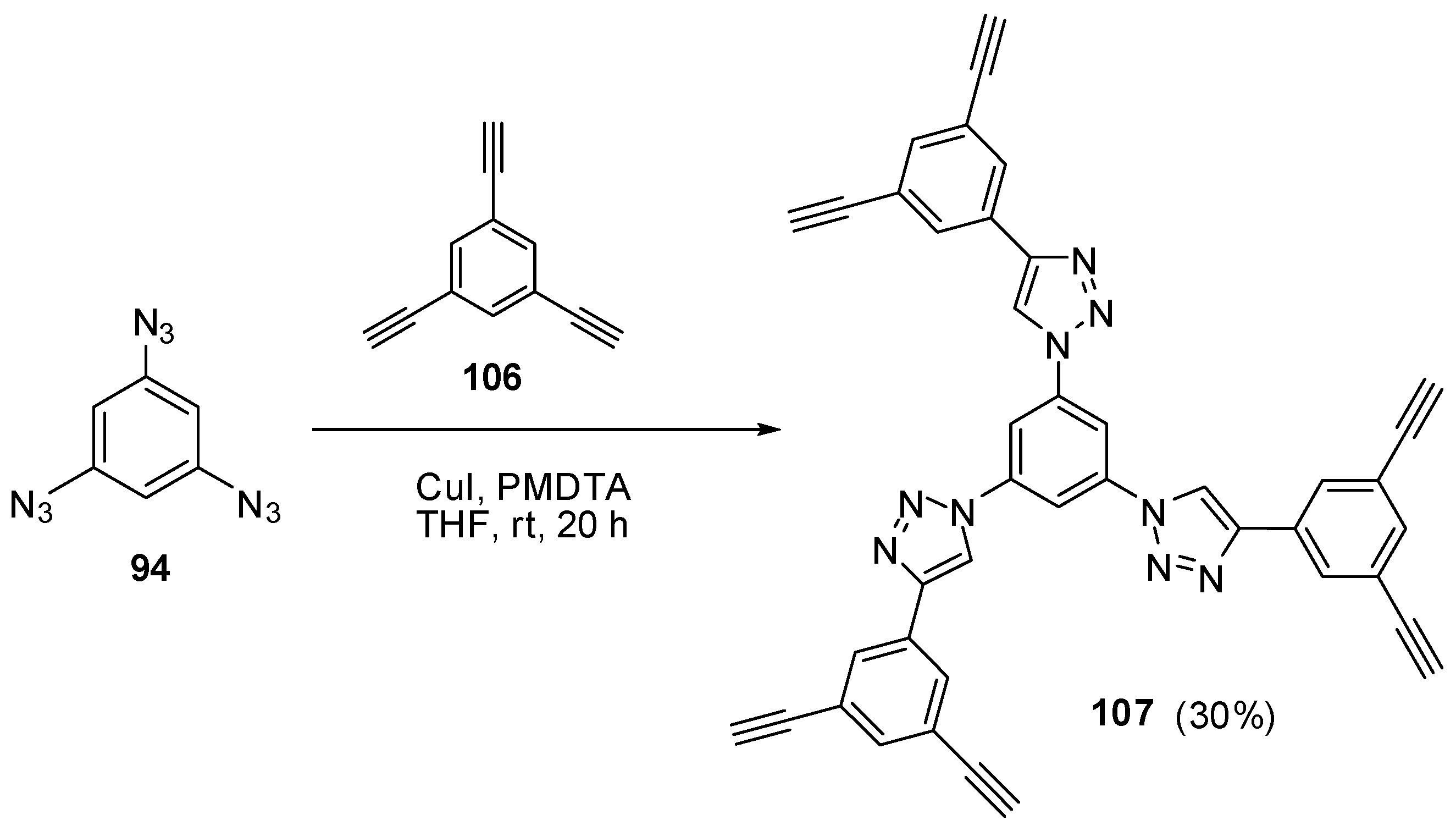
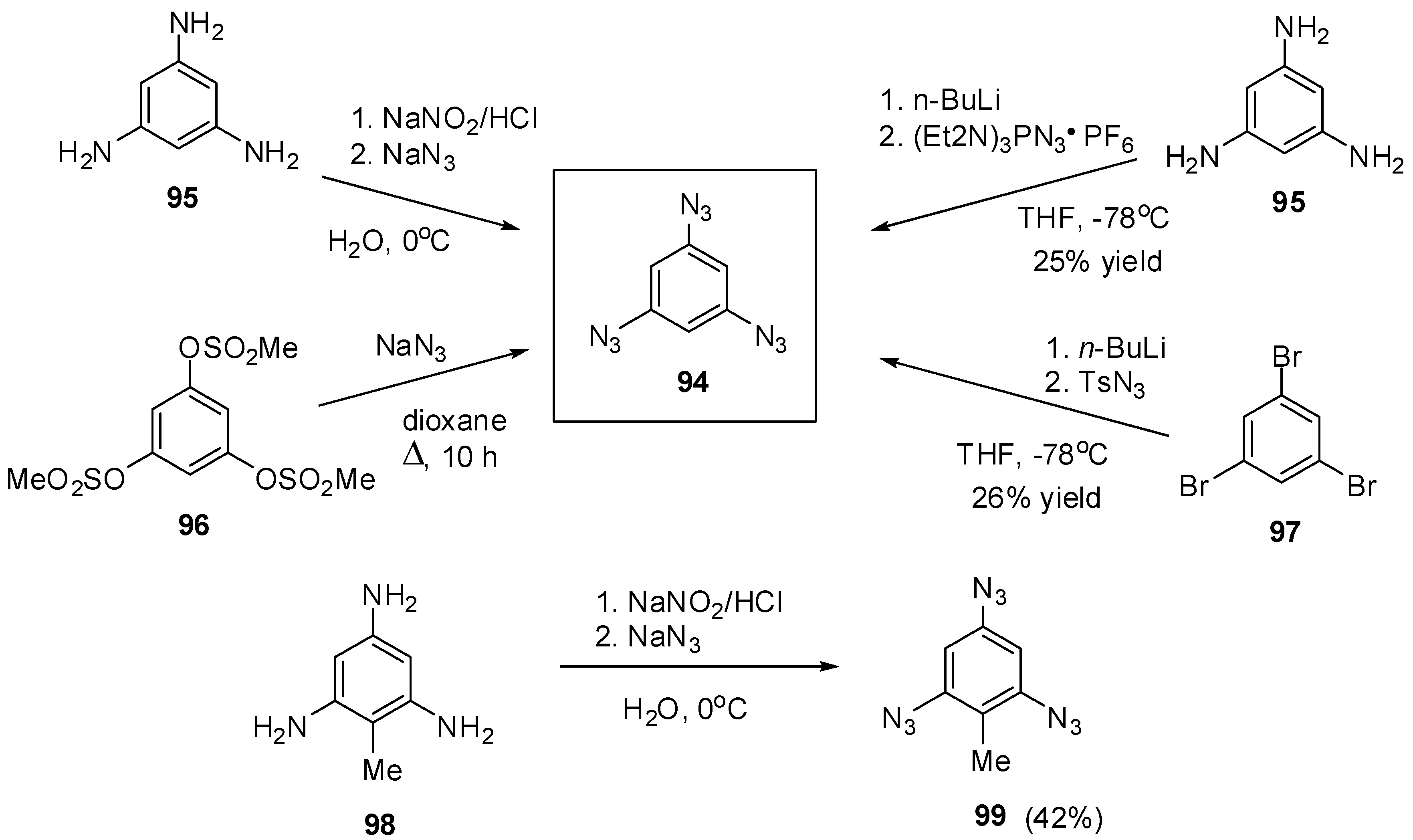
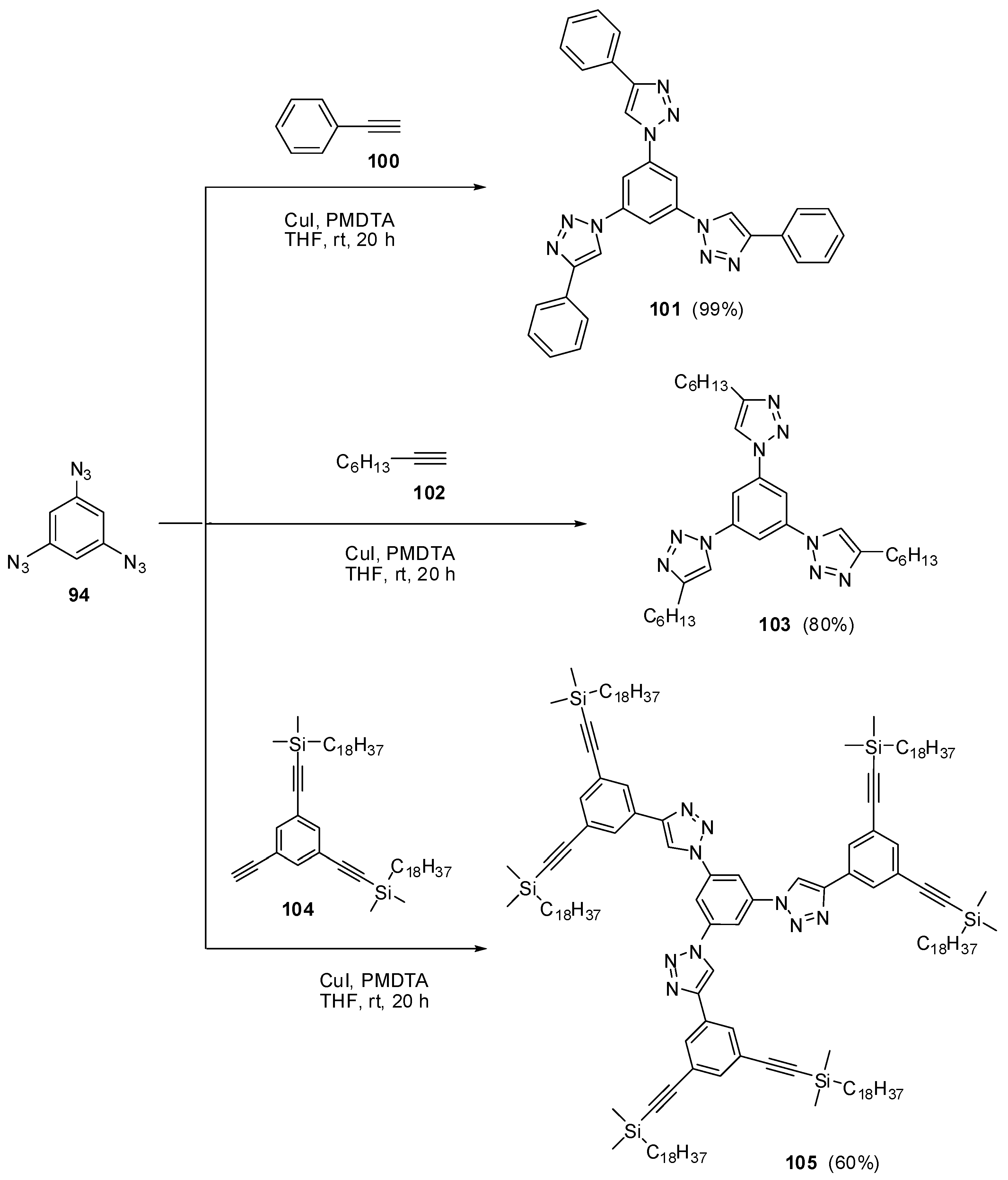
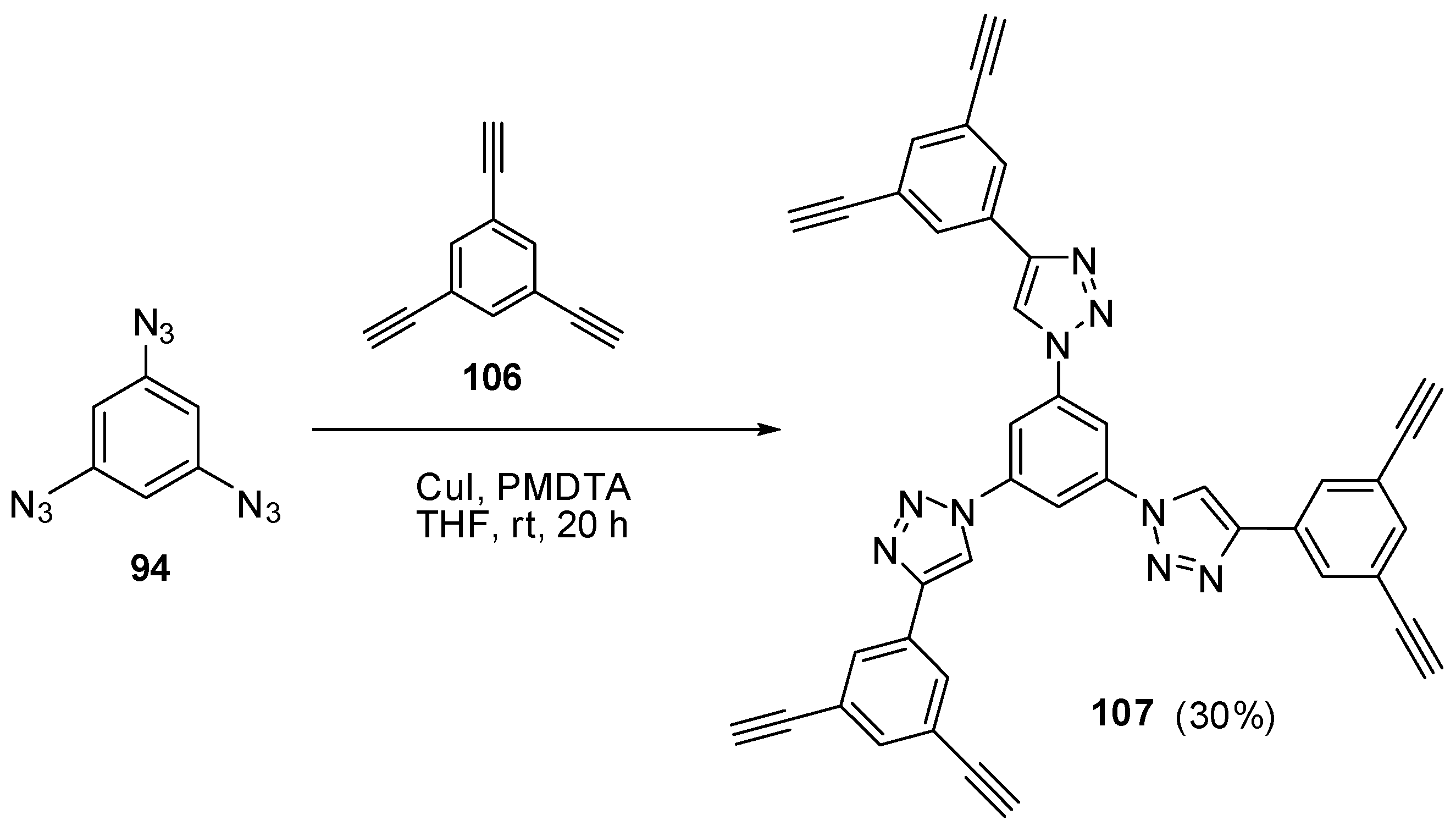
The most contradictory data were reported on the synthesis and properties of triazide 94. In 1967, Breslow and Marcantonio patented the preparation of 94, based on diazotization of triamine 95 (Scheme 25) [131]. However, neither the protocol of preparation nor the yield and melting point of 94 were reported. Later, Kim and Lee have described the preparation of 94 by azidation of ester 96 in boiling dioxane [132]. Again, neither the yield nor melting point of 94 was reported. It was just mentioned that NMR spectra of triazide 94 display a signal of protons at δ 7.30 ppm and a signal of carbons at δ 128.0 ppm. Very recently, the synthesis of triazide 94 from various precursors has been investigated by Juriček [133]. He found that triamine 95 is very unstable in acidic media and cannot be converted in triazide 94, using the diazotization reaction. The highest yield (26%) of triazide 94 was achieved in the reaction of tribromobenzene 97 with n-butyllithium followed by the diazo-transfer reaction from tosylazide to intermediate trilithiobenzene (Scheme 25). The stepwise treatment of triamine 95 at first with n-butyllithium and then with (Et2N)3PN3·PF6 salt as a diazo-transfer reagent gave triazide 94 in 25% yield (Scheme 25) [133]. Triazide 94 was obtained as a yellow solid that was insensitive toward impact and friction. NMR spectra of triazide 94 displayed a signal of protons at δ 6.45 ppm and two signals of carbon atoms at δ 106.1 (CH) and 143.2 (C–N3) ppm. These spectroscopic characteristics agreed well with 1H- and 13C-NMR spectra of triazide 99 obtained in 42% yield by diazotization of triamine 98 (Scheme 25) [24]. Similarly to triazide 94, triazide 99 was obtained as a yellow solid with a melting point of 103–104 °C. NMR spectra of triazide 99 displayed two signals of protons at δ 1.93 (Me) and 6.48 (CH) ppm and four signals of carbon atoms at δ 10.0 (Me), 103.8 (CH), 117.2 (C–Me), 138.4 (C4–N3) and 140.3 (C2,6–N3) ppm. Similarly to triazide 94, triazide 99 is also insensitive toward impact and friction. Both triazides 94 and 99 are efficient cross-linking agents for polymer chemistry [131,134]. These triazides are also promising starting materials for supramolecular chemistry. Thus, the copper-catalyzed azide-alkyne cycloadditions (CuAAC- or click-reactions) of triazide 94 with acetylenes 100, 102 and 104 afforded tris-adducts 101, 103 and 105 (Scheme 26) [133]. The more unpredictable reaction of triazide 94 with tris-acetylene 106 gave tris-adduct 107 in just 30% yield (Scheme 27) [133]. The latter can be obtained in 99% yield by alkali hydrolysis of tris-adduct 105 [133]. Both hexaacetylenes 105 and 107 possess a huge synthetic potential and are of considerable interest as building-blocks for synthetic chemistry.
Scheme 25.
Synthesis of triazides 94 and 99.
Recent X-ray diffraction studies have shown that the azido groups of triazides 81 and 88 are rotated out of the C6 plane in order to reduce steric repulsion [124,128]. Due to this effect, all three azido groups of 81 and 88 are structurally nonequivalent and slightly differ in the C–Nα, Nα–Nβ and Nβ–Nγ bond distances and the C–Nα–Nβ and Nα–Nβ–Nγ angles. These parameters of triazides 81 and 88 are very close to that of the γ-azido groups of triazidopyridines 55b and 55c [102,103,104]. It is therefore not surprising that the γ-azido groups of triazidopyridines 55b and 55c and the azido groups of triazides 92a and 92b display similar chemical shifts in 15N-NMR spectra [73,130]. In 13C-NMR spectra of D3h symmetric triazides 88, 92a, 92b and 94, the signals of the C–N3 atoms are the most deshielded and manifested in the δ 134–150 ppm region, while the signals of the C–R atoms are usually observed in the δ 95–122 ppm region [128,130,133].
Scheme 26.
Some click-reactions of triazide 94.
Scheme 27.
Сlick-reaction of triazide 94 with tris-acetylene 106.
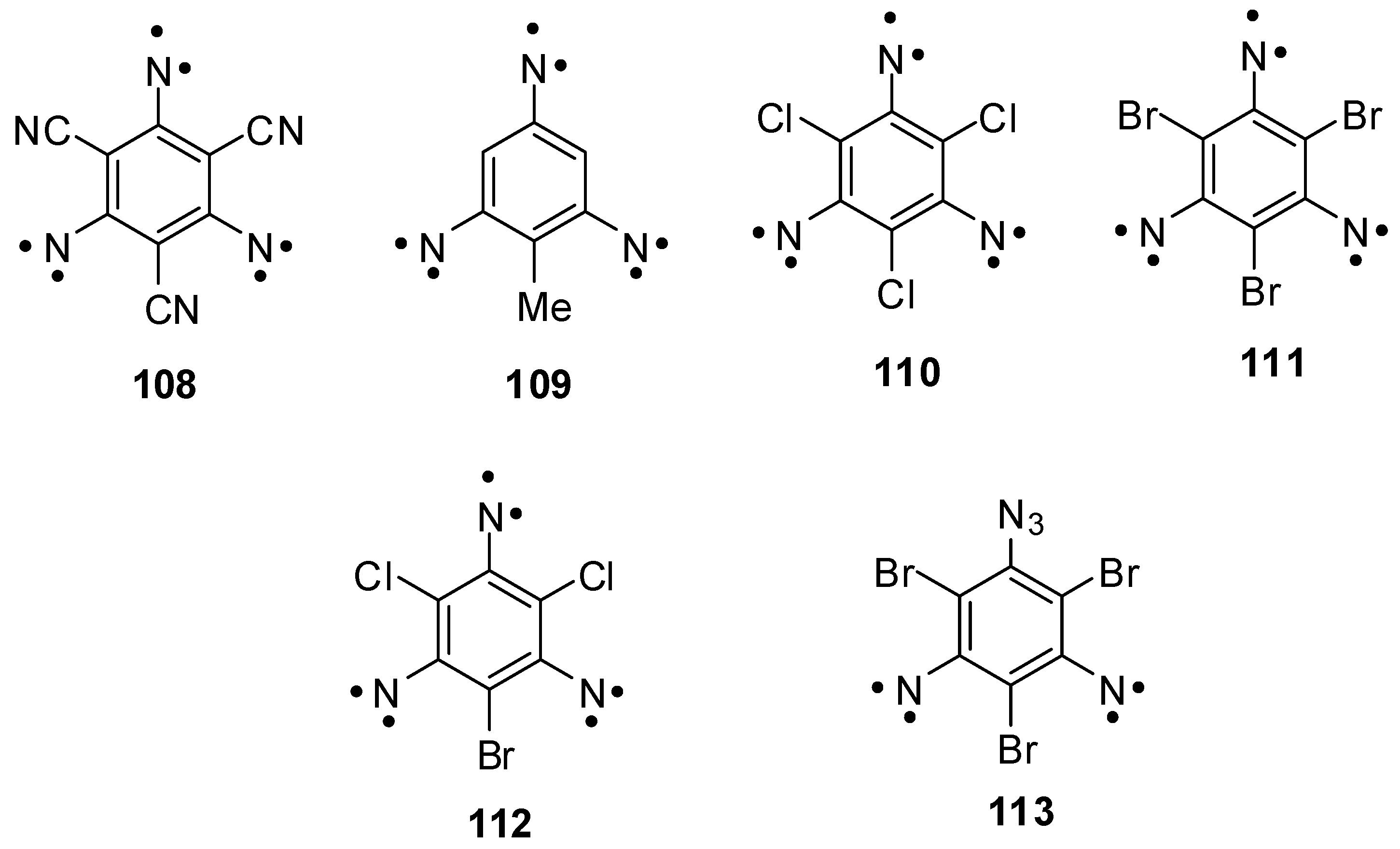
Triazides 88, 92a–c and 99 have played a historical role in investigations of high-spin organic molecules. Trinitrene 108 obtained by the photolysis of triazide 88 in frozen organic solutions became the first organic septet hexaradical that was detected with X-band EPR spectroscopy (Scheme 28) [10]. Based on intuitive assignment of signals in EPR spectrum, the magnetic parameter D ≈ −0.055 cm−1 of trinitrene 108 was calculated. For many years, this value of D served as a benchmark in investigations of many other high-spin organic molecules [117]. Only recently, owing to the appearance of modern computer line-shape EPR spectral simulation programs, it was established that trinitrene 108 has D = −0.092 cm−1 [29]. Similar values of D were also determined for trinitrenes 109 (D = −0.0934 cm−1) and 110 (D = −0.0957 cm−1) obtained by the photolysis of triazides 99 and 92a, respectively [24,29]. These studies showed that septet 1,3,5-trinitrenobenzenes have relatively low spin densities on the nitrene units and, as a result, relatively weak dipolar spin-spin interactions and magnetic properties among septet trinitrenes. On the other hand, the introduction of heavy atoms in positions 2, 4 and 6 of septet 1,3,5-trinitrenobenzenes allows the preparation of organic hexaradicals possessing the record values of magnetic anisotropy. Thus, trinitrene 111 obtained by the photolysis of triazide 92b shows D = −0.203 cm−1 [33]. In such molecules, the large negative value of D arises due to very strong anisotropic spin-orbit interactions. As a result, the D value of trinitrene 111 exceeds almost two times the D value of septet 2,4,6-trinitreno-s-triazine 17 (see Scheme 5) possessing the strongest dipolar spin-spin interactions among septet trinitrenes. Photochemical studies of triazidobenzenes provide also important information about the effect of subtle structural changes in the molecules of these compounds on magnetic properties of organic polyradicals. Thus, it was found that, in contrast to trinitrenes 108, 109, 110 and 111, trinitrene 112 has a positive sign of D = 0.124 cm−1 and does not possess the magnetism [32]. Since the presence of only one heavy bromine atom in the molecule, the principal magnetic Z-axis of trinitrene 112 lies in the molecular plane perpendicularly to the C–Br bond, and the principal values Dxx, Dyy and Dzz of the total tensor D have such magnitudes and signs for which the total parameter D is large in magnitude and positive in sign. Another unexpected result was obtained during W-band EPR studies of quintet dinitrene 113 formed in the photolysis of triazide 92b [33]. In contrast to all known quintet dinitrenes, dinitrene 113 showed negative sign of D = −0.306 cm−1 due to the effect of three heavy bromine atoms.
Scheme 28.
High-spin nitrenes obtained by photolysis of triazides 88, 92a–c and 99.
7. Tetraazides
Six-membered aromatic tetraazides represent one of the least studied classes of organic compounds. To the moment, the synthesis of only four such tetraazides has been reported, and only one of them was characterized with modern spectroscopic methods. The first representative of six-membered aromatic tetraazides, 1,2,4,5-tetrazido-3,6-dihydroxybenzene (115), was prepared by Sorm in 1939, using the reduction of tetraazidoquinone 114 with potassium iodide in acidic media (Scheme 29) [135]. During this reaction, deep-blue quinone 114 was turned into hydroquinone 115 as insoluble in water white solid. The latter was metastable in the air and quickly oxidized to the starting quinone 114. On comparison with very sensitive tetraazidoquinone 114, tetraazidohydroquinone 115 was much less sensitive to impact, friction and spark. The second representative of this class of compounds, tetrazidophthalic acid 117, was prepared in 95% yield by the reaction of tetrachlorophthalic anhydride 116 with sodium azide in dimethylsulfoxide at room temperature (Scheme 29) [136]. Tetraazide 117 was obtained as a white solid with a melting point of 113 °C. Since its very high sensitivity to mechanical stimuli, no any further studies of 117 were carried out. Finally, Pannell in 1975 has reported the synthesis of tetraazide 119 by the reaction of tetrachloride 118 with sodium azide in dimethylformamide at 80 °C (Scheme 29) [137,138]. Recently, this tetraazide was obtained by azidation of tetrachloride 118 with sodium azide in aqueous acetone at room temperature in 96% yield [91].
Scheme 29.
Synthesis of tetraazides 115, 117 and 119.
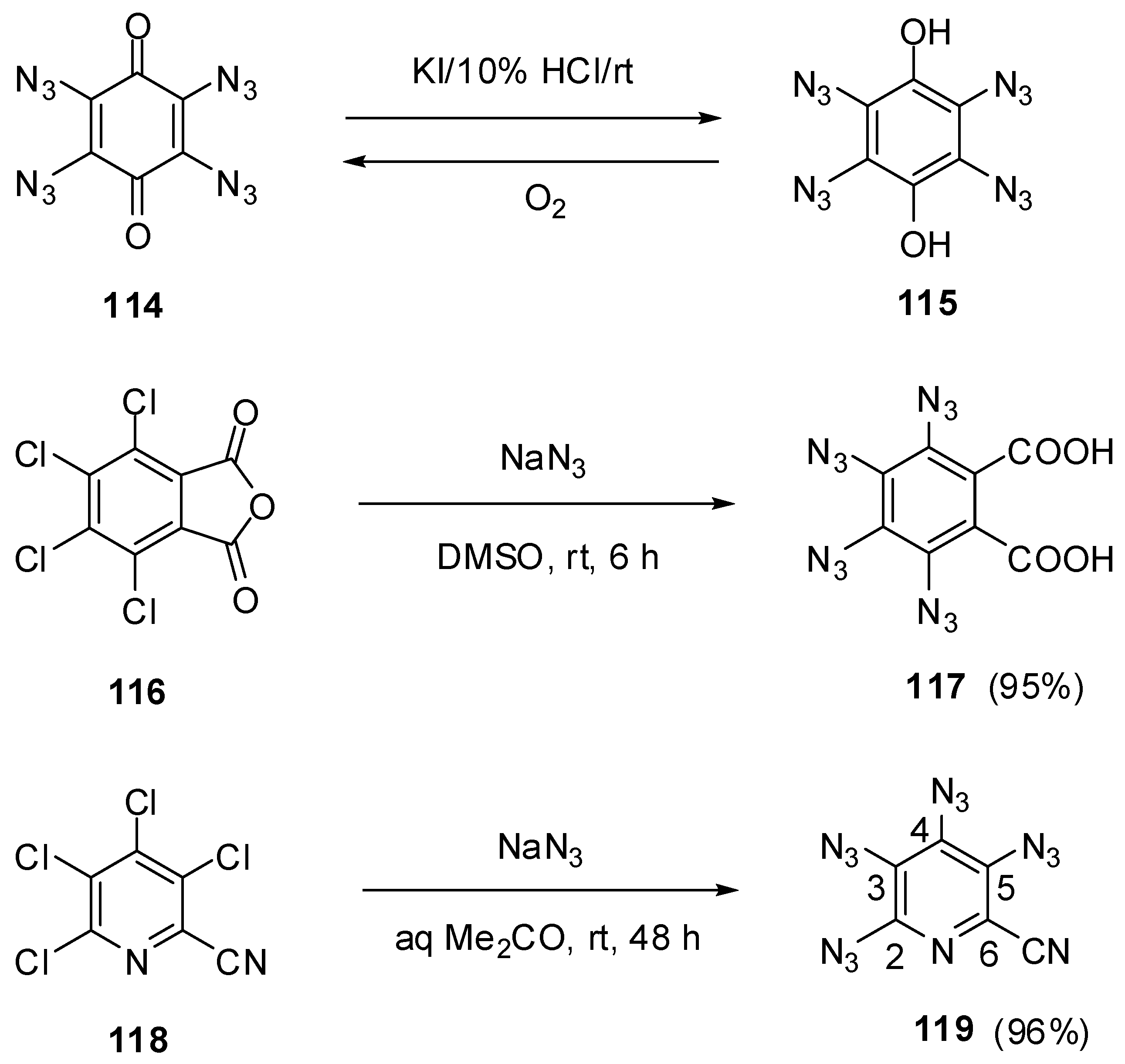
On crystallization from ethanol, tetraazide 119 is obtained as a brown solid with a melting point of 103 °C [92]. This tetraazide is very sensitive to impact and friction and violently explodes on quick heating at 23 °C. On working with it, one should always handle 119 only with plastic spatulas and use thick gloves behind a blast shield. Due to interatomic interactions of the neighboring azido groups in the molecule, the rate of the thermal decomposition of 119 is 1000 times higher than that of 2,4,6-triazido-3,5-dicyanopyridine (55c) [139]. Most likely, the thermal decomposition of 119 follows the chain-mechanism, yielding molecular nitrogen and C3N4 carbon nitrides as the final products. The IR and UV spectra of tetraazide 119 are very similar to those of triazide 55c [92]. The 13C-NMR spectrum of tetraazide 119 shows six signals of the carbon atoms at δ 113.4 (CN), 118.2 (C-6), 122.8 (C-3), 129.7 (C-5), 133.1 (C-4) and 145.1 ppm (C-2) [91].
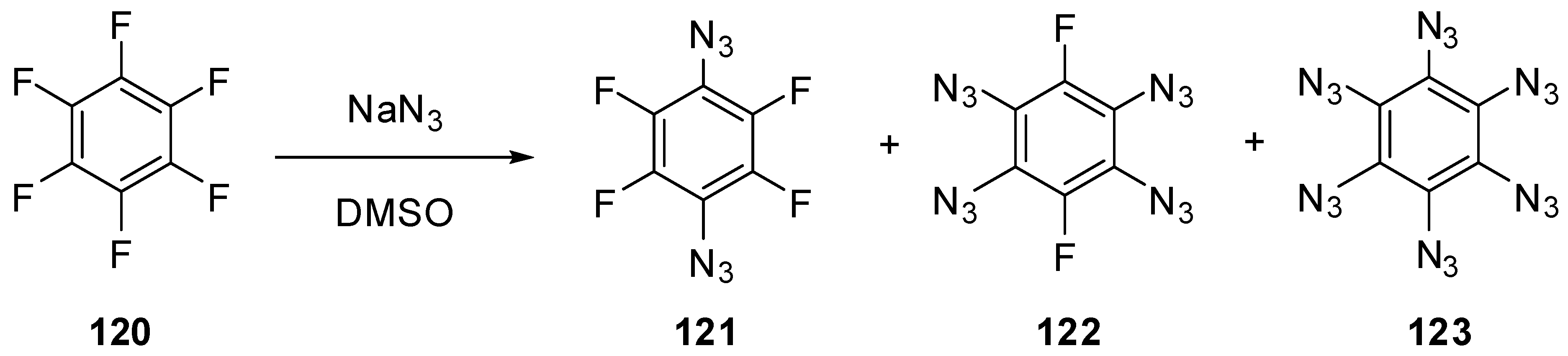
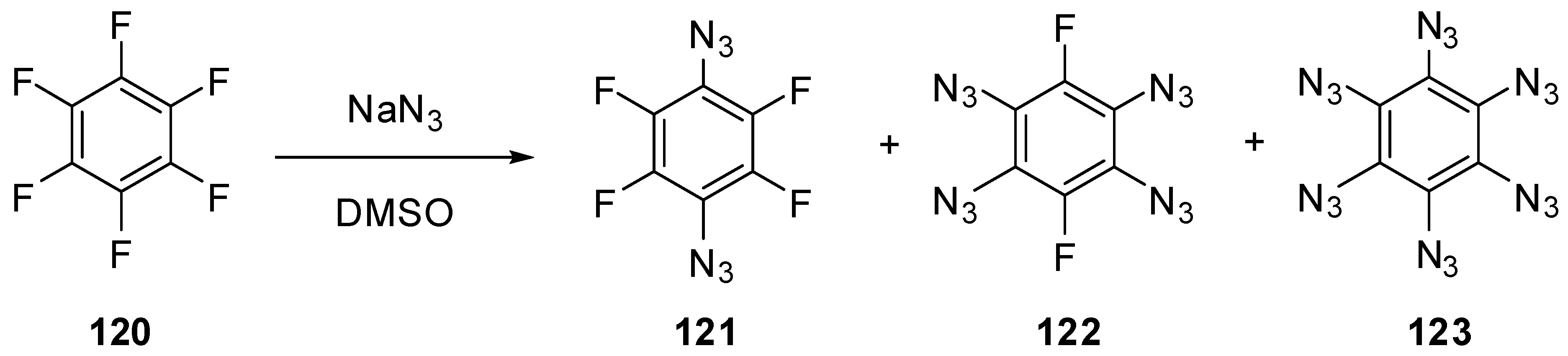
To the moment, nothing is known about the properties of six-membered aromatic compounds with five and six azido groups in the ring. It has just been reported that azidation of hexafluorobenzene (120) with sodium azide in hot dimethylsulfoxide led to the formation of a mixture of diazide 121 and tetraazide 122 as the major products as well as hexaazide 123 as a minor product (Scheme 30) [140]. However, during the next five decades, no any other information on the synthesis, properties and use of tetraazide 122 and hexaazide 123 appeared in the literature. So far, hexaazide 123 as well as pentaazidopyridine and tetraazidopyrimidine can be considered only as hypothetical molecules in theoretical studies [141,142,143].
Scheme 30.
Azidation of hexafluorobenzene 120.
8. Summary
Of the twenty five six-membered aromatic triazides known to date, eighteen have been synthesized and spectrally characterized only in the last two decades. Many of these triazides are prepared in one step and in high yield from commercially available starting halides. Moreover, most of these triazides are rather safety compounds and can explode only under very specific conditions. Owing to these features, six-membered aromatic triazides may be of considerable interest as starting materials for synthetic chemistry and photochemistry as well as cross-linking agents for polymer chemistry and microelectronics. The high antitumor activity of 2,4,6-triazidopyrimidine against Sarcoma 180, Piss lymphosarcoma and Guerin carcinoma indicates that some of aromatic triazides may possess interesting biological properties. However, the most promising application of aromatic triazides may be their use as starting materials in click-chemistry aimed at the design of new polyfunctional supramolecular systems possessing useful chemical, physical and biological properties. Thus, for instance, just the successive cycloaddition of different acetylenes to such triazides already provides unique opportunities to synthesize a great variety of new organic compounds, using relatively simple experimental procedures.
Conflicts of Interest
The author declares no conflict of interest.
References
- Sutton, T.C. Structure of cyanuric triazide, (C3N3)(N3)3. Philos. Mag. 1933, 15, 1001–1018. [Google Scholar] [CrossRef]
- Bragg, W.H. Structure of the azide group [crystal structure of cyanuric triazide]. Nature 1934, 134, 138. [Google Scholar] [CrossRef]
- Hughes, E.W. Crystal structure of cyanuric triazide. J. Chem. Phys. 1935, 3, 1–5. [Google Scholar] [CrossRef]
- Knaggs, J.E. Structure of cyanuric triazide. J. Chem. Phys. 1935, 3, 241. [Google Scholar] [CrossRef]
- Knaggs, E. The crystal structure of cyanuric triazide. Proc. Roy. Sci. Lond. 1935, 150, 576–602. [Google Scholar] [CrossRef]
- Kroke, E.; Schwarz, M.; Buschmann, V.; Miehe, G.; Fuess, H.; Riedel, R. Nanotubes formed by detonation of C/N precursors. Adv. Mater. 1999, 11, 158–161. [Google Scholar] [CrossRef]
- Gillan, E.G. Synthesis of nitrogen-rich carbon nitride networks from an energetic molecular azide precursor. Chem. Mater. 2000, 12, 3906–3912. [Google Scholar] [CrossRef]
- Wang, J.; Gillan, E.G. Low-temperature deposition of carbon nitride films from a molecular azide, (C3N3)(N3)3. Thin Solid Films 2002, 422, 62–68. [Google Scholar] [CrossRef]
- Utschig, T.; Schwarz, M.; Miehe, G.; Kroke, E. Synthesis of carbon nanotubes by detonation of 2,4,6-triazido-1,3,5-triazine in the presence of transition metals. Carbon 2004, 42, 823–828. [Google Scholar] [CrossRef]
- Wasserman, E.; Schueller, K.; Yager, W.A. EPR detection of the septet ground state of a trinitrene. Chem. Phys. Lett. 1968, 2, 259–260. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Kuhn, A.; Wong, M.W.; Wentrup, C. Mono-, di- and trinitrenes in the pyridine series. J. Am. Chem. Soc. 2000, 122, 1572–1579. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Walton, R.; Sanborn, J.A.; Lahti, P.M. Quintet and septet state systems based on pyridylnitrenes: Effect of substitution on open-shell high-spin states. J. Am. Chem. Soc. 2000, 122, 1580–1588. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Electronic absorption spectra of quintet and septet pyridylnitrenes. Mendeleev Commun. 2002, 12, 168–170. [Google Scholar] [CrossRef]
- Sato, T.; Narazaki, A.; Kawaguchi, Y.; Niino, H.; Bucher, G. Dicyanocarbodiimide and trinitreno-s-triazine generated by consecutive photolysis of triazido-s-triazine in a low-temperature nitrogen matrix. Angew. Chem. Int. Ed. 2003, 42, 5206–5209. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Narazaki, A.; Kawaguchi, Y.; Niino, H.; Bucher, G.; Grote, D.; Wolff, J.J.; Wenk, H.H.; Sander, W. Generation and photoreactions of 2,4,6-trinitreno-1,3,5-triazine, a septet trinitrene. J. Am. Chem. Soc. 2004, 126, 7846–7852. [Google Scholar] [CrossRef] [PubMed]
- Kuzina, S.I.; Mikhailov, A.I.; Chapyshev, S.V. Radiolysis and photolysis of crystalline 2,4,6-triazido-3,5-dichloropyridine: Generation of quintet dinitrenes. High Energy Chem. 2007, 41, 245–250. [Google Scholar] [CrossRef]
- Kuzina, S.I.; Mikhailov, A.I.; Chapyshev, S.V. Effect of microwave radiation on the magnetic properties of quintet dinitrenes. Dokl. Phys. Chem. 2007, 412, 34–37. [Google Scholar] [CrossRef]
- Morgunov, R.B.; Berdinskii, V.L.; Kirman, M.V.; Tanimoto, Y.; Chapyshev, S.V. Photochemical magnetism of crystalline 2,4,6-triazido-3,5-dichloropyridine. High Energy Chem. 2007, 41, 33–36. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Kuzina, S.I.; Mikhailov, A.I. Generation of quintet dinitrenes by γ-radiolysis of crystalline 2,4,6-triazido-3,5-dicyanopyridine. Mendeleev Commun. 2007, 17, 207–208. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Grote, D.; Finke, C.; Sander, W. Matrix isolation and EPR spectroscopy of septet 3,5-difluoropyridyl-2,4,6-trinitrene. J. Org. Chem. 2008, 73, 7045–7051. [Google Scholar] [CrossRef] [PubMed]
- Misochko, E.Y.; Akimov, A.V.; Chapyshev, S.V. High resolution EPR spectroscopy of septet 3,5-dichloropyridyl-2,4,6-trinitrene in solid argon. Fine-structure parameters of six electron-spin cluster. J. Chem. Phys. 2008, 129, 174510. [Google Scholar] [CrossRef] [PubMed]
- Chapyshev, S.V.; Neuhaus, P.; Grote, D.; Sander, W. Matrix isolation and magnetic parameters of septet 3,5-dicyanopyridyl-2,4,6-trinitrene. J. Phys. Org. Chem. 2010, 23, 340–346. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Korchagin, D.V.; Neuhaus, P.; Sander, W. High-spin intermediates of the photolysis of 2,4,6-triazido-3-chloro-5-fluoropyridine. Beilstein J. Org. Chem. 2013, 9, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Chapyshev, S.V.; Misochko, E.Y.; Akimov, A.V.; Dorokhov, V.G.; Neuhaus, P.; Grote, D.; Sander, W. Molecular structure and magnetic parameters of septet 2,4,6-trinitrenotoluene. J. Org. Chem. 2009, 74, 7238–7244. [Google Scholar] [CrossRef] [PubMed]
- Misochko, E.Y.; Korchagin, D.V.; Bozhenko, K.V.; Chapyshev, S.V.; Aldoshin, S.M. A density functional theory study of the zero-field splitting in high-spin nitrenes. J. Chem. Phys. 2010, 133, 064101. [Google Scholar] [CrossRef] [PubMed]
- Misochko, E.Y.; Akimov, A.V.; Masitov, A.A.; Korchagin, D.V.; Yakushchenko, I.K.; Chapyshev, S.V. High-spin organic molecules with dominant spin-orbit contribution and unprecedentedly large magnetic anisotropy. J. Chem. Phys. 2012, 137, 064308. [Google Scholar] [CrossRef] [PubMed]
- Misochko, E.Y.; Akimov, A.V.; Mazitov, A.A.; Korchagin, D.V.; Chapyshev, S.V. Magnetic anisotropy parameters of matrix-isolated septet 1,3,5-trinitreno-2,4,6-trichlorobenzene. Russ. Chem. Bull. 2012, 61, 2218–2224. [Google Scholar] [CrossRef]
- Kuzina, S.I.; Tokarev, S.V.; Korchagin, D.V.; Kolpakov, G.A.; Khudyakov, D.V.; Chapyshev, S.V.; Mikhailov, A.I.; Nadtochenko, V.A.; Aldoshin, S.M. Molecular magnetic structures based on high-spin intermediates of low-temperature radiolysis of azido derivatives and possibilities of their use in undulator systems. Russ. Chem. Bull. 2013, 62, 255–264. [Google Scholar] [CrossRef]
- Misochko, E.Y.; Akimov, A.V.; Masitov, A.A.; Korchagin, D.V.; Aldoshin, S.M.; Chapyshev, S.V. Matrix isolation ESR spectroscopy and magnetic anisotropy of D3h symmetric septet trinitrenes. J. Chem. Phys. 2013, 138, 204317. [Google Scholar] [CrossRef] [PubMed]
- Chapyshev, S.V.; Ushakov, E.N.; Neuhaus, P.; Sander, W. Matrix isolation, zero-field splitting parameters, and photoreactions of 2,4,6-trinitrenopyrimidines. J. Org. Chem. 2014, 79, 6047–6053. [Google Scholar] [PubMed]
- Misochko, E.Y.; Akimov, A.V.; Mazitov, A.A.; Korchagin, D.V.; Chapyshev, S.V. Magnetic anisotropy parameters of matrix-isolated septet 1,3,5-trinitreno-2,4,6-tribromobenzene. Russ. Chem. Bull. 2015, 64, 87–91. [Google Scholar] [CrossRef]
- Misochko, E.Y.; Masitov, A.A.; Akimov, A.V.; Korchagin, D.V.; Chapyshev, S.V. Heavy atom effect on magnetic anisotropy of matrix-isolated monobromine substituted septet trinitrene. J. Phys. Chem. A 2015, 119, 2413–2419. [Google Scholar] [CrossRef] [PubMed]
- Akimov, A.; Masitov, A.; Korchagin, D.; Chapyshev, S.; Misochko, E.; Savitsky, A. W-band EPR studies of high-spin nitrenes with large spin-orbit contribution to zero-field splitting. J. Chem. Phys. 2015, 143, 084313. [Google Scholar] [CrossRef] [PubMed]
- Chapyshev, S.V. Selective reactions on the azido groups of aromatic polyazides. Synlett 2009, 2009, 1–8. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Aromatic polyazides and high-spin nitrenes. Russ. Chem. Bull. 2011, 60, 1274–1285. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Schilling, C.; Jung, N.; Bräse, S. Cycloaddition reactions with azides: An overview. In Organic Azides: Synthesis and Applications; Bräse, S., Banert, K., Eds.; Wiley: Chichester, UK, 2010; pp. 269–284. [Google Scholar]
- Juwarker, H.; Lenhardt, J.M.; Pham, D.M.; Craig, S.L. 1,2,3-Triazole CH···Cl- contacts guide anion binding and concomitant folding in 1,4-diaryl triazole oligomers. Angew. Chem. Int. Ed. 2008, 47, 3740–3743. [Google Scholar] [CrossRef] [PubMed]
- Meudtner, R.M.; Hecht, S. Helicity inversion in responsive foldamers induced by achiral halide ion guests. Angew. Chem. Int. Ed. 2008, 47, 4926–4930. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Flood, A.H. Strong, Size-selective, and electronically tunable C-H···Halide binding with steric control over aggregation from synthetically modular, shape-persistent[34]triazolophanes. J. Am. Chem. Soc. 2008, 130, 12111–12122. [Google Scholar] [CrossRef] [PubMed]
- Megiatto, J.D., Jr.; Schuster, D.I. General method for synthesis of functionalized macrocycles and catenanes utilizing “click” chemistry. J. Am. Chem. Soc. 2008, 130, 12872–12873. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pink, M.; Karty, J.A.; Flood, A.H. Dipole-promoted and size-dependent cooperativity between pyridyl-containing triazolophanes and halides leads to persistent sandwich complexes with iodide. J. Am. Chem. Soc. 2008, 130, 17293–17295. [Google Scholar] [CrossRef] [PubMed]
- Juwarker, H.; Lenhardt, J.M.; Castillo, J.C.; Zhao, E.; Krishnamurthy, S.; Jamiolkowski, R.M.; Kim, K.; Craig, S.L. Anion binding of short, flexible aryl triazole oligomers. J. Org. Chem. 2009, 74, 8924–8934. [Google Scholar] [CrossRef] [PubMed]
- Latyshev, G.V.; Baranov, M.S.; Kazantsev, A.V.; Averin, A.D.; Lukashev, N.V.; Beletskaya, I.P. Copper-catalyzed [1,3]-dipolar cycloaddition for the synthesis of macrocycles containing acyclic, aromatic and steroidal moieties. Synthesis 2009, 41, 2605–2615. [Google Scholar]
- Zheng, L.; Huang, X.; Shen, Y.; Cheng, Y. Click chemistry approach to fluorescence-based polybinaphthyls incorporating a triazole moiety for Hg2+ recognition. Synlett 2010, 2010, 453–456. [Google Scholar]
- Huang, X.; Dong, Y.; Meng, J.; Cheng, Y.; Zhu, C. Fluorescence polymer incorporating triazole and benzo[2,1,3]thiadiazole moieties for Ni2+ detection. Synlett 2010, 2010, 1841–1844. [Google Scholar]
- Hua, Y.; Flood, A.H. Flipping the switch on chloride concentrations with a light-active foldamer. J. Am. Chem. Soc. 2010, 132, 12838–12840. [Google Scholar] [CrossRef] [PubMed]
- Megiatto, J.D., Jr.; Schuster, D.I. Introduction of useful peripheral functional groups on [2]catenanes by combining Cu(I) template synthesis with “click” chemistry. New J. Chem. 2010, 34, 276–286. [Google Scholar] [CrossRef]
- Juricek, M.; Felici, M.; Contreras-Carballada, P.; Lauko, J.; Bou, S.R.; Kouwer, P.H.J.; Brouwer, A.M.; Rowan, A.E. Triazole-pyridine ligands: A novel approach to chromophoric iridium arrays. J. Mater. Chem. 2011, 21, 2104–2111. [Google Scholar] [CrossRef]
- Plietzsch, O.; Schilling, C.I.; Grab, T.; Grage, S.L.; Ulrich, A.S.; Comotti, A.; Sozzani, P.; Muller, T.; Brase, S. Click chemistry produces hyper-cross-linked polymers with tetrahedral cores. New J. Chem. 2011, 35, 1577–1581. [Google Scholar] [CrossRef]
- Zornik, D.; Meudtner, R.M.; El Malah, T.; Thiele, C.M.; Hecht, S. Designing structural motifs for clickamers: Exploiting the 1,2,3-triazole moiety to generate conformationally restricted molecular architectures. Chem. Eur. J. 2011, 17, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dong, Y.; Li, J.; Huang, X.; Cheng, Y.; Zhu, C. A Highly selective and sensitive polymer-based fluorescence sensor for Hg2+-ion detection via click reaction. Chem. Asian J. 2011, 6, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hajipour, A.R.; Abrishami, F. Synthesis and characterization of novel polyimides containing triazoles units in the main chain by click chemistry. J. Appl. Polym. Chem. 2012, 124, 1757–1763. [Google Scholar]
- You, L.; Chen, S.; Zhao, X.; Liu, Y.; Lan, W.; Zhang, Y.; Lu, H.; Cao, C.; Li, Z. C-H···O hydrogen bonding induced triazole foldamers: Efficient halogen bonding receptors for organohalogens. Angew. Chem. Int. Ed. 2012, 51, 1657–1661. [Google Scholar]
- Hua, Y.; Liu, Y.; Chen, C.; Flood, A.H. Hydrophobic collapse of foldamer capsules drives picomolar-level chloride binding in aqueous acetonitrile solutions. J. Am. Chem. Soc. 2013, 135, 14401–14412. [Google Scholar] [CrossRef] [PubMed]
- Merckx, T.; Verwilst, P.; Dehaen, W. Preorganization in bistriazolyl anion receptors. Tetrahedron Lett. 2013, 54, 4237–4240. [Google Scholar] [CrossRef]
- Wu, C.; Li, Z.; Xu, X.; Zhao, Z.; Zhao, X.; Wang, R.; Li, Z. Folding-induced folding: The assembly of aromatic amide and 1,2,3-triazole hybrid helices. Chem. Eur. J. 2014, 20, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Gallagher, N.M.; Bie, F.; Li, Q.; Che, Y.; Wang, Y.; Jiang, H. Aromatic triazole foldamers induced by C-H···X (X = F, Cl) intramolecular hydrogen bonding. J. Org. Chem. 2014, 79, 5134–5144. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hua, Y.; Flood, A.H. β-Sheet-like hydrogen bonds interlock the helical turns of a photoswitchable foldamer to enhance the binding and release of chloride. J. Org. Chem. 2014, 79, 8383–8396. [Google Scholar] [CrossRef] [PubMed]
- Merckx, T.; Haynes, C.J.E.; Karagiannidis, L.E.; Clarke, H.J.; Holder, K.; Kelly, A.; Tizzard, G.J.; Coles, S.J.; Verwilst, P.; Gale, P.A.; et al. Anion binding and transport properties of cyclic 2,6-bis(1,2,3-triazol-1-yl)pyridines. Org. Biomol. Chem. 2015, 13, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Finger, H. Über Abkömmlinge des Cyanurs. J. Prakt. Chem. 1907, 75, 103–104. [Google Scholar]
- Ott, E.; Ohse, E. Zur kenntnis einfachen Cyan- und Cyanurverbindungen. II. Über das Cyanurtriazid (C3N12). Ber. Dtsch. Chem. Ges. 1921, 54B, 179–186. [Google Scholar] [CrossRef]
- Keßenich, E.; Klapötke, T.M.; Knitzek, J.; Nöth, H.; Schultz, A. Characterization, crystal structure of 2,4-bis(triphenylphosphanimino)tetrazolo[5,1-a]-[1,3,5]triazine, and improved crystal structure of 2,4,6-triazido-1,3,5-triazine. Eur. J. Inorg. Chem. 1998, 1998, 2013–2016. [Google Scholar] [CrossRef]
- Kessenich, E.; Polborn, K.; Schultz, A. Reaction of 2,4,6-triazido-1,3,5-triazine with triphenylphosphane. Synthesis and characterization of the novel 2-triphenylphosphanimino-4-azidotetrazolo[5,1-a]-[1,3,5]triazine and 2,4,6-tris(triphenylphosphanimino)-1,3,5-triazine. Inorg. Chem. 2001, 40, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.V. Carbonic acid azides. J. Am. Chem. Soc. 1928, 50, 1922–1930. [Google Scholar] [CrossRef]
- Allschwil, E.N.; Stein, L.E. Azido-1,3,5-triazines. Patent NL 6413689, 28 May 1965. [Google Scholar]
- Beck, W.; Bauder, M. Reactions of hydridoplatinum(II) complexes with organic azides: Amido complexes. Chem. Ber. 1970, 103, 583–589. [Google Scholar] [CrossRef]
- Pochinok, V.Y.; Avramenko, L.F.; Grigorenko, T.F.; Pochinok, A.V.; Sidorenko, I.A.; Bovchalyuk, L.N. Synthesis of 2,4,6-triazidopyrimidine, 2,4,6-triazido-s-triazine, and triazenes produced from them. Ukr. Khim. Zh. 1979, 45, 975–978. [Google Scholar]
- Thust, R.; Schneider, M.; Wagner, U.; Schreiber, D. Structure/activity investigations in eight arylalkyltriazenes comparison of chemical stability mode of decomposition and SCE induction in Chinese hamster V79-E cells. Cell Biol. Toxicol. 1991, 7, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Kirkwood, J.M. Re-evaluating the role of dicarbazine in metastatic melanoma: What have we learned in 30 years? Eur. J. Cancer 2004, 40, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Kölmel, D.K.; Jung, N.; Bräse, S. Azides—Diazonium ions—Triazenes: Versatile nitrogen-rich functional groups. Aust. J. Chem. 2014, 67, 328–336. [Google Scholar] [CrossRef]
- Kesting, W. Über Cyanurphosphinimines und über pyrogene Spaltungsversuche an Äthyl- und Methylester der normalen Cyanursäure. J. Prakt. Chem. 1923, 105, 242–250. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Ushakov, E.N.; Chernyak, A.V. 15N-NMR spectra and reactivity of 2,4,6-triazidopyridines, 2,4,6-triazidopyrimidine and 2,4,6-triazido-s-triazine. Magn. Reson. Chem. 2013, 51, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M.; Rienacker, C.M. Drop hammer test investigations on some inorganic and organic azides. Propellants Explos. Pyrotech. 2001, 26, 43–47. [Google Scholar] [CrossRef]
- Huynh, M.H.; Hiskey, M.; Pollard, C.; Montoya, D.; Hartline, E.; Gilardi, R. 4,4′,6,6′-Tetrasubstituted hydrazo- and azo-1,3,5-triazines. J. Energ. Mater. 2004, 22, 217–229. [Google Scholar] [CrossRef]
- Miller, D.R.; Swenson, D.C.; Gillan, E.G. Synthesis and structure of 2,5,8-triazido-s-heptazine: An energetic and luminescent precursor to nitrogen-rich carbon nitrides. J. Am. Chem. Soc. 2004, 126, 5372–5373. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.H.; Hiskey, M.; Hartline, E.; Montoya, D.; Gilardi, R. Polyazido high-nitrogen compounds: Hydrazo- and azo-1,3,5-triazine. Angew. Chem. Int. Ed. 2004, 43, 4924–4928. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Gao, H.; Boatz, J.A.; Drake, G.W.; Twamley, B.; Shreeve, J.M. Polyazidopyrimidines: High-energy compounds and precursors to carbon nanotubes. Angew. Chem. Int. Ed. 2006, 45, 7262–7265. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M. Chemistry of High-Energy Materials; Walter de Gruyter: Berlin, Germany, 2011; pp. 135–140. [Google Scholar]
- Matyáš, R.; Pachman, J. Primary Explosives; Springer Science & Business Media: Berlin, Germany, 2013; pp. 71–130. [Google Scholar]
- Moriarty, R.M.; Rahman, M.; King, G.J. Organic nitrenes in single crystals. Observation of hyperfine structure in the E.S.R. (electron spin resonance). J. Am. Chem. Soc. 1966, 88, 842–843. [Google Scholar] [CrossRef]
- Nakai, T.; Sato, K.; Shiomi, D.; Takui, T.; Itoh, K.; Kozaki, M.; Okada, K. High-spin nitrenes with s-triazine skeleton. Mol. Cryst. Liq. Cryst. 1999, 334, 157–166. [Google Scholar] [CrossRef]
- Banks, R.E.; Prakash, A.; Venayak, N.D. Studies in azide chemistry. Part IX. Investigations involving fluorinated azidopyrimidines and 4-azido-3-chloro-2,5,6-trifluoropyridine. J. Fluor. Chem. 1980, 16, 325–338. [Google Scholar] [CrossRef]
- Pochiniok, A.V.; Sharykina, N.I.; Bakhtiarova, T.A. Antitumor activity of 2,4,6-triazidopyrimidine. Fiziol. Akt. Veshchestva 1990, 22, 76–78. [Google Scholar]
- Pochinok, A.V.; Smirnov, V.A.; Brichkin, S.B.; Pochinok, V.Y.; Avramenko, L.F.; Grigorenko, T.F. Photolysis of azido derivatives of pyrimidine. Khim. Vys. Energ. 1982, 16, 151–154. [Google Scholar]
- Cernicharo, J.; Guelin, M.; Pardo, J.R. Detection of the linear radical HC4N in IRC + 10216. Astrophys. J. Lett. 2004, 615, L145. [Google Scholar] [CrossRef]
- Allan, R.D.; Greenwood, J.R.; Hambley, T.W.; Hanrahan, J.R.; Hibbs, D.E.; Itani, S.; Tran, H.W.; Turner, P. Studies on pyridazine azide cyclisation reactions. Org. Biomol. Chem. 2004, 2, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Moshchitskii, S.D.; Zeikan, A.A.; Pavlenko, A.F.; Kukhar, V.P. Synthesis and reactions of azidopolybromopyridines. Chem. Heterocycl. Compd. 1979, 15, 1197–1200. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Chernyak, A.V.; Yakushchenko, I.K. Chemoselective Staudinger-phosphite reaction on the azido-groups of 2,4,6-triazido-3,5-dibromopyridine. J. Heterocycl. Chem. 2015. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Synthesis and regioselective cycloaddition reactions of 2,4,6-triazido-3,5-dichloropyridine. Mendeleev Commun. 1999, 9, 164–166. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Bergstrasser, U.; Regitz, M. Effect of electronic factors on 1,3-dipolar cycloaddition of 2,4,6-triazidopyridines to tert-butylphosphaacetylene. Chem. Heterocycl. Compd. 1996, 32, 59–63. [Google Scholar] [CrossRef]
- Mikhailov, Yu. M.; Chapyshev, S.V.; Nedel’ko, V.V. Synthesis, thermal stability, heats of formation and explosive properties of cyano-substituted derivatives of di-, tri- and tetraazidopyridines. Russ. Chem. Bull. 2009, 58, 2097–2102. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Ibata, T. Synthesis of highly substituted pyridines through nucleophilic substitution of tetrachloro-3-cyanopyridine. Heterocycles 1993, 36, 2185–2190. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Synthesis and regioselective cycloaddition of 2,4,6-triazido-3-chloro-5-cyanopyridine with norbornene. Chem. Heterocycl. Compd. 1993, 29, 1426–1427. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Korchagin, D.V.; Shilov, G.V.; Aldoshin, S.M. Synthesis and structure of asymmetric 2,4,6-triazidopyridines. Chem. Heterocycl. Compd. 2011, 47, 817–825. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Chernyak, A.V. Synthesis of 2,4,6-triazidopyridine and its 3,5-diiodo derivative. Synthesis 2012, 44, 3158–3160. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Synthesis of 2,4,6-triazido-3,5-difluoropyridine. Chem. Heterocycl. Compd. 1999, 35, 632. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Synthesis, thermolysis, and mass-spectrometry of perfluorinated di- and triazidopyridines. Chem. Heterocycl. Compd. 2001, 37, 968–975. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Di- and triazidation of 3-chlorotetrafluoropyridine. J. Fluor. Chem. 2011, 132, 991–994. [Google Scholar] [CrossRef]
- Finke, C.; Grote, D.; Seidel, R.W.; Chapyshev, S.V.; Sander, W. Matrix isolation and IR spectroscopic characterization of 3,5-difluoropyridyl-2,4,6-trinitrene. J. Phys. Org. Chem. 2012, 25, 486–492. [Google Scholar] [CrossRef]
- Ganin, Y.V.; Chapyshev, S.V.; Russian Academy of Sciences, Chernohgolovka, Russian Federation. Unpublished work. 2015.
- Kuzina, S.I.; Mikhailov, A.I.; Chapyshev, S.V.; Korchagin, D.V.; Shilov, G.V.; Aldoshin, S.M. Properties of quintet dinitrenes in 2,4,6-triazido-3,5-dichloropyridine crystals. Russ. J. Phys. Chem. A 2008, 82, 1870–1877. [Google Scholar] [CrossRef]
- Kuzina, S.I.; Korchagin, D.V.; Shilov, G.V.; Chapyshev, S.V.; Mikhailov, A.I.; Aldoshin, S.M. Generation of quintet dinitrenes by low-temperature radiolysis of crystalline 2,4,6-triazido-3,5-dicyanopyridine. Dokl. Phys. Chem. 2008, 418, 7–12. [Google Scholar] [CrossRef]
- Aldoshin, S.M.; Korchagin, D.V.; Bozhenko, K.V.; Shilov, G.V.; Chapyshev, S.V. A study of molecular and crystalline structure of 2,4,6-triazido-3-chloro-5-trifluoromethylpyridine and the rotation barrier of the γ-azido group around the С-N bond. Bull. Russ. Acad. Sci. Phys. 2008, 72, 1556–1561. [Google Scholar] [CrossRef]
- Gronowitz, S.; Zanirato, P. Preparation, reactivity, NMR properties and semiempirical calculations of 2-azido- and 3-azido-selenophene. J. Chem. Soc. Perkin Trans. 1994, 2, 1815–1819. [Google Scholar] [CrossRef]
- Budyka, M.F.; Zyubina, T.S. Theoretical investigation of the azido group dissociation in aromatic azides. J. Mol. Struct. (Theochem) 1997, 419, 191–199. [Google Scholar] [CrossRef]
- Alajarin, M.; Conesa, C.; Rzepa, H.S. Ab initio SCF-MO study of the Staudinger phosphorylation reaction between a phosphane and an azide to form phosphazene. J. Chem. Soc. Perkin Trans. 2 1999, 1811–1814. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Selective derivatization of 2,4,6-triazidopyridines by the Staudinger reaction. Mendeleev Commun. 1999, 9, 166–167. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Platz, M.S. Selective reduction of the azido groups of 2,4,6-triazidopyridines. Mendeleev Commun. 2001, 11, 56–57. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Anisimov, V.M. Stereo- and regioselective cycloaddition of norbornene to 2,4,6-triazidopyridines. Chem. Heterocycl. Compd. 1997, 33, 1315–1325. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Regioselective and regiospecific cycloaddition of acetylenes to 2,4,6-triazido-pyridines. Chem. Heterocycl. Compd. 2000, 36, 1289–1299. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Bergstrasser, U.; Regitz, M. 1,3-Dipolar tris-cycloaddition of tert-butylphospha-acetylene to 2,4,6-triazido-3-chloro-5-cyanopyridine. Russ. Chem. Bull. 1996, 45, 242–243. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Anisimov, V.M. Quantum-chemical study of the nature of regioselectivity in reactions of 2,4,6-triazidopyridines with tert-butylphosphaacetylene. Chem. Heterocycl. Compd. 1997, 33, 587–596. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Regioselective cycloaddition of the dimethyl ester of acetylenedicarboxylic acid to 2,4,6-triazidopyridines. Chem. Heterocycl. Compd. 2001, 37, 861–866. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Nucleophilic replacement of the azido groups by amines in 2,4,6-triazido-3-chloro-5-cyanopyridine. Mendeleev Commun. 2007, 17, 287–288. [Google Scholar] [CrossRef]
- Chapyshev, S.V. Selective thermolysis of the azido groups of 2,4,6-triazidopyridines. Chem. Heterocycl. Compd. 2003, 39, 83–86. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Walton, R.; Lahti, P.M. Orbital control in the selective photolysis of the azido groups of 2,4,6-triazidopyridines. Mendeleev Commun. 2000, 10, 187–188. [Google Scholar] [CrossRef]
- Wetner, W., Jr. Magnetic Atoms and Molecules; Dover Publications: New York, NY, USA, 1989; p. 294. [Google Scholar]
- Chapyshev, S.V.; Korchagin, D.V.; Neuhaus, P.; Costa, P.; Sander, W.; Russian Academy of Sciences, Chernohgolovka, Russian Federation. Unpublished work. 2015.
- Sugisaki, K.; Toyota, K.; Sato, K.; Shiomi, D.; Kitagawa, M.; Takui, T. An ab initio MO study of heavy atom effects on the zero-field splitting tensors of high-spin nitrenes: How the spin-orbit contributions are affected. Phys. Chem. Chem. Phys. 2014, 16, 9171–9181. [Google Scholar] [CrossRef] [PubMed]
- Turek, O. 2,4,6-Trinitro-1,3,5-tristriazobenzene, a new explosive for detonators. Chim. Ind. 1931, 26, 781–794. [Google Scholar]
- Bailey, A.S.; Case, J.R. 4,6-Dinitrobenzofuroxan, nitrobenzodifuroxan, and benzotrifuroxan. A new series of complex-forming reagents for aromatic hydrocarbons. Tetrahydron 1958, 3, 113–131. [Google Scholar] [CrossRef]
- Adam, D.; Holl, G.; Klapötke, T.M. Nitrophenyl azides: A combined experimental and theoretical study. Heteroat. Chem. 1999, 10, 548–553. [Google Scholar] [CrossRef]
- Adam, D.; Karaghiosoff, K.; Klapötke, T.M.; Holl, G.; Kaiser, M. Triazidotrinitro benzene: 1,3,5-(N3)3-2,4,6-(NO2)3C6. Propellants Explos. Pyrotech. 2002, 27, 7–11. [Google Scholar] [CrossRef]
- Namba, K.; Yamashita, T. Nitroazidobenzenes. I. Synthesis of nitroazidobenzenes from nitrochlorobenzenes and sodium azide. Kogyo Kayaku Kyokaishi 1958, 19, 86–89. [Google Scholar]
- Bailey, A.S. Improvement in the preparation of benzotrifuroxan—Further examples of complex formation by this reagent. J. Chem. Soc. 1960, 4710–4712. [Google Scholar]
- Wallenfels, K.; Witzler, F.; Friedrich, K. 1,3,5-Trichlor-2,4,6-tricyan-benzol und 1,3,5-trifluor-2,4,6-tricyan-benzol. Tetrahedron 1967, 23, 1845–1855. [Google Scholar] [CrossRef]
- Becker, M.; Voss, K.; Villenger, A.; Schulz, A. An efficient route to 1,3,5-triazido-2,4,6-tricyanobenzene. Z. Naturforsh. 2012, 67b, 643–649. [Google Scholar] [CrossRef]
- Boiko, V.N.; Gogoman, I.V.; Shchupak, G.M.; Yagupol’skii, L.M. Nucleophilic substitution in aromatic compounds with fluorine-containing substituents. X. Reactions of 2,4,6-trichloro- and 2,6-dichloro-3,5-bis[(trifluoromethyl)sulfonyl]nitrobenzenes with nucleophiles. Zh. Org. Khim. 1987, 23, 2586–2591. [Google Scholar]
- Chapyshev, S.V.; Chernyak, A.V. Triazidation of 2,4,6-trifluorobenzenes. J. Fluorine Chem. 2013, 156, 303–306. [Google Scholar] [CrossRef]
- Breslow, D.S.; Marcantonio, A.F. Polyazide Crosslinking Agents. U.S. Patent 3297674, 10 January 1967. [Google Scholar]
- Kim, H.J.; Lee, W.S. Anion Receptors, and Electrolyte Using the Same. WO Patent 2007126262, 8 November 2007. [Google Scholar]
- Juriček, M. Triazole Materials: Towards Extending Aromaticity. Ph.D. Thesis, Radboud University, Nijmegen, The Netherlands, 2011; pp. 114–139. [Google Scholar]
- Chapyshev, S.V. Synthesis of 2,4,6-Triazidotoluene for Use as Photoaffinic and Crosslinking Agent. RU Patent 2430080, 27 September 2011. [Google Scholar]
- Sorm, F. The preparation of tetraazido-1,4-benzoquinone. Chem. Obz. 1939, 14, 37–39. [Google Scholar]
- Marburg, S.; Grieco, P.A. Reaction of cyclic anhydrides with sodium azide in polar aprotic media. Tetrahedron Lett. 1966, 1303–1309. [Google Scholar] [CrossRef]
- Pannell, C.D. Pyridyl azides and their Derivatives. U.S. Patent 3773774, 20 November 1973. [Google Scholar]
- Pannell, C.D. Pyridyl Azides and Derivatives. U.S. Patent 3883542, 13 May 1975. [Google Scholar]
- Nedel’ko, V.V.; Korsunskii, B.L.; Larikova, T.S.; Mikhailov, Yu. M.; Chapyshev, S.V.; Chukanov, N.V. The thermal decomposition of azidopyridines. Russ. J. Phys. Chem. B 2011, 5, 244–249. [Google Scholar] [CrossRef]
- Morse, J.G.; Kuhn, L.P. The Reaction of Hexafluorobenzene with Sodium Azide; Defense Technical Information Center: Fort Belvoir, USA, 1970; pp. 1–20. [Google Scholar]
- Liu, X.F.; Xu, W.G.; Lu, S.X. DFT theoretical study on nitrogen-rich compounds C6H(6–n)(N3)n (n = 1–6). Gaodeng Xuexiao Huaxue Xuebao 2009, 30, 1406–1409. [Google Scholar]
- Du, H.; Xu, X.; Liu, X.; Wang, F.; Zhang, J.; Gong, X. Theoretical studies on the nitro and azido derivatives of benzene. Xuaxue Xuebao 2011, 69, 269–276. [Google Scholar]
- Du, H.C.; Wang, Y. The first-principle study on the crystal structures of azido derivatives of benzene. J. Energ. Mater. 2014, 32, 162–171. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chapyshev, S.V. Six-Membered Aromatic Polyazides: Synthesis and Application. Molecules 2015, 20, 19142-19171. https://doi.org/10.3390/molecules201019142
AMA Style
Chapyshev SV. Six-Membered Aromatic Polyazides: Synthesis and Application. Molecules. 2015; 20(10):19142-19171. https://doi.org/10.3390/molecules201019142
Chicago/Turabian StyleChapyshev, Sergei V. 2015. "Six-Membered Aromatic Polyazides: Synthesis and Application" Molecules 20, no. 10: 19142-19171. https://doi.org/10.3390/molecules201019142