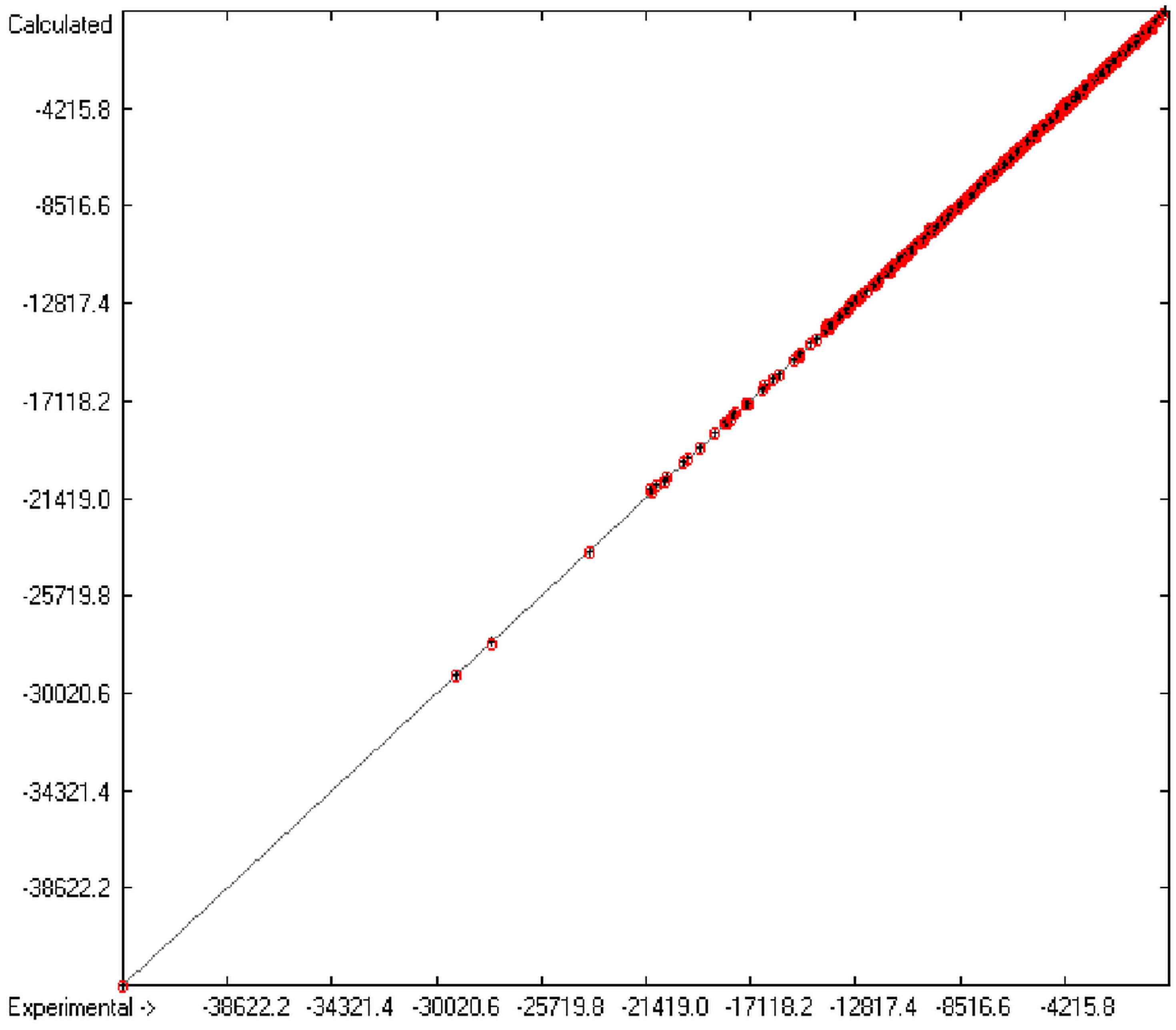
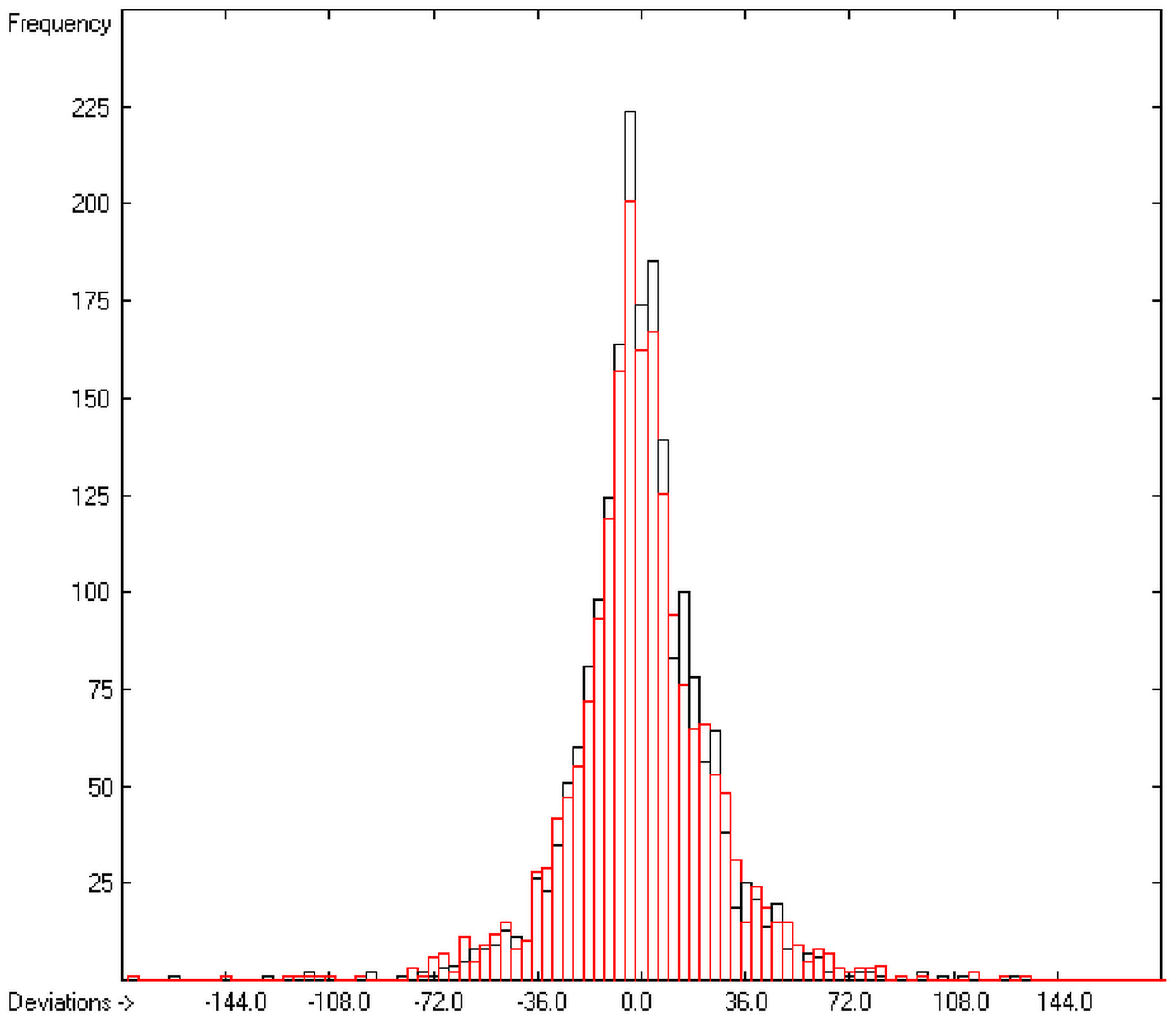
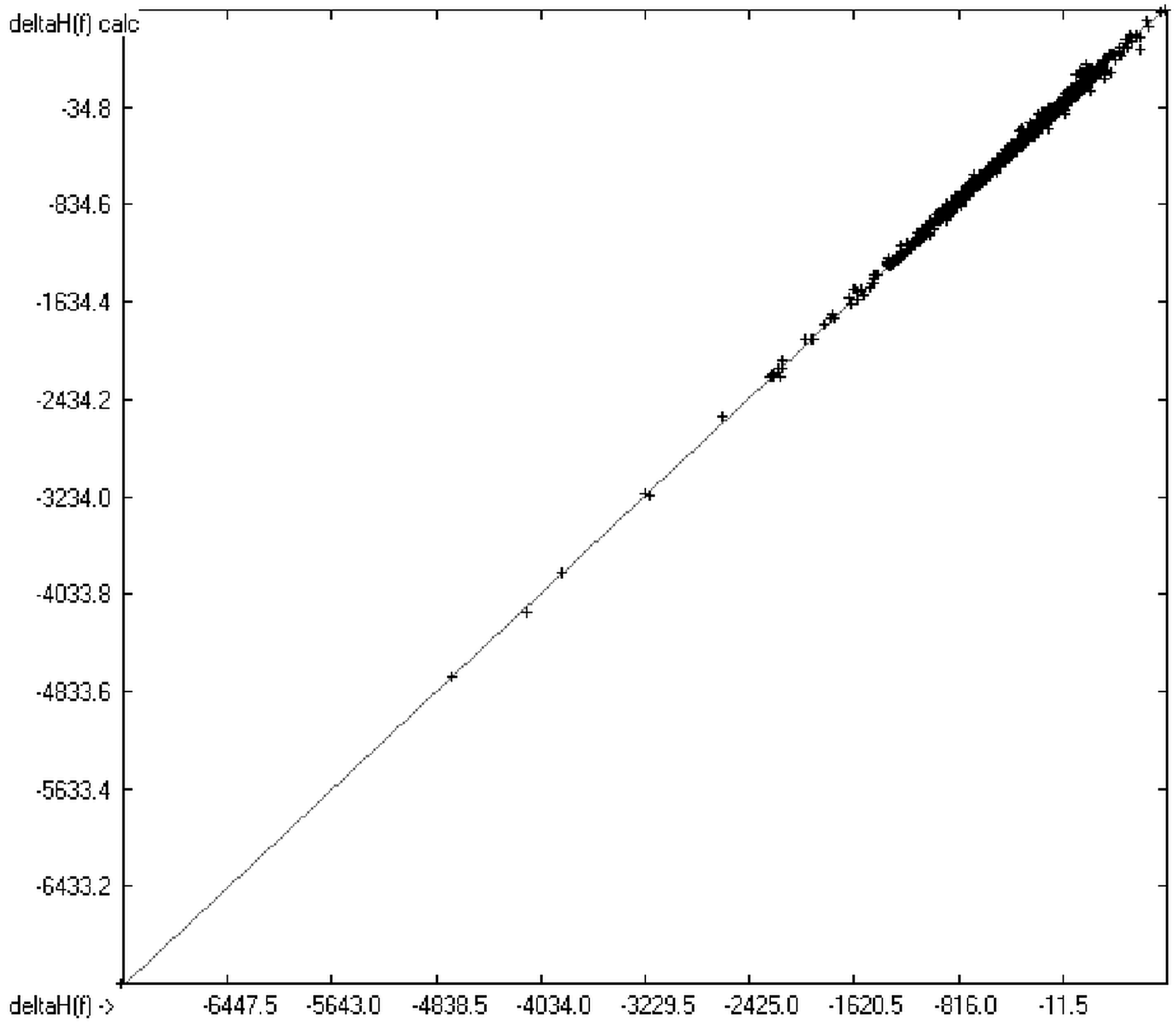
1. Introduction
The published methods for the calculation of a molecular descriptor, if based on a given set of experimental data for known molecules, usually cannot be generalized, be it that they are based on certain molecular fragment parameters such as bond energies [
1,
2,
3], only applicable for thermodynamic properties, be it that they are founded on simple atom contribution methods [
4], referring to the atoms’ properties themselves or on substituents [
5], which are also of limited viability. Hence, the goal was to find a method which would overcome all of these limitations and, beyond this, would allow the development of a general computer algorithm for the reliable calculation of as many molecular descriptors as possible which utilises the molecular structures and properties as available from a given compounds database.
The most promising approach was described by Ghose and Crippen for the calculation of the logP
O/W values [
6,
7], where the molecules are broken down into a set of up to 110 atom types, for which the hydrophobicity contribution was calculated from experimental data using the group-additivity model and least-squares technique. Analogously, the authors used this approach for the evaluation of the molar refractivity [
8]. The standard fitting procedure for the latter, however, was replaced by a quadratic programming algorithm, arguing that the “physical concept of molar refractivity is the volume of the molecule or atom, which cannot have a negative value”, which is not guaranteed if the standard procedure is applied.
Furthermore, K. J. Miller [
9,
10] applied the group additivity method for the calculation of the molecular polarizability using atomic hybrid components and atomic hybrid polarizabilites, an approach which differs from the present one in that the type of the neighbourhood atoms is ignored.
Klopman, Wang and Balthasar [
11] tried a similar method to Ghose and Crippen’s for the estimation of the aqueous solubility of organic compounds, deriving their own experience on the applicability of the group-additivity method for the calculation of the logP values. Analogously, H. Sun [
12] developed a universal group-additivity system for the prediction of logP, solubility logS, logBB (to which will be referred to later) and human intestinal absorption.
Earlier methods for the calculation of the heat of combustion have either been derived from the additivity of bond energies as suggested by Pauling [
1], Klages [
2] and Wheland [
3], or are based on various empirical relations between certain features of a series of molecules, such as the percentage of carbon [
13] or hydrogen [
14], and their heat of combustion. Further attempts [
15] have been made using group contributions, which are based on theoretical assumptions and the “heats of atomization”. Another approach has been chosen by Kharash [
16,
17] in that his method of calculation depends on the number of electrons in a molecule, multiplied by the combustion value of each electron and the result corrected for structural and functional features. There are many more publications suggesting various empirical methods for the calculation of the heat of combustion from experimental data (short abstracts of which have been given by Handrick [
18]), however, in all these cases they are limited to specific classes of molecules. In 1956, Handrick [
18] published a method which is “based on adequate experimental evidence that the molar heat of combustion of any organic homologous series bearsa straight-line relation to the number of atoms of oxygen lacking in the molecule which are required to burn the compounds to carbon dioxide, water, nitrogen, HX, and sulfur dioxide.” He called this number “molecular oxygen balance”. For the calculation he used this parameter together with a number of rules for various functional groups and applying paraffin as a base. Evidently, none of the methods described so far provides a straightforward pathway to a simple algorithm for the calculation of the heat of combustion, which is generally applicable for any kind of complexities of molecules. Nevertheless, Handrick’s observation of the rigid relation between starting material and combustion products clearly indicated that a generalizable approach for the calculation of the heat of combustion is achievable.
For the calculation of the heat of formation there are many highly sophisticated quantum-theoretical methods on the market nowadays, (see, e.g., Ohlinger
et al. [
19]). However, these methods have a few disadvantages in that they are usually progressively time-consuming and thus expensive for routine evaluations and limited to relatively small molecules. Beyond this, the accuracy of their results is by no means better than the one achieved by group-additivity methods. Therefore, the latter approach, as described in 1993 by Cohen and Benson [
20] for enthalpy-of-formation calculations, has still found its justification in that it is very fast and its parameters are based on experimental data.
A particularly difficult field in computer chemistry is the prediction of the biological activity of molecules, because in most cases their mode of action is unknown and even varies from molecule to molecule. Therefore, studies dealing with the calculation of bioactivity descriptors based on a series of experimental data usually do not, or only summarily, discuss the reason as to why a certain set of molecular parameters has been applied. Typical examples are the descriptors toxicity and the blood-brain barrier described in the following.
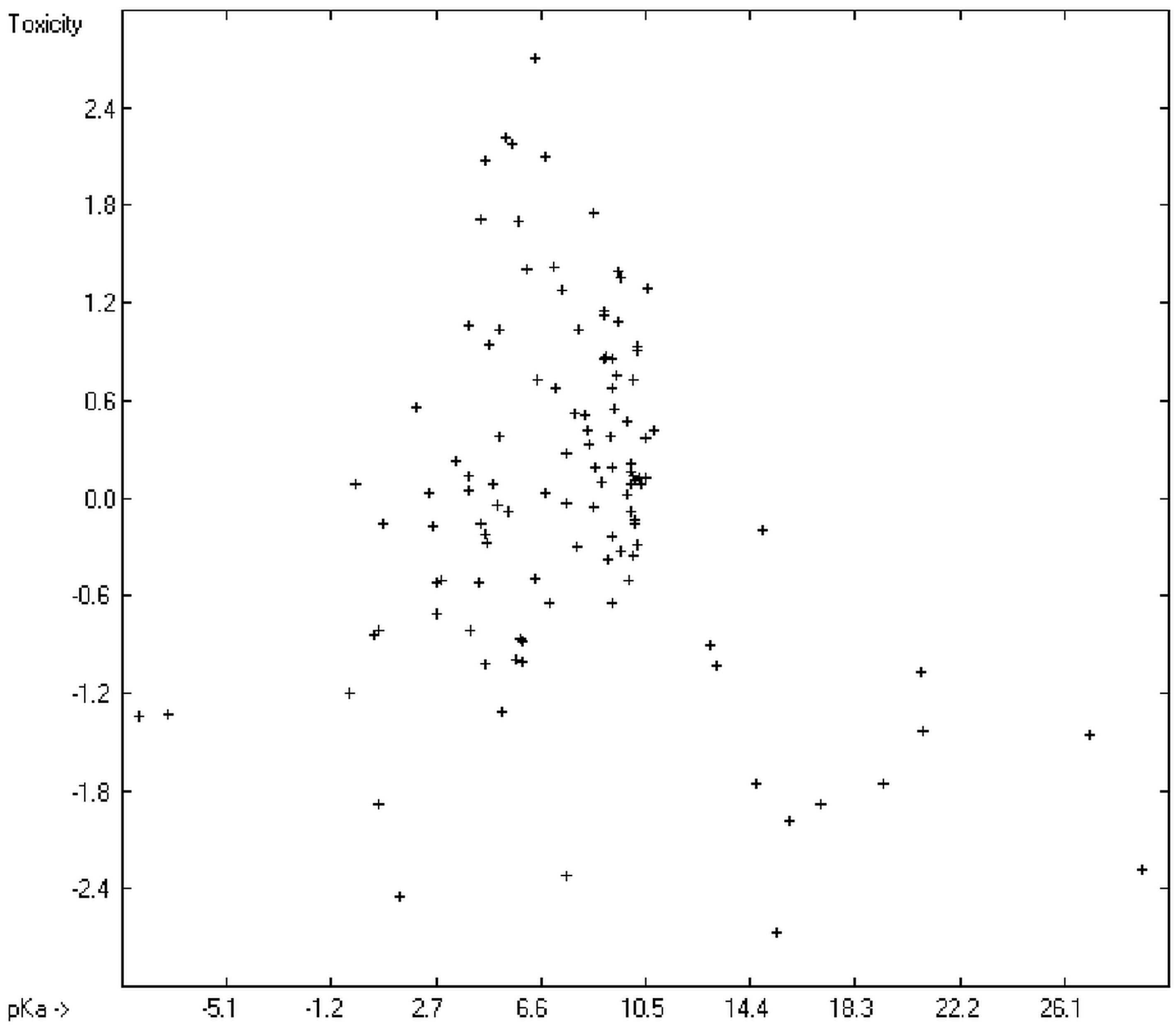
Prediction of the toxicity of organic compounds in water has become another important area for QSAR studies. In most cases the experimental data for a series of commonly used compounds have been determined by their effects on the protozoan
Tetrahymena pyriformis. Various methods have been applied to predict this descriptor: recently, Schultz [
21] derived the toxicity of a series of substituted benzenes from the hydrophobicity, determined as logP
O/W, plus the electrophilic reactivity, quantified by the maximum superdelocalizability S
max; Duchowicz
et al. [
22] filtered out seven parameters from a set of 1338 topological, geometrical and electronic molecular descriptors, feeding them into an artificial neural network to evaluate the toxicity of 250 phenol derivatives; similarly, Melagraki
et al. [
23] used the hydrophobicity (logP
O/W), the acidity constant (pKa), the HOMO and LUMO orbital energies and the hydrogen bond donor number (N
hdon) and applying an ANN method based on the radial basis function architecture for the prediction of the toxicity of 221 phenols and compared the data to standard multiple linear regression models; Ellison [
24] reduced the number of parameters to the hydrophobicity logP
O/W itself plus a constant to derive the toxicity of alcohols, esters, ketones and cyanides, defining for each of these groups a structural range of applicability; density functional theory as well as other semiempirical Hamiltonian methods have been used by Pasha [
25] to evaluate—besides the molecular weight—the hardness, chemical potential, total energy and electrophilic index, which are then introduced into a multiple linear regression analysis and various other regression calculations for the evaluation of the toxicity of 50 phenol derivatives. A preliminary attempt, induced by Ellison’s work, to directly correlate logP
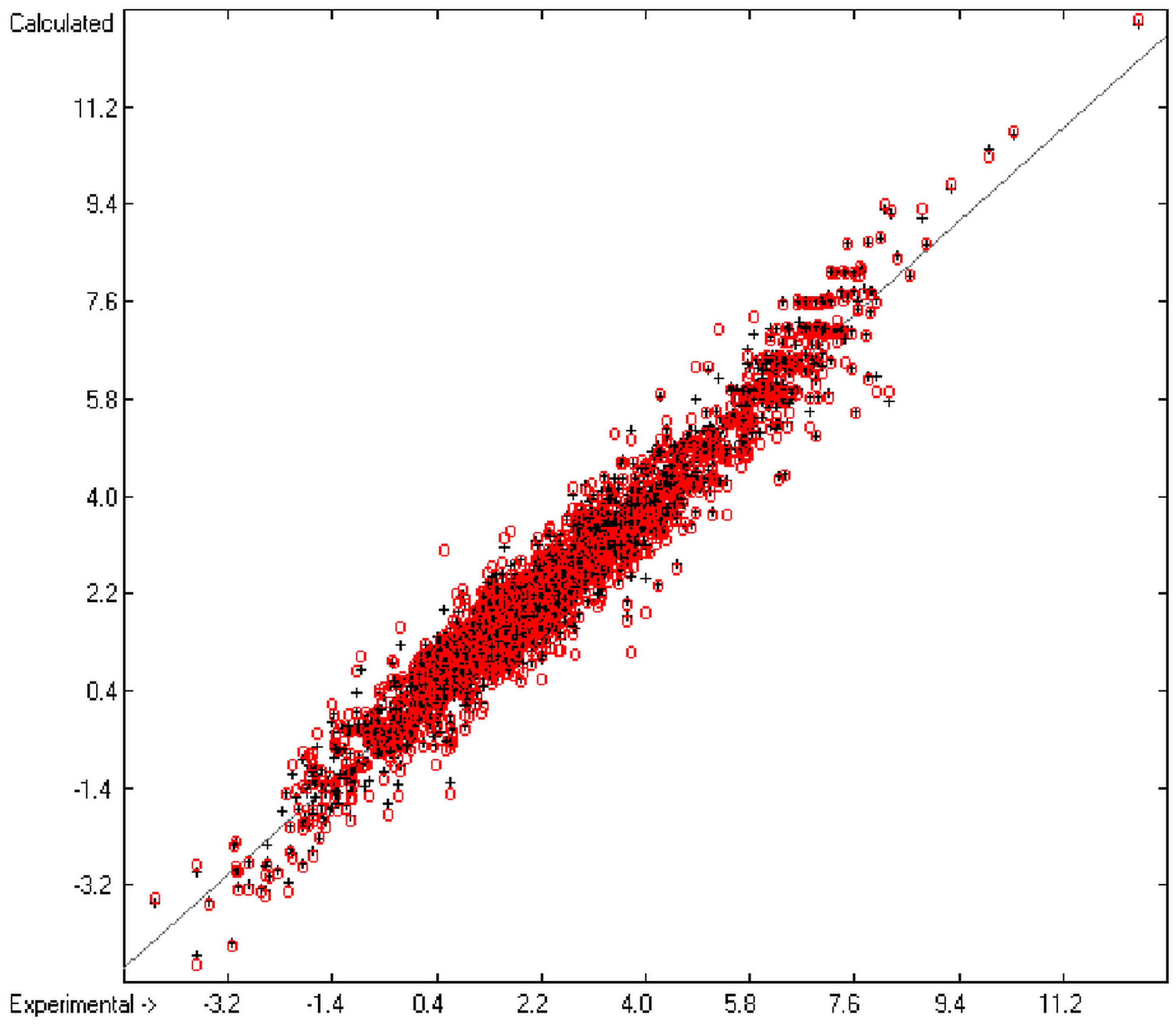
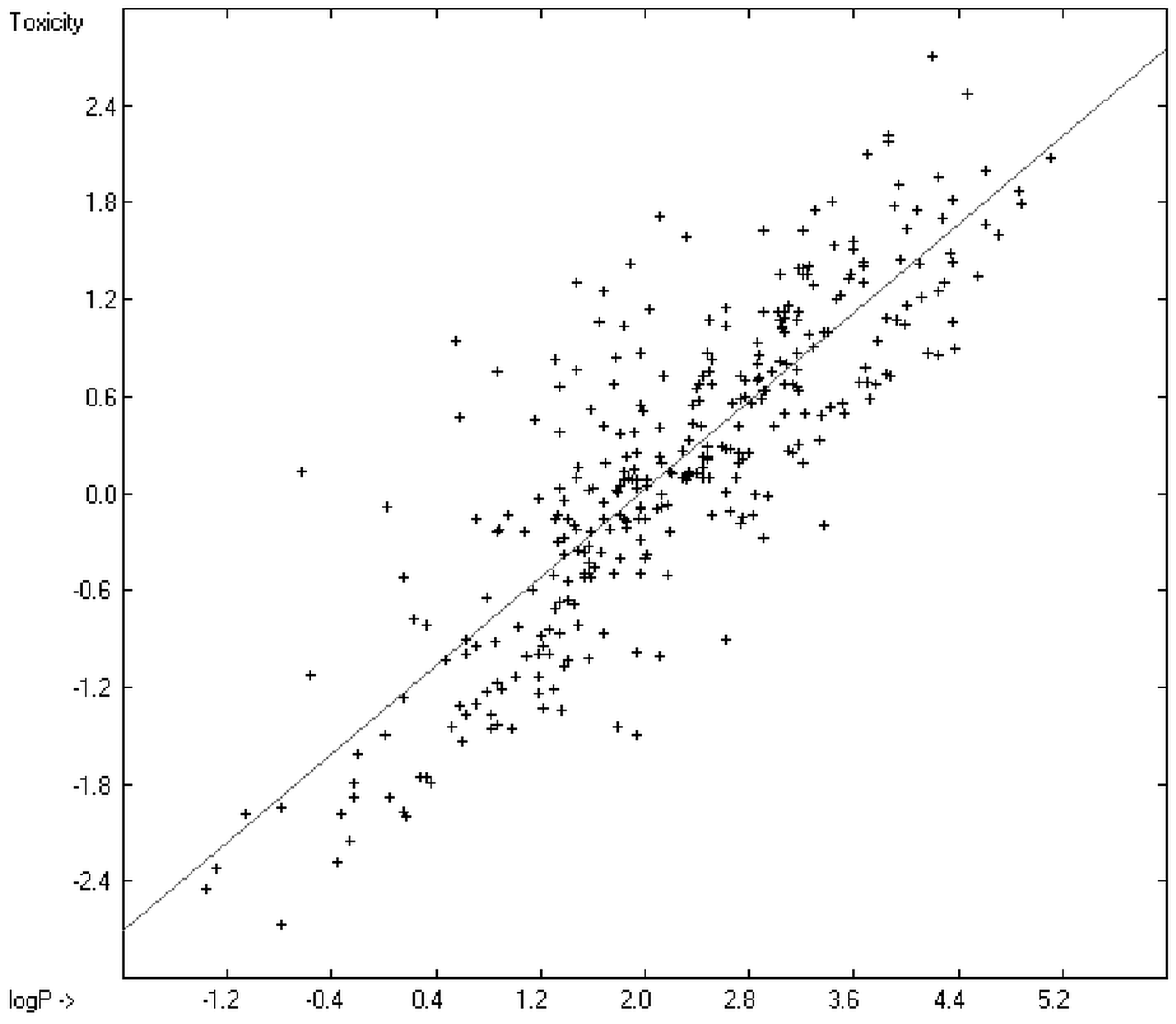
O/W with toxicology data of 335 compounds for which both experimental data are known and which encompass the whole range of chemical structures mentioned above yielded a correlation coefficient R
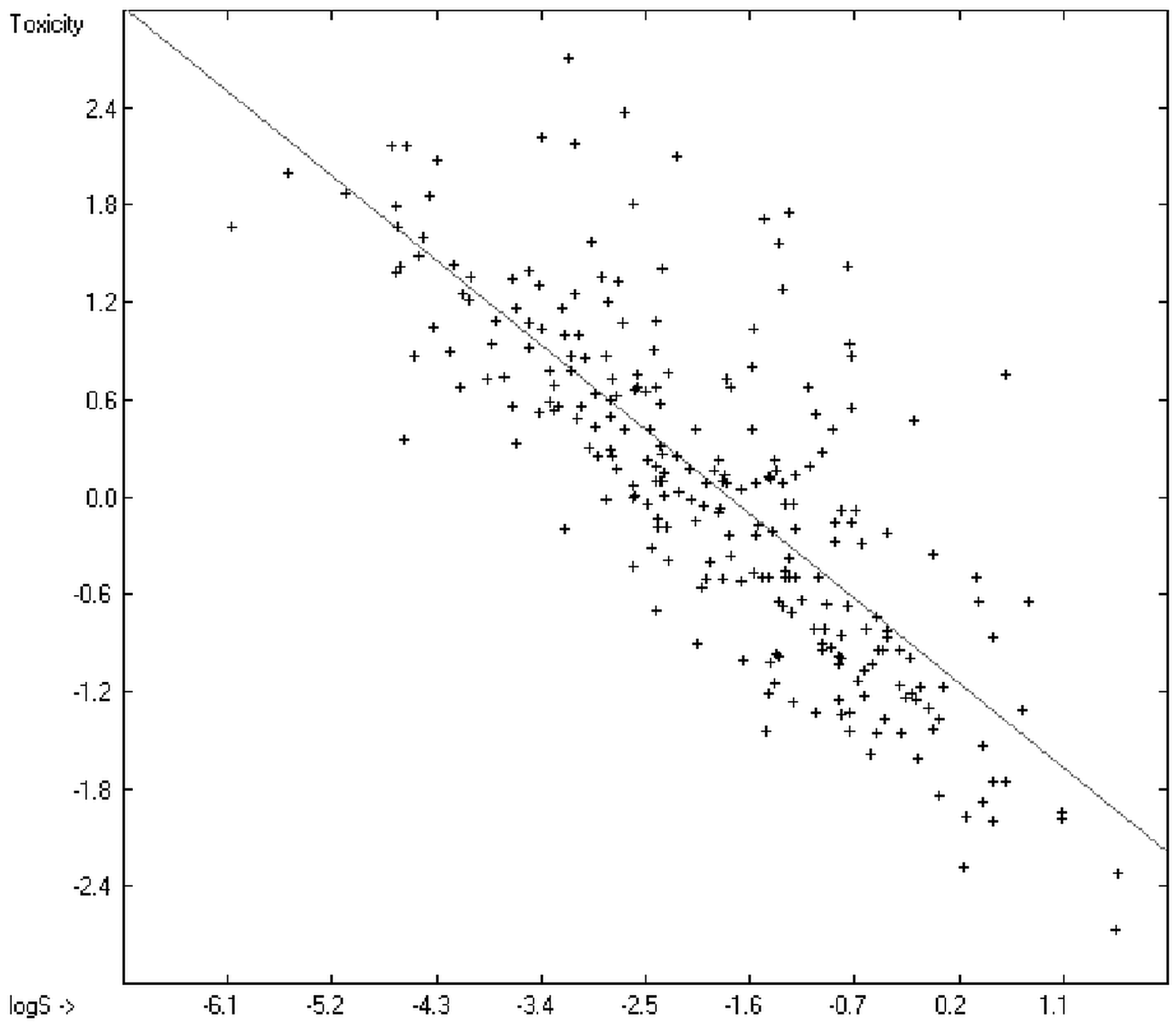
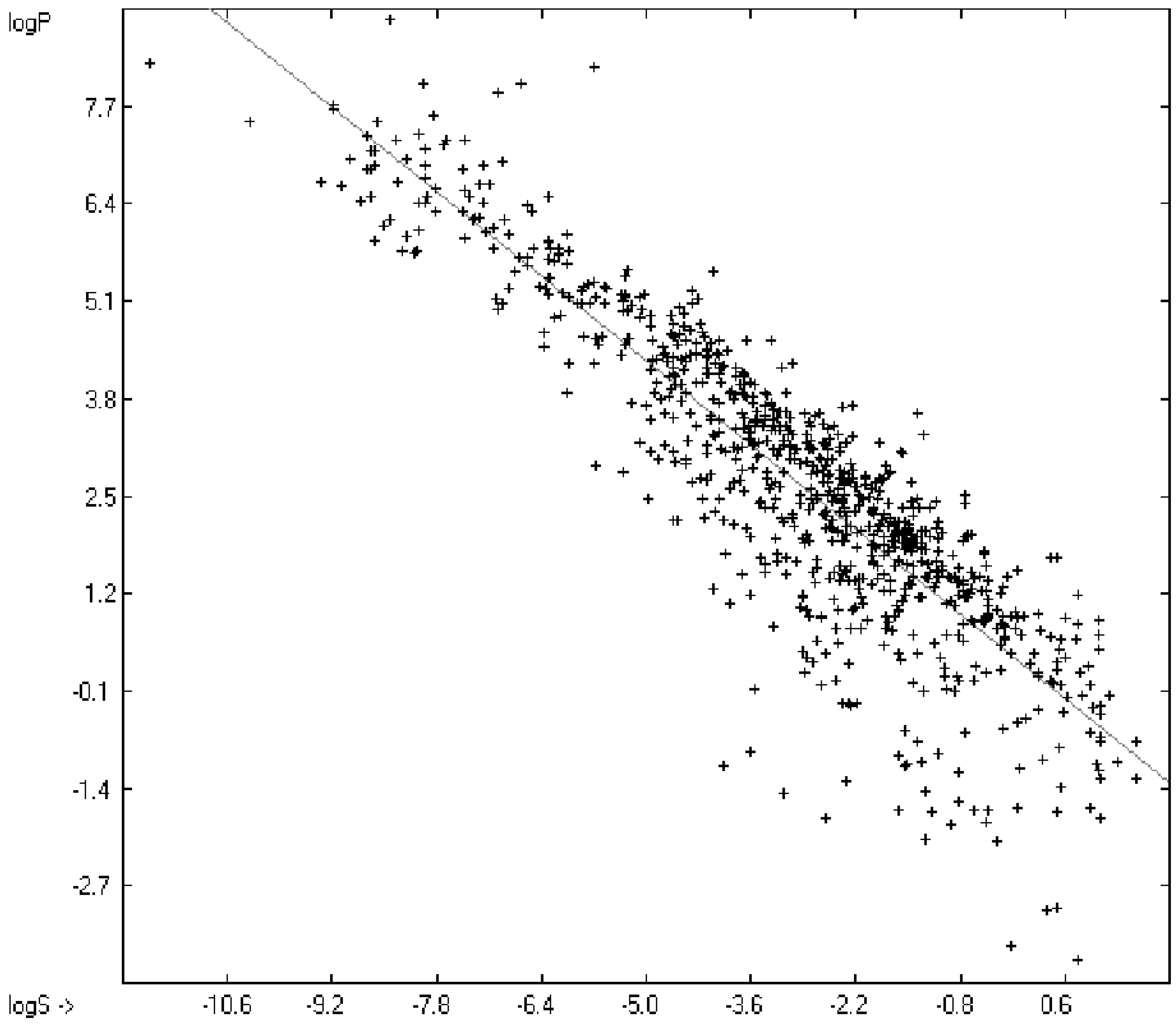
2 of 0.7043 (the correlation diagram of which is shown further down). This encouraging result gave reason to try to apply the group-contribution method itself for the calculation of a compound’s toxicology value, based on the experimental data of the entire spectrum of chemical structures as far as their experimental data were available.
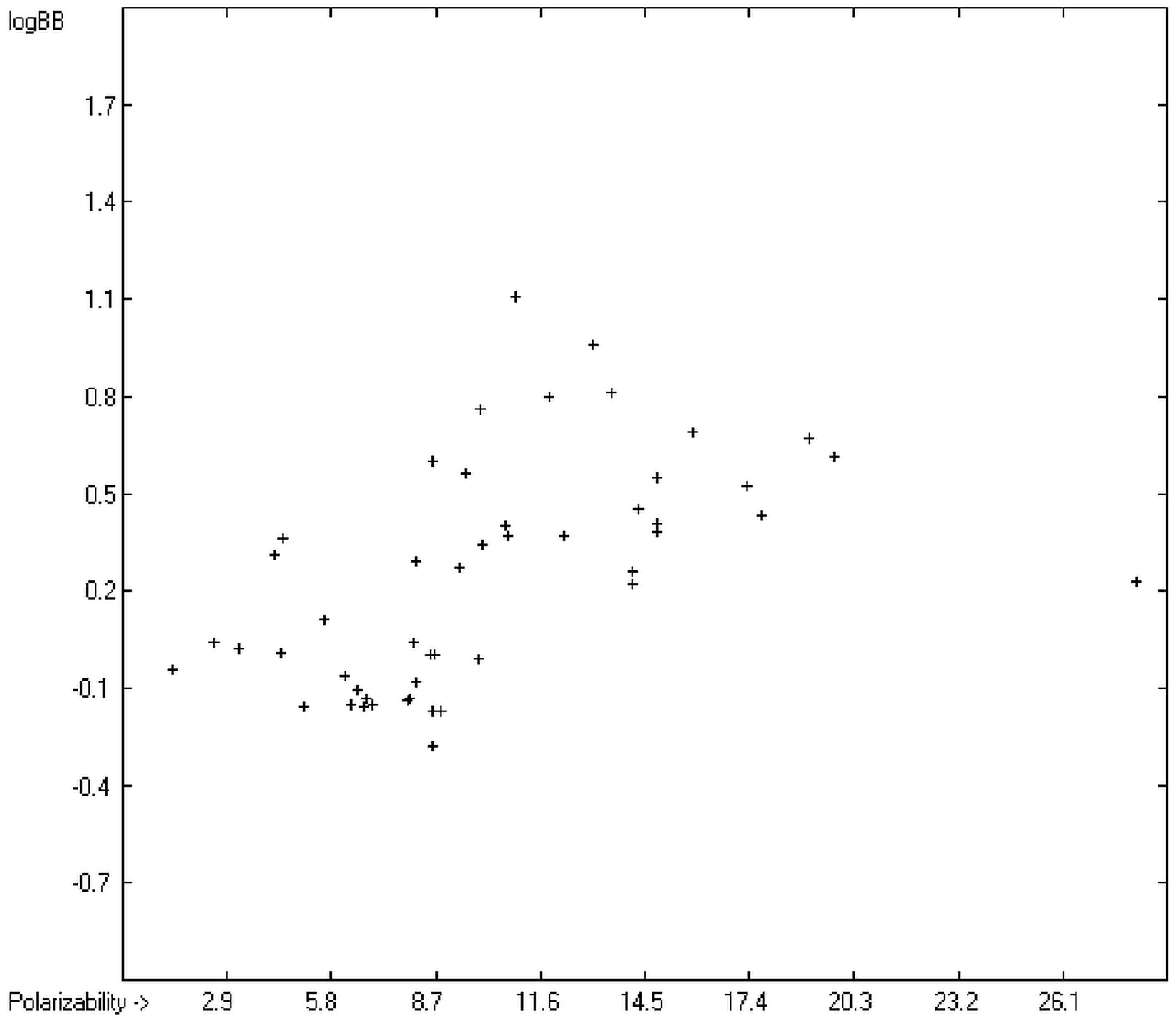
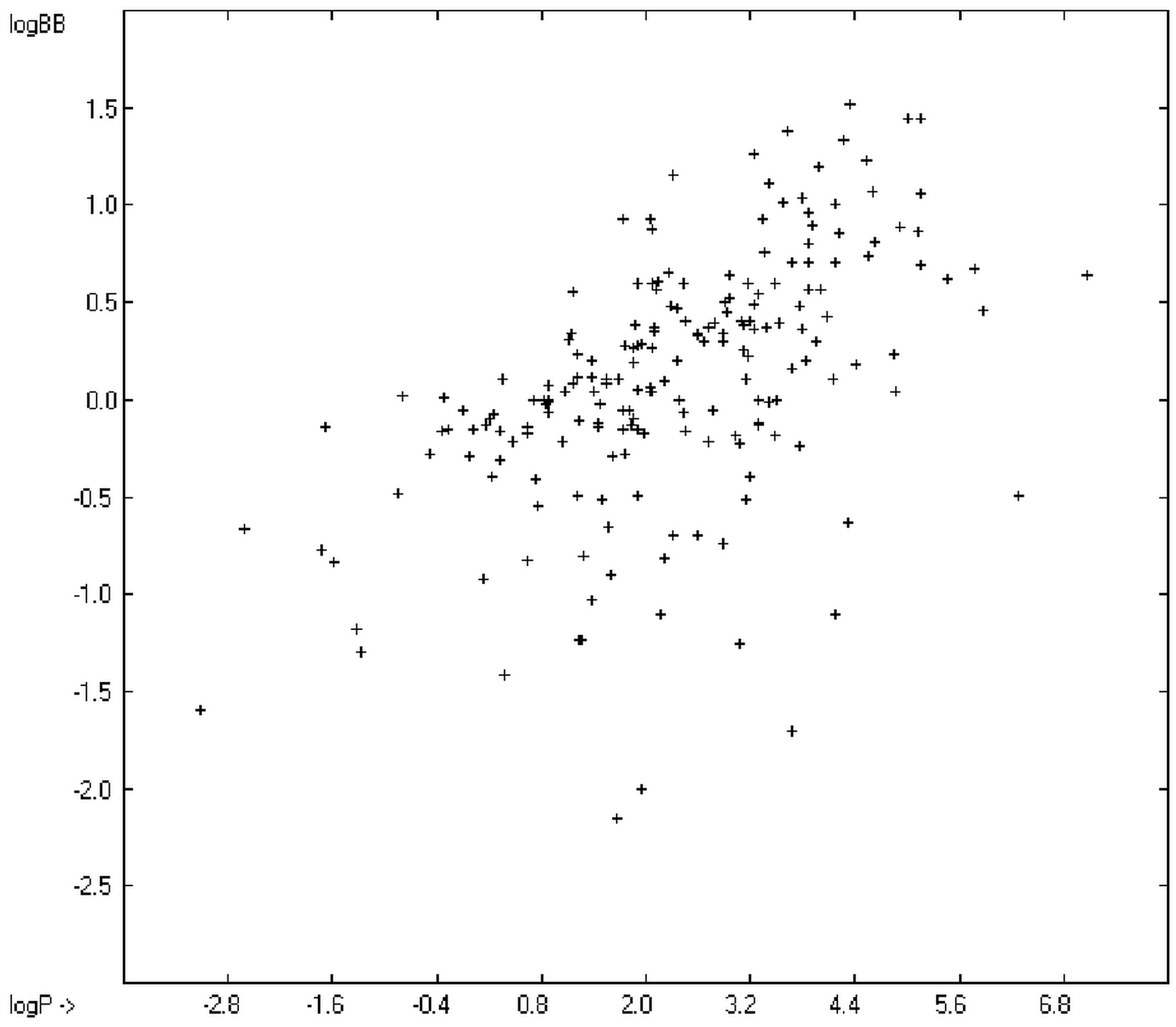
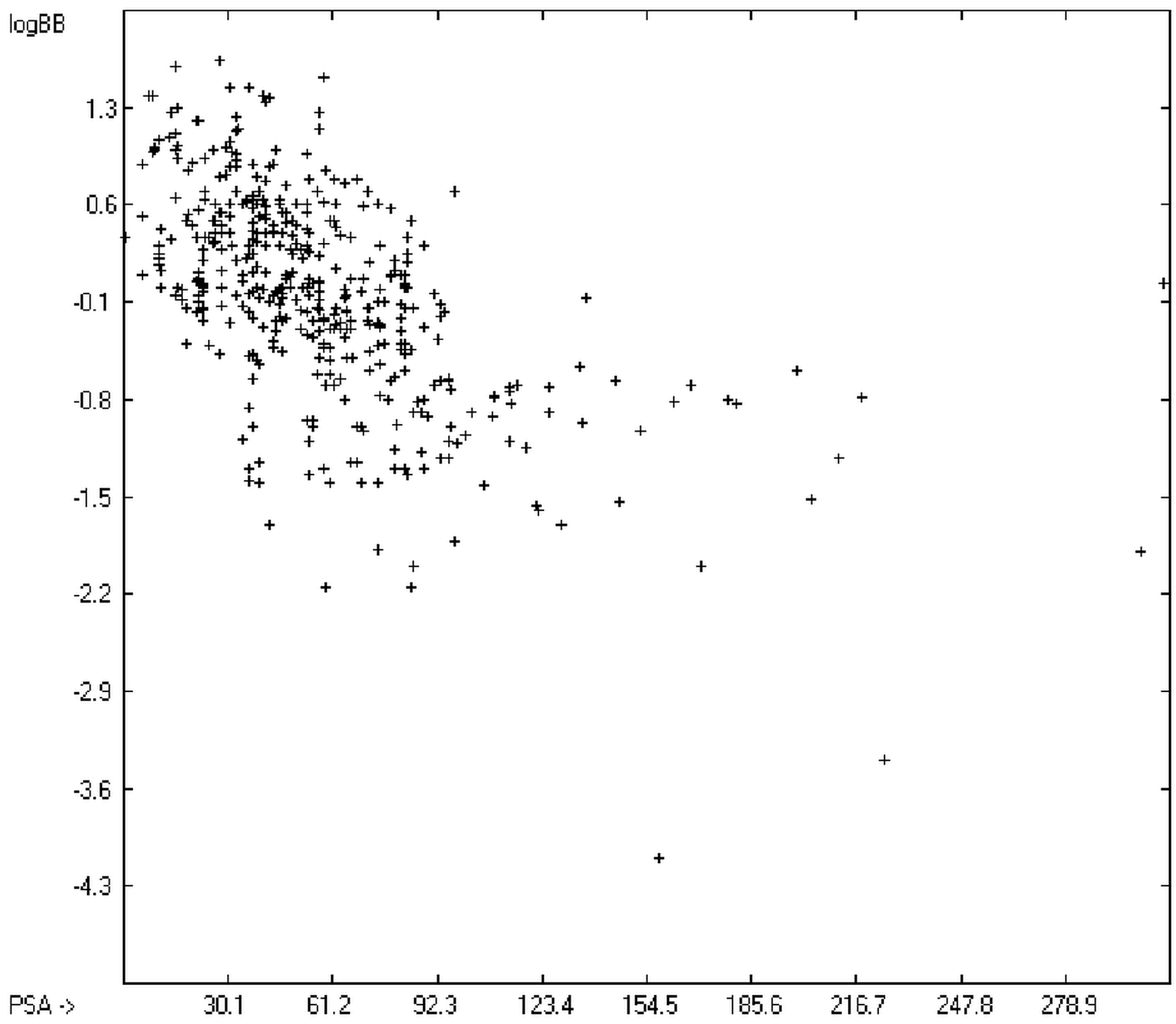
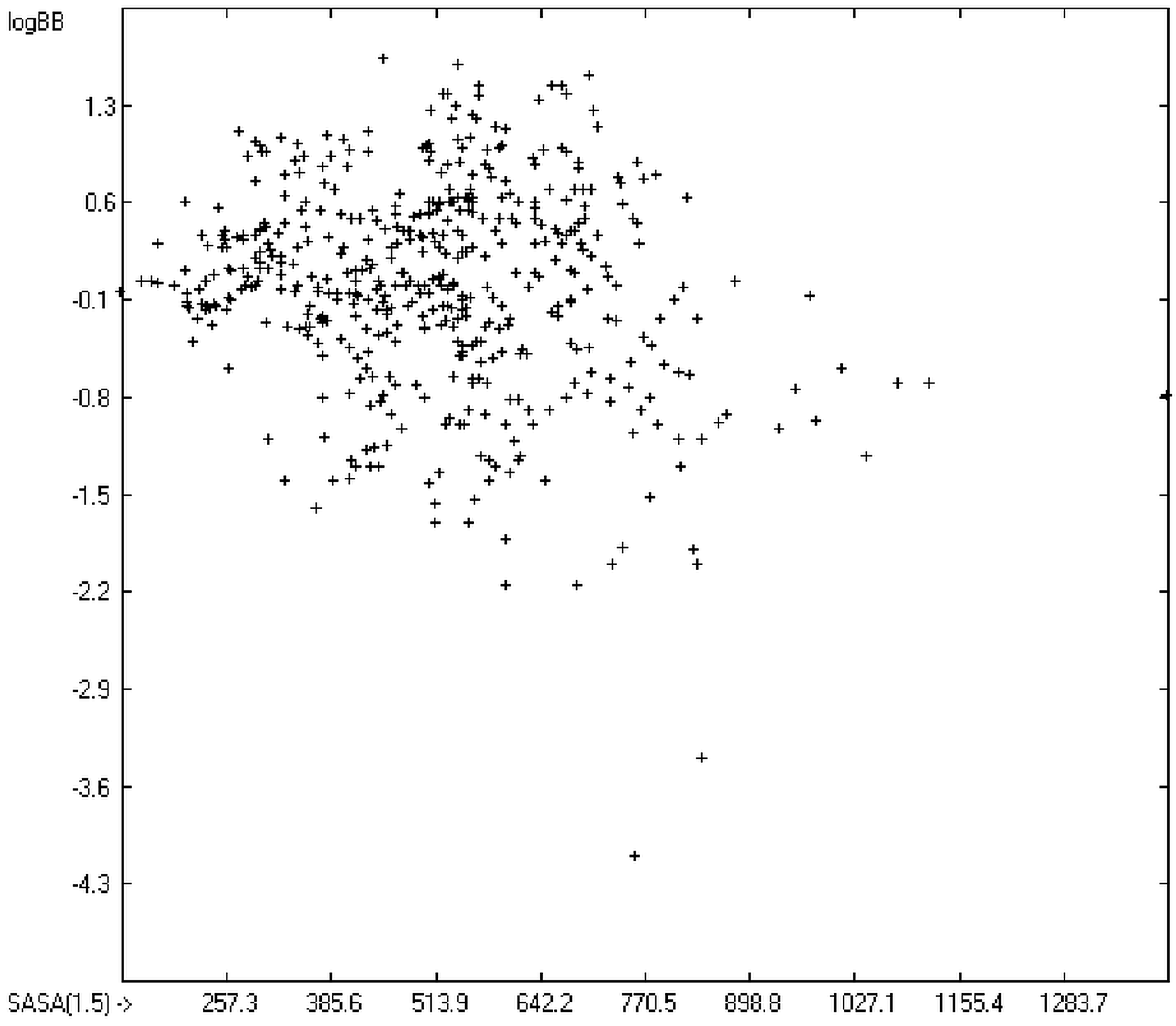
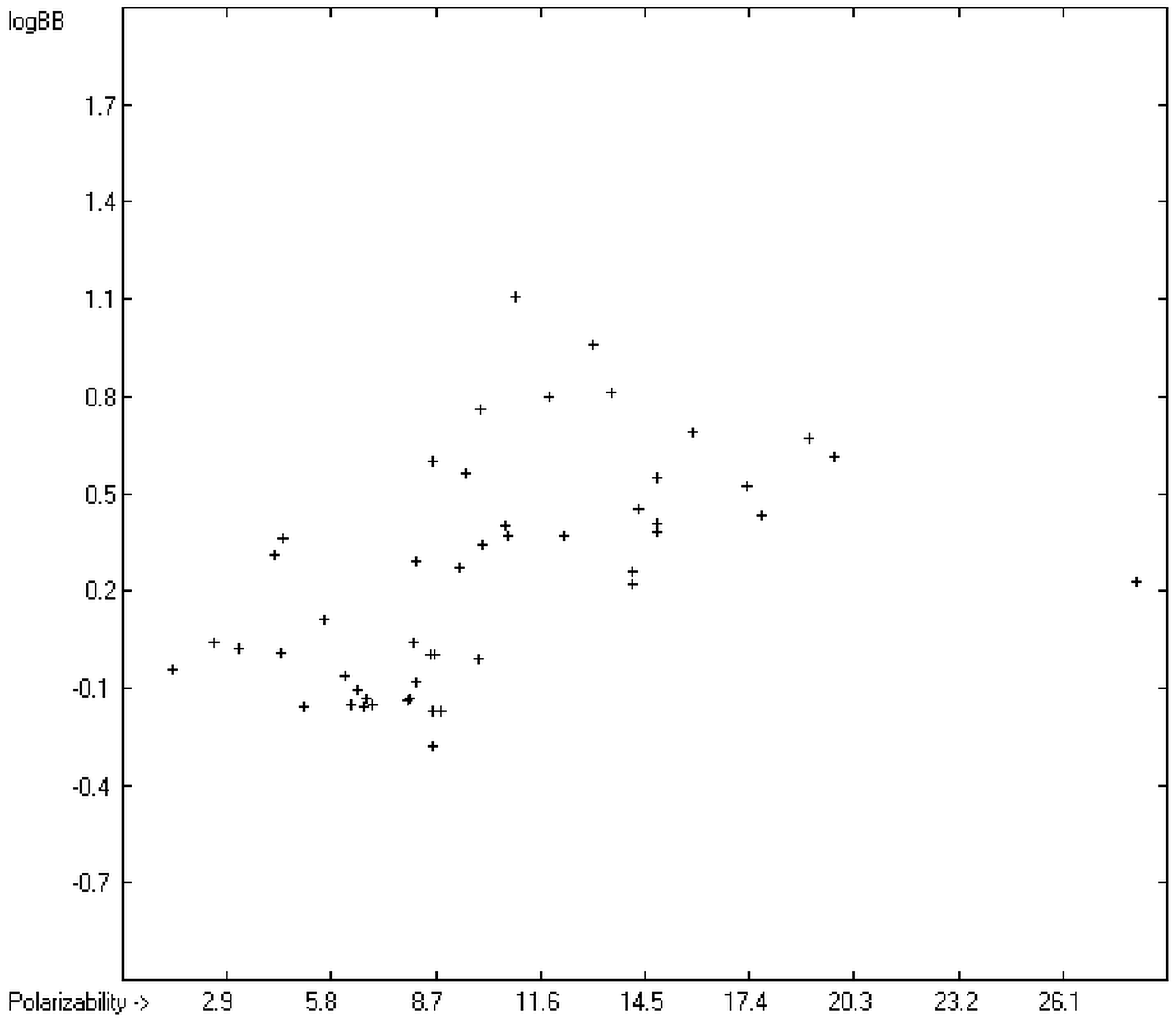
The blood-brain barrier (BBB) is a very efficient cellular system to protect the brain from unwanted content in the surrounding blood stream. In most cases, this may be desirable to prevent CNS-related side-effects of drugs. Logically, however, this barrier also tries to prevent intrusion of therapeutic chemicals for treatment of cerebral diseases. Fortunately, at least in the therapeutic sense, this barrier is not completely insurmountable, but the experimental determination of the barrier penetration of a new drug is time-consuming and expensive. Therefore, many attempts to predict the degree of BBB penetration, defined as the steady-state brain/blood distribution ratio logBB, have been published: Luco [
26] used topological descriptors in partial least-squares analysis for the modeling logBB of 61 compounds; Fu
et al. [
27] based their model on the molecular volume and polar surface area of 79 compounds; the electrotopological states of the constituting atoms of 106 molecules was used by Rose
et al. [
28]. Thermodynamic calculations, such as the evaluation of the free solvation energy by Keserü and Molnar [
29] as well as molecular dynamics simulations, e.g., by Carpenter
et al. [
30], have been applied to predict logBB, based on a very limited number of examples. Genetic algorithms have been used by Hou and Xu [
31] on a series of 27 descriptors calculated from 96 structurally diverse compounds in order to select the statistically most significant groups of linear models with up to three or four descriptors. They concluded from the best-fitting models that logP and the partial negative solvent-accessible surface area play a crucial role in the BBB permeability. Similarly, Chen
et al. [
32] also observed the importance of the polar surface area and logP, using an artificial neural network model. On the other hand, P. Garg and J. Verma [
33], also based on an ANN model, concluded that the order of importance in the evaluation of the BBB permeability is the molecular weight, followed by the polar surface area, logP, the number of H-bond acceptors and the number of H-bond donors. Quantum chemical descriptors (dipole moment, polarizability, equalized molecular electronegativity, molecular hardness, molecular softness, molecular electrophilicity, charges, charge separations, covalent H-bond acidity and basicity as well as electrostatic potential derived properties), calculated by an ab initio method, have been put together by van Damme
et al. [
34] with a series of classical descriptors encompassing logP, molecular weight, polar surface area and further structure- and shape-related properties in a model of finally eight parameters. Again, it turned out that loP and the polar surface area, besides the Mulliken charge-related descriptors, seem to be essential attributes of the model to reproduce the logBB data best, which they ascribe to the assumption that “logBB is a function of the lipophilicity and electronic properties of the molecule” [
34]. Several further authors carried out logBB calculations based on the two parameters logP and polar surface area of the molecules, either on these parameters alone such as Clark [
35] or together with the polarizabilty (De Sä
et al. [
36]), or including the number of acidic or basic atoms (Vilar
et al. [
37]), or only logP together with the molecular mass or the isolated atomic energy (Bujak
et al. [
38]). Interestingly however, Lanevskij
et al. [
39] observed that there is no direct correlation between logP
O/W and logBB at all (a fact which is confirmed in the present work), indicating “that logBB is not a measure of lipophilicity-driven BBB permeability” [
39]. They found that replacement of the experimental logBB values by the ratios of total brain to unbound plasma concentrations (which meant to correct logBB by the amount of protein binding in the plasma) considerably improved correlation with logP. Sun [
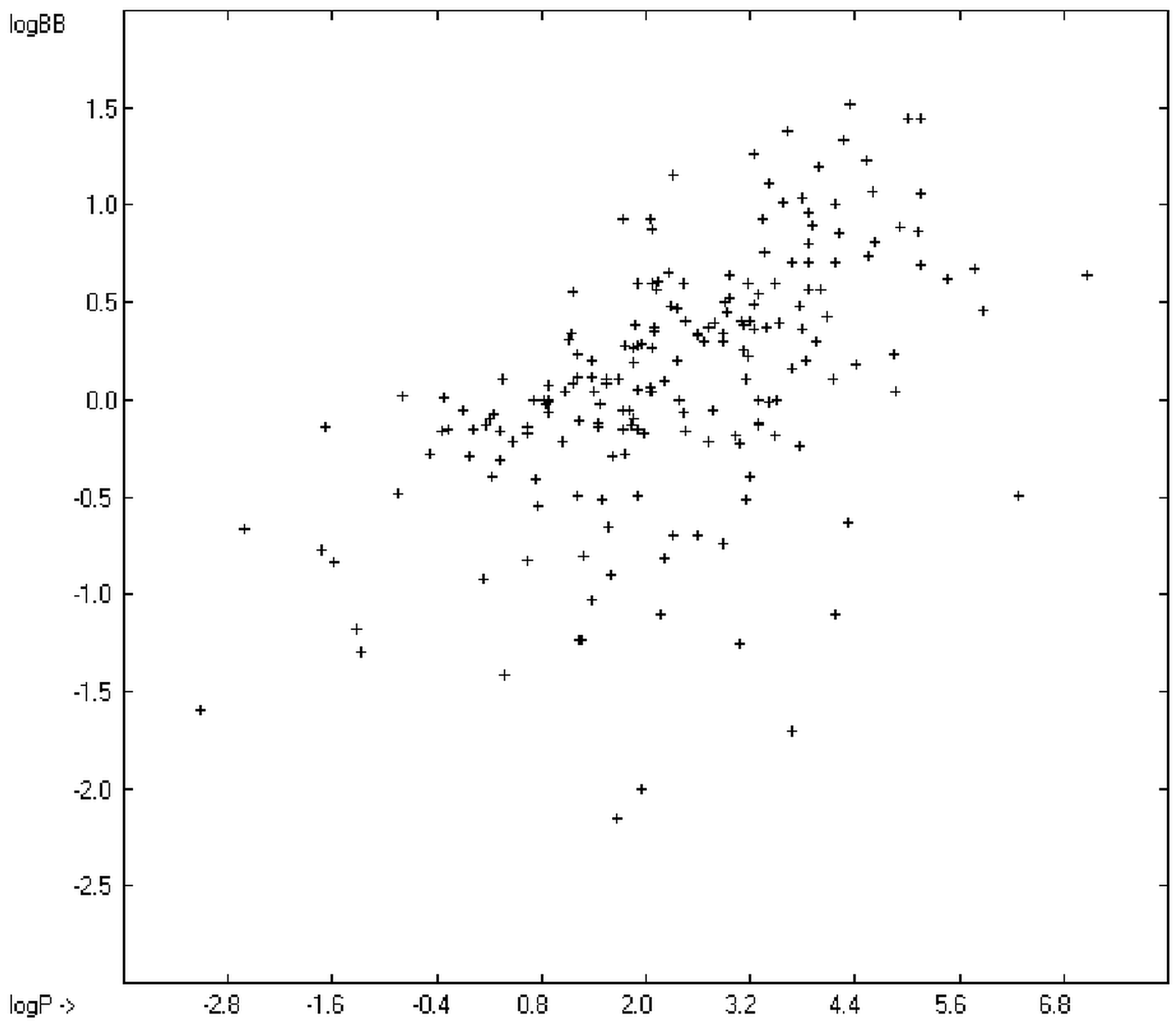
12] tried a direct approach to evaluate logBB by applying a number of atom type descriptors, which is very similar to the present group-additivity method, characterizing 57 compounds, representing a limited structural diversification set.
In view of the many different—successful but mostly elaborate—attempts to reliably evaluate all the molecular descriptors mentioned above it seemed unrealistic to propose a general and simple computer algorithm which would be able to calculate all the descriptors at once. However, as will be shown here, the present algorithm lifts all the limitations discussed above and is not only suitable for the calculation of thermodynamic (heat of combustion and—indirectly-formation), solubility-related (logP and logS), optical (molar refractivity), electrical (molecular polarizability) as well as biological (toxicology and potentially CNS-related) properties of a molecule at once, but also delivers reliable results and, beyond this, has the advantage of being easily extendable to compounds with structural features for which as yet no parameters are known without the need to readjust the computer algorithm.
2. General Procedure
The general algorithm for the calculation of the mentioned molecular descriptors is founded on the principle of atom group contributions in analogy to the method described by Ghose and Crippen [
6,
7], extended in some cases by a few specific terms which will be outlined later on.
2.1. Definition of the Atom Groups
The present calculation procedure takes advantage of a knowledge database of presently more than 20,000 compounds, stored in geometry-optimized three-dimensional form, wherein—fulfilling the first requirement—for a certain number of molecules the experimental values for the molecular descriptors considered here are known and included in the database, each by a specific term known to the computer algorithm.
The second requirement for the calculation of the contributions of the atom groups is their definition. Since in the present approach, which should be equally applicable for the calculation of various molecular descriptors which have nothing in common but the molecular structure as a whole, no prior assumption was allowed as to the method of partitioning the molecule into its fragments. Therefore, in a potentially naive attempt, the molecular structures are broken down into their lowest-possible but still distinguishable fragments,
i.e., into the constituting atoms and their immediate neighbourhood as was suggested by Cohen and Benson [
20,
40]. Under this prerequisite, in principle, the definition of the group terms and their setup in a table could have been taken over by a computer algorithm, which would make use of the structural information of all the molecules in the database for which the requested experimental data are known, but in order to maintain a certain logic in the table order, the group terms have been generated manually and set up in a general table, which then should serve as a “mother” table for the individual parameters tables.
The above-mentioned fragmentation principle made it easy to define the atom groups in a standardized way enabling it to be set up into a programmable algorithm: each group consists of a central atom and its immediate neighbour atoms. The central atom, called “backbone atom”, is bound to at least two other atoms and is characterized by its atom name, its atom type being defined by either its orbital hybridization or bond type or its number of bonds, where required for distinction, and by its charge, if not zero. The neighbour atoms are collected in a term which lists all the neighbours following the order H > B > C > N > O > S > P > Si > F > Cl > Br > I and for each encompasses—in this order—the bond type of its bond with the backbone atom (if not single), its atom name and its number of occurrences (if >1). (For better readability of a neighbours term containing iodine its symbol is written as J.) Additionally, if the total net charge of the neighbour atoms is non-zero, the charge is appended to the neigbour term by a “(+)” or “(−)”, respectively.
Finally, for N with three single bonds (atom type “N sp3”) and O and S with two single bonds (atom types “O” and “S2”, respectively), where neighbour atoms are part of a conjugated moiety, the neighbour term is further supplemented by the terms “(pi)”, “(2pi)” or “(3pi)”, respectively. This is to take account of the increased strength of a group’s bonds due to the π-orbital conjugation of the backbone atom’s lone-pair electrons with conjugated neighbour moieties.
Hence, an atom group is uniquely defined by the term for the backbone-atom type and the term for its neighbours, which is easily interpretable as shown in the examples
Table 1. For clarity the backbone atom is pronounced in the “meaning” column in boldface.
Table 1.
Group examples and their meaning.
Table 1.
Group examples and their meaning.
| Atom Type | Neighbours | Meaning | Atom Type | Neighbours | Meaning |
|---|
| C sp3 | H3C | C–CH3 | N sp3 | H2C | C–NH2 |
| C sp3 | H3N | N–CH3 | N sp3 | H2C(pi) | C–N*H2 |
| C sp3 | H2C2 | C–CH2–C | N sp3 | C2N(2pi) | C–N*(N)–C |
| C sp3 | H2CO | C–CH2–O | N sp2 | H=C | C=NH |
| C sp3 | HC3 | C–CH(C)–C | N sp2 | C=N | N=N–C |
| C sp3 | HC2Cl | C–CH(Cl)–C | N sp2 | =CO | C=N–O |
| C sp3 | HCO2 | C–CH(O)–O | N(+) sp3 | H3C | C–NH3+ |
| C sp3 | C3N | C–C(C)2–N | N(+) sp3 | H2C2 | C–NH2+–C |
| C sp3 | C2F2 | C–CF2–C | N(+) sp2 | CO=O(−) | O=N+(O−)–C |
| C sp2 | H2=C | C=CH2 | N aromatic | :C2 | C:N:C |
| C sp2 | HC=C | C=CH–C | N(+) sp | =N2(−) | N=N+=N(−) |
| C sp2 | HC=N | N=CH–C | O | HC | C–OH |
| C sp2 | H=CN | C=CH–N | O | HC(pi) | C–O*H |
| C sp2 | HN=O | O=CH–N | O | Si2 | Si–O–Si |
| C sp2 | C2=O | O=C(C)–C | P3 | C3 | C–P(C)–C |
| C sp2 | C=CN | C=C(C)–N | P4 | CO2=O | O=P(O2)–C |
| C sp2 | =CNO | C=C(N)–O | P4 | N2O=O | O=P(O)(N)–N |
| C sp2 | N=NO | N=C(N)–O | S2 | HC(pi) | C–S*H |
| C sp2 | NO=O | O=C(N)–O | S2 | CS | C–S–S |
| C aromatic | H:C2 a | C:CH:C | S4 | CO=O2 | C–S(=O)2–O |
| C aromatic | H:C:N | C:CH:N | S4 | O2=O | O–S(=O)–O |
| C aromatic | :CN:N | C:C(N):N | Si | C2Cl2 | C–SiCl2–C |
| C sp | H#C b | C#CH | Si | OCl3 | O–SiCl3 |
| C sp | C#N | N#C–C | | | |
| C sp | #CN | C#C–N | | | |
| C sp | =C2 | C=C=C | | | |
| C sp | =C=O | C=C=O | | | |
It is evident that this radical break down of molecules into the atom groups as shown does not reflect any knowledge about the molecules’ three-dimensional structure. Yet, it is well known that structural peculiarities such as buttressing effects, ring strains, gauche bond interactions or internal hydrogen bonds have a distinct influence on the values of the molecules’ heat of formation and combustion.
In the case of the calculation of logP values, Klopman
et al. [
41], using a different group-additivity method, found that for pure saturated and unsaturated hydrocarbons inclusion of a correction factor per carbon atom clearly improved conformance with experiments. They also added a correction parameter for non-branched (CH
2)
n chains on (hetero)aromatics with a polar end group X where n is greater than 1. Although the atom group fragmentation method in the present case is more detailed, the suggested correction factors have been included here as well (and in the case of the non-branched CH
2 chains without restrictions). They indeed caused some improvement as will be outlined later.
In order to take account of these specific steric interactions and hydrophobic effects, the table of atom groups has been extended by some groups for which the terms “atom type” and “neighbours” are not rigorously applicable, but which are treated in the calculation of the group contributions in exactly the same way as ordinary atom groups. In
Table 2, the definitions of these special groups and their explanation are given.
Table 2.
Special Groups and their Meaning.
Table 2.
Special Groups and their Meaning.
| Atom Type | Neighbours | Meaning |
|---|
| H | H Acceptor | Intramolecular H bridge between acidic H (on O, N or S) and basic acceptor (O, N or F) |
| H | H | Intramolecular H–H distance <2 Angstroms |
| H | H | Intramolecular H–H distance 2–2.3 Angstroms |
| Angle60 | | Bond angle <60 deg |
| Angle90 | | Bond angle between 60 and 90 deg |
| Angle102 | | Bond angle between 90 and 102 deg |
| Alkane | No of C atoms | Correction factor per carbon atom in pure alkanes |
| Unsaturated HC | No of C atoms | Correction factor per carbon atom in pure aromatics, olefins and alkynes |
| X(CH2)n | No of CH2 groups | Correction factor per CH2 group in CH2 chains with end group X = CH3, NH2, OH, SH or halogen |
The present detailed fragmentation of the molecules clearly bears positive and negative consequences. On the positive side lies the stronger “individualization” of the atom groups leading to better conformance with experimental data. This is particularly evident when dealing with molecules which can acquire various prototropic forms, e.g., ordinary amino acids, the equilibrium of which usually lies on the zwitterionic side. This paper will show that the differences between the calculated and experimental values of certain properties immediately answer the question concerning these equilibria. A second advantage of the present fragmentation method is the easy extendability of the number of atom groups if required for the inclusion of further molecules with known experimental descriptors data without the need to alter the computer algorithm. In fact, it is the applied parameters table itself instructing the computer program which atomic and special groups are to be taken into account for the calculations of the contributions and subsequently the descriptor data.
The negative side of this detailed molecule break-down, however, already shows up at the time of evaluating the group-contribution values: the number of molecules carrying a specific atom group can decrease to figures, which are no longer representative to confirm the final contribution value. In the extreme case of only one molecule for a given atom group, its calculated contribution value is merely the “last” summand to exactly fit the experimental descriptor value. The present work took account of this in that in all the consecutive calculations of molecular descriptors only atom groups were considered which were represented by at least three independent training molecules.
An obvious consequence of these conditions is apparent when entering a new molecule for which not all of the atom groups it contains are found—or if found are represented by less than three training molecules—in the parameters table. In that case the corresponding molecular descriptor can simply not be evaluated. This consequently requires that the first step of an automated calculation algorithm is to check if all these conditions are met.
2.2. Calculation of the Group Contributions
The algorithm for the evaluation of the atom group contributions for each of the title descriptors is identical. The only difference is given by the input data: the first step is the extraction from the database of a list of molecules with the known experimental value of the descriptor in question. For each molecule of this list the atom groups are then defined and counted following the rules given above.
The further proceeding is then ruled by the content of the manually set-up “mother”-parameters table of atomic and special groups: this mother table initially covers all possible combinations of “backbone” atom types and neighbourhoods. For a specific descriptor, however, always a certain—and for each descriptor different—surplus number of atom groups remains which is not represented in any molecule of the applied molecules list. These atom groups are removed before proceeding further, thus leaving an individual parameters table for a particular descriptor. This table is finally complemented with those special groups shown in
Table 2 as required for this descriptor.
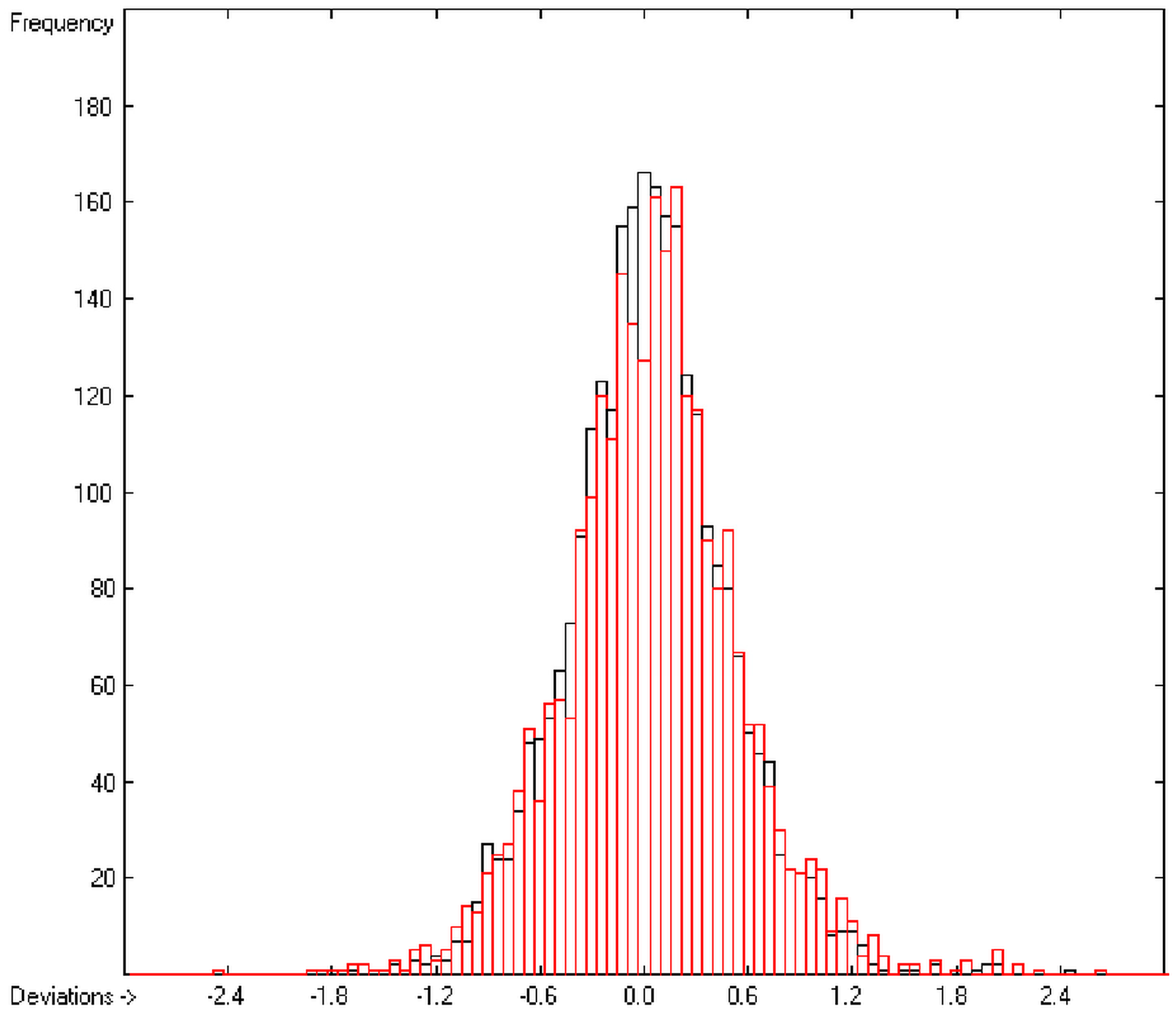
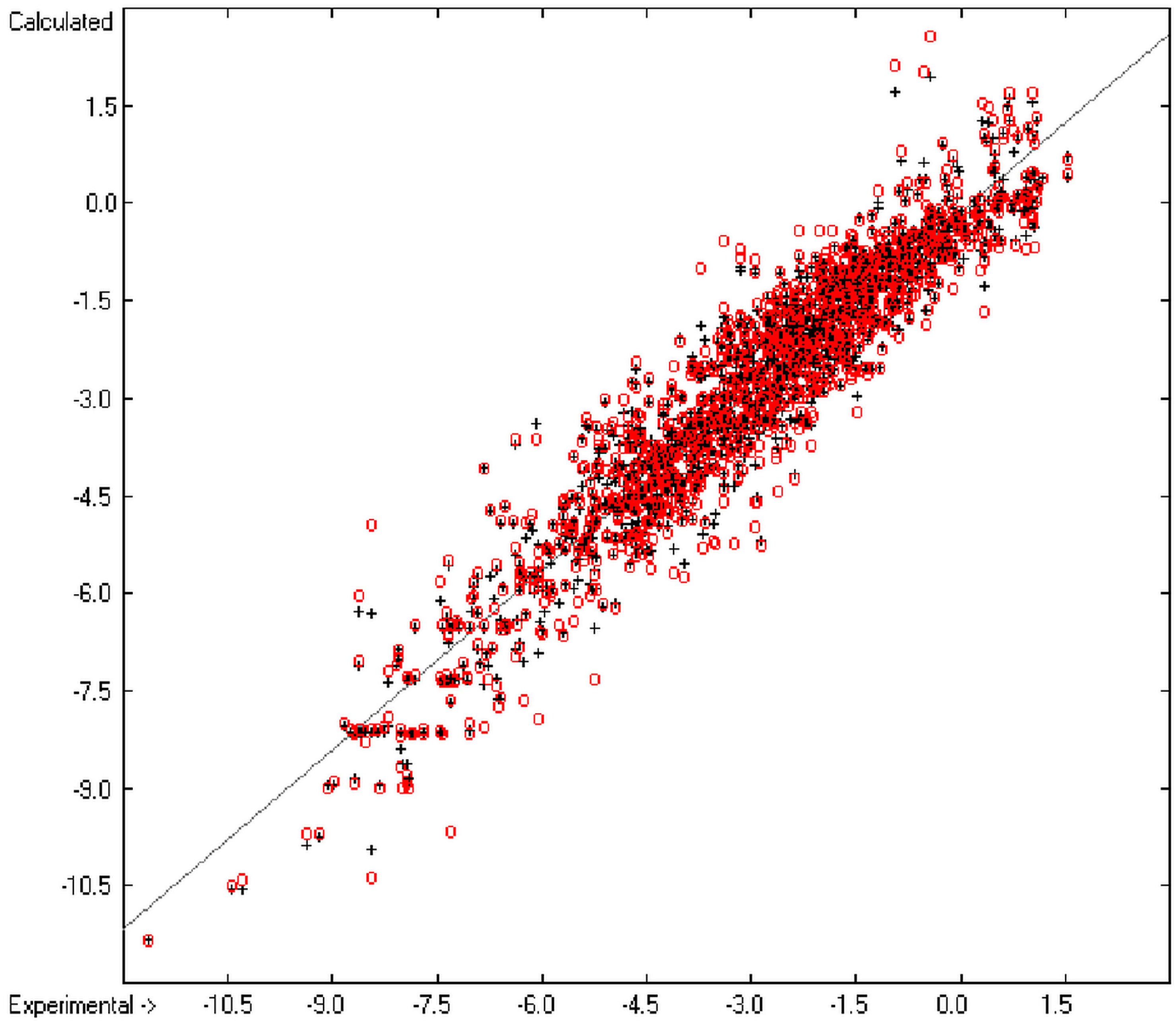
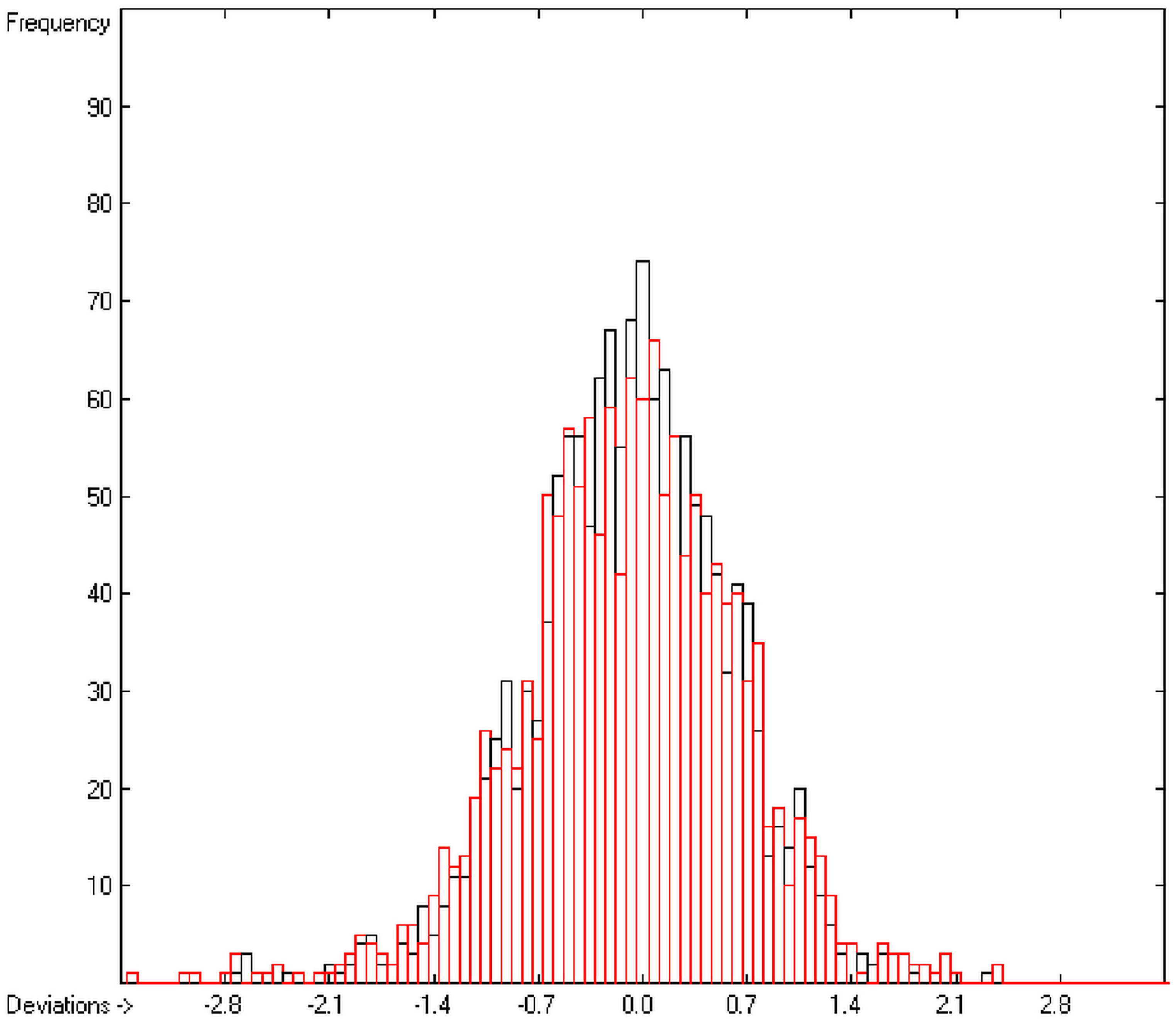
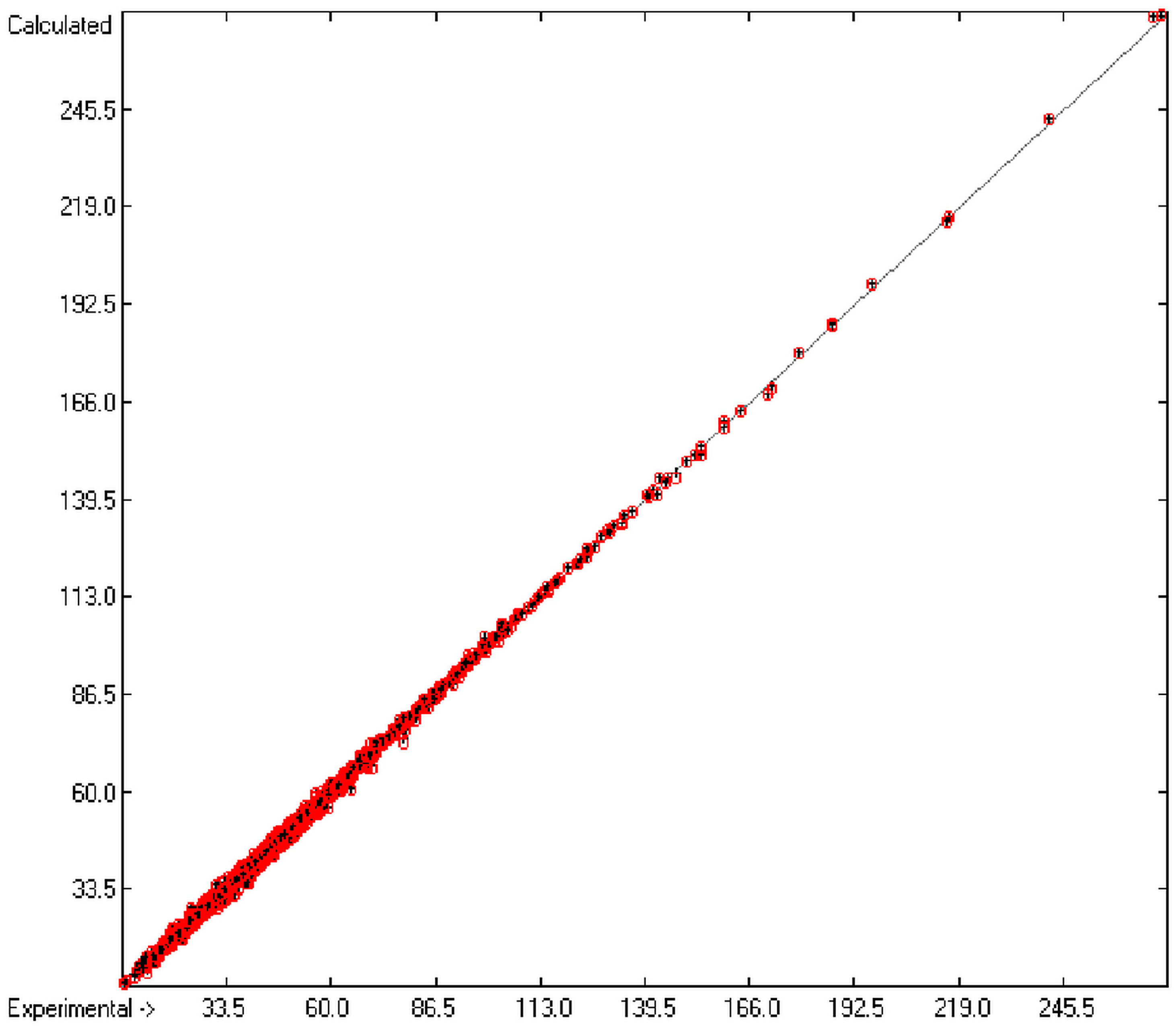
The resulting data set is then translated into an M × (N + 1) matrix where M is the number of molecules and (N + 1) the number of atomic and special groups plus an element for the experimental value. Each matrix element (i,j) then receives the number of occurrences of the jth atomic or special group in the ith molecule. After normalization of this matrix into an Ax = B matrix equation and its equalization by means of the Gauss-Seidel calculus, the resulting group-contribution values are entered into the corresponding parameters table. Additionally, to each atomic and special group the number of its occurrences (its frequency) and the number of molecules containing it are added. Next, the parameters table receives the information about the goodness of fit (R2), the average and standard deviation and the total number of molecules on which the calculation is based.
2.3. Calculation of the Descriptors
Once the group contributions are set up in the corresponding parameters tables, the computation of any of the descriptors’ values Y is a mere summing up of the contributions of the atom groups found in a molecule following the general Equation 1
wherein
ai and
bj are the contribution values, listed in the respective parameters table,
Ai is the number of occurrences of the
ith atom group,
Bj is the number of occurrences of the special groups and C is a constant. However, as was mentioned earlier, this calculation is limited to molecules for which each atom group it contains (not special group!) the corresponding one is present in the corresponding parameters table and its value is confirmed by at least three training molecules. Hence, a computer algorithm has to start with the definition and counting of all the molecule’s atom groups (applying the same procedure as in the second step for the calculation of the group contributions), then check for any atom group that is missing (or is not confirmed) in the parameters table and then either continue using the above formula if all groups are found or reject further calculation. Calculation of all the title descriptors at once on a notebook is done in a split second, once the compound’s three-mensional structure is generated and added to the molecules database (see
Appendix).
2.4. Cross-Validation Calculations
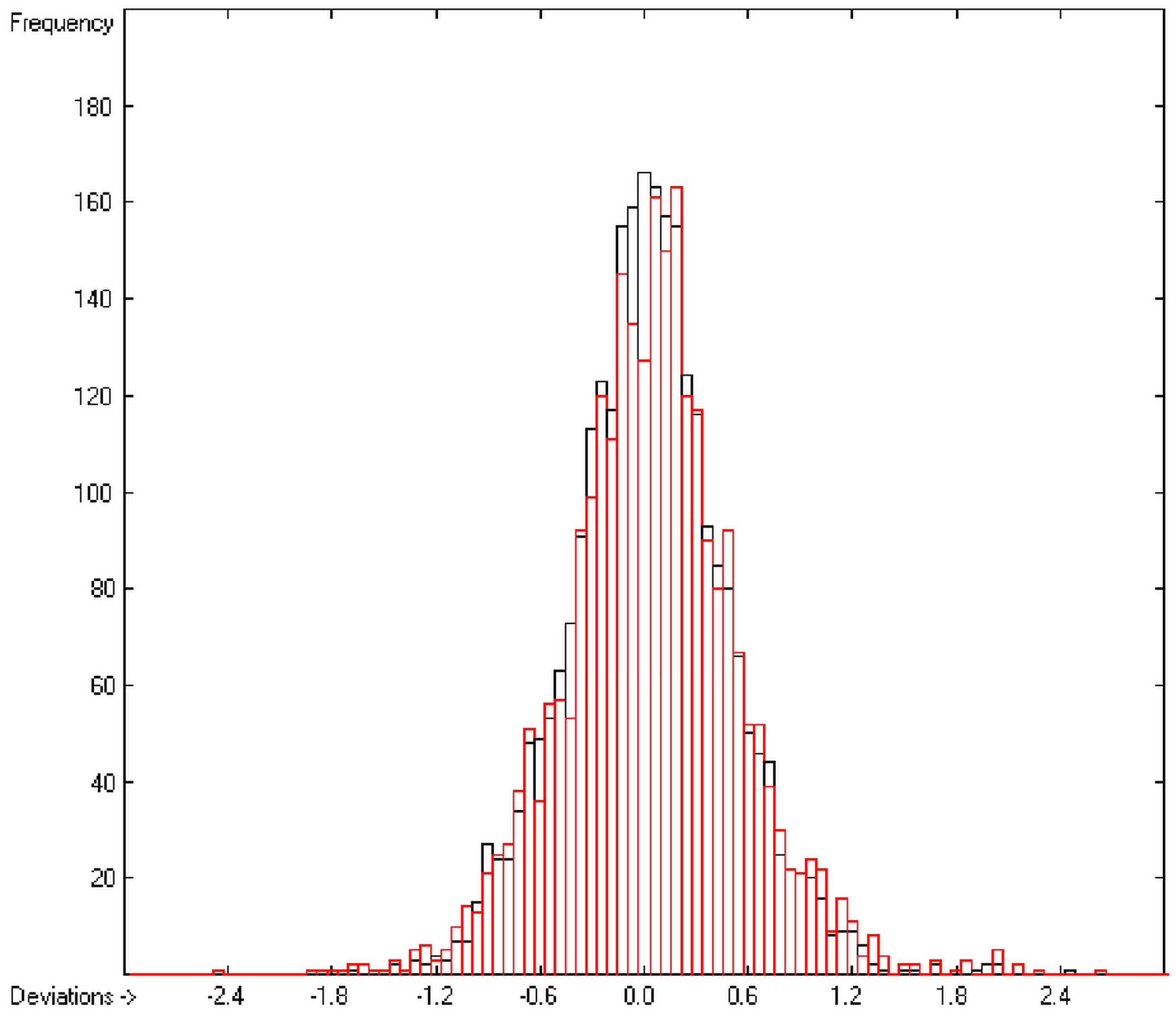
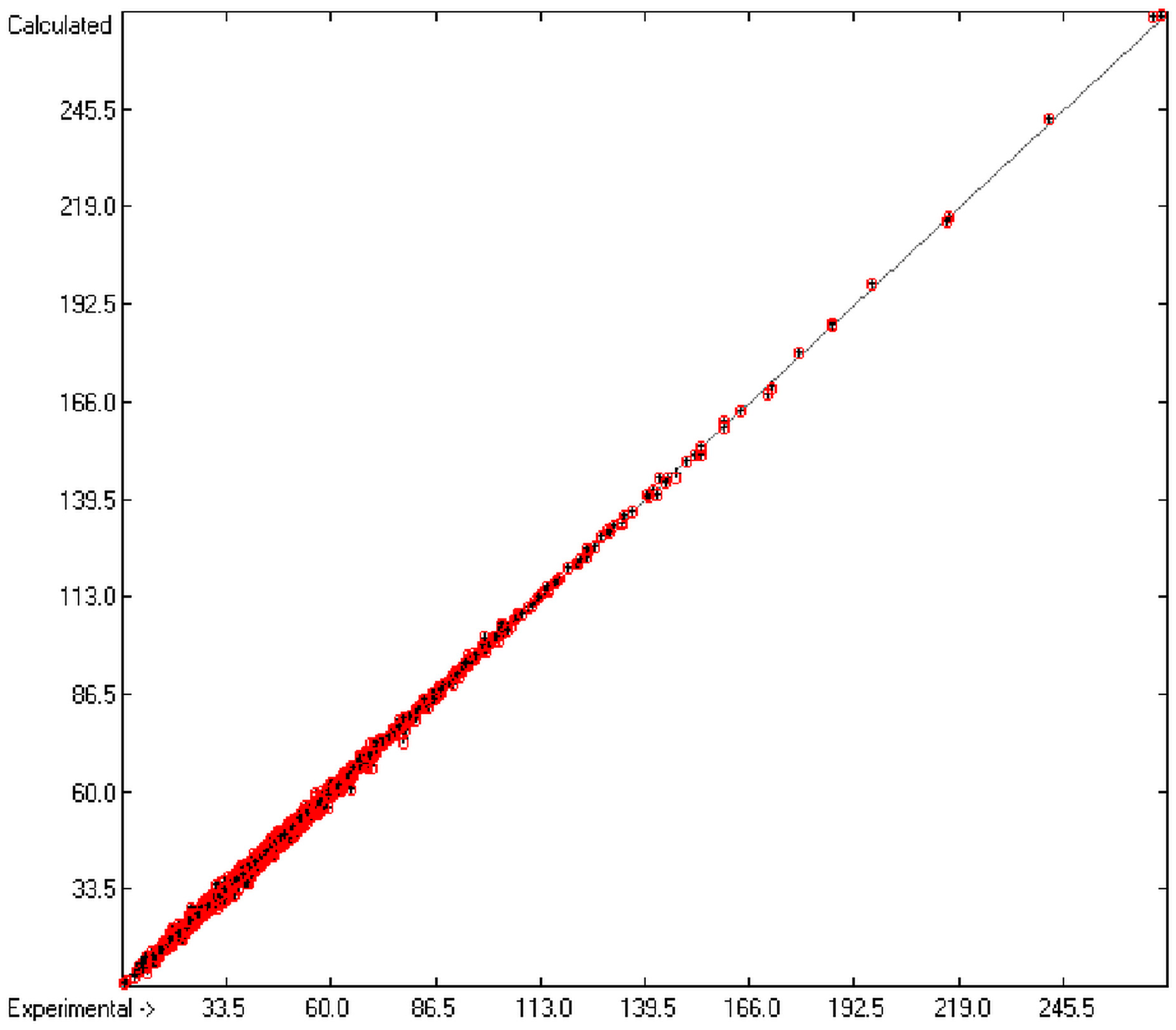
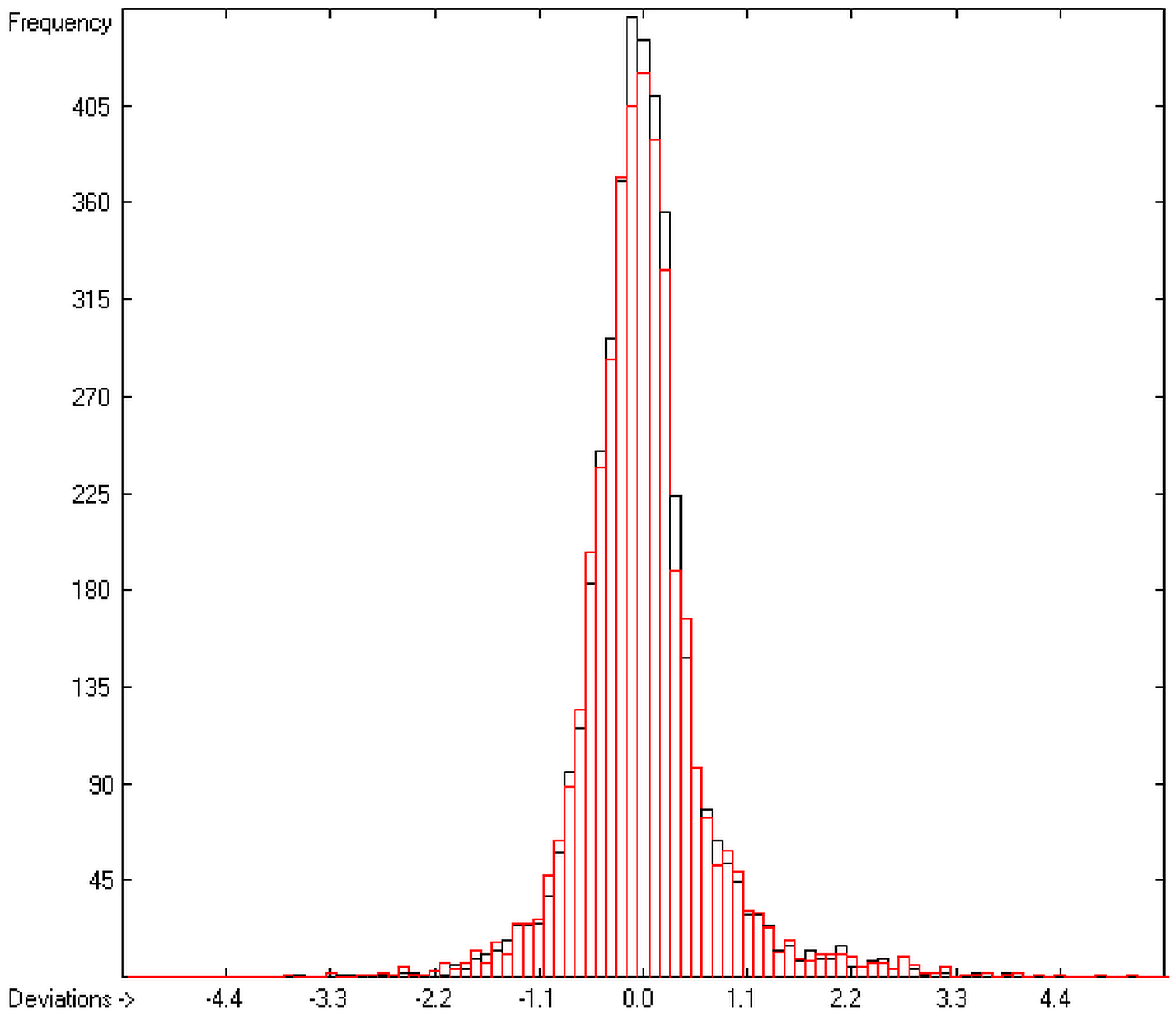
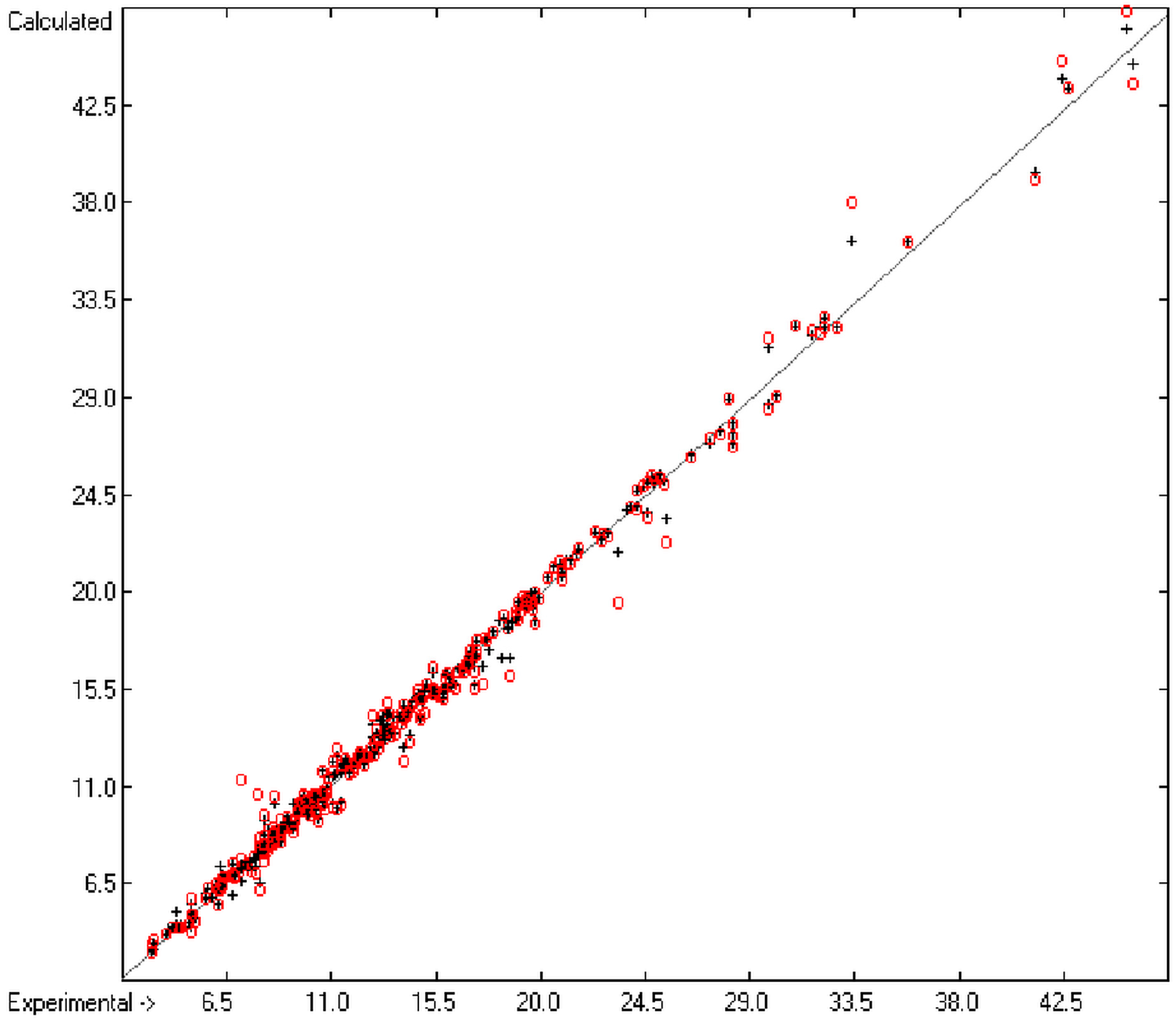
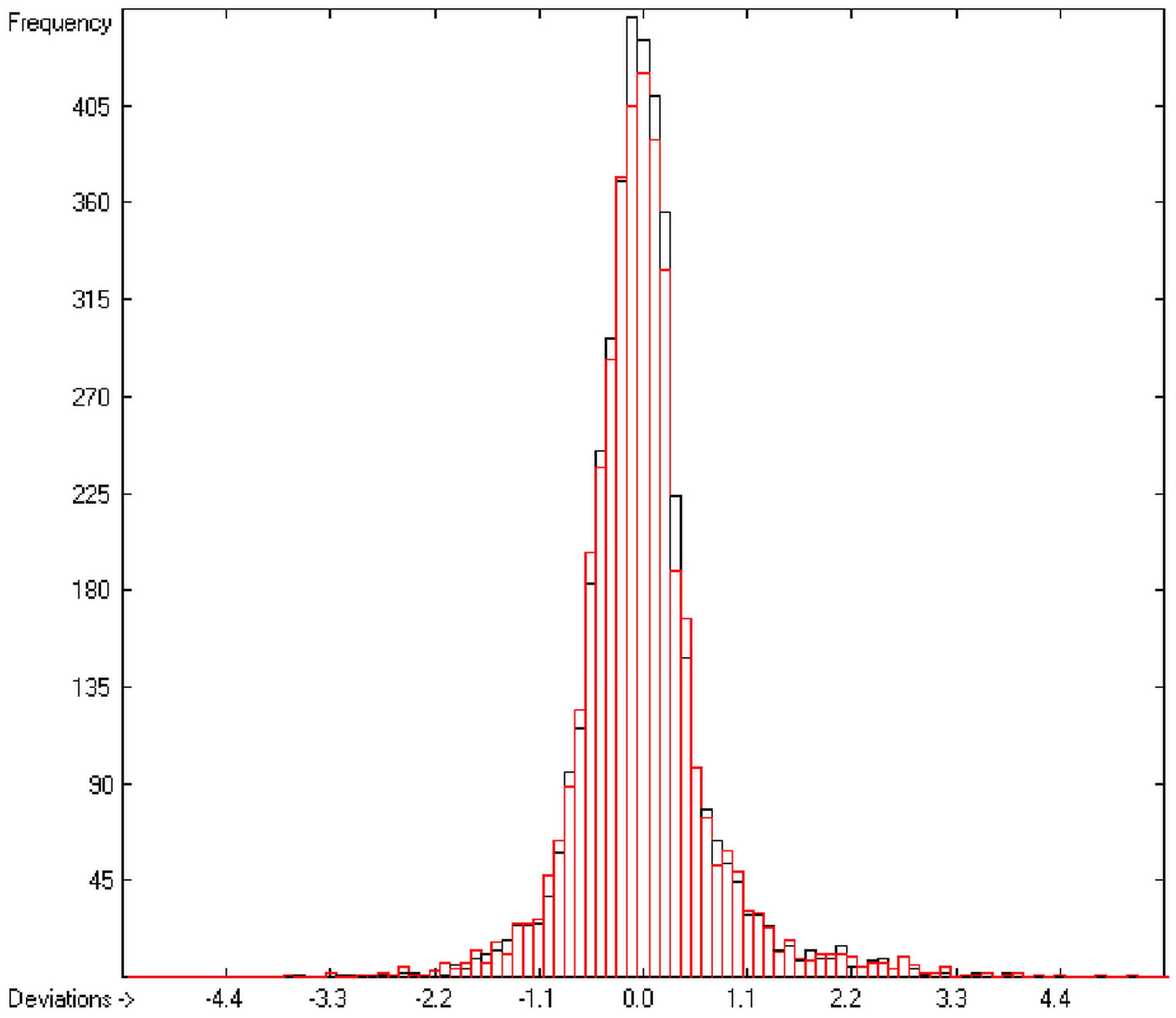
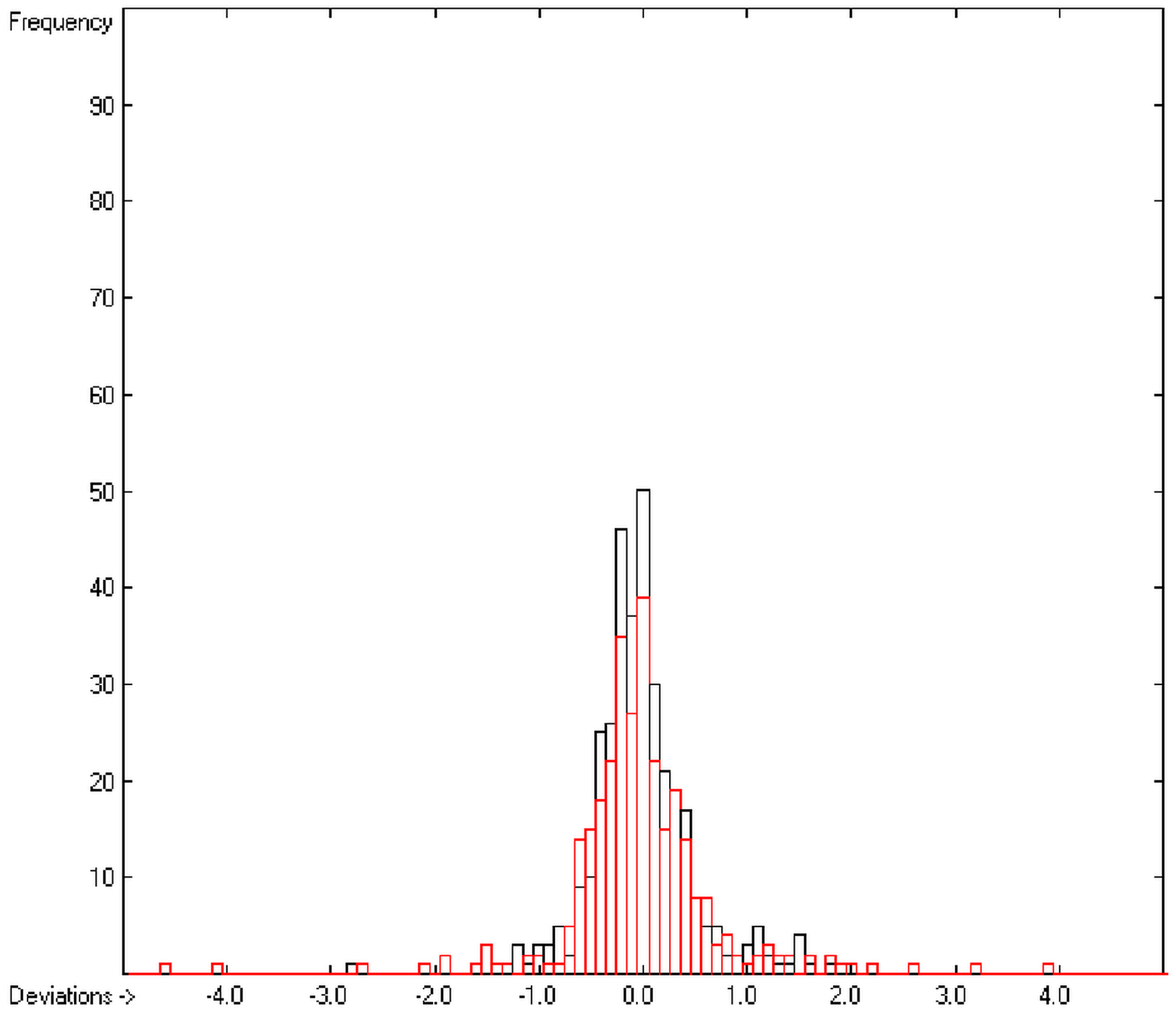
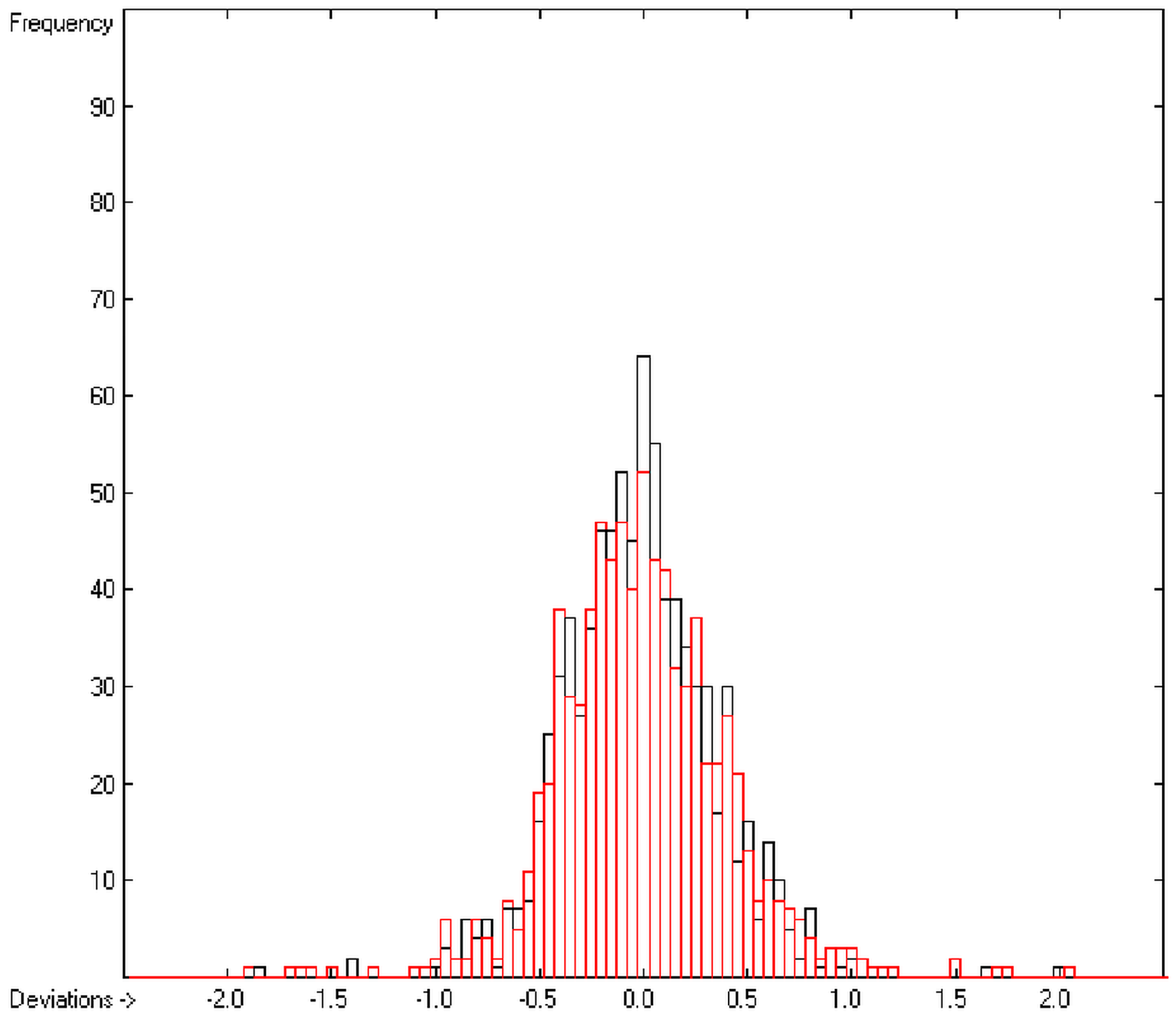
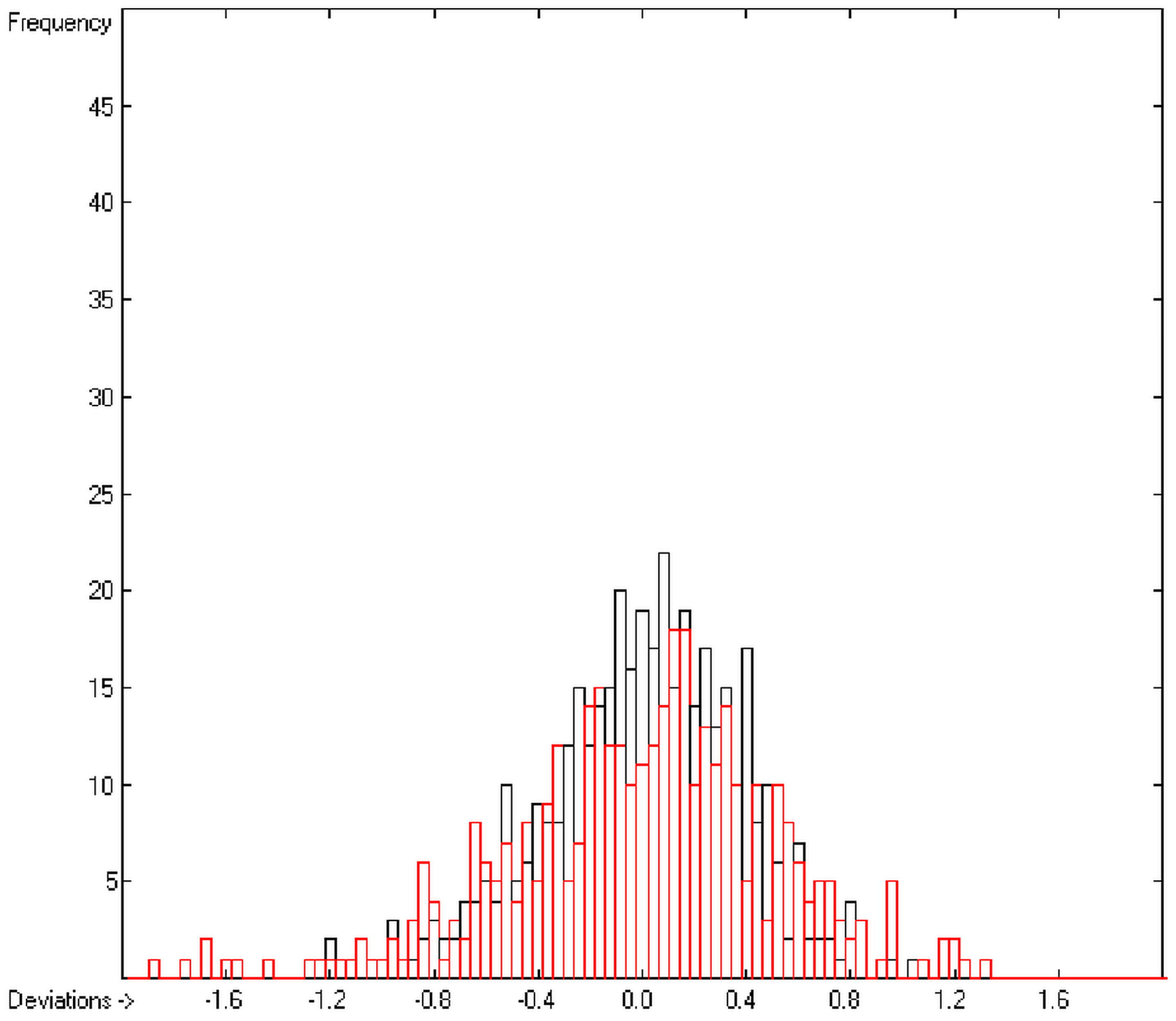
In order to check the plausibility of the results of the group-additivity method for the prediction of the molecular descriptors, in each case a k-fold cross-validation calculation is carried out, whereby, after a few tentative calculations with various k values, k is in all cases chosen to be 10. Accordingly, the complete list of compounds holding a particular experimental descriptor value is first copied into a training set, wherefrom a test set is extracted by the transfer of every k-th, i.e., every 10th compound, thus producing a training set containing 90% of the molecules of the original list and the remaining 10% as test set. In a next step, the training set is used to calculate the atom groups parameters set and then, by means of these parameters, the prediction value is evaluated for each molecule of the test set and added to its properties list. This procedure is repeated k (=10) times, each time shifting the extraction process for the test-set from the re-setup training set by the repetition run-time number, this way making sure that each compound is used exactly once as a test molecule and that no inadvertent clusters of certain structures are extracted from the training sets. Finally, the collected prediction data of all the test molecules are used to evaluate the cross-validated regression coefficient Q2 and the corresponding average and standard deviation. These data are finally entered at the end of each parameters table. The number of compounds on which these cross-validation calculations are founded is in general smaller than the number of compounds used for the evaluation of the correlation coefficient R2, because due to the exclusion of the test compounds in the atom group parameters calculations certain atom groups may no be longer represented by enough molecules and, thus, test compounds having these atom groups are excluded from the prediction calculation.
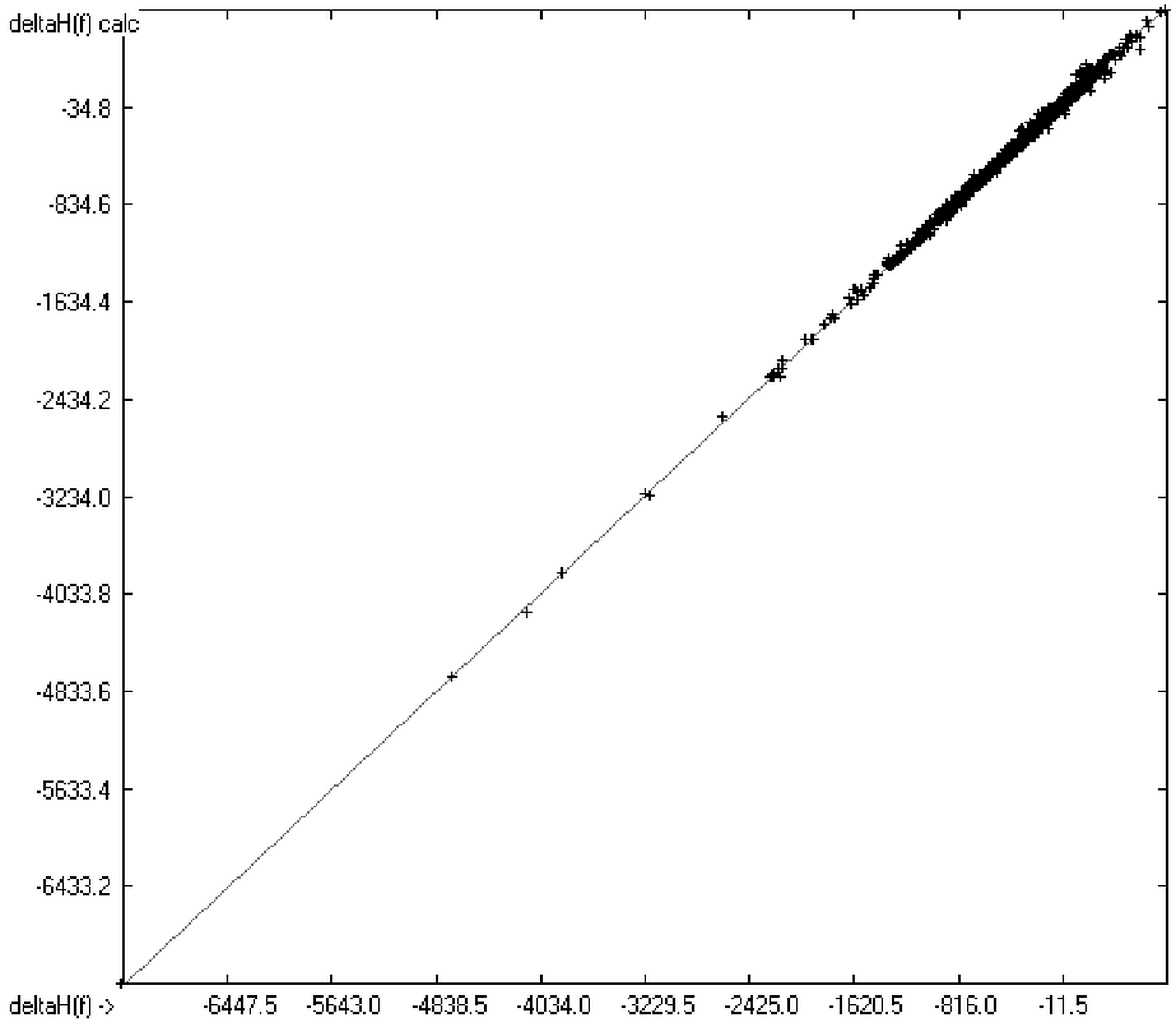
4. Conclusions
A generally applicable computer algorithm based on the well-established group-additivity method has been presented and has been applied for the calculation of the seven molecular descriptors heat of combustion, logP, logS, molar refractivity, molecular polarizability, aqueous toxicity and logBB. An eighth descriptor, the heat of formation, was calculated indirectly using the calculated value of the heat of formation. The definition of the atom groups has been set up in a way that allowed a straightforward program code of the computer algorithm except for the special groups for which, however, code development could take advantage of the information of the 3D-molecular structures stored in the molecules database. The complete algorithm, realized in ChemBrain IXL, thus enables the computation of the contributions of all the atom groups as well as all the described special groups for descriptor evaluations; their inclusion, however, is governed by their presence or absence in the respective parameters tables. Within this context it is worth mentioning that for the prediction of the refractivity, molecular polarizability and toxicity in principle a 3D geometry is not required.
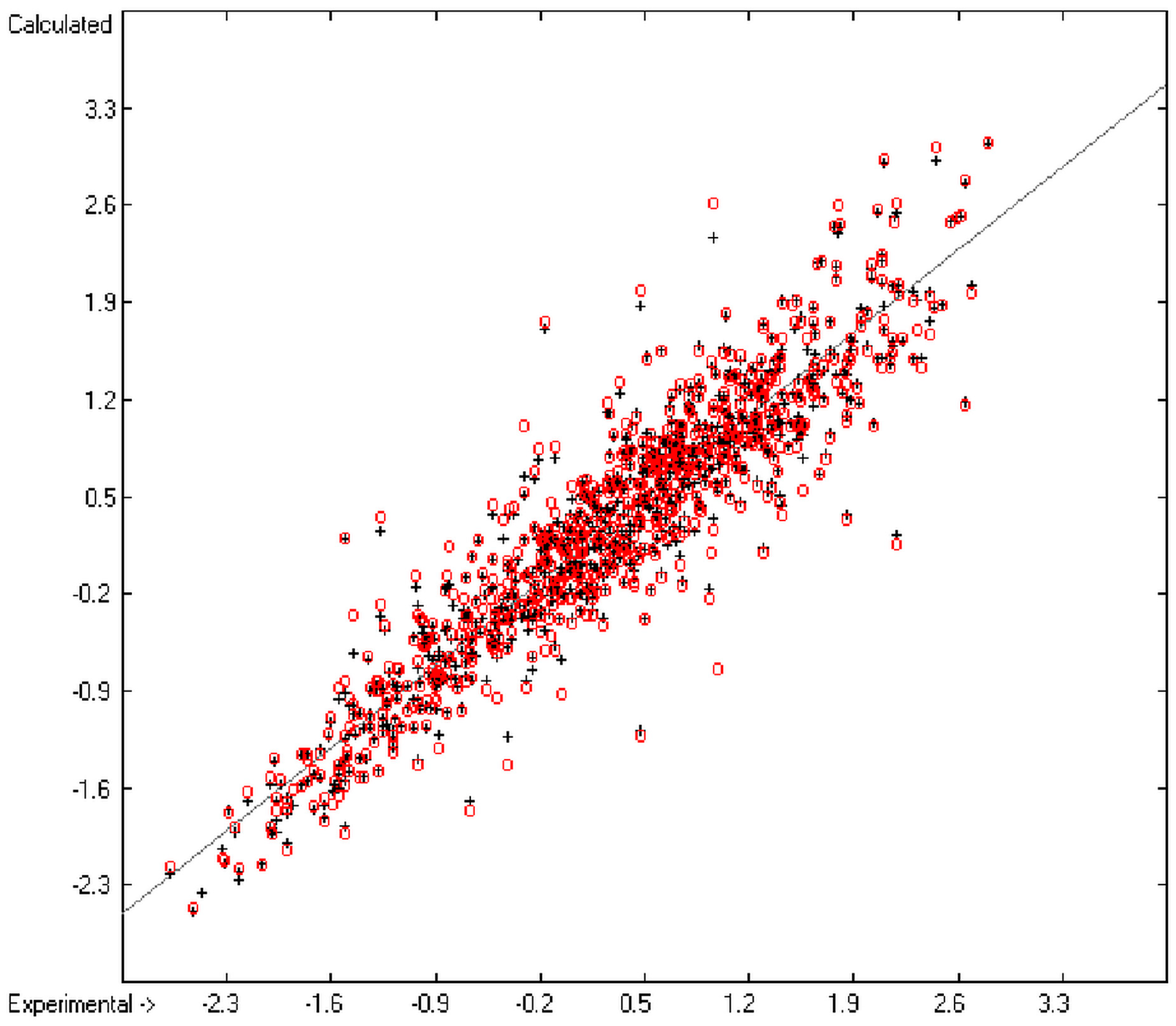
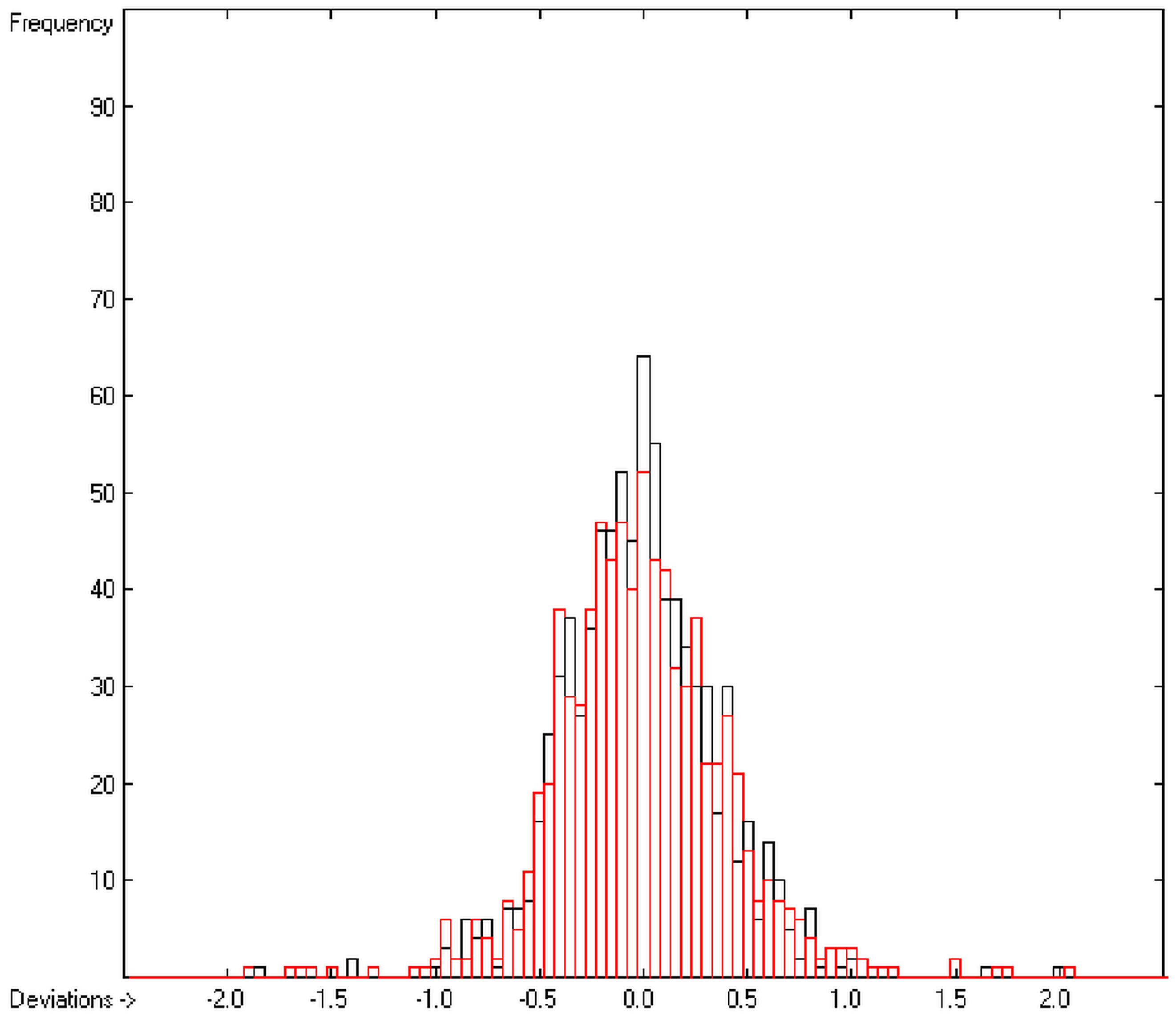
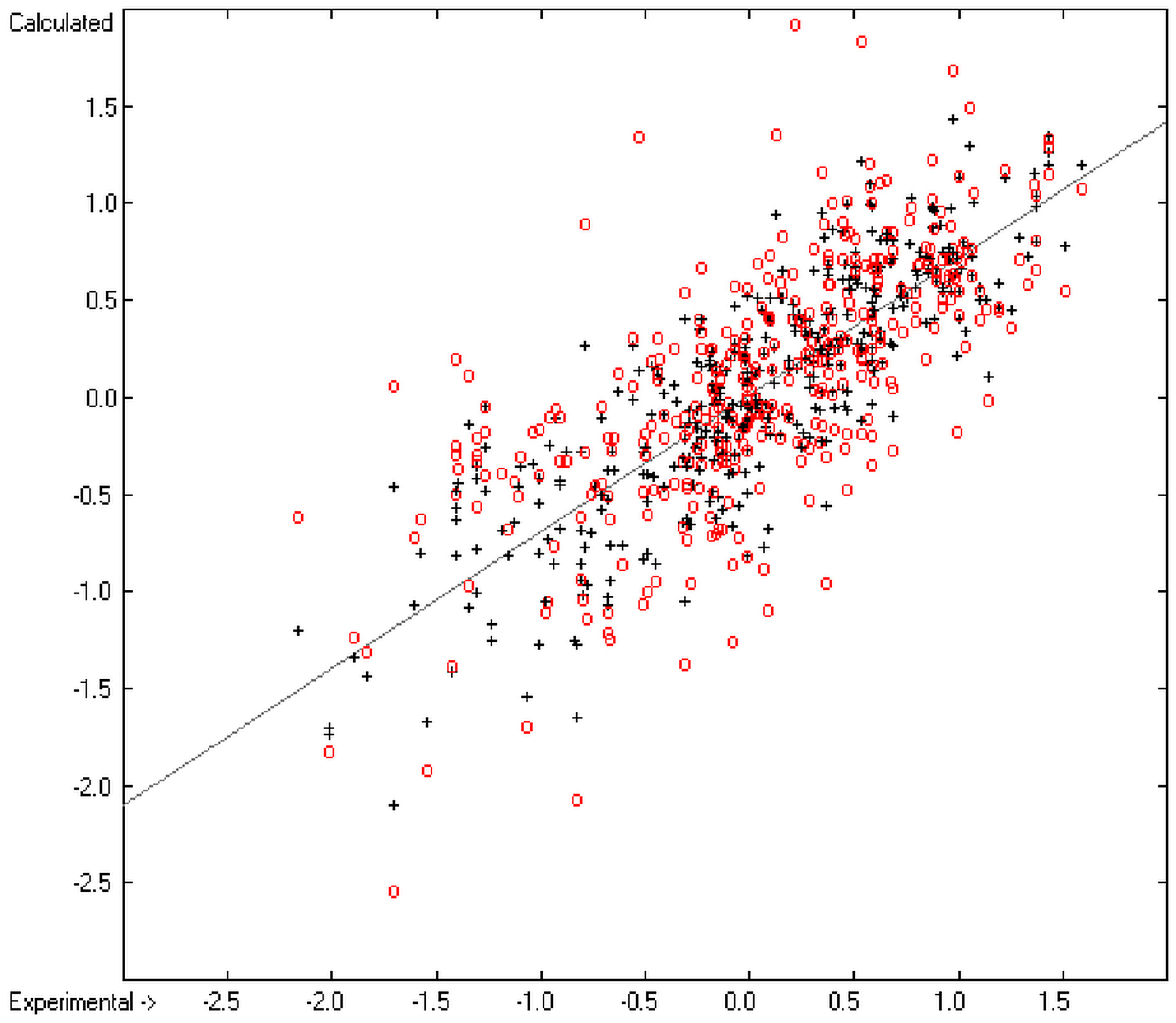
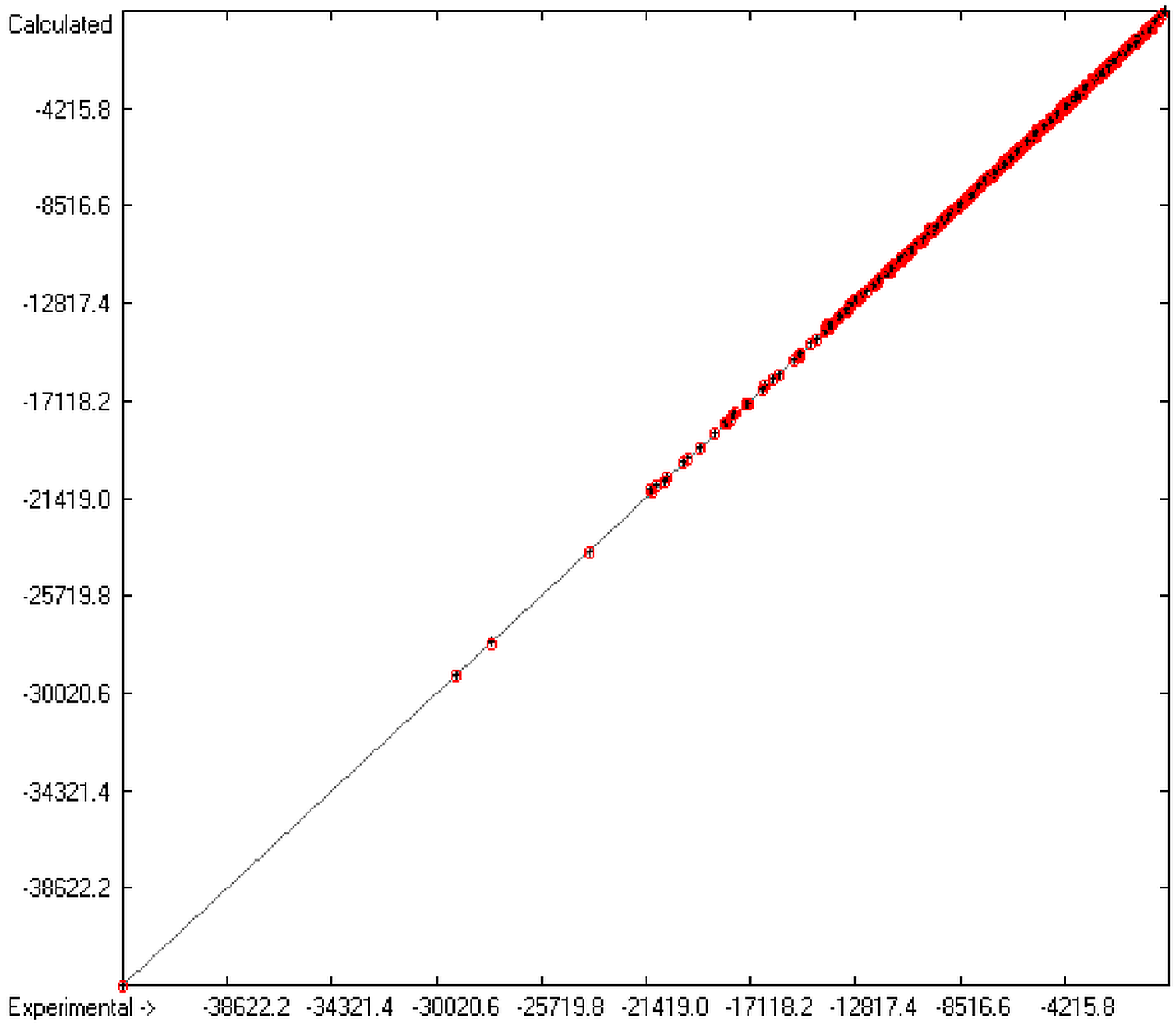
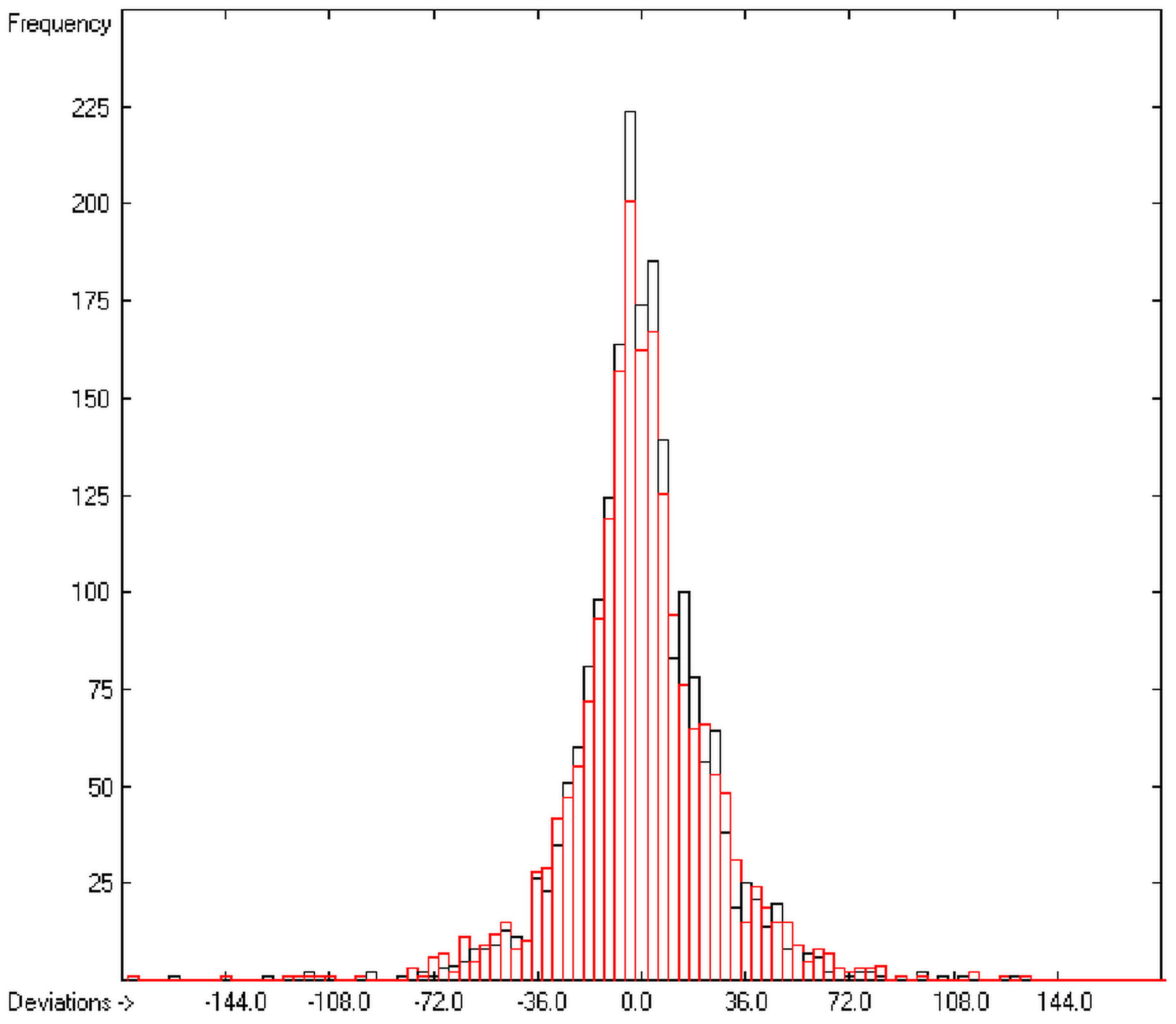
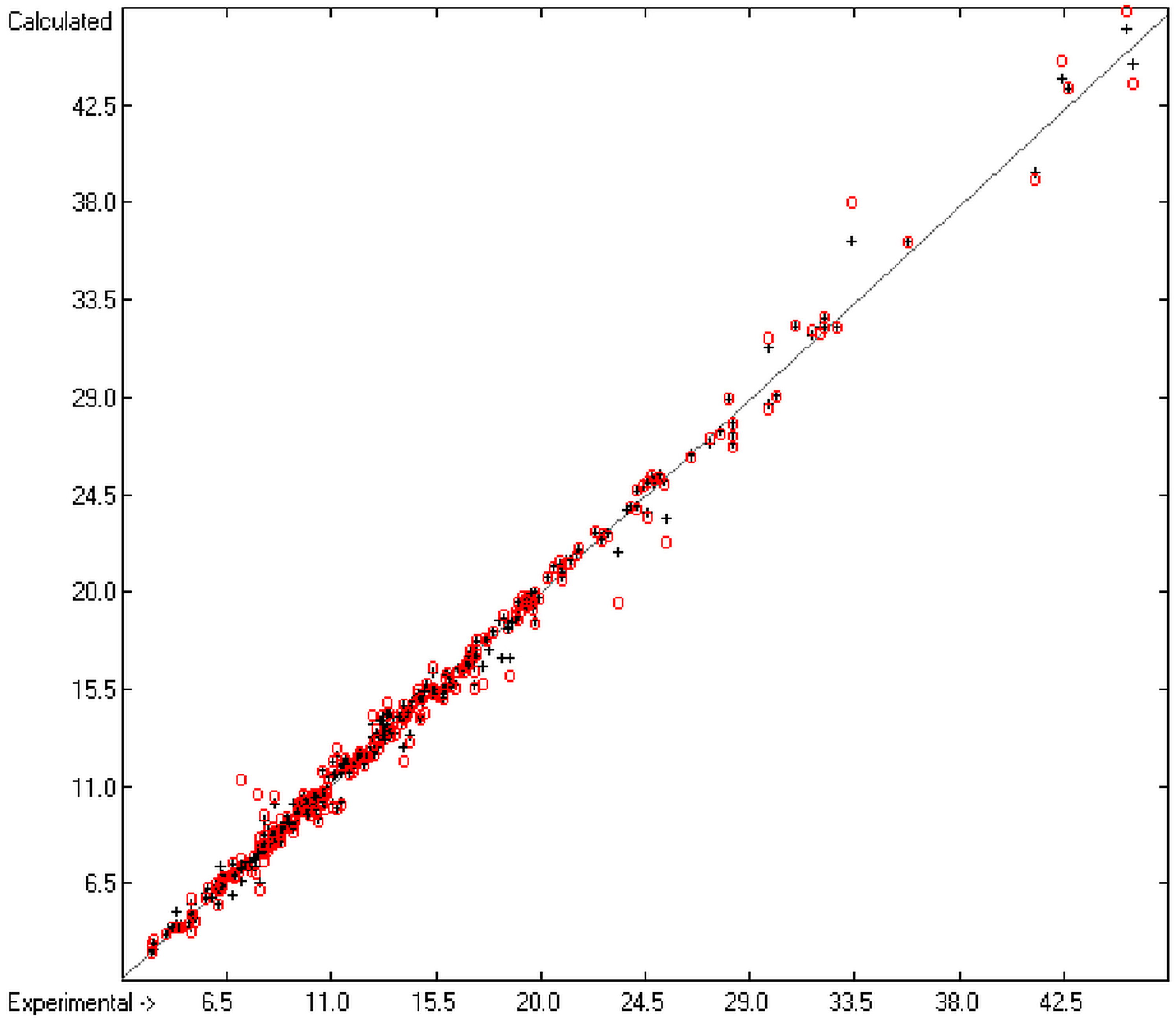
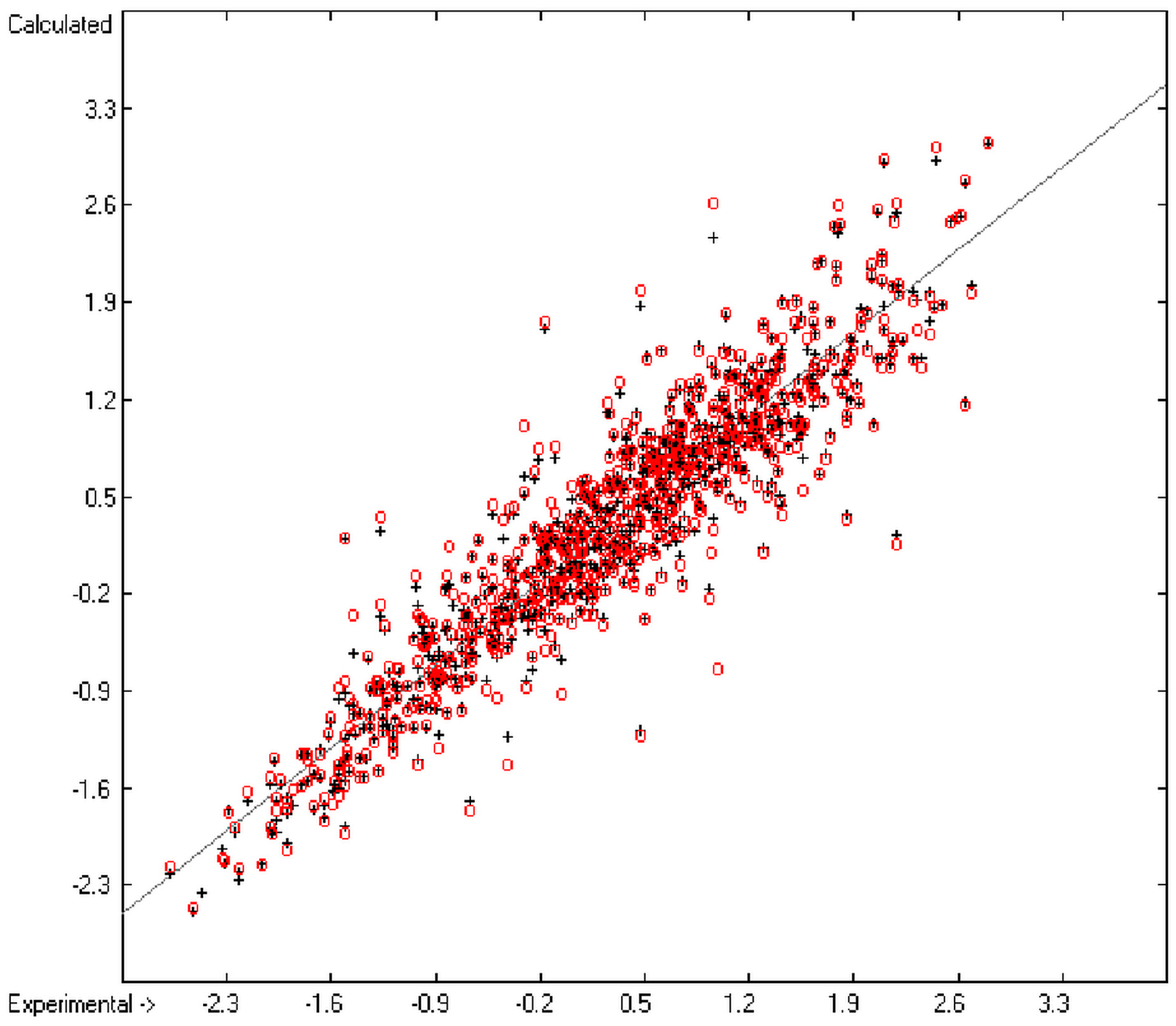
The present group-additivity algorithm has shown its versatility in that it is capable of producing results at once that are in good to excellent agreement with experimental data for six of the seven title descriptors. The present study has also shown the limits of the group-additivity method as such in an area where too many unknown or incalculable factors influence the experimental data as has been exemplified for logBB.
The number of molecules in the database—at present about 20,700—which encompasses a representative collection of organic and metal-organic compounds of commercial as well as scientific relevance and which has all the referenced data stored, and the amount of compounds for which the title descriptors could be evaluated under the given constraints provides an accountable estimate of the scope of applicability of each of the presented tables of group contributions. For the heat of combustion and formation it is ca. 75%, for logP ca. 84%, for logS ca. 73%, for the molecular polarizability ca. 42%, for the refractivity ca. 75% and for the toxicity ca. 41%. These percentage numbers evidently reflect the number of experimental data available at present. There is no doubt, however, that even with a larger database of compounds for the calculation of the group contributions there is a limit to the improvement of the accuracy of the predictions on the basis of this method, not only because there is little hope that the existing experimental databases and their deficiencies will be re-examined in the laboratories but also because of influences on the results that can principally not be dealt with by this method, as there are non-neighbouring effects (e.g., gauche or cis), intramolecular charge effects or non-bonded interactions.
In view of these facts there is truth in the words which Cohen and Benson [
10] stated in their closing remarks saying that the atom group additivity method is “a useful tool for making rapid property estimates or for checking the likely reliability of existing measurements”.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}