
Synthesis and X-ray Structural Studies of a Substituted 2,3,4,5-Tetrahydro-1H-3-benzazonine and a 1,2,3,5-Tetrahydro-4,3-benzoxazonine

Abstract
: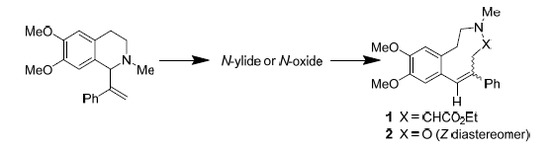
1. Introduction
2. Results and Discussion
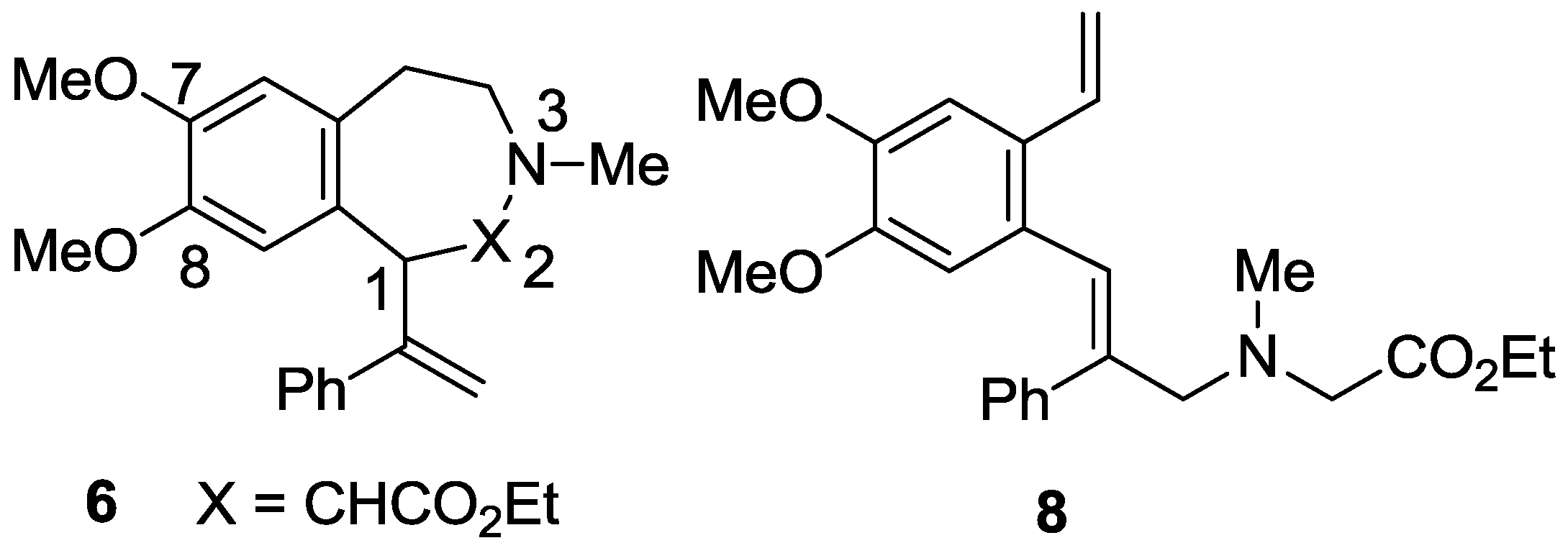
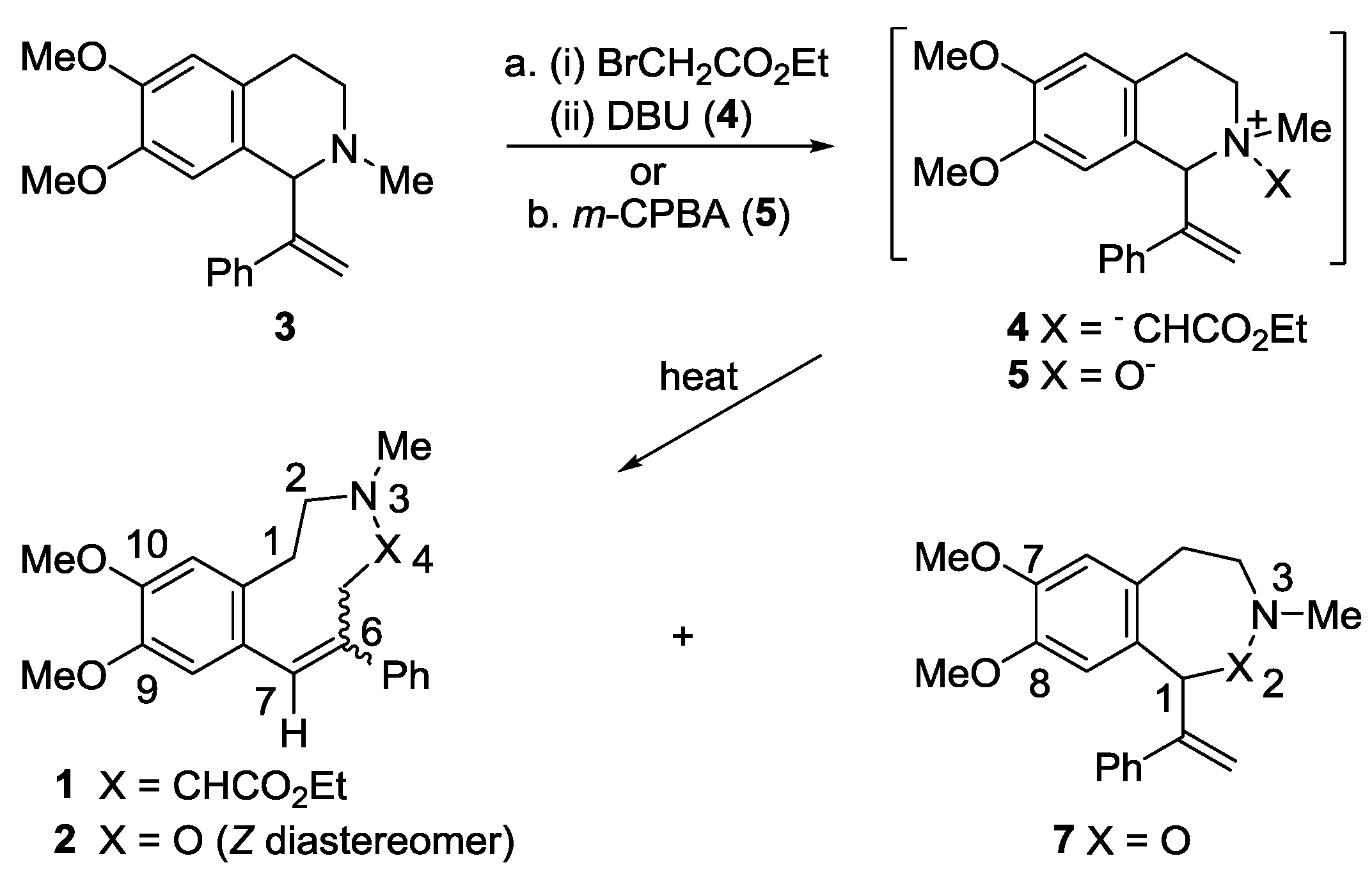
2.1. Synthesis
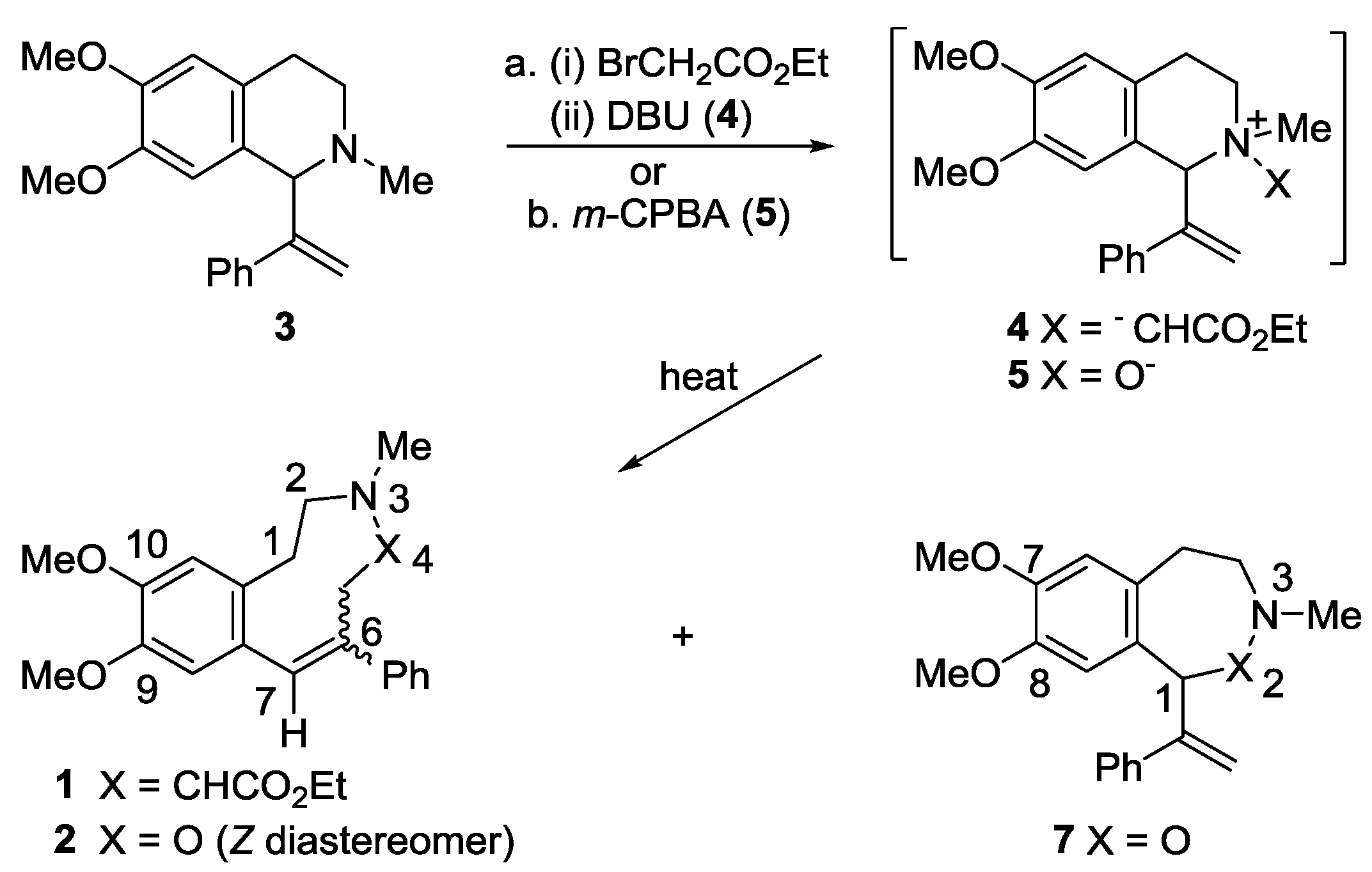
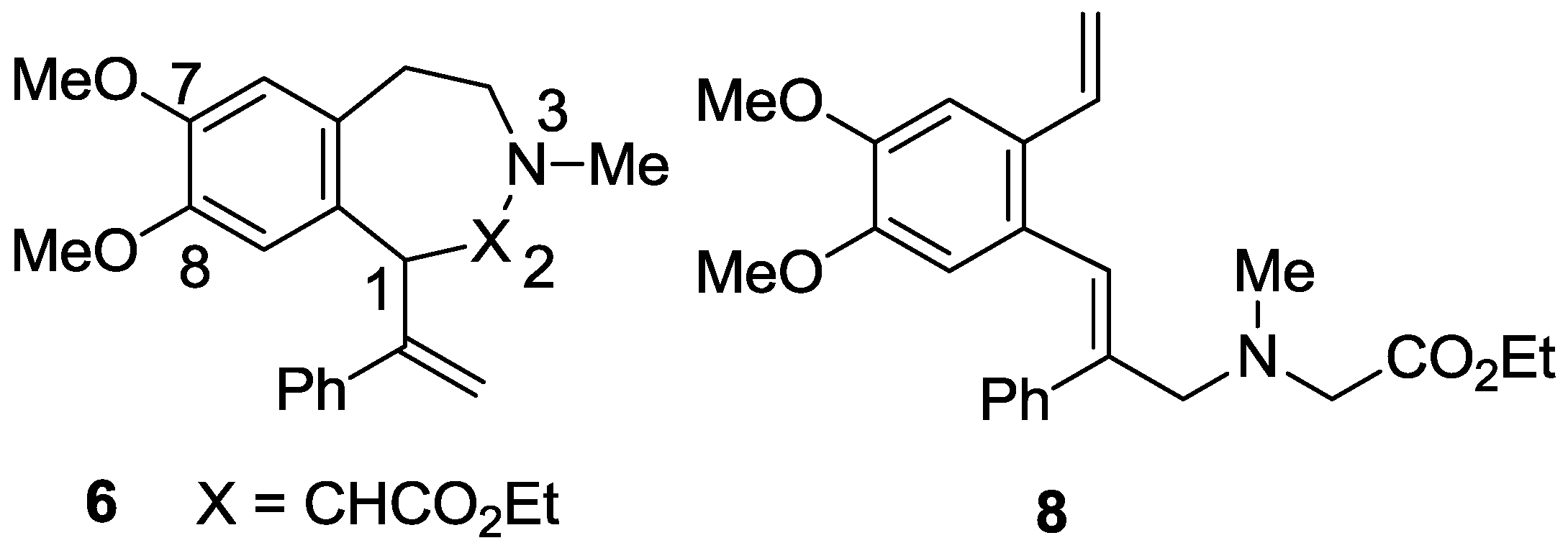
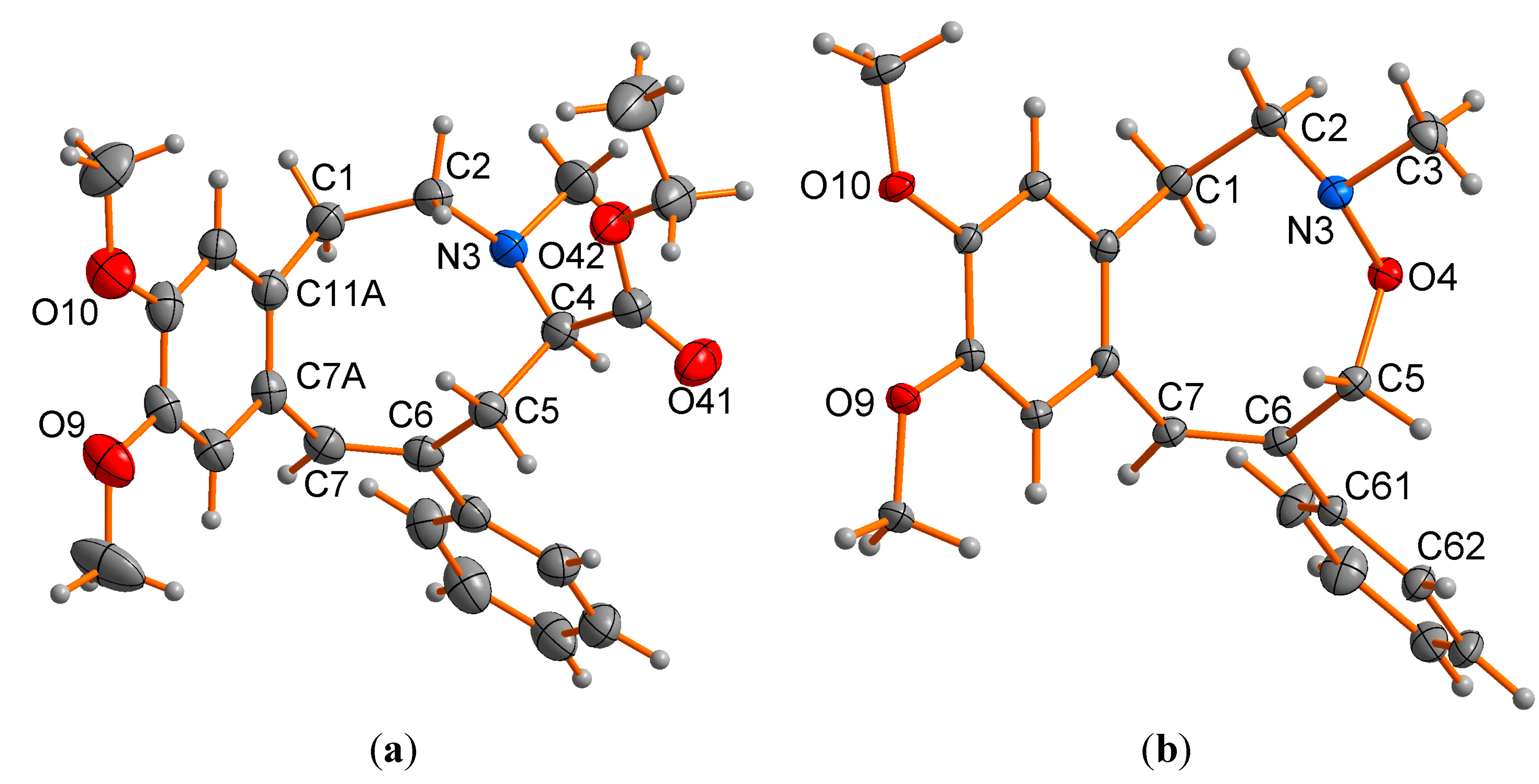
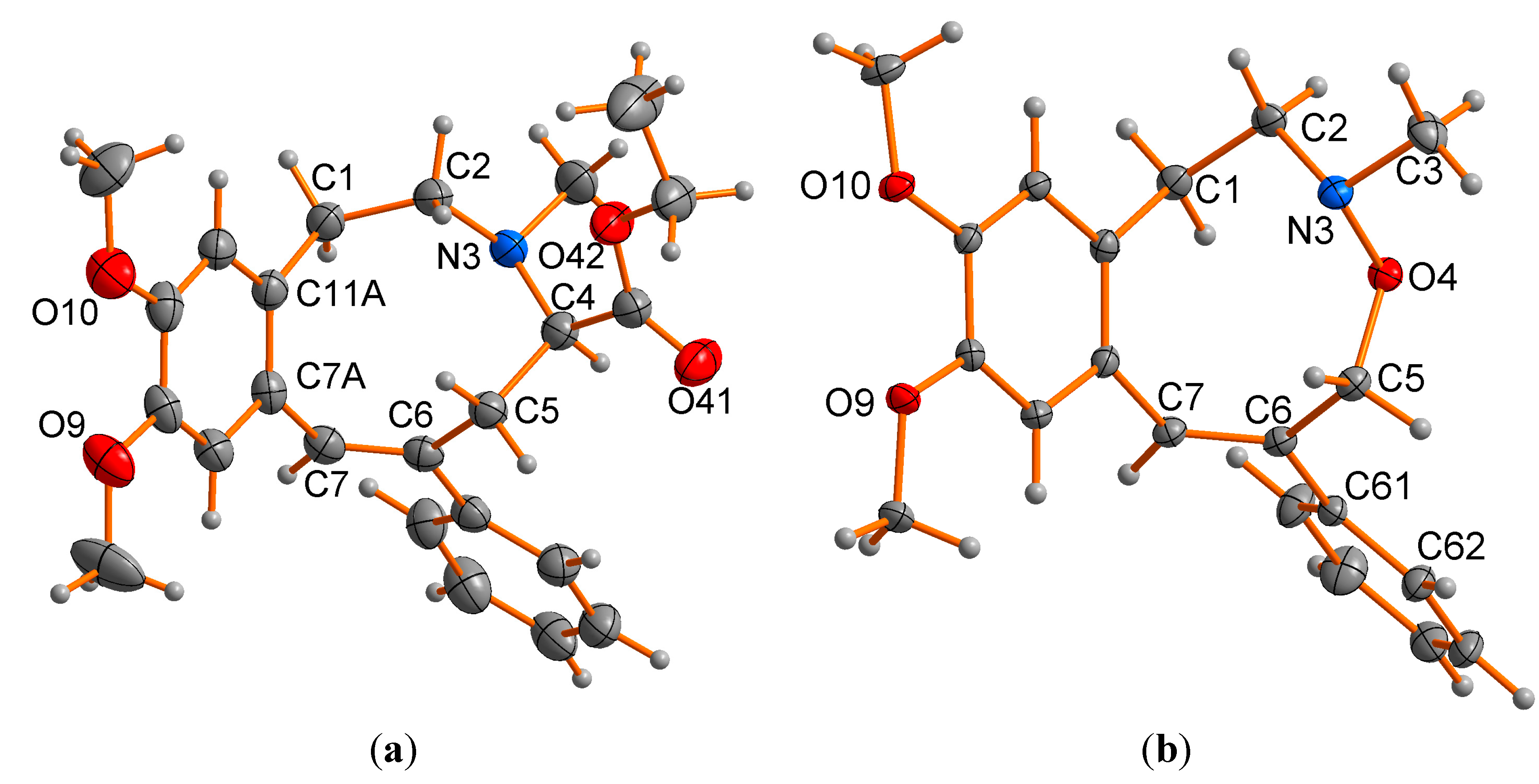
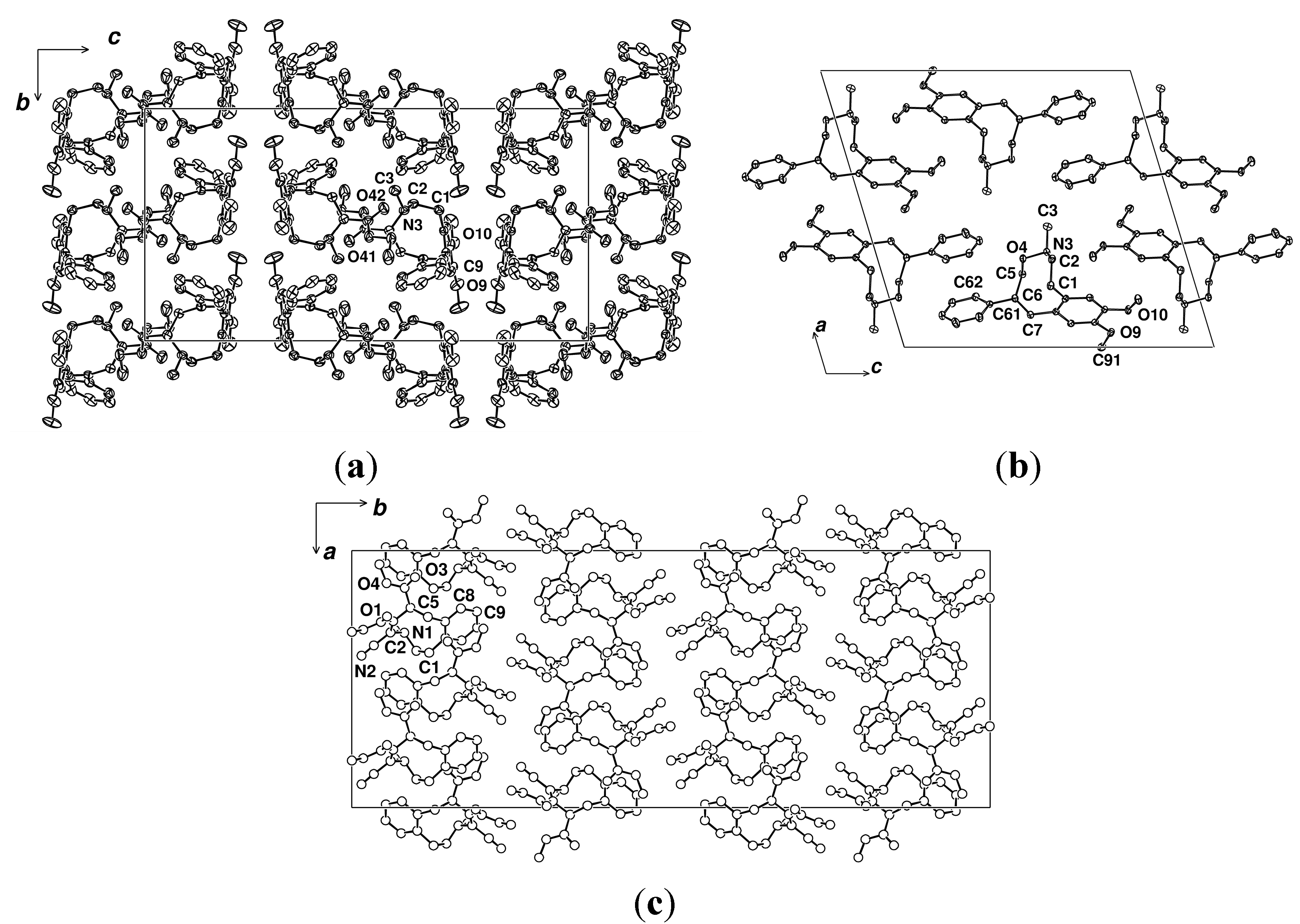
2.2. X-ray Structural Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E-1 | 2 | AYEKOF [26] | SELXUC [27] |
|---|---|---|---|---|
| 11a-1-2-3 | 105.2 (2) | 52.56 (12) | −68.4 (2) | −43.3 (3) |
| 1-2-3-4 | −105.0 (2) | 63.90 (11) | −64.1 (2) | 75.3 (3) |
| 2-3-4-5 | 55.8 (2) | −122.48 (8) | 104.4 (2) | 50.8 (2) |
| 3-4-5-6 | 59.0 (2) | 113.25 (9) | 2.8 (3) | −109.4 (2) |
| 4-5-6-7 | −118.1 (2) | −90.31 (12) | −96.1 (2) | 54.2 (3) |
| 5-6-7-7a | 0.9 (2) | 4.0 (2) | 110.4 (2) | −72.1 (3) |
| 6-7-7a-11a | 70.0 (2) | 69.59 (14) | −76.7 (2) | 105.4 (3) |
| 7-7a-11a-1 | 2.1 (2) | −3.32 (15) | −2.4 (3) | 4.3 (3) |
| 7a-11a-1-2 | −91.6 (2) | −109.39 (11) | 104.4 (2) | −60.4 (3) |
| Atoms | Parameter (Macrocycle) | Atoms | Parameter (Phenyl Ring) |
|---|---|---|---|
| Distances (Å) | |||
| C(1)-C(2) | 1.544(2), 1.5259(15), 1.522(3), 1.539(3) | C(7a)-C(8) | 1.409(2), 1.4105(14), 1.401(3), 1.389(4) |
| C(1)-C(11a) | 1.512(2), 1.5129(14), 1.513(3), 1.521(2) | C(7a)-C(11a) | 1.389(2), 1.3918(13), 1.405(3), 1.406(4) |
| C(2)-N(3) | 1.475(2), 1.4588(14), 1.483(3), 1.504(3) | C(8)-C(9) | 1.370(2), 1.3834(13), 1.389(3), 1.357(4) |
| N(3)-C,O(4) | 1.450(2),1.4544(10), 1.431(3), 1.518(4) | C(9)-C(10) | 1.396(2), 1.4101(13), 1.383(3), 1.370(5) |
| N(3)-C(3) | 1.459(2), 1.4590(14), 1.470(3), 1.491(3) | C(10)-C(11) | 1.380(2), 1.3836(13), 1.388(3), 1.381(4) |
| C,O(4)-C(5) | 1.533(2),1.4453(12), 1.346(3), 1.517(4) | C(11)-C(11a) | 1.407(2), 1.4094(13), 1.397(3), 1.390(3) |
| C(5)-C(6) | 1.509(2), 1.5106(13), 1.505(3), 1.522(4) | C(9)-O(9) | 1.371(2), 1.3683(11), -, - |
| C(6)-C(7) | 1.340(2), 1.3431(13), 1.555(3), 1.534(4) | C(10)-O(10) | 1.370(2), 1.3651(11), -, - |
| C(7)-C(7a) | 1.483(2), 1.4876(13), 1.511(3), 1.515(3) | ||
| C(6)-C(61) | 1.488(2), 1.4892(13), 1.524(3), - | ||
| Angles (degrees) | |||
| C(11a)-C(1)-C(2) | 115.71(13), 113.11(8), 113.9(2), 117.2(2) | C(7)-C(7a)-C(8) | 117.16(14), 118.12(8), 118.5(2), 118.6(3) |
| C(1)-C(2)-N(3) | 113.79(13), 111.51(8), 113.5(2), 116.3(2) | C(7)-C(7a)-C(11a) | 123.75(14), 122.46(9), 122.7(2), 123.4(2) |
| C(2)-N(3)-C,O(4) | 117.61(13), 106.90(7), 112.0(2), 115.8(2) | C(8)-C(7a)-C(11a) | 119.09(14), 119.29(8), 118.8(2), 117.9(2) |
| C(2)-N(3)-C(3) | 113.73(13), 111.35(9), 110.0(2), 111.6(2) | C(7a)-C(8)-C(9) | 121.82(16). 121.48(9), 121.4(2), 122.6(3) |
| C,O(4)-N(3)-C(3) | 112.03(12), 105.62(8), 112.3(2), 111.2(2) | C(8)-C(9)-O(9) | 125.26(16), 125.56(9), -, - |
| N(3)-C,O(4)-C(5) | 112.08(12), 108.87(7), 121.5(2), 113.7(2) | C(10)-C(9)-O(9) | 115.41(15), 115.25(8), -, - |
| C,O(4)-C(5)-C(6) | 113.93(13), 112.02(8), 120.9(2), 118.1(2) | C(8)-C(9)-C(10) | 119.32(15), 119.19(9), 119.7(2), 119.6(3) |
| C(5)-C(6)-C(7) | 121.28(14), 121.43(9), 113.3(2), 118.3(3) | C(9)-C(10)-C(11) | 119.51(15), 119.44(9), 119.5(2), 119.9(2) |
| C(5)-C(6)-C(61) | 118.25(13), 118.33(8), 111.3(2), - | C(9)-C(10)-O(10) | 114.75(15), 115.29(8), -, - |
| C(61)-C(6)-C(7) | 120.31(14), 120.24(9), 112.3(2), - | C(11)-C(10)-O(10) | 125.73(16), 125.27(9), -, - |
| C(6)-C(7)-C(7a) | 124.86(14), 126.88(9), 112.44(14), 110.8(2) | C(10)-C(11)-C(11a) | 121.66(16), 121.54(9), 121.6(2), 121.1(3) |
| C(11)-C(11a)-C(7a) | 118.57(15), 119.00(9), 119.0(2), 118.9(2) | ||
| C(11)-C(11a)-C(1) | 118.73(14), 118.00(8), 118.0(2), 117.6(2) | ||
| C(7a)-C(11a)-C(1) | 122.70(14), 122.98(8), 123.0(2), 123.5(2) | ||
| C(9)-O(9)-C(91) | 115.93(14), 116.82(8), -, - | ||
| C(10)-O(10)-C(101) | 117.91(15), 116.86(8), -, - | ||
| Compound | E-1 | 2 |
|---|---|---|
| Formula | C24H29NO4 | C20H23NO3 |
| Fw (Da) | 395.5 | 325.4 |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | Pbca (# 61) | P21/n (#14 (variant)) |
| a (Å) | 12.321(1) | 16.5518(6) |
| b (Å) | 13.400(1) | 6.1433(2) |
| c (Å) | 25.698(3) | 17.8034(6) |
| β (°) | 106.931(4) | |
| V (Å3) | 4243(1) | 1731.8(1) |
| T (K) | 150 | 100 |
| Dc (gcm−3) | 1.238 | 1.248 |
| Z (f.u.) | 8 | 4 |
| μ (mm−1) | 0.084 | 0.083 |
| Specimen (mm3) | 0.36 × 0.15 × 0.10 | 0.43 × 0.16 × 0.15 |
| Tmin/max | 0.77 | 0.97 |
| 2θmax (deg.) | 58 | 60 |
| Nt | 50344 | 17112 |
| N | 5582 (0.030) | 5052 (0.022) |
| No (I > 2σ(I)) | 3101 | 4391 |
| R1 (I > 2σ(I)) | 0.053 | 0.043 |
| wR2 (a(b)) | 0.15 (0.088) | 0.111 (0.053, 0.58) |
| |Δρmax| (eÅ−3) | 0.28 | 0.44 |
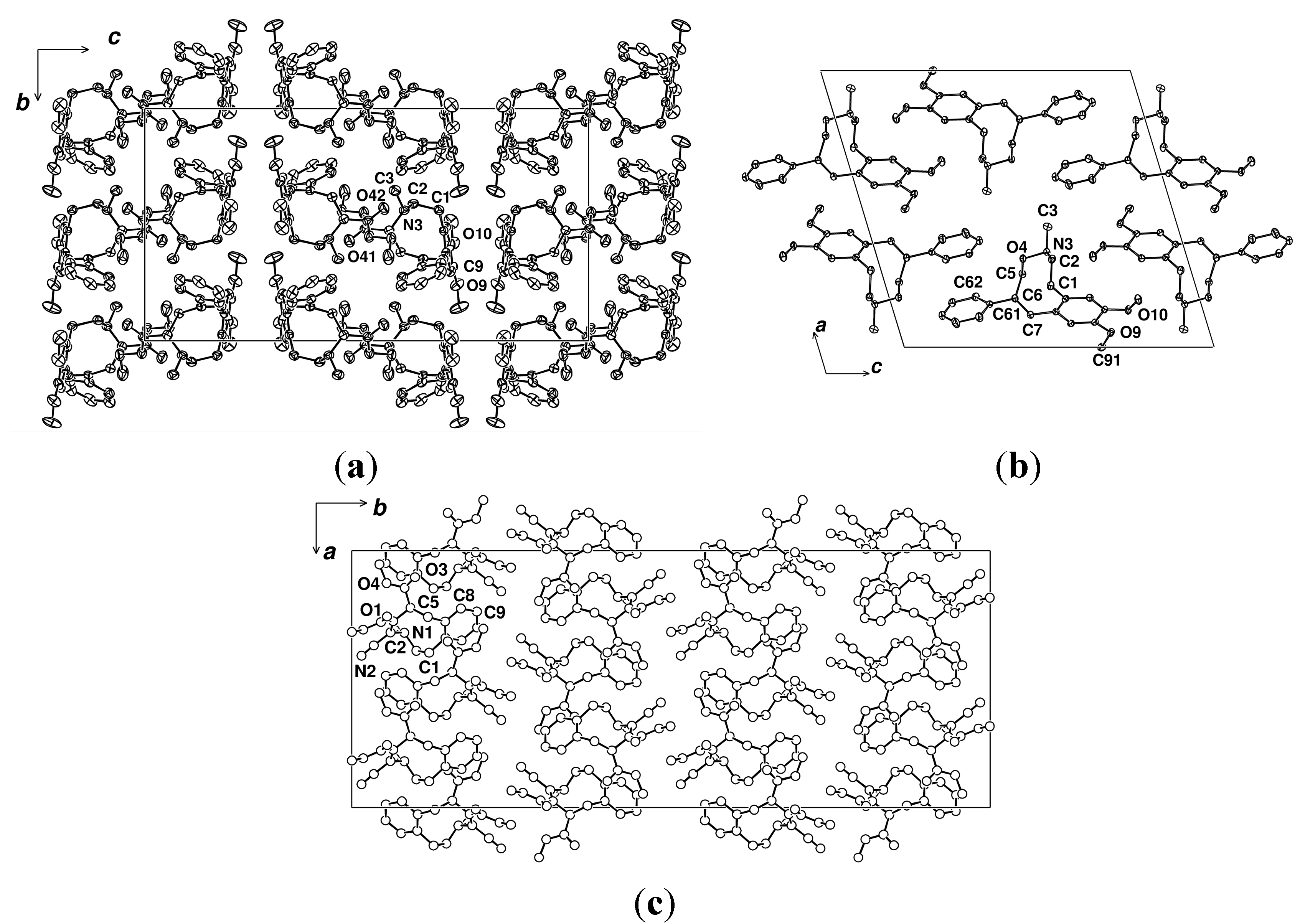
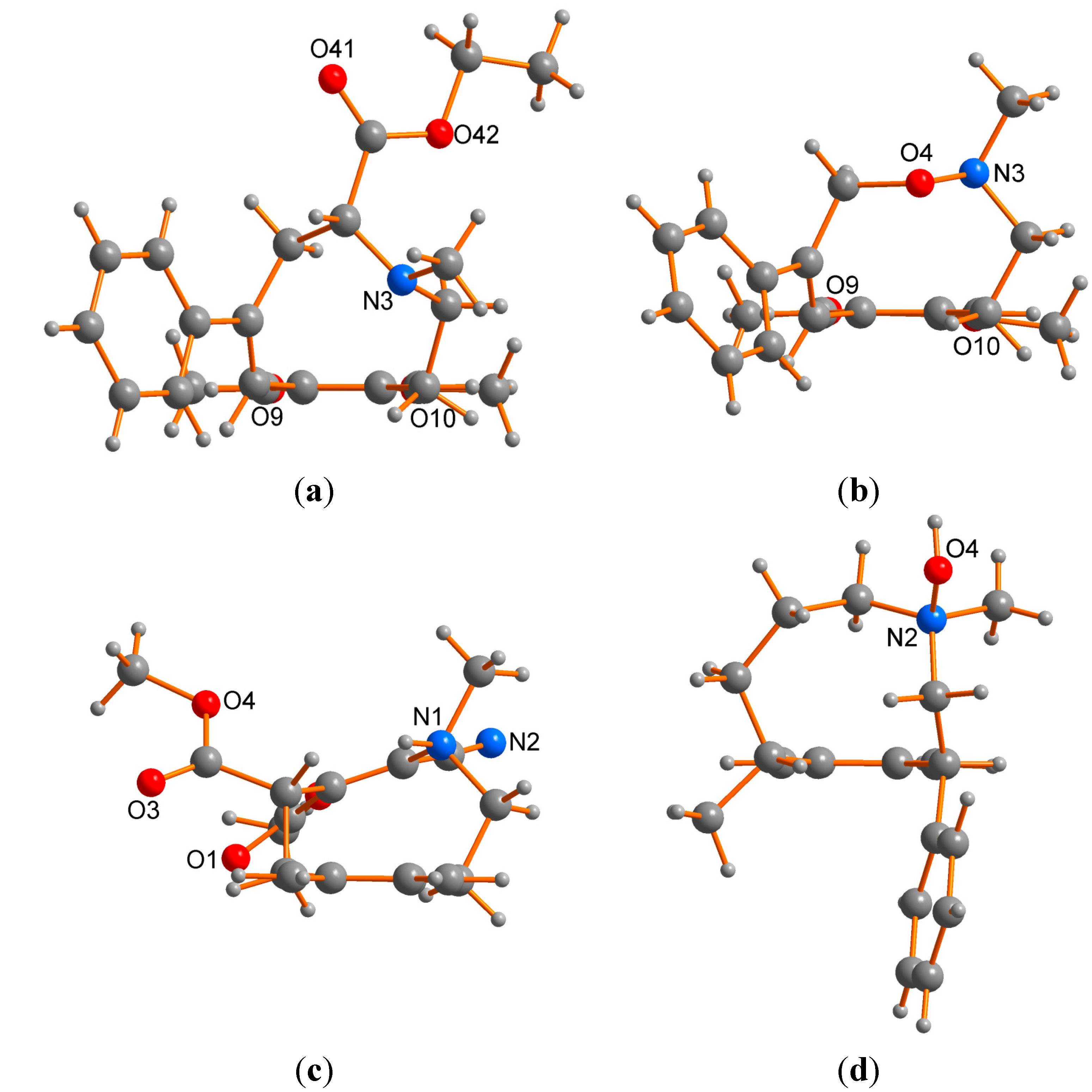
3. Experimental Section
3.1. General
3.2. Synthesis of 6,7-Dimethoxy-2-methyl-1-(1-phenylethenyl)-1,2,3,4-tetrahydroisoquinoline (3)
3.3. Ethyl (E and Z)-9,10-Dimethoxy-3-methyl-6-phenyl-2,3,4,5-tetrahydro-1H-3-benzazonine-4-carboxylate (E-1 and Z-1)
3.4. 6,7-Dimethoxy-2-methyl-1-(1-phenylethenyl)-1,2,3,4-tetrahydroisoquinoline N-oxide (5)
3.5. (Z)-9,10-Dimethoxy-3-methyl-6-phenyl-1,2,3,5-tetrahydro-4,3-benzoxazonine (2) and 7,8-Dimethoxy-3-methyl-1-(1-phenylethenyl)-1,3,4,5-tetrahydro-2,3-benzoxazepine (7)
3.6. X-ray Structure Determinations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maas, G.; Reinhard, R.; Herz, H.-G. Two-carbon ring enlargement of five-, six-, and seven-membered 1-aza-2-vinylcycloalk-2-enes with dimethyl acetylenedicarboxylate and subsequent thermal isomerization reactions. Z. Naturforschung B J. Chem. Sci. 2006, 61, 385–395. [Google Scholar]
- Majhi, T.P.; Achari, B.; Chattopadhyay, P. Advances in the synthesis and biological perspectives of benzannulated medium ring heterocycles. Heterocycles 2007, 71, 1011–1052. [Google Scholar] [CrossRef]
- Roy, B.; De, R.N.; Hazra, S. Synthesis of novel isoxazolidines and medium-ring heterocycles oxazocines and oxazonines. Lett. Org. Chem. 2011, 8, 391–400. [Google Scholar] [CrossRef]
- Ramig, K. Stereodynamic properties of medium-ring benzo-fused nitrogenous heterocycles: Benzodiazepines, benzazepines, benzazocines, and benzazonines. Tetrahedron 2013, 69, 10783–10795. [Google Scholar] [CrossRef]
- El-Subbagh, H.; Wittig, T.; Decker, M.; Elz, S.; Nieger, M.; Lehmann, J. Dopamine/serotonin receptor ligands. Part IV [1]: synthesis and pharmacology of novel 3-benzazecines and 3-benzazonines as potential 5-HT2A and dopamine receptor ligands. Arch. Pharm. Pharm. Med. Chem. 2002, 335, 443–448. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Narayana, V.V.; Prasad, M.S.; Ramakumar, K. High-yielding synthesis of Nefopam analogues (functionalized benzoxazocines) by sequential one-pot cascade operations. Org. Biomol. Chem. 2009, 7, 3372–3378. [Google Scholar] [CrossRef] [PubMed]
- Seto, S.; Tanioka, A.; Ikeda, M.; Izawa, S. Design, synthesis, and evaluation of novel 2-substituted-4-aryl-6,7,8,9-tetrahydro-5H-pyrimido[4,5-b][1,5]oxazocin-5-ones as NK1 antagonists. Bioorg. Med. Chem. 2005, 13, 5717–5732. [Google Scholar] [CrossRef] [PubMed]
- Shirai, N.; Sumiya, F.; Sato, Y.; Hori, M. Rearrangement of 1-methyl-2-(substituted-phenyl)piperidinium 1-methylides in a neutral medium. J. Org. Chem. 1989, 54, 836–840. [Google Scholar] [CrossRef]
- Kitano, T.; Shirai, N.; Sato, Y. Rearrangement of 3-aryl-4-methylmorpholinium-4-methylides. Synthesis 1991, 11, 996–998. [Google Scholar] [CrossRef]
- Clark, J.S.; Hodgson, P.B.; Goldsmith, M.D.; Blake, A.J.; Cooke, P.A.; Street, L.J. Rearrangement of ammonium ylides produced by intramolecular reaction of catalytically generated metal carbenoids. Part 2. Stereoselective synthesis of bicyclic amines. J. Chem. Soc. Perkin Trans. 1 2001, 3325–3337. [Google Scholar]
- Klapars, A.; Parris, S.; Anderson, K.W.; Buchwald, S.L. Synthesis of medium ring nitrogen heterocycles via a tandem copper-catalyzed C-N bond formation—Ring-expansion process. J. Am. Chem. Soc. 2004, 126, 3529–3533. [Google Scholar] [CrossRef] [PubMed]
- Gibson (née Thomas), S.E.; Guillo, N.; Middleton, R.J.; Thuilliez, A.; Tozer, M.J. Synthesis of conformationally constrained phenylalanine analogues via 7-, 8- and 9-endo Heck cyclizations. J. Chem. Soc. Perkin Trans. 1 1997, 447–455. [Google Scholar] [CrossRef]
- Yang, Y.-K.; Tae, J. Synthesis of heterocycles containing an N-O bond by ring-closing metathesis of dienes tethered by hydroxylamine. Synlett 2003, 7, 1043–1045. [Google Scholar]
- Potapov, V.V.; Fetisova, N.A.; Nikitin, A.V.; Ivachtchenko, A.V. A convenient synthesis of heterocyclic compounds containing 11-oxo-6,11,12,13-tetrahydrodibenzo[b,g][1,5]oxazonine fragment. Mendeleev Commun. 2009, 19, 287–289. [Google Scholar] [CrossRef]
- Bremner, J.B.; Winzenberg, K.N. Photosolvolysis of bridgehead quaternary ammonium salts. I. Synthesis of some 3-benzazonine derivatives. Aust. J. Chem. 1984, 37, 1203–1215. [Google Scholar] [CrossRef]
- Bremner, J.B.; Winzenberg, K.N. Photosolvolysis of bridgehead quaternary ammonium salts. II. Synthesis of some 2,5-benzoxazonine derivatives and attempted synthesis of the 1,2,4,5,6,7-hexahydro-3,5-benzoxazonine system. Aust. J. Chem. 1984, 37, 1659–1676. [Google Scholar] [CrossRef]
- Bailey, T.S.; Bremner, J.B.; Carver, J.A. [2,3] Sigmatropic rearrangement of 1-vinylic tetrahydroisoquinoline N-ylides and N-oxides. Tetrahedron Lett. 1993, 34, 3331–3334. [Google Scholar] [CrossRef]
- Bailey, T.S.; Bremner, J.B.; Hockless, D.C.; Skelton, B.W.; White, A.H. Ring enlargement of 1-cyclopropyl and 1-(trans-2'-phenylcyclopropyl) tetrahydroisoquinoline N-oxide derivatives. Tetrahedron Lett. 1994, 35, 2409–2412. 1994, 35, 2409–2412. [Google Scholar]
- Zhang, X. DFT studies on Meisenheimer rearrangement. Comput. Theor. Chem. 2011, 966, 383–390. [Google Scholar] [CrossRef]
- Vanecko, J.A.; Wan, H.; West, F.G. Recent advances in the Stevens rearrangement of ammonium ylides, Application to the synthesis of alkaloid natural products. Tetrahedron 2006, 62, 1043–1062. [Google Scholar] [CrossRef]
- Sweeney, J.B. Sigmatropic rearrangement of “onium” ylids. Chem. Soc. Rev. 2009, 38, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Soldatenkov, A.T.; Soldatova, S.A.; Mamyrbekova-Bekro, J.A.; Gimranova, G.S.; Malkova, A.V.; Polyanskii, K.B.; Kolyadina, N.M.; Khrustalev, V.N. Synthesis and molecular structure of 2,3,4,5-tetrahydro-1H-3-benzazepine derivatives and dimethyl 4-cyano-2,3,6,7-tetrahydro-1H-3-benzazonine-5,6-dicarboxylate. Chem. Heterocycl. Compd. 2012, 48, 1332–1339. [Google Scholar] [CrossRef]
- Whaley, W.M.; Meadow, M. The synthesis and resolution of (±)-corlumine. J. Chem. Soc. 1953, 1067–1070. [Google Scholar] [CrossRef]
- Bersch, H.-W.; Hoff, D.; Schon, D. Synthesen und umlagerungen von tertiären α-arylallylaminen (Synthesis and rearrangements of tertiary α-arylallylamines). Arch. Pharm. (Weinheim) 1978, 311, 1029–1042. [Google Scholar] [CrossRef]
- Bersch, H.-W.; Rißmann, R.; Schon, D. Isomerisierungen von tertiaren allylaminen und ringerweiternde umlagerungen durch quaternisierung (Isomerization of tertiary allylamines and ring expanding rearrangement by quaternization). Arch. Pharm. (Weinheim) 1982, 315, 749–760. [Google Scholar] [CrossRef]
- Soldatenkov, A.T.; Soldatova, S.A.; Suleimanov, R.R.; Kolyadina, N.M.; Khrustalev, V.N. Synthesis of the first representative of the 2,3,6,7-tetrahydro-1H-benzo[d]azonine system from 1-cyanomethyl-2-methyl-1,2,3,4-tetrahydroisoquinolinium chloride and dimethyl acetylenedicarboxylate. Chem. Heterocycl. Compd. 2010, 46, 245–247. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Parvez, M.; Olofson, R.A. Stereochemistry in a medium-sized ring. Highly diasteroselective N-oxidation of a substituted 3-benzazonine. X-ray crystal structure of an unusual complex between an amine N-oxide and saccharin. J. Org. Chem. 1990, 55, 760–764. [Google Scholar] [CrossRef]
- Glaser, R.; Shiftan, D.; Levi-Roso, G.; Ergaz, I.; Geresh, S.; Drouin, M. cis-Cyclononene conformational families and a crystallographic example of a skew-chair-boat type-2 conformation. J. Org. Chem. 2002, 67, 5486–5496. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Bao, H.; Qi, X.; Tambar, U.K. Stereoselective [2,3]-rearrangements of amine N-oxides. Synlett 2011, 2011, 1789–1792. [Google Scholar] [CrossRef]
- Bao, H.; Qi, X.; Tambar, U.K. Catalytic enantioselective [2,3]-rearrangements of amine N-oxides. J. Am. Chem. Soc. 2011, 133, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, T.S.; Bremner, J.B.; Skelton, B.W.; White, A.H. Synthesis and X-ray Structural Studies of a Substituted 2,3,4,5-Tetrahydro-1H-3-benzazonine and a 1,2,3,5-Tetrahydro-4,3-benzoxazonine. Molecules 2015, 20, 487-502. https://doi.org/10.3390/molecules20010487
Bailey TS, Bremner JB, Skelton BW, White AH. Synthesis and X-ray Structural Studies of a Substituted 2,3,4,5-Tetrahydro-1H-3-benzazonine and a 1,2,3,5-Tetrahydro-4,3-benzoxazonine. Molecules. 2015; 20(1):487-502. https://doi.org/10.3390/molecules20010487
Chicago/Turabian StyleBailey, Timothy S., John B. Bremner, Brian W. Skelton, and Allan H. White. 2015. "Synthesis and X-ray Structural Studies of a Substituted 2,3,4,5-Tetrahydro-1H-3-benzazonine and a 1,2,3,5-Tetrahydro-4,3-benzoxazonine" Molecules 20, no. 1: 487-502. https://doi.org/10.3390/molecules20010487